

Abstract
Theadenovirus type 5 (Ad5) E1B-55K and E4orf6 proteins are required together to stimulate viral late nuclear mRNA export to the cytoplasm and to restrict host cell nuclear mRNA export during the late phase of infection. Previous studies have shown that these two viral proteins interact with the cellular proteins elongins B and C, cullin 5, RBX1, and additional cellular proteins to form an E3 ubiquitin-protein ligase that polyubiquitinates p53 and probably one or more subunits of the MRE11-RAD50-NBS1 (MRN) complex, directing their proteasomal degradation. The MRN complex is required for cellular DNA double-strand break repair and induction of the DNA damage response by adenovirus infection. To determine if the ability of E1B-55K and E4orf6 to stimulate viral late mRNA nuclear export requires the ubiquitin-protein ligase activity of this viral ubiquitin-protein ligase complex, we designed and expressed a dominant-negative mutant form of cullin 5 in HeLa cells before infection with wild-type Ad5 or the E1B-55K null mutant dl1520. The dominant-negative cullin 5 protein stabilized p53 and the MRN complex, indicating that it inhibited the viral ubiquitin-protein ligase but had no effect on viral early mRNA synthesis, early protein synthesis, or viral DNA replication. However, expression of the dominant-negative cullin 5 protein caused a decrease in viral late protein synthesis and viral nuclear mRNA export similar to the phenotype produced by mutations in E1B-55K. We conclude that the stimulation of adenovirus late mRNA nuclear export by E1B-55K and E4orf6 results from the ubiquitin-protein ligase activity of the adenovirus ubiquitin-protein ligase complex.
Two adenovirus type 5 (Ad5) early proteins, E1B-55K and E4orf6, function to stimulate nuclear export and translation of viral late mRNAs, inhibit host cell mRNA nuclear export, and inhibit the functions of p53 and the MRE11-RAD50-NBS1 (MRN) DNA double-strand break repair complex during wild-type (WT) Ad5 infection (3, 4, 6, 11, 18, 30, 35, 47, 50, 64, 83). Initial insight into how these two proteins are involved in these diverse processes came from characterizing adenoviral mutant forms of E1B-55K and/or E4orf6. Mutants defective in expressing either E1B-55K or E4orf6 or with a mutation in genes for both show similar phenotypes: They all have a defect in viral late nuclear message export to the cytoplasm and cannot inhibit host cell mRNA nuclear export to the cytoplasm, and synthesis of viral late proteins is reduced compared to that of WT Ad5 (3, 4, 18, 29, 30, 47, 64). These mutants are also unable to induce the degradation of p53 or subunits of the MRN complex (56, 65, 73, 78).
Much information has accumulated that is relevant to the functions of E1B-55K and E4orf6. The two viral proteins associate in vivo (68) and colocalize in Ad5-infected cell nuclei at viral DNA replication-transcription centers, as well as other regions of the nucleoplasm and cytoplasm during the late phase of infection (59). Both E1B-55K and E4orf6 have nuclear export signals and nuclear localization signals required for shuttling between the nucleus and the cytoplasm (22, 32, 82). E1B-55K also has RNA binding activity in vitro, although no specificity was observed for binding either host cell or viral mRNA (26). These results suggested that E1B-55K and E4orf6 might be directly involved in redirecting both viral and cytoplasmic mRNA nuclear export in infected cells with the viral proteins directly binding viral mRNPs and transporting them through nuclear pore complexes.
A major advance in understanding how E1B-55K and E4orf6 inactivate p53 during adenovirus infection came from the discovery that these two proteins associate with the cellular proteins elongins B and C, cullin 5, RBX1, and additional cellular proteins to form an ∼800-kDa ubiquitin-protein ligase complex (often referred to as E3) that polyubiquitinates p53 (37, 65). This adenoviral ubiquitin-protein ligase complex (Ad-Ub protein ligase) is also implicated in the degradation of MRN subunits since MRN complexes, in addition to p53, are not degraded by proteasomes in cells infected with a mutant altered in either E1B-55K or E4orf6 (78). Also, E1B-55K binds the MRN complex even in the absence of E4orf6, indicating that, as for p53, E1B-55K is the primary substrate-binding subunit of the viral ubiquitin-protein ligase (10, 37, 50).
In the absence of E4orf6 and E4orf3, the cellular MRN complex is recruited to viral DNA replication centers (10, 24). The linear double-stranded adenoviral genomes act as targets for the MRN complex (10, 24, 53, 75, 78, 81). This repair complex, when uninhibited, functions with additional nonhomologous end-joining DNA-double-strand-break repair proteins to concatemerize adenoviral genomes, making them too long to package into adenovirus capsids (24, 53, 78, 81). The MRN complex also interferes with viral DNA replication even when concatemerization of the viral genomes is blocked in mutant cells with defects in other cellular proteins required for double-strand break repair (24, 75). This may be because exo- and endonuclease activities associated with the MRN complex (19, 62) inhibit initiation of viral DNA synthesis at the termini of the viral genome. E1B-55K inactivates the MRN complex in two ways. First, it binds the nuclear MRN complex, causing it to be exported from the nucleus into cytoplasmic centers of protein ubiquitination and proteosomal degradation called aggresomes (50). Araujo et al. (1) reported that E4orf3 also induces nuclear export of the MRN complex to aggresomes. Second, the Ad-Ub protein ligase complex targets MRN subunits for degradation by 26S proteasomes (10, 50, 78). These recent results suggest alternative possible explanations for why E1B-55K and E4orf6 associate with viral DNA replication centers and shuttle between the nucleus and cytoplasm. The viral proteins may associate with viral DNA replication centers because of their association with MRN complexes recruited to viral DNA replication centers by binding to the ends of viral DNA. Nucleus-cytoplasmic shuttling (21, 22, 45) may be a consequence of their function in exporting MRN complexes and p53 from the nucleus to cytoplasmic aggresomes (1, 50).
The observation that >95% of the E1B-55K in Ad5-infected cells early in the late phase of infection is associated with the ∼800-kDa ubiquitin-protein ligase complex (37) suggests that all of the E1B-55K and E4orf6 functions, including stimulation of viral late mRNA nuclear export and translation and inhibition of host mRNA nuclear export, as well as degradation of p53 and MRN complex subunits, are due to the viral ubiquitin-protein ligase activity. Consistent with this hypothesis, treatment of Ad5-infected cells with a protease inhibitor results in a decrease in viral late protein synthesis similar to that observed during infection with an E1B-55K or E4orf6 mutant, although the accompanying decrease in viral DNA replication complicated the interpretation of these results (14). To test this hypothesis further, we analyzed the influence of a dominant-negative mutant form of CUL5, the host cell cullin incorporated into the SCF-like Ad-Ub protein ligase, during the course of adenovirus infection. We find that expression of dominant-negative CUL5 under conditions that inhibit the degradation of p53 and MRE11 produces a phenotype for viral late mRNA nuclear export and translation similar to that of an E1B-55K deletion mutant. These results strongly support the hypothesis that E1B-55K and E4orf6 influence late mRNA nuclear export and translation by functioning as an SCF-like ubiquitin-protein ligase.
MATERIALS AND METHODS
Cells and viruses.
HeLa (71, 72), 293 (33), and A549 (46) cells were grown in Dulbecco's modified Eagle's medium (DMEM) supplemented with 100 U/ml penicillin, 100 μg/ml streptomycin, and 10% fetal bovine serum (FBS) at 37°C in 5% CO2. 293Cre4 cells (12) used in the construction of the E1-substituted Ad5 vectors were grown in the same supplemented DMEM plus 400 μg/ml G418. HEK293 cells were used for plaque titer determination. 293 and HeLa spinner cells were grown in Joklik's modified Eagle minimal essential medium (SMEM)plus 5% FBS with 100 U/ml penicillin, 100 μg/ml streptomycin, and 2 mM l-glutamine. Propagation of viruses was in 293 spinner cells in SMEM with 2% FBS. Ad5, dl1520, and Ad5 vector stock titers were determined by plaque assay in duplicate on 293 cells.
Construction of E1-substituted Ad5 vectors.
A modified pAdlox shuttle plasmid and Ad5 ψ5 vector were used to construct E1-substituted Ad5 recombinants by Cre-mediated recombination between loxP sites in the ψ5 vector and the pAdlox plasmid (38). To enhance the expression of an inserted gene, a simian virus 40 (SV40) small-T intron and woodchuck hepatitis virus posttranscriptional regulatory element (51, 85) were inserted into the pAdlox multiple cloning site of pAdlox. Inserted coding regions for CUL5 NTD or CUL5 FL were inserted into the modified pAdlox (pAdlox*) plasmid so that transcripts from the cytomegalovirus immediate-early promoter include the FL or NTD coding region followed by an SV40 small-T intron, the woodchuck hepatitis virus posttranscriptional regulatory element, and an SV40 poly(A) site. A 2-μg portion of pAdlox*-CUL5 NTD or pAdlox*-CUL5 FL linearized with SfiI plus 1 μg ψ5 vector DNA was transfected into 293Cre4 cells (38) with QIAGEN Effectene. Recombinant ψ5 vectors were plaque purified and propagated in 293 cells and assayed by plaque titration on 293 cells.
Dominant-negative cullin 5 (CUL5 NTD).
CUL5 cDNA was cloned by reverse transcription (RT)-PCR from HeLa cytoplasmic RNA with primers encoding a Flag epitope at the amino terminus, as well as a SalI site preceding the start codon and a BamHI site following the stop codon. For the CUL5 NTD, a second reverse primer was used with a stop codon following amino acid 393. The full-length CUL5 (FL) and CUL5 NTD sequences were TA cloned into pCR2.1 (Invitrogen) before transfer to pAdlox*. The mutations V341R and L344D were introduced into pAdlox*-NTD (Stratagene site-directed mutagenesis kit). The CUL5 coding regions of pAdlox*-FL and pAdlox*-NTD were sequenced before introduction into ψ5 vectors (Table 2).
TABLE 2.
