

Abstract
Human immunodeficiency virus (HIV) infection is often accompanied by infection with other pathogens that affect the clinical course of HIV disease. Here, we identified another virus, human herpesvirus 7 (HHV-7) that interferes with HIV type 1 (HIV-1) replication in human lymphoid tissue, where critical events of HIV disease occur. Like the closely related HHV-6, HHV-7 suppresses the replication of CCR5-tropic (R5) HIV-1 in coinfected blocks of human lymphoid tissue. Unlike HHV-6, which affects HIV-1 by upregulating RANTES, HHV-7 did not upregulate any CCR5-binding chemokine. Rather, the inhibition of R5 HIV-1 by HHV-7 was associated with a marked downregulation of CD4, the cellular receptor shared by HHV-7 and HIV-1. HHV-7-induced CD4 downregulation was sufficient for HIV-1 inhibition, since comparable downregulation of CD4 with cyclotriazadisulfonamide, a synthetic macrocycle that specifically modulates expression of CD4, resulted in the suppression of HIV infection similar to that seen in HHV-7-infected tissues. In contrast to R5 HIV-1, CXCR4-tropic (X4) HIV-1 was only minimally suppressed by HHV-7 coinfection. This selectivity in suppression of R5 and X4 HIV-1 is explained by a suppression of HHV-7 replication in X4- but not in R5-coinfected tissues. These results suggest that HIV-1 and HHV-7 may interfere in lymphoid tissue in vivo, thus potentially affecting the progression of HIV-1 disease. Knowledge of the mechanisms of interaction of HIV-1 with HHV-7, as well as with other pathogens that modulate HIV-1 replication, may provide new insights into HIV pathogenesis and lead to the development of new anti-HIV therapeutic strategies.
Human herpesvirus 7 (HHV-7) is a member of the Betaherpesvirinae subfamily and, with HHV-6, is classified in the Roseolovirus genus (1, 2). HHV-7 was initially isolated from CD4+ T cells of a healthy individual (7), and CD4 is a critical component of its cellular entry receptor (19). Similar to HHV-6, primary HHV-7 infection occurs in early childhood with very high prevalence and is occasionally associated with roseola infantum. Thus far, HHV-7 has not been directly linked to any disease in immunocompetent adults, and it is considered a low-morbidity virus (23). However, like other members of the Herpesviridae family, HHV-7 may act as an opportunistic agent in immunocompromised individuals (18). In particular, postmortem studies of the lymph nodes from AIDS patients supports the hypothesis of an extensive HHV-7 reactivation from latency during the course of HIV-1 infection (17) although, in the peripheral blood of these patients the detection of HHV-7 is infrequent, most likely as a consequence of the reduced number of target CD4+ T cells (3, 4, 6).
To understand the complex interactions of HHV-7 and HIV-1 in coinfected individuals, we investigated here the interactions of HHV-7 with HIV-1 in the context of human lymphoid tissue ex vivo where critical events of viral pathogenesis occur in vivo. In this system which supports productive viral infection without exogenous activation (8), we found that, like HHV-6, HHV-7 suppresses the replication of CCR5-tropic (R5) HIV-1 but only mildly inhibits the replication of CXCR4-tropic (X4) HIV-1. However, we found that the molecular mechanisms of these phenomena are different: whereas HHV-6 affects HIV-1 by upregulating RANTES (12), HHV-7-induced HIV-1 suppression is mediated by downregulation of CD4, the cellular receptor shared by HHV-7 and HIV-1.
MATERIALS AND METHODS
Human tonsillar tissue culture.
Tonsils obtained in routine tonsillectomy performed at the Children's Hospital NMC (Washington, DC) were received under an Institutional Review Board-approved protocol within a few hours after surgery. Tonsils were dissected in blocks of approximately 2 mm and placed on top of collagen sponge gels floating in six-well plates as described earlier (8). The culture medium was changed every 3 days.
Viral stocks.
HHV-7, strain AL (21), was expanded in primary human cord blood mononuclear cells. The culture supernatants were clarified by centrifugation, and the viral stocks were characterized for infectivity and frozen at −80°C until use. The R5 HIV-1 variant SF162 and X4 variant LAI.04 viral stocks were obtained from the Rush University Virology Quality Assurance Laboratory (Chicago, IL). The 50% tissue culture infectious dose was 2.59 × 105 IU/ml for both the R5 SF162 and X4 LAI.04 viral stock as determined in peripheral blood mononuclear cell cultures.
HIV and HHV-7 infection.
Tissue blocks were infected simultaneously with HHV-7 or HIV-1 (LAI.04 or SF162) as described elsewhere (8). Specifically, 5 μl of clarified virus-containing medium were placed on top of each of 27 blocks for each experimental condition obtained from the tonsil tissue of one donor. Experiments were repeated with tissues of n donors, where n is specified throughout the text below. In the case of HIV, each block was inoculated with approximately 0.5 ng of p24. For HHV-7 infection, each tissue block was inoculated with approximately 2 × 106 genome equivalents. We assessed productive HIV-1 infection by measuring p24 concentration in culture medium during the 3 days between successive medium changes using an enzyme-linked immunosorbent assay (ELISA) commercial kit (Perkin-Elmer, Boston, MA). We assessed HHV-7 infection by real-time PCR as described below.