Sequences of primers used in this study
| Primer use and name | Sequence 5′ to 3′ | Amplicon size (bp) |
|---|---|---|
| Quantitative RT-PCR | ||
| L5 Forward | CGGAGACAAAACTAAACCTGTAACAC | 102 |
| L5 Reverse | TCCCATGAAAATGACATAGAGTATGC | |
| L3 Forward | CGCTTTCCAGGCTTTGTTTC | 63 |
| L3 Reverse | GCGACCGGCCGTATTG | |
| L2 Forward | CGGTCTTTGTGGTTCTTGCA | 79 |
| L2 Reverse | TGCATTCTTCCTCGGAATCC | |
| 5′ EF2 promoter | CCTTCGCAGCGCAGTCACA | 146 |
| 3′ EF2 promoter | GGCGGTGGATTCTCCCAGGT | |
| Kanamycin forward | CGAGTGATTTTGATGACGAGCGT | 93 |
| Kanamycin reverse | CGACTGAATCCGGTGAGAATGG | |
| Site-directed mutagenesis | ||
| FwdVtoRLtoD | CTGACTCTGAGAAATACAGAGAGCAGGATCTTACACTATTTAATAG | |
| RevVtoRandLtoD | CTATTAAATAGTGTAAGATCCTGCTCTCTGTATTTCTCAGAGTCAG | |
| Fwd Stop Cul5 | GCTACCATATTTAAATAATAATTACCTTTGAAGCAGAAGGGGGTGGG | |
| Rev Stop Cul5 | CCCACCCCCTTCTGCTTCAAAGGTAATTATTATTTAAATATGGTAGC |
Western blotting analysis.
Cells were washed once in cold phosphate-buffered saline (PBS). Cells were lysed and extracted with a modified radioimmunoprecipitation assay buffer (50 mM Tris, pH 7.4, 1 mM EDTA, 150 mM NaCl, 0.25% sodium deoxycholate, 1% NP-40) with addition of a Complete protease inhibitor cocktail EDTA-free mini tablet (Roche) per 10 ml of radioimmunoprecipitation assay buffer for 30 min on ice with gentle vortexing, followed by three freeze-thaw cycles in an ethanol-dry ice bath. Cellular debris was removed by centrifugation at maximum speed for 10 to 15 min at 4°C in a microcentrifuge. These protein extracts or immunoprecipitates prepared from them were subjected to sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) on 10 or 12% gels, electroblotted to nitrocellulose membranes, blocked by incubation in PBS-0.05% Tween 20-5% nonfat dry milk (blocking buffer), and incubated in fresh blocking buffer with appropriate dilutions of the antibodies or antisera listed in Table 1. At room temperature, membranes were washed three times in PBS-0.05% Tween 20 (wash buffer) for 10 min each wash, incubated with the appropriate horseradish peroxidase-conjugated goat anti-mouse or anti-rabbit antibody (Bio-Rad) in blocking buffer for 1 h, and washed again in wash buffer under the same conditions mentioned above, and proteins were visualized with the ECL-Plus kit (GE Healthcare). Images were obtained by exposure to either KODAK X-ray film or a Bio-Rad VersaDoc image reader.
TABLE 1.
Antibodies used in immunoprecipitation and Western blotting
| Antibody | Company or source | Catalog no. or reference |
|---|---|---|
| Ad5 virion, rabbit antiserum raised against purified, disrupted Ad5 virions | NAa | This paper |
| Adenovirus 72-kDa DNA-binding protein, (M186)B6 mouse monoclonal antiserum | NA | 66 |
| Adenovirus EIA, M73 mouse monoclonal antiserum | NA | 39 |
| Adenovirus E1B-55K (Ad2 or Ad5), 58K2A6 mouse monoclonal antiserum | NA | 70 |
| Adenovirus E4orf6-E4orf6/7, RSA#3 M45 mouse monoclonal antiserum | Patrick Hearing | 58 |
| β-Actin, mouse monoclonal | Abcam Inc. | mABcam 8226 |
| Cand-1, rabbit polyclonal | Oncogene Research Products | PC745 |
| CUL5, rabbit polyclonal against amino-terminal peptide | NA | 37 |
| Elongin C/SIII p15, mouse monoclonal | BD Transduction Laboratories | 610760 |
| Flag M2, mouse monoclonal | Sigma | F 3165 |
| Ku86, rabbit polyclonal (H-300) | Santa Cruz Biotechnology, Inc. | sc-9034 |
| MRE11, mouse monoclonal 12D7 | GeneTex, Inc. | MS-MRE-PX1 |
| p53, mouse monoclonal (DO-1) | Santa Cruz Biotechnology, Inc. | sc-126 |
| p53, rabbit polyclonal (FL-393) | Santa Cruz Biotechnology, Inc. | sc-6243 |
| ROC1 (RBX1), rabbit polyclonal | NeoMarkers | RB-069-P0 |
NA, not applicable.
Immunoprecipitation.
HeLa spinner cells were infected with one of the ψ5 vectors at 10 PFU/cell. At 12 h later, the same cells were infected again (superinfected) with Ad5 or dl1520 at 100 PFU/cell. At 18 h postsuperinfection, cells were harvested by low-speed centrifugation, washed twice with ice-cold PBS, and lysed with 50 mM Tris HCl (pH 7.4)-1% NP-40-150 mM NaCl-1 mM EDTA (lysis buffer) with rotation at 4°C for 30 min. Lysates were freeze-thawed several times and centrifuged at maximum speed in a Beckman Coulter microcentrifuge at 4°C for 15 min, and supernatants were centrifuged at 100,000 × g in a VTi65 rotor for 1.5 h at 4°C. Protein lysates were quantified by Bradford assay (Bio-Rad). A 2.4-mg portion of protein per immunoprecipitation was precleared with Amersham Biosciences Sepharose A beads (50 μl packed beads) and Santa Cruz Biotechnology mouse immunoglobulin G (IgG)-conjugated beads (10 μl packed beads) for 4 to 7 h. The suspension was centrifuged at 8,000 rpm in a Beckman Coulter microcentrifuge for 30 s, and the supernatant was removed and incubated on a Nutator overnight at 4°C with 2A6-Sepharose A, Sigma M2 anti-Flag-conjugated beads, or mouse IgG-conjugated beads as a control (25 to 30 μl packed beads). The suspension was centrifuged as before, and beads were washed two times with lysis buffer and two more times with wash buffer (50 mM Tris HCl, pH 7.4, 150 mM NaCl). M2 elution was performed as described by the manufacturer (Sigma), with 3× Flag peptide for elution. 2A6-Sepharose A and mouse IgG-conjugated beads were eluted by incubation at 100°C for 10 min in 3× Laemmli sample buffer.
Analysis of viral nucleic acids and proteins in superinfected cells.
Subconfluent 10-cm HeLa plates were infected with the respective ψ5 vectors at 50 PFU/cell (preinfection). At 16 h later, the cells were infected again (superinfected) with Ad5 or dl1520 at 40 PFU/cell. At 16, 24, 36, or 48 h postsuperinfection, cells were washed once with PBS and trypsinized with GIBCO 0.5% trypsin. Trypsin was inactivated with 10% FBS DMEM, and the cells were gently pelleted, washed again with ice-cold PBS, resuspended in 1 ml ice-cold PBS, and aliquoted as follows: 200 μl for DNA isolation, 400 μl for protein extraction, and 400 μl for nuclear and cytoplasmic RNA extraction.
DNA isolation.
Cells were lysed in 1 ml NP-40 lysis buffer (10 mM Tris, pH 8.0, 150 mM NaCl, 1.5 mM MgCl2, 0.65% NP-40) for 30 min to 1 h on ice. Nuclei were pelleted at 2,000 rpm in a microcentrifuge for 2 to 3 min. Nuclei were washed once in 1 ml NP-40 lysis buffer and pelleted as before. DNA was isolated with the QIAGEN DNeasy kit with addition of RNase A in the first step of isolation.
Cytoplasmic and nuclear RNA isolation.
Cells were lysed in buffer RLN (50 mM Tris, pH 8.0, 140 mM NaCl, 1.5 mM MgCl2, 0.5% NP-40, 1,000 U/ml RNase inhibitor) for 5 min on ice. Nuclei were pelleted at 2,000 rpm for 3 min at 4°C in a microcentrifuge. RNA was isolated from the supernatant (cytoplasmic fraction) with the QIAGEN RNeasy kit, and cytoplasmic RNA was eluted in 100 μl with the provided RNase-free water. Nuclei were washed with 1 ml RLN buffer and pelleted as before. The supernatant was carefully aspirated, and nuclear RNA was extracted with Invitrogen TRIZOL reagent as described by the manufacturer. The nuclear RNA pellet was dissolved in 30 μl diethyl pyrocarbonate-treated water.
RT.
Two micrograms of RNA was first treated with Invitrogen amplification grade DNase I as described by the manufacturer. The final volume after addition of EDTA was 11 μl. Six microliters was used for transcription with the Invitrogen Superscript III kit, while 5 μl was saved for the RT negative control. To control for transcription efficiency, 4 × 1010 copies of the Promega 1.2-kb kanamycin polyadenylated RNA control (catalog no. C1381) was spiked into each sample prior to DNase I digestion. RT was primed with oligo(dT) to select for processed mRNA. RT was performed as described by the manufacturer with the kanamycin primer sequence provided by the manufacturer.
Quantitative PCR.
An Applied Biosystems 7500 real-time PCR system was used for the quantitative RT-PCR. ABI SYBR green PCR master mix was used. A 2.5-μmol portion of each primer was added to the reaction mixture, followed by 5 μl of diluted template in a total reaction volume of 25 μl. Quantitative PCR was done in triplicate for each sample by denaturing at 95°C for 10 min, followed by 40 cycles of 95°C for 15 s and 60°C for 45 s. For absolute quantification, a standard curve was generated for the cellular translation elongation factor EF2 promoter region cloned into pCDNA3.1 (Invitrogen TA Cloning Kit). ψ5 vector DNA isolated from purified virions was used for the standard curves of L2, L3, and L5. Sets of primers (Table 2) for individual quantitativeRT-PCR of L2, L3, and L5 cDNAs were designed to amplify the sequence from the common 3′ end of all processed RNAs from each of these late regions. Raw numbers were normalized to equal internal control kanamycin RNA. Standard deviations were calculated by the best value of the sum or quotient.
For each independent experiment, each treatment was done in duplicate. Transcripts are presented as the number per cell, while viral genomes are presented as the total number of adenovirus genomes per cellular EF2 gene.
In vivo 35S labeling.
Subconfluent 60 mM HeLa plates were infected as already described. At ∼2 h before harvesting, cells were washed twice with PBS and then 770 μl of GIBCO (−)l-cysteine, (−)l-methionine, (−)l-glutamine medium was added to each plate, which was incubated for 15 min before addition of 100 μCi of Tran35S-Label (MP, formerly ICN Biomedicals, Inc.) and incubation for 2 h. Cells were washed twice again in PBS, and protein lysates were harvested as already described. Protein concentration was determined, and equal amounts of protein were analyzed by SDS-PAGE in a 10% gel. Gels were dried, counted, and imaged with a Molecular Dynamics PhosphorImager.
RESULTS
Dominant-negative cullin 5 inhibits the Ad5 viral ubiquitin-protein ligase.
To specifically inhibit Ad5 ubiquitin-protein ligase activity without affecting most other cellular protein ubiquitin ligases, we expressed a dominant-negative mutant form of CUL5, the cellular cullin in the Ad-Ub protein ligase. Currently, seven human cullin family members are known, CUL1, -2, -3, -4A, -4B, -5, and -7, as well as two human proteins that contain a cullin homology domain, the APC2 subunit of the anaphase-promoting complex/cyclosome, and PARC, the p53 cytoplasmic anchor protein (63). Multiple sequence alignments between CUL5 and the other human cullins demonstrated that CUL5 is most similar to CUL1; they share significant similarity (57) and 30.3% identity (Fig. 1B). To target CUL5-containing complexes specifically, we designed and expressed a dominant-negative CUL5 mutant based on the structure of CUL1 in the SCF ubiquitin-protein ligase complex (90; Fig. 1A) and the similarity between the structures of SCF and elongin B- and C-containing ubiquitin ligases (13, 65; Fig. 1B). The C-terminal domain of CUL1 binds RBX1, the small polypeptide that recruits an E2 ubiquitin-conjugating enzyme that conjugates ubiquitin to the substrate specified by the ubiquitin-protein ligase complex (Fig. 1A).
FIG. 1.

Design of dominant-negative CUL5 mutant. (A) Model of the structure of the SCF E3 ubiquitin protein ligase complex bound to an E2-conjugating enzyme that passes ubiquitin from the indicated Cys (cyan) to a substrate bound to Skp2 (90). (B) Models of the similarity between CUL2-based VHL E3 and CUL5-based Ad5 E3 with the determined structure of SCF (65). (C) Alignment of the N-terminal domains of CUL1 (amino acids 1 to 410) and CUL5. Regions of similarity were scored (57) as indicated in the color key at the top. The third line indicates amino acid identity (★), high similarity (:), and similarity (.). Black arrows point to the conserved hydrophobic residues changed. (D) Cul1 NTD-CTD interface: Val367 and Leu371 side chains shown in yellow (90).
We reasoned that deletion of the C-terminal domain of CUL5 would generate a dominant-negative form of CUL5 that would bind to the complex of E1B-55K, E4orf6, and elongins B and C via an interaction with the elongin C subunit (Fig. 1B) but would not be able to bind RBX1 and consequently could not ubiquitinate substrates bound by the E1B-55K/E4orf6 complex. In this way, the E3 ubiquitin-protein ligase activity of the complex would be specifically inhibited without inhibiting other potential functions of E1B-55K or E4orf6.
Using an Ad5 E1A-E1B-deleted expression vector, Ψ5 (38), we expressed a Flag-tagged N-terminal region of CUL5 that terminated following residue 384, homologous to residue 410 in CUL1. Pavletich and coworkers had expressed residues 1 to 410 of CUL1 in Escherichia coli in their work on the structure of human SCF (90). In preliminary immunoprecipitation experiments, we found that the residue 1 to 384 CUL5 N-terminal region did not interact well with an E1B-55K/E4orf6/elongin BC complex in HeLa cells coinfected with WT Ad5 and that much of the truncated CUL5 expressed in HeLa cells appeared to be insoluble. Pavletich and coworkers reported that the equivalent N-terminal fragment of CUL1 expressed in E. coli also has poor solubility (90). To increase solubility sufficiently for purification and crystallography, Zheng et al. (89) constructed an N-terminal domain V367D L371D double mutant in residues on the surface of the N-terminal domain that interact with hydrophobic residues in the C-terminal domain at the interface between the two domains (Fig. 1D). Consequently, we constructed a V341D L344D double mutant of the CUL5 N-terminal domain at CUL5 residues that align best with equivalent CUL1 residues V367 and V370 (henceforth called CUL5 NTD or simply NTD).
We reasoned that CUL5 NTD would have to be expressed at a high concentration relative to endogenous CUL5 to compete for binding to the viral ubiquitin ligase. To determine if this can be achieved with these vectors, HeLa cells were infected with the NTD vector (Ψ5-NTD) or, as controls, with the empty Ψ5 vector (Ψ5) or the same Ψ5 vector expressing Flag-tagged full-length WT human CUL5 (Ψ5-FL). Extracts were prepared at 36 h postinfection (hpi) and subjected to Western blotting with an anti-CUL5 antiserum (Fig. 2A). The full-length CUL5 vector produced approximately 10 times the endogenous level observed in the extract from empty-vector-infected cells (Fig. 2A, a band of full-length CUL5 in the vector lane was apparent at longer exposure). The NTD was also expressed at a far higher level than endogenous CUL5. It was observed as a doublet in the Western blot, probably because of partial proteolysis at the N terminus since antibody to the N-terminal Flag epitope detected only one NTD band. (The doublet probably is not due to NEDD8 conjugation to the NTD, since NEDD8 is bound to the C-terminal domains of cullins [61].)
FIG. 2.

CUL5 NTD assembles into Ad5 E3. (A) Western blot assay of FL and NTD CUL5 expression in Ψ5-FL- or Ψ5-NTD-infected HeLa cells (multiplicity of infection = 10) at 32 hpi. (B) FL and NTD CUL5 interactions. Extracts from HeLa cells preinfected with Ψ5-FL or ΨT-NTD (multiplicity of infection = 10) for 12 h and then superinfected with either Ad5 or dl1520 (multiplicity of infection = 100) for 18 h were immunoprecipitated (IP) with anti-E1B-55K monoclonal antibody 2A6 (α-55K) or anti-Flag M2 monoclonal antibody (α-flag) as indicated. FL and NTD CUL5 were detected with either anti-CUL5 rabbit antiserum or anti-Flag M2 monoclonal antibody. (C) CUL5 NTD inhibits Ad5 E3. Duplicate HeLa cell cultures were preinfected with the Ψ5 vector alone, Ψ5-NTD, or Ψ5-FL (multiplicity of infection = 50) or mock infected for 16 h and then superinfected with WT Ad5 (multiplicity of infection = 40) as indicated. Extracts were prepared 48 hpi with Ad5 and subjected to Western blotting. CAND1 and β-actin were used as loading controls.
To determine whether the CUL5 NTD can form a complex with elongins B and C, E4orf6, and E1B55K (Fig. 1B), HeLa cells were infected with Ψ5-NTD or Ψ5-FL for 12 h to allow expression of Flag-tagged NTD or FL CUL5, and then the same cells were infected again (superinfected) with either WT Ad5 or the E1B-55K null mutant dl1520 (5). Since the Ψ5 vector is deleted for E1A and E1B, no E1B-55K was expressed during the initial 12 h, and because of the absence of E1A, the expression of other viral genes was expected to be greatly diminished and delayed compared to infection with WT Ad5 (7, 42). Following superinfection, E1A expressed from the superinfecting WT Ad5 or dl1520 genome was expected to stimulate transcription from both the Ψ5 vector and the superinfecting genome, leading to the replication of both viral genomes and expression of late viral proteins from both the Ψ5 vector and superinfecting genome. Superinfection with dl1520 should have resulted in expression of the same viral genes as in cells superinfected with WT Ad5, except for the absence of E1B-55K (28). Extracts prepared when the superinfected cells reached the late phase of infection (18 hpi with WT Ad5), were subjected to immunoprecipitation and Western blotting. As expected, anti-E1B-55K monoclonal antibody 2A6 (α-55K) precipitated E1B-55K, E4orf6, elongin C, full-length CUL5, and RBX1 from cells superinfected with Ψ5-FL (Fig. 2B). None of these proteins were immunoprecipitated from cells infected with Ψ5-FL and dl1520 because of the absence of E1B-55K. Significantly, E4orf6, elongins B and C, and the CUL5 NTD but no detectable RBX1 coprecipitated with anti-E1B-55K from cells preinfected with Ψ5-NTD (Fig. 2B, α-55K). When immunoprecipitation was done with antibody to the Flag epitope on overexpressed FL or NTD CUL5 (α-Flag), high levels of Flag-FL CUL5 were immunoprecipitated along with high levels of RBX1, which is intimately associated with the C-terminal domain of cullins (Fig. 1A). As expected, no RBX1 coimmunoprecipitated with the Flag-tagged NTD protein (Fig. 2B). Both the FL and NTD CUL5 proteins bind elongin C, E4orf6, and E1B-55K, although the levels of all of these proteins were lower in the α-Flag immunoprecipitates from cells expressing NTD CUL5 compared to FL CUL5. These results may suggest that FL CUL5 with bound RBX1 binds the elonginBC/E4orf6/E1B-55K complex with higher affinity than the NTD protein does. Finally, both FL and NTD CUL5 bound elongin C and E4orf6 in the absence of E1B-55K (cells superinfected with dl1520), as observed in insect cells coinfected with baculovirus expression vectors for these proteins (65). These results are precisely those expected if the NTD binds to the elonginBC/E4orf6/E1B-55K complex observed earlier (65; Fig. 1B); however, binding by NTD CUL5 may be reduced compared to binding by FL CUL5.
To determine if expression of CUL5 NTD inhibits the ubiquitin ligase activity of the Ad5-Ub ligase, we investigated whether overexpression of the NTD inhibited the degradation of host cell p53 and MRE11. p53 is the one well-established substrate of the Ad5-Ub ligase because it is polyubiquitinated by the purified complex in vitro (37, 65). Proteasomal degradation of the MRE11 and RAD50 subunits of the MRN complex requires both E1B-55K and E4orf6, indicating that at least one of these proteins is very likely a second substrate of the Ad5-Ub ligase (78). Duplicate cultures of HeLa cells were initially infected with the Ψ5-NTD, Ψ5-FL, or empty Ψ5 vector. Sixteen hours later, the six cultures were all superinfected with WT Ad5. At 48 hpi with WT Ad5, extracts were prepared from these and uninfected cells cultured in parallel and Western blotted for p53 and MRE11, as well as CAND1 (a cellular protein that binds and inactivates cullins (89) and β-actin as loading controls (Fig. 2C).
In HeLa cells, the endogenous level of p53 is low to undetectable by Western blotting because this human cervical carcinoma cell line expresses the human papillomavirus type 18 (HPV18) E6 protein from the integrated viral genome (72). E6 forms a ubiquitin-p53 ligase with host cell protein E6AP (55). However, upon infection with Ad12, p53 is initially stabilized and then degraded (34). In HeLa cells infected with an E4orf6 mutant that does not assemble the Ad5-Ub ligase, p53 is bound by E1B-55K and accumulates to a high level in both the nucleus and cytoplasmic aggresomes, as it does in Ad5-transformed cells that lack early region 4 (50, 88). This is probably because E1B-55K, HPV18 E6, and the principal cellular ubiquitin-p53 ligase MDM2 (8) compete for binding to the N-terminal region of p53 (43, 49). In the absence of E4orf6, E1B-55K binds and stabilizes p53 (48), probably by sequestering it from the HPV E6 and MDM2 ubiquitin-p53 ligases. p53 also accumulated to a high level in the cultures preinfected with the NTD vector (Fig. 2C). However, it was not detectable in cells preinfected with the empty vector or the FL vector. Presumably, p53 in cells infected with the empty and FL CUL5 vectors was polyubiquitinated by both the HPV E6- and Ad-Ub ligases and degraded by proteasomes. However, inhibition of the Ad-Ub protein ligase by overexpression of the NTD resulted in p53 stabilization rather than degradation, as in cells infected with an E4orf6 mutant that expresses E1B-55K but cannot assemble the Ad-Ub ligase in the absence of E4orf6. Similarly, MRE11 was degraded in cells preinfected with the empty and FL vectors but was only partially reduced in cells preinfected with the NTD vector (Fig. 2C). These results indicate that high-level expression of the CUL5 NTD inhibits Ad-Ub protein ligase activity.
Viral genome replication and early viral protein expression are not inhibited by CUL5 NTD.
Early viral mRNAs, proteins, and viral DNA replication are near WT Ad5 levels in E1B-55K mutant-infected HeLa cells (25). Similarly, we found that E1A, the 72-kDa DNA-binding protein, and E1B-55K were expressed at similar levels at early times after Ad5 infection in cells preinfected with the Ψ5-NTD, Ψ5-FL, and empty Ψ5 vectors (Fig. 3A). In this experiment, E4orf6 accumulated to a higher level in cells preinfected with the NTD vector, but in most experiments E4orf6 was expressed at similar levels in cells preinfected with the empty Ψ5 vector, Ψ5-NTD, or Ψ5-FL (data not shown). Viral DNA also accumulated to similar levels in cells preinfected with each of these Ψ5 vectors and then analyzed at 16, 24, and 36 h post Ad5 superinfection, as determined by quantitative PCR (Fig. 3B).
FIG. 3.

CUL5 NTD does not inhibit early Ad5 gene expression or viral DNA replication. (A) Ad5 early protein expression in HeLa cells preinfected with the indicated Ψ5 vectors determined by Western blotting. (B) Viral DNA replication in HeLa cells preinfected with Ψ5 vectors. Values were normalized to one copy of the cellular EF2 gene assayed in the same sample, and means and standard deviations of results from duplicate cultures assayed in triplicate are shown.
CUL5 NTD inhibits viral late protein expression.
The phenotype of E1B-55K mutants that correlates best with the ability of Ad5 to replicate in various tumor cell lines is the level of late viral proteins synthesized during the late phase of infection (25, 36, 60). Expression of late viral proteins is decreased severalfold in HeLa cells infected with E1B-55K mutants compared to WT Ad5 (25). In the case of dl1520, this results in an approximately 2-log decrease in the production of PFU in HeLa cells (5, 86). The defect in viral late protein synthesis is due in part to decreased nuclear export of viral late mRNAs in E1B-55K mutant-infected cells (25, 35, 64). Moreover, in WT Ad5-infected HeLa cells, host cell mRNAs are retained in nuclei in the late phase of infection while mRNAs transcribed from the viral genome are preferentially exported to the cytoplasm, independent of their sequence (6, 25). This switch in the preferential export of viral compared to cellular mRNAs during the late phase of Ad5 infection does not occur following infection with E1B-55K mutants (25).
To determine if the E1B-55K/E4orf6 function that stimulates viral late protein synthesis in HeLa cells requires the ubiquitination activity of the Ad-Ub protein ligase complex, duplicate cultures of HeLa cells were infected with the empty Ψ5 vector, Ψ5-NTD, or Ψ5-FL. At 16 hpi with these vectors, all cultures were superinfected with WT Ad5 and extracts were prepared at 24 hpi with WT Ad5 and subjected to Western blotting. Western blots were probed with a rabbit antiserum raised against purified, disrupted Ad5 virions and CAND1 as an internal control (Fig. 4A). For comparison, HeLa cells were infected with WT Ad5 or dl1520 and extracts were prepared 24 h after infection and Western blotted with the same anti-Ad5 virion serum and anti-β-actin as a loading control (Fig. 4B). The level of virion proteins expressed in HeLa cells preinfected with the NTD vector to inhibit the ubiquitin ligase activity of the Ad-Ub protein ligase was reduced comparably to that observed in dl1520-infected compared to WT Ad5-infected HeLa cells. Similar results were also observed in A549 cells and in HeLa cells preinfected at 32°C for 16 h with Ψ5 vectors and 32 h postsuperinfection with Ad5 (data not shown). In multiple repeats of this experiment, the reduction of viral proteins was quantitated by performing serial dilutions of the sample from cells preinfected with the empty Ψ5 vector on the same Western blot as the sample from cells preinfected with the Ψ5-NTD vector. Western blots were also analyzed with a quantitative imaging system (see Materials and Methods). Late viral proteins were reduced from twofold to eightfold, depending on the viral late protein, pVII generally being decreased the most and hexon the least. In some experiments, preinfection with the Ψ5-FL vector inhibited viral late protein expression to a small extent compared to cells infected with the empty vector (Fig. 4A), but this was not observed in most experiments. It might be due to a modest disturbance in elongin B- and C-based ubiquitin-protein ligase function resulting from CUL5 overexpression and the resulting change in stoichiometry of CUL5 to its interacting partners. But the reduction in viral late protein expression was reproducibly much greater in cells preinfected with the Ψ5-NTD vector than in cells preinfected with the Ψ5-FL vector.
FIG. 4.
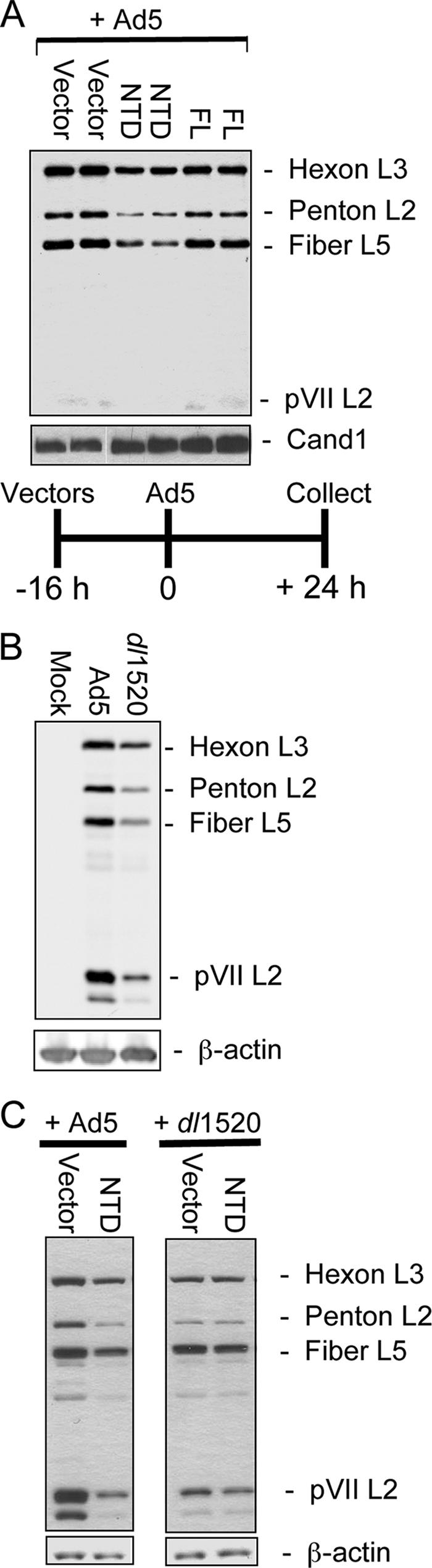
CUL5 NTD inhibits viral late gene expression. (A) Duplicate independent cultures of HeLa cells were preinfected with the indicated Ψ5 vectors (multiplicity of infection = 50) and superinfected with Ad5. Extracts prepared 24 h later were Western blotted with anti-Ad5 serum. Major virion proteins are shown. (B) Extracts prepared from HeLa cells 24 hpi with Ad5 or dl1520 were analyzed as in panel A. (C) Four cultures of HeLa cells were preinfected with the empty Ψ5 vector or Ψ5-NTD for 16 h and superinfected with either Ad5 or dl1520 for 24 h as indicated at the top. Cell extracts were prepared and analyzed as in panel A.
These results raised the question of whether the decrease in viral late gene expression in HeLa cells preinfected with the NTD vector might be due to a generalized defect in cellular protein synthesis rather than specific inhibition of the Ad5-Ub ligase. For example, sequestration of a large fraction of cellular elongins B and C in inactive complexes might produce a complex phenotype that affected viral late protein synthesis indirectly. To test this possibility, HeLa cells were coinfected with the Ψ5 vectors as before and then superinfected with either WT Ad5 or dl1520. We reasoned that if the decreased virion protein synthesis in cells preinfected with the NTD vector was due to a generalized defect in cellular protein synthesis, we should observe a decrease in the low level of virion protein synthesis in dl1520-infected cells relative to cells preinfected with the empty vector. Further, the magnitude of this difference would be comparable to the decrease in late gene expression observed in WT Ad5 superinfection. Alternatively, if the decreased virion protein synthesis in Ψ5-NTD-preinfected cells superinfected with WT Ad5 was due to specific inhibition of the Ad-Ub ligase and not a nonspecific inhibition of protein synthesis, we should observe no significant further decrease in virion protein expression in cells superinfected with dl1520 because the Ad-Ub ligase is not expressed in cells infected with this E1B-55K null mutant.
In fact, no significant further decrease in virion protein expression occurred in HeLa cells preinfected with the Ψ5-NTD vector compared to the empty vector or Ψ5-FL and superinfected with dl1520 (Fig. 4C). In the control empty vector preinfected cells, late viral protein expression was decreased in dl1520-superinfected compared to WT Ad5-superinfected cells, as expected, because of the absence of E1B-55K. In the dl1520-superinfected cells, the level of viral late protein was not significantly diminished further by preinfection with Ψ5-NTD as it was in cells superinfected with WT Ad5 (Fig. 4C). Similar results were observed in three independent experiments. The same result was observed again in the pulse-labeling experiment of Fig. 6. Consequently, preinfection with the Ψ5-NTD vector does not reduce viral late gene expression through some nonspecific inhibitory effect of the CUL5 NTD. These results strongly support the model in which E1B-55K and E4orf6 stimulate viral late protein synthesis by functioning as a CUL5-based protein-ubiquitin ligase.
FIG. 6.

Inhibition of host cell protein synthesis is only partially dependent on Ad5 E3. HeLa cells were preinfected with Ψ5 vectors and superinfected with WT Ad5 or dl1520 as indicated at the top. Cells were pulse-labeled with Tran35S-label from 36 to 38 hpi, and cell extracts were prepared and subjected to SDS-PAGE and autoradiography (A). Total counts in the lanes in the area indicated by the line to the right of the gel are plotted in panel B.
Expression of CUL5 NTD inhibits viral late mRNA nuclear export.
To determine if the reduced virion protein expression caused by the CUL5 NTD resulted from a reduced viral late mRNA concentration in the cytoplasm, viral late cytoplasmic RNAs were analyzed by quantitative RT-PCR. HeLa cells were preinfected with the two Ψ5 vectors or the empty vector as before and superinfected with WT Ad5 16 h later. Cytoplasmic RNA was isolated at 24 hpi with WT Ad5 and analyzed with primers for the 3′ ends of the L2, L3, and L5 mRNAs (74) (Fig. 5A). For comparison, HeLa cells were also infected with WT Ad5 and dl1520 without vector preinfection; RNA was isolated at 24 hpi and analyzed in the same way (Fig. 5B).
FIG. 5.

CUL5 NTD inhibits viral late mRNA nuclear export. Cytoplasmic (cyto) and nuclear RNA was isolated from the same cells analyzed in Fig. 4A and B. (A and B) Numbers of cytoplasmic transcripts per cell determined as described in Materials and Methods. (C and D) Numbers of cytoplasmic or nuclear transcripts per cell.
As reported earlier, the L2, L3, and L5 mRNAs were expressed at lower levels in E1B-55K mutant-infected HeLa cells compared to WT Ad5-infected HeLa cells (Fig. 5B). Expression of the dominant-negative CUL5 NTD by preinfection of cells with the Ψ5-NTD vector caused a similar reduction in viral cytoplasmic late RNAs compared to control cells preinfected with the empty Ψ5 vector (Fig. 5A), although the decrease in viral late cytoplasmic RNA expression was not as great in dl1520-infected as in WT Ad5-infected cells (compare Fig. 5A with B). This may be because degradation of target proteins is not completely blocked by overexpression of the CUL5 NTD, as observed from the partial degradation of MRE11 compared to mock-infected cells (Fig. 2C). In contrast, there is no activity of the absent Ad-Ub ligase in dl1520-infected cells. In dl1520-infected cells, where there is no E1B-55K, L2 cytoplasmic RNA was not decreased relative to that in WT Ad5-infected cells as much as the L3 and L5 cytoplasmic RNAs (Fig. 5B and D). We do not know why the inhibition of late mRNA expression is greater for L3 and L5 than for L2 (Fig. 5B) and L4 (data not shown; also evident in reference 64). However, the decreased effect of the NTD on L2 expression is consistent with the phenotype of an E1B-55K mutant, although less severe, presumably because of residual Ad-Ub ligase activity.
Earlier work has shown that the decrease in viral late mRNAs in E1B-55K mutant-infected HeLa cells is due at least in part to decreased mRNA nuclear export compared to that observed in WT Ad5-infected cells (6, 25, 35, 64). This phenotype can be observed as a decrease in the ratio of cytoplasmic viral RNA to nuclear viral RNA in E1B-55K mutant-infected HeLa cells (30), although interpretation of this simple ratio is complicated by the fact that fully processed nuclear mRNAs that are not exported are subject to degradation (23).
To determine if a similar defect in viral late mRNA nuclear export results from overexpression of the CUL5 NTD, nuclear RNA was also isolated from the same cells from which cytoplasmic RNA was analyzed in Fig. 5A and B. Nuclear L2, L3, and L5 viral RNAs were assayed by quantitative RT-PCR, and the number of cytoplasmic RNA molecules per cell was divided by the number of nuclear RNA molecules per cell. In WT Ad5-infected HeLa cells at 24 hpi, this ratio was 2 to 3. In HeLa cells infected with the E1B-55K mutant dl1520, the ratio in L3 and L5 dropped to 0.5 to 1 but no significant change in the ratio of cytoplasmic to nuclear viral late RNA was observed for L2 RNA (Fig. 5D). This difference for L2 compared to L3 and L5 has not been described previously but may be due to differences in details of the experimental conditions used (e.g., 32°C versus 37°C) that may affect processes such as the rate of degradation of nuclear RNAs that are not exported (23) or the rate of degradation of mRNAs in the cytoplasm, both of which would influence the ratio of cytoplasmic to nuclear RNA.
Precisely the same pattern of changes in the ratio of cytoplasmic to nuclear viral late RNA was observed in HeLa cells preinfected with the Ψ5-NTD vector compared to the empty and Ψ5-FL vectors (Fig. 5C and D). The ratio was lower for L3 and L5 but not significantly changed for L2 (Fig. 5D). These results strongly support the hypothesis that the stimulation of viral late mRNA nuclear export produced by E1B-55K and E4orf6 requires the ubiquitin-protein ligase activity of the Ad-Ub protein ligase built from basic cellular components of a major class of cellular ubiquitin-protein ligases (the CUL5 class), with E4orf6/E1B-55K substituted for cellular substrate-targeting subunits such as Skp2/Cks1 in SCF (Fig. 1A and B).
Host protein synthesis inhibition is only partially dependent on the viral ubiquitin-protein ligase.
E1B-55K mutants fail to inhibit host protein synthesis late in infection, as is observed with WT Ad5 (25). To analyze the influence of the Ad-Ub ligase activity on this E1B-55K function, the same infection scheme used in Fig. 5 was repeated, except that cells were pulse-labeled with [35S]methionine-cysteine 34 to 36 h postsuperinfection. Total cell protein was isolated, the same amount of protein from each infection was subjected to SDS-PAGE, and the dried gel was counted with a phosphorimager (Fig. 6A). Consistent with analysis of late virion protein expression by Western blotting (Fig. 4A and data not shown for times earlier and later postinfection), pulse-labeling showed that preinfection with Ψ5-NTD inhibited the rate of viral late protein synthesis compared to cells preinfected with the empty Ψ5 vector (Fig. 6A, compare lanes 4 and 5). Overexpression of FL CUL5 in Ψ5-FL-preinfected cells (lane 6) caused much less inhibition. Also, in cells superinfected with the dl1520 E1B-55K mutant instead of WT Ad5, viral late protein synthesis was inhibited only minimally and equally by overexpression of either CUL5 FL or NTD (Fig. 6A, lanes 7 to 9). These results once again strongly support the model in which Ad5 ubiquitin-protein ligase activity stimulates viral late protein synthesis (by an unknown mechanism; see Discussion).
Preinfection of HeLa cells with the Ψ5 empty vector (Fig. 6A, lane 4) resulted in a slightly higher rate of translation of viral late proteins in Ad5-superinfected HeLa cells than observed in cells that were mock preinfected and then superinfected with WT Ad5 (lane 2). This increase was even greater comparing dl1520-superinfected cells that were mock preinfected or preinfected with the empty Ψ5 vector (lane 3 versus lane 7). This may have resulted from early entry and uncoating of the E1A-E1B deleted Ψ5 vector genome in the preinfected cells. This vector DNA would be available for transcription and replication as soon as E1A was expressed in cells superinfected with either WT Ad5 or dl1520. This may have led to a somewhat faster time course of infection when empty-vector-preinfected cells were superinfected compared to cells that did not contain the genome of a vector with E1A-E1B deleted in their nuclei before infection with WT Ad5 or dl1520. Indeed, we also observed a slightly lower viral DNA concentration in mock-preinfected cells than in empty Ψ5 vector-infected cells at 16, 24, and 36 h postsuperinfection (data not shown). However, this effect of preinfection with Ψ5 vectors on viral late protein synthesis was much less than the effect of NTD expression observed by comparing cells preinfected with Ψ5-NTD and the most appropriate control, cells preinfected with the empty Ψ5 vector (Fig. 6A, compare lanes 4 and5).
The inhibition of host protein synthesis by WT Ad5 at 36 hpi is evident in the greatly decreased number of counts in host proteins that migrate to positions throughout the length of the lane in the mock-infected cell lane compared to the neighboring Ad5-infected cell lane (Fig. 6A). To quantitate host protein synthesis, total counts were determined in the region of each lane between the band at the top of the resolving gel and hexon at ∼120 kDa, the largest and most abundantly synthesized viral protein (bracket in Fig. 6A, counts in Fig. 6B). By this assay, WT Ad5 inhibited cellular protein synthesis to ∼20% of the rate in mock-infected cells. dl1520 infection inhibited cellular protein synthesis less than WT Ad5 infection (to 40 to 50% of that in mock-infected cells), but significant inhibition was nonetheless evident in the absence of E1B-55K. In cells preinfected with Ψ5 vectors (Fig. 6B, lanes 4 to 9), host protein synthesis was reduced to 20 to 25% of that in mock-infected cells under all conditions and overexpression of CUL5 NTD had little effect. As discussed below, these results suggest that the decreased inhibition of host cell protein synthesis in cells infected with E1B-55K mutants (Fig. 6B) is an indirect effect of decreased expression of late proteins, including 100K, the Ad5 late protein responsible for inhibition of host protein synthesis (16, 84). Indeed, L4 cytoplasmic RNA, which encodes 100K, is reduced in dl1520-infected cells to 25% of the concentration observed in WT Ad5-infected HeLa cells (data not shown).
DISCUSSION
The adenovirus E1B-55K protein has multiple functions (25). It can cooperate with the small E1A protein to oncogenically transform cells (20) by binding to and inactivating p53 (50, 69, 86, 88). In Ad5-infected cells (as opposed to transformed cells), it associates with the E4orf6 protein encoded at the other end of the viral genome (68), which in turn binds to components of a major class of cellular ubiquitin-protein ligases: elongins B and C, CUL5, and RBX1 (Fig. 1B) (37, 65). Several additional cellular proteins join these, forming a complex of ∼800 kDa that polyubiquitinates p53 in vitro (37). This complex also likely polyubiquitinates subunits of the MRN complex (78) required for initiating a cellular DNA damage response after infection by Ad5 mutants that fail to inactivate the MRN complex (10). During the late phase of infection, both E1B-55K and E4orf6 are also required to stimulate viral late protein synthesis in HeLa cells (5, 18, 35, 83). This is due in part to the stimulation of viral late mRNA nuclear export at the expense of host cell mRNA nuclear export (3, 4, 6, 25, 35, 64).
How can the E1B-55K and E4orf6 proteins influence processes as diverse as cell cycle control, apoptosis, mRNA nuclear export, and translation? The results presented here strongly support the model in which, as for the degradation of p53 and the MRN complex during Ad5 infection, the influence of E1B-55K and E4orf6 on mRNA nuclear export requires the ubiquitin-protein ligase activity of the Ad-Ub protein ligase complex they form. An earlier paper reached the same conclusion in studies with a proteasome inhibitor (14). However, the inhibition of viral DNA synthesis by the proteasome inhibitor complicated the interpretation of these experiments. In our study, a more specific inhibition of the Ad5 ubiquitin ligase was achieved by overexpression of a dominant-negative CUL5 mutant. Under these conditions, there was no inhibition of viral DNA replication (Fig. 3B). We also analyzed viral late nuclear and cytoplasmic RNAs. The results strongly support the model in which the viral E1B-55K/E4orf6 E3 ubiquitin ligase stimulates viral late mRNA nuclear export. The conclusion that all E1B-55K functions derive from its function in the Ad-Ub protein ligase complex also fits with the observation that >95% of the cellular E1B-55K present in HeLa cells during the late phase of infection is in the ∼800-kDa viral ubiquitin ligase complex (37). Also, the Ad-Ub protein ligase complex complex and its substrates MRN and p53 (to which it is presumably bound) are exported out of the nucleus to cytoplasmic aggresomes (1, 50), cellular degradation centers for misfolded proteins where components of the ubiquitin-proteasome system are enriched (27, 44).
How might the degradation of a cellular (or viral) protein stimulate viral mRNA nuclear export and inhibit host cell mRNA export? In one possible model, the viral ubiquitin ligase directs the degradation of a protein required for export of most host cell mRNPs but not viral mRNPs. This would inhibit host mRNP export and might stimulate viral mRNP export by reducing competition for export factors by host mRNPs. For example, RNA export factor binds most mRNPs and facilitates binding of the principal mRNP transporter, the NXF1 (TAP)-Ntr1 (p15) heterodimer (79). But some retroviruses contain a constitutive transport element, a structured region in the 3′ untranslated region of the viral mRNAs that binds NXF1 directly (17). The 201-base 5′ tripartite leader associated with the >20 mRNAs transcribed from the major late promoter stimulates Ad5 late mRNA nuclear export (41). Perhaps, in addition to its function in viral late protein synthesis (16), the tripartite leader also functions similarly to retroviral constitutive transport elements. In that case, inactivation of the RNA export factor might stimulate viral mRNA export (Fig. 7, model 1).
FIG. 7.

Models for stimulation of viral late mRNA export by degradation of a host cell protein. REF, RNA export factor; CBC, cap-binding complex.
In a second model consistent with our results and earlier observations, viral infection induces an antiviral response that inhibits mRNA nuclear export. In WT Ad5-infected cells, according to this model, some essential component of the antiviral response would be targeted for proteasomal degradation by the viral ubiquitin-protein ligase (Fig. 7, model 2). The antiviral response would not be blocked in E1B-55K mutant-infected cells because of the absence of Ad-Ub protein ligase complex. In WT Ad5-infected cells, the mechanism leading to preferential export of viral late mRNAs may involve simple competition for the NXF1-Ntx1 nuclear exporter between cellular mRNAs and the prodigious amount of viral late mRNAs produced during the late phase of infection. In addition, as mentioned earlier, the tripartite leader stimulates nuclear export during the late phase of infection (41).
What is the substrate of the Ad5 ubiquitin-protein ligase whose degradation or, potentially, monoubiquitination (77) stimulates viral late mRNA nuclear export at the expense of cellular mRNA export in HeLa cells? Two substrates are known, p53 and the MRN complex. The critical target cannot be p53 because the defect in viral late gene expression is also observed in p53−/− cells (31, 36, 67, 80).
Could the failure of an E1B-55K mutant to stimulate viral late mRNA nuclear export be due to a failure to inactivate the MRN complex and hence a failure to inhibit the DNA damage response (10)? Of potential relevance to this question, two components of the nonsense-mediated mRNA decay (NMD) pathway were recently shown also to participate in the DNA damage response (2). Components of the exon junction recognition complex that function in NMD also participate in mRNA nuclear export (54). Consequently, NMD components may function in both mRNA and DNA damage surveillance (2), providing a possible mechanism for the DNA damage response to influence nuclear mRNA export. SMG1, required for NMD, is an ATM/R-related protein kinase that phosphorylates ATM/R sites in p53 and is required for a full p53 response (9). In response to DNA damage, SMG1 and ATR phosphorylate UPF1, a component of the exon junction recognition complex proposed to induce mRNA association with cytoplasmic P bodies, where translation is suppressed, and the mRNA is degraded (76). UPF1 is both an RNA- and a DNA-helicase whose depletion by shorthairpin RNA induces an ATR-dependent DNA damage response and cell cycle arrest in early S phase (2). Perhaps a branch of the DNA damage response resulting from SMG1 and ATM/R phosphorylation of NMD components leads to inhibition of viral late mRNA nuclear export in cells infected with E1B-55K or E4orf6 mutants. Further studies are necessary to determine the extent to which the DNA damage response contributes to the inhibition of viral nuclear mRNA export in HeLa cells infected with E1B-55K and E4orf6 mutants.
While two substrates of the Ad5 ubiquitin-protein ligase are known currently, the example of HPV E6 raises the possibility that there may be additional, as-yet-unknown, substrates. HPV-E6 was first identified as a component of an E3 ubiquitin ligase by its ability to stimulate polyubiquitination of p53 (55). However, subsequent studies suggest that it has multiple cellular substrates (55). In addition to the DNA damage response, infection with E1B-55K and E4orf6 mutants may induce other cellular antiviral responses that inhibit viral mRNA nuclear export. Perhaps a component of an as-yet-undiscovered antiviral response is also targeted for proteasomal degradation by the Ad-Ub protein ligase.
Shutoff of host cell protein synthesis during the late phase of infection was only partially dependent on E1B-55K function (Fig. 6A and B, compare lanes 1 to 3) and was not significantly inhibited by overexpression of the dominant-negative CUL5 NTD (Fig. 6A and B, compare lanes 4 to 6). These results suggest that inhibition of host protein synthesis is not a direct function of E1B-55K. Rather, E1B-55K may influence host cell protein synthesis shutoff indirectly by stimulating viral late protein synthesis. One of the late viral proteins is 100K, a multifunctional protein that (i) acts as a chaperone required for proper folding of the hexon trimers that make up the faces of the virion icosahedron (40), (ii) stimulates translation of mRNAs with a 5′ tripartite leader common to viral mRNAs transcribed from the major late transcription unit, and (iii) inhibits host protein synthesis (15, 16, 84). 100K binds to the eIF4G scaffold subunit of the eIF4 translation initiation factor, displacing the MNK1 kinase. This leads to hypophosphorylation of the eIF4E cap-binding subunit of the eIF4 complex and inhibition of cap-dependent mRNA translation (15). However, 100K also contains RNA-binding domains that bind specifically to mRNAs containing the 5′ tripartite leader, stimulating their translation in the face of hypophosphorylated eIF4E by a specialized translational initiation mechanism called ribosome shunting (87). Mutation of E1B-55K may partially reverse inhibition of host cell protein synthesis indirectly by causing decreased 100K expression (25, 60).
McCormick and colleagues have proposed that dl1520 (also known as ONYX015) might be an effective anticancer therapy by replicating in p53−/− tumor cells while being blocked from replication in p53+ cells (52). However, several studies indicated that the ability of E1B-55K mutants to replicate in various tumor cell lines in culture does not correlate well with p53 status (31, 36, 67, 80). Nonetheless, results of clinical trials indicate that ONYX015 is partially effective in treating head and neck squamous cell carcinomas in some patients but not in others. Perhaps tumor cells that support high-level replication by E1B-55K mutants have inactivated or repressed DNA and RNA surveillance mechanisms, in addition to the p53 pathway, that are inactivated by the Ad5 ubiquitin-protein ligase but not by E1B-55K mutants. Understanding how the Ad5 ubiquitin-protein ligase contributes to the stimulation of viral late mRNA export in HeLa cells may allow determination of which tumors might be treated effectively by adenovirus E1B-55K mutants.
Acknowledgments
We thank Carol Eng for invaluable technical assistance in cell culture maintenance, Seán Gallaher for initial assistance with the setup of the quantitative PCR of genomes and scientific input, and the many members of the Berk lab for comments.
This research was supported by grant CA64799 from the NIH, USPHS. J.L.W. was the recipient of a Genetics Mechanisms training grant from NIH USPHS National Research Service award GM07104.
Footnotes
Published ahead of print on 1 November 2006.
REFERENCES
- 1.Araujo, F. D., T. H. Stracker, C. T. Carson, D. V. Lee, and M. D. Weitzman. 2005. Adenovirus type 5 E4orf3 protein targets the Mre11 complex to cytoplasmic aggresomes. J. Virol. 79:11382-11391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Azzalin, C. M., and J. Lingner. 2006. The human RNA surveillance factor UPF1 is required for S phase progression and genome stability. Curr. Biol. 16:433-439. [DOI] [PubMed] [Google Scholar]
- 3.Babiss, L. E., and H. S. Ginsberg. 1984. Adenovirus type 5 early region 1b gene product is required for efficient shutoff of host protein synthesis. J. Virol. 50:202-212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Babiss, L. E., H. S. Ginsberg, and J. E. Darnell, Jr. 1985. Adenovirus E1B proteins are required for accumulation of late viral mRNA and for effects on cellular mRNA translation and transport. Mol. Cell. Biol. 5:2552-2558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Barker, D. D., and A. J. Berk. 1987. Adenovirus proteins from both E1B reading frames are required for transformation of rodent cells by viral infection and DNA transfection. Virology 156:107-121. [DOI] [PubMed] [Google Scholar]
- 6.Beltz, G. A., and S. J. Flint. 1979. Inhibition of HeLa cell protein synthesis during adenovirus infection. Restriction of cellular messenger RNA sequences to the nucleus. J. Mol. Biol. 131:353-373. [DOI] [PubMed] [Google Scholar]
- 7.Berk, A. J., F. Lee, T. Harrison, J. Williams, and P. A. Sharp. 1979. Pre-early adenovirus 5 gene product regulates synthesis of early viral messenger RNAs. Cell 17:935-944. [DOI] [PubMed] [Google Scholar]
- 8.Brooks, C. L., and W. Gu. 2006. p53 ubiquitination: Mdm2 and beyond. Mol. Cell 21:307-315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Brumbaugh, K. M., D. M. Otterness, C. Geisen, V. Oliveira, J. Brognard, X. Li, F. Lejeune, R. S. Tibbetts, L. E. Maquat, and R. T. Abraham. 2004. The mRNA surveillance protein hSMG-1 functions in genotoxic stress response pathways in mammalian cells. Mol. Cell 14:585-598. [DOI] [PubMed] [Google Scholar]
- 10.Carson, C. T., R. A. Schwartz, T. H. Stracker, C. E. Lilley, D. V. Lee, and M. D. Weitzman. 2003. The Mre11 complex is required for ATM activation and the G2/M checkpoint. EMBO J. 22:6610-6620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cathomen, T., and M. D. Weitzman. 2000. A functional complex of adenovirus proteins E1B-55kDa and E4orf6 is necessary to modulate the expression level of p53 but not its transcriptional activity. J. Virol. 74:11407-11412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chen, L., M. Anton, and F. L. Graham. 1996. Production and characterization of human 293 cell lines expressing the site-specific recombinase Cre. Somat. Cell Mol. Genet. 22:477-488. [DOI] [PubMed] [Google Scholar]
- 13.Conaway, R. C., C. S. Brower, and J. W. Conaway. 2002. Emerging roles of ubiquitin in transcription regulation. Science 296:1254-1258. [DOI] [PubMed] [Google Scholar]
- 14.Corbin-Lickfett, K. A., and E. Bridge. 2003. Adenovirus E4-34kDa requires active proteasomes to promote late gene expression. Virology 315:234-244. [DOI] [PubMed] [Google Scholar]
- 15.Cuesta, R., Q. Xi, and R. J. Schneider. 2000. Adenovirus-specific translation by displacement of kinase Mnk1 from cap-initiation complex eIF4F. EMBO J. 19:3465-3474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cuesta, R., Q. Xi, and R. J. Schneider. 2001. Preferential translation of adenovirus mRNAs in infected cells. Cold Spring Harbor Symp. Quant. Biol. 66:259-267. [DOI] [PubMed] [Google Scholar]
- 17.Cullen, B. R. 2003. Nuclear mRNA export: insights from virology. Trends Biochem. Sci. 28:419-424. [DOI] [PubMed] [Google Scholar]
- 18.Cutt, J. R., T. Shenk, and P. Hearing. 1987. Analysis of adenovirus early region 4-encoded polypeptides synthesized in productively infected cells. J. Virol. 61:543-552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.D'Amours, D., and S. P. Jackson. 2002. The Mre11 complex: at the crossroads of DNA repair and checkpoint signalling. Nat. Rev. Mol. Cell. Biol. 3:317-327. [DOI] [PubMed] [Google Scholar]
- 20.Debbas, M., and E. White. 1993. Wild-type p53 mediates apoptosis by E1A, which is inhibited by E1B. Genes Dev. 7:546-554. [DOI] [PubMed] [Google Scholar]
- 21.Dobbelstein, M., J. Roth, W. T. Kimberly, A. J. Levine, and T. Shenk. 1997. Nuclear export of the E1B 55-kDa and E4 34-kDa adenoviral oncoproteins mediated by a rev-like signal sequence. EMBO J. 16:4276-4284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dosch, T., F. Horn, G. Schneider, F. Kratzer, T. Dobner, J. Hauber, and R. H. Stauber. 2001. The adenovirus type 5 E1B-55K oncoprotein actively shuttles in virus-infected cells, whereas transport of E4orf6 is mediated by a CRM1-independent mechanism. J. Virol. 75:5677-5683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Erkmann, J. A., and U. Kutay. 2004. Nuclear export of mRNA: from the site of transcription to the cytoplasm. Exp. Cell Res. 296:12-20. [DOI] [PubMed] [Google Scholar]
- 24.Evans, J. D., and P. Hearing. 2005. Relocalization of the Mre11-Rad50-Nbs1 complex by the adenovirus E4 ORF3 protein is required for viral replication. J. Virol. 79:6207-6215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Flint, S. J., and R. A. Gonzalez. 2003. Regulation of mRNA production by the adenoviral E1B 55-kDa and E4 Orf6 proteins. Curr. Top. Microbiol. Immunol. 272:287-330. [DOI] [PubMed] [Google Scholar]
- 26.Gabler, S., H. Schutt, P. Groitl, H. Wolf, T. Shenk, and T. Dobner. 1998. E1B 55-kilodalton-associated protein: a cellular protein with RNA-binding activity implicated in nucleocytoplasmic transport of adenovirus and cellular mRNAs. J. Virol. 72:7960-7971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Garcia-Mata, R., Y. S. Gao, and E. Sztul. 2002. Hassles with taking out the garbage: aggravating aggresomes. Traffic 3:388-396. [DOI] [PubMed] [Google Scholar]
- 28.Gaynor, R. B., and A. J. Berk. 1983. cis-acting induction of adenovirus transcription. Cell 33:683-693. [DOI] [PubMed] [Google Scholar]
- 29.Gonzalez, R., W. Huang, R. Finnen, C. Bragg, and S. J. Flint. 2006. Adenovirus E1B 55-kilodalton protein is required for both regulation of mRNA export and efficient entry into the late phase of infection in normal human fibroblasts. J. Virol. 80:964-974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gonzalez, R. A., and S. J. Flint. 2002. Effects of mutations in the adenoviral E1B 55-kilodalton protein coding sequence on viral late mRNA metabolism. J. Virol. 76:4507-4519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Goodrum, F. D., and D. A. Ornelles. 1998. p53 status does not determine outcome of E1B 55-kilodalton mutant adenovirus lytic infection. J. Virol. 72:9479-9490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Goodrum, F. D., T. Shenk, and D. A. Ornelles. 1996. Adenovirus early region 4 34-kilodalton protein directs the nuclear localization of the early region 1B 55-kilodalton protein in primate cells. J. Virol. 70:6323-6335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Graham, F. L., J. Smiley, W. C. Russell, and R. Nairn. 1977. Characterization of a human cell line transformed by DNA from human adenovirus type 5. J. Gen. Virol. 36:59-72. [DOI] [PubMed] [Google Scholar]
- 34.Grand, R. J., D. Owen, S. M. Rookes, and P. H. Gallimore. 1996. Control of p53 expression by adenovirus 12 early region 1A and early region 1B 54K proteins. Virology 218:23-34. [DOI] [PubMed] [Google Scholar]
- 35.Halbert, D. N., J. R. Cutt, and T. Shenk. 1985. Adenovirus early region 4 encodes functions required for efficient DNA replication, late gene expression, and host cell shutoff. J. Virol. 56:250-257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Harada, J. N., and A. J. Berk. 1999. p53-independent and -dependent requirements for E1B-55K in adenovirus type 5 replication. J. Virol. 73:5333-5344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Harada, J. N., A. Shevchenko, A. Shevchenko, D. C. Pallas, and A. J. Berk. 2002. Analysis of the adenovirus E1B-55K-anchored proteome reveals its link to ubiquitination machinery. J. Virol. 76:9194-9206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hardy, S., M. Kitamura, T. Harris-Stansil, Y. Dai, and M. L. Phipps. 1997. Construction of adenovirus vectors through Cre-lox recombination. J. Virol. 71:1842-1849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Harlow, E., B. R. Franza, Jr., and C. Schley. 1985. Monoclonal antibodies specific for adenovirus early region 1A proteins: extensive heterogeneity in early region 1A products. J. Virol. 55:533-546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hong, S. S., E. Szolajska, G. Schoehn, L. Franqueville, S. Myhre, L. Lindholm, R. W. Ruigrok, P. Boulanger, and J. Chroboczek. 2005. The 100K-chaperone protein from adenovirus serotype 2 (subgroup C) assists in trimerization and nuclear localization of hexons from subgroups C and B adenoviruses. J. Mol. Biol. 352:125-138. [DOI] [PubMed] [Google Scholar]
- 41.Huang, W., and S. J. Flint. 1998. The tripartite leader sequence of subgroup C adenovirus major late mRNAs can increase the efficiency of mRNA export. J. Virol. 72:225-235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jones, N., and T. Shenk. 1979. An adenovirus type 5 early gene function regulates expression of other early viral genes. Proc. Natl. Acad. Sci. USA 76:3665-3669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kao, C. C., P. R. Yew, and A. J. Berk. 1990. Domains required for in vitro association between the cellular p53 and the adenovirus 2 E1B 55K proteins. Virology 179:806-814. [DOI] [PubMed] [Google Scholar]
- 44.Kopito, R. R. 2000. Aggresomes, inclusion bodies and protein aggregation. Trends Cell Biol. 10:524-530. [DOI] [PubMed] [Google Scholar]
- 45.Krätzer, F., O. Rosorius, P. Heger, N. Hirschmann, T. Dobner, J. Hauber, and R. H. Stauber. 2000. The adenovirus type 5 E1B-55K oncoprotein is a highly active shuttle protein and shuttling is independent of E4orf6, p53 and Mdm2. Oncogene 19:850-857. [DOI] [PubMed] [Google Scholar]
- 46.Lehman, T. A., W. P. Bennett, R. A. Metcalf, J. A. Welsh, J. Ecker, R. V. Modali, S. Ullrich, J. W. Romano, E. Appella, J. R. Testa, et al. 1991. p53 mutations, ras mutations, and p53-heat shock 70 protein complexes in human lung carcinoma cell lines. Cancer Res. 51:4090-4096. [PubMed] [Google Scholar]
- 47.Leppard, K. N., and T. Shenk. 1989. The adenovirus E1B 55 kd protein influences mRNA transport via an intranuclear effect on RNA metabolism. EMBO J. 8:2329-2336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Levine, A. J. 1990. The p53 protein and its interactions with the oncogene products of the small DNA tumor viruses. Virology 177:419-426. [DOI] [PubMed] [Google Scholar]
- 49.Lin, J., J. Chen, B. Elenbaas, and A. J. Levine. 1994. Several hydrophobic amino acids in the p53 amino-terminal domain are required for transcriptional activation, binding to mdm-2 and the adenovirus 5 E1B 55-kD protein. Genes Dev. 8:1235-1246. [DOI] [PubMed] [Google Scholar]
- 50.Liu, Y., A. Shevchenko, A. Shevchenko, and A. J. Berk. 2005. Adenovirus exploits the cellular aggresome response to accelerate inactivation of the MRN complex. J. Virol. 79:14004-14016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Loeb, J. E., W. S. Cordier, M. E. Harris, M. D. Weitzman, and T. J. Hope. 1999. Enhanced expression of transgenes from adeno-associated virus vectors with the woodchuck hepatitis virus posttranscriptional regulatory element: implications for gene therapy. Hum. Gene Ther. 10:2295-2305. [DOI] [PubMed] [Google Scholar]
- 52.McCormick, F. 2003. Cancer-specific viruses and the development of ONYX-015. Cancer Biol. Ther. 2:S157-S160. [PubMed] [Google Scholar]
- 53.Mohammadi, E. S., E. A. Ketner, D. C. Johns, and G. Ketner. 2004. Expression of the adenovirus E4 34k oncoprotein inhibits repair of double strand breaks in the cellular genome of a 293-based inducible cell line. Nucleic Acids Res. 32:2652-2659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Moore, M. J. 2005. From birth to death: the complex lives of eukaryotic mRNAs. Science 309:1514-1518. [DOI] [PubMed] [Google Scholar]
- 55.Münger, K., and P. M. Howley. 2002. Human papillomavirus immortalization and transformation functions. Virus Res. 89:213-228. [DOI] [PubMed] [Google Scholar]
- 56.Nevels, M., S. Rubenwolf, T. Spruss, H. Wolf, and T. Dobner. 2000. Two distinct activities contribute to the oncogenic potential of the adenovirus type 5 E4orf6 protein. J. Virol. 74:5168-5181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Notredame, C., D. G. Higgins, and J. Heringa. 2000. T-Coffee: a novel method for fast and accurate multiple sequence alignment. J. Mol. Biol. 302:205-217. [DOI] [PubMed] [Google Scholar]
- 58.Obert, S., R. J. O'Connor, S. Schmid, and P. Hearing. 1994. The adenovirus E4-6/7 protein transactivates the E2 promoter by inducing dimerization of a heteromeric E2F complex. Mol. Cell. Biol. 14:1333-1346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ornelles, D. A., and T. Shenk. 1991. Localization of the adenovirus early region 1B 55-kilodalton protein during lytic infection: association with nuclear viral inclusions requires the early region 4 34-kilodalton protein. J. Virol. 65:424-429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.O'Shea, C. C., C. Soria, B. Bagus, and F. McCormick. 2005. Heat shock phenocopies E1B-55K late functions and selectively sensitizes refractory tumor cells to ONYX-015 oncolytic viral therapy. Cancer Cell 8:61-74. [DOI] [PubMed] [Google Scholar]
- 61.Pan, Z. Q., A. Kentsis, D. C. Dias, K. Yamoah, and K. Wu. 2004. Nedd8 on cullin: building an expressway to protein destruction. Oncogene 23:1985-1997. [DOI] [PubMed] [Google Scholar]
- 62.Petrini, J. H., and T. H. Stracker. 2003. The cellular response to DNA double-strand breaks: defining the sensors and mediators. Trends Cell Biol. 13:458-462. [DOI] [PubMed] [Google Scholar]
- 63.Petroski, M. D., and R. J. Deshaies. 2005. Function and regulation of cullin-RING ubiquitin ligases. Nat. Rev. Mol. Cell. Biol. 6:9-20. [DOI] [PubMed] [Google Scholar]
- 64.Pilder, S., M. Moore, J. Logan, and T. Shenk. 1986. The adenovirus E1B-55K transforming polypeptide modulates transport or cytoplasmic stabilization of viral and host cell mRNAs. Mol. Cell. Biol. 6:470-476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Querido, E., P. Blanchette, Q. Yan, T. Kamura, M. Morrison, D. Boivin, W. G. Kaelin, R. C. Conaway, J. W. Conaway, and P. E. Branton. 2001. Degradation of p53 by adenovirus E4orf6 and E1B55K proteins occurs via a novel mechanism involving a Cullin-containing complex. Genes Dev. 15:3104-3117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Reich, N. C., P. Sarnow, E. Duprey, and A. J. Levine. 1983. Monoclonal antibodies which recognize native and denatured forms of the adenovirus DNA-binding protein. Virology 128:480-484. [DOI] [PubMed] [Google Scholar]
- 67.Rothmann, T., A. Hengstermann, N. J. Whitaker, M. Scheffner, and H. zur Hausen. 1998. Replication of ONYX-015, a potential anticancer adenovirus, is independent of p53 status in tumor cells. J. Virol. 72:9470-9478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Sarnow, P., P. Hearing, C. W. Anderson, D. N. Halbert, T. Shenk, and A. J. Levine. 1984. Adenovirus early region 1B 58,000-dalton tumor antigen is physically associated with an early region 4 25,000-dalton protein in productively infected cells. J. Virol. 49:692-700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Sarnow, P., Y. S. Ho, J. Williams, and A. J. Levine. 1982. Adenovirus E1b-58kd tumor antigen and SV40 large tumor antigen are physically associated with the same 54 kd cellular protein in transformed cells. Cell 28:387-394. [DOI] [PubMed] [Google Scholar]
- 70.Sarnow, P., C. A. Sullivan, and A. J. Levine. 1982. A monoclonal antibody detecting the adenovirus type 5-E1b-58Kd tumor antigen: characterization of the E1b-58Kd tumor antigen in adenovirus-infected and -transformed cells. Virology 120:510-517. [DOI] [PubMed] [Google Scholar]
- 71.Scheffner, M., K. Munger, J. C. Byrne, and P. M. Howley. 1991. The state of the p53 and retinoblastoma genes in human cervical carcinoma cell lines. Proc. Natl. Acad. Sci. USA 88:5523-5527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Schwarz, E., U. K. Freese, L. Gissmann, W. Mayer, B. Roggenbuck, A. Stremlau, and H. zur Hausen. 1985. Structure and transcription of human papillomavirus sequences in cervical carcinoma cells. Nature 314:111-114. [DOI] [PubMed] [Google Scholar]
- 73.Shen, Y., G. Kitzes, J. A. Nye, A. Fattaey, and T. Hermiston. 2001. Analyses of single-amino-acid substitution mutants of adenovirus type 5 E1B-55K protein. J. Virol. 75:4297-4307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Shenk, T. 2001. Adenoviruses. Lippincott Williams & Wilkins, Philadelphia, Pa.
- 75.Shepard, R. N., and D. A. Ornelles. 2004. Diverse roles for E4orf3 at late times of infection revealed in an E1B 55-kilodalton protein mutant background. J. Virol. 78:9924-9935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Sheth, U., and R. Parker. 2006. Targeting of aberrant mRNAs to cytoplasmic processing bodies. Cell 125:1095-1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Sigismund, S., S. Polo, and P. P. Di Fiore. 2004. Signaling through monoubiquitination. Curr. Top. Microbiol. Immunol. 286:149-185. [DOI] [PubMed] [Google Scholar]
- 78.Stracker, T. H., C. T. Carson, and M. D. Weitzman. 2002. Adenovirus oncoproteins inactivate the Mre11-Rad50-NBS1 DNA repair complex. Nature 418:348-352. [DOI] [PubMed] [Google Scholar]
- 79.Stutz, F., and E. Izaurralde. 2003. The interplay of nuclear mRNP assembly, mRNA surveillance and export. Trends Cell Biol. 13:319-327. [DOI] [PubMed] [Google Scholar]
- 80.Turnell, A. S., R. J. Grand, and P. H. Gallimore. 1999. The replicative capacities of large E1B-null group A and group C adenoviruses are independent of host cell p53 status. J. Virol. 73:2074-2083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Weiden, M. D., and H. S. Ginsberg. 1994. Deletion of the E4 region of the genome produces adenovirus DNA concatemers. Proc. Natl. Acad. Sci. USA 91:153-157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Weigel, S., and M. Dobbelstein. 2000. The nuclear export signal within the E4orf6 protein of adenovirus type 5 supports virus replication and cytoplasmic accumulation of viral mRNA. J. Virol. 74:764-772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Weinberg, D. H., and G. Ketner. 1986. Adenoviral early region 4 is required for efficient viral DNA replication and for late gene expression. J. Virol. 57:833-838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Xi, Q., R. Cuesta, and R. J. Schneider. 2005. Regulation of translation by ribosome shunting through phosphotyrosine-dependent coupling of adenovirus protein 100k to viral mRNAs. J. Virol. 79:5676-5683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Xu, Z. L., H. Mizuguchi, T. Mayumi, and T. Hayakawa. 2003. Woodchuck hepatitis virus post-transcriptional regulation element enhances transgene expression from adenovirus vectors. Biochim. Biophys. Acta 1621:266-271. [DOI] [PubMed] [Google Scholar]
- 86.Yew, P. R., and A. J. Berk. 1992. Inhibition of p53 transactivation required for transformation by adenovirus early 1B protein. Nature 357:82-85. [DOI] [PubMed] [Google Scholar]
- 87.Yueh, A., and R. J. Schneider. 2000. Translation by ribosome shunting on adenovirus and hsp70 mRNAs facilitated by complementarity to 18S rRNA. Genes Dev. 14:414-421. [PMC free article] [PubMed] [Google Scholar]
- 88.Zantema, A., J. A. Fransen, A. Davis-Olivier, F. C. Ramaekers, G. P. Vooijs, B. DeLeys, and A. J. Van der Eb. 1985. Localization of the E1B proteins of adenovirus 5 in transformed cells, as revealed by interaction with monoclonal antibodies. Virology 142:44-58. [DOI] [PubMed] [Google Scholar]
- 89.Zheng, J., X. Yang, J. M. Harrell, S. Ryzhikov, E. H. Shim, K. Lykke-Andersen, N. Wei, H. Sun, R. Kobayashi, and H. Zhang. 2002. CAND1 binds to unneddylated CUL1 and regulates the formation of SCF ubiquitin E3 ligase complex. Mol. Cell 10:1519-1526. [DOI] [PubMed] [Google Scholar]
- 90.Zheng, N., B. A. Schulman, L. Song, J. J. Miller, P. D. Jeffrey, P. Wang, C. Chu, D. M. Koepp, S. J. Elledge, M. Pagano, R. C. Conaway, J. W. Conaway, J. W. Harper, and N. P. Pavletich. 2002. Structure of the Cul1-Rbx1-Skp1-F boxSkp2 SCF ubiquitin ligase complex. Nature 416:703-709. [DOI] [PubMed] [Google Scholar]