Quantification of HHV-7 DNA by calibrated real-time PCR.
HHV-7 viral load was determined by measurement of the number of viral genome equivalents present in the tissue culture medium. The DNA was purified with the QIAamp kit (QIAGEN GmbH, Hilden, Germany) according to the manufacturer's instructions. Quantification was performed by a quantitative calibrated real-time PCR TaqMan assay on an ABI Prism 7000 sequence detector (PE Applied Biosystems, Foster City, CA). The specific primer set used to detect HHV-7 was as follows: HHV-7 forward, 5′-AGCGGTACCTGTAAAATCATCCA-3′; and HHV-7 reverse, 5′-AACAGAAACGCCACCTCGAT-3′. The probe 5′-ACCAGTGAGAACATCGCTCTAACTGGATCA-3′ was covalently linked at the 5′ end to the reporter dye 6-carboxyfluorescein and at the 3′ end to the quencher dye 6-carboxy-tetramethyl-rhodamine. The reaction mixture contained TaqMan PCR master mix, 300 nM concentrations of each primer, and 5 μl of DNA template. After activation of the AmpliTaq Gold for 10 min at 95°C, 40 cycles of 15 s at 95°C and 1 min at 60°C were carried out. We obtained a reference standard curve using serially diluted plasmids containing the target genes and normalized the results using a synthetic DNA calibrator molecule (106 copies per sample) added to the samples before the extraction step, allowing us to control for intersample extraction efficiency and to monitor PCR artifacts (5). The primers and the calibrator probe were not cross-reactive with the HHV-7 sequences.
Flow cytometry.
Flow cytometry analysis was performed on single-cell suspensions (cells were mechanically isolated from control and ex vivo-infected blocks of human lymphoid tissue 12 days postinfection). Lymphocytes were identified according to their light-scattering properties and then analyzed for the expression of lymphocyte-specific markers. The cells were washed three times and initially stained with a combination of the following monoclonal antibodies (MAbs; all from Caltag Laboratories, Burlingame, CA, unless specified otherwise): anti-CD3-Cy7-PE, anti-CD4-Cy5.5-PE, anti-CD8-Tricolor, anti-CCR5-APC-Cy7, and anti-CXCR4-APC (the latter two MAb are from BD Pharmingen, San Diego, CA). Prior to cell surface staining, counting beads (Caltag) were added to each tube to quantify cell depletion. Values were normalized for the total tissue weight at day 12 for tissue blocks from each donor. In several experiments CD8+ T cells were used as a normalization basis since the numbers of these cells were not changed by HIV-1 (13) or HHV-7 (see results) in ex vivo infected human lymphoid tissue. To identify cells productively infected with HHV-7 or HIV-1, we permeabilized the cells with Fix&Perm reagent (Caltag) and stained them with anti-HHV-7 tegumental protein pp85 (Advanced Biothecnology, Inc., Columbia, MD, and also kindly provided by Gabriella Campadelli-Fiume) directly coupled in our laboratory with Alexa 488 (Molecular Probes, Carlsbad, CA) or with anti-p24 KC57-RD1 (Beckman Coulter, Inc., Fullerton, CA), respectively. Infection of non-naive versus naive cells was determined as described above, with the antibody combination of CD3 Cy7-PE, CD4 Cy5.5-PE, CD8 PE, CD45RO-APC, CD45RA-TriColor, CD62L-Cy7-APC, and anti-HHV-7 pp85-Alexa 488 for intracellular staining. The data were acquired with a LSRII flow cytometer (BD Biosciences, San Jose, CA) using 532, 488, and 638 laser lines and Diva 4.12 software. The data were analyzed with the FLOWJO software (Tree Star, San Carlos, CA).
Downregulation of CD4 was evaluated by the downshift of fluorescence intensity of cells stained for CD4, relative to matched uninfected tissues. First, we determined the median of the distribution of this fluorescence in control uninfected samples. By definition, the numbers of cells with a fluorescence intensity above and below this median are equal and their ratio is equal to 1. Upon downregulation of CD4, the number of cells with a fluorescence intensity below the median value in the untreated control increased, and the number of cells with fluorescence intensity above this value decreased. The change in the ratio of cell numbers (percent) in these two subsets was used as a measure of CD4 downregulation (DCD4). Thus, DCD4 = (1 − Flhinf/Fllinf) × 100, where Fllinf and Flhinf are the numbers of cells in infected tissues with fluorescence below and above, respectively, the median in the matched control.
Chemokine production.
The levels of interleukin-1α (IL-1α), IL-1β, IL-2, IL-4, IL-6, IL-7, IL-8, IL-12, IL-15, IL-16, macrophage inflammatory protein 1α (MIP-1α), MIP-1β, RANTES, MIG, granulocyte-macrophage colony-stimulating factor, IP-10, gamma interferon, tumor necrosis factor alpha, and SDF-1β were evaluated by using a multiplex bead array assay performed on a Bioplex (Luminex; Bio-Rad). All of the antibodies and cytokine standards were purchased as antibody pairs from R&D Systems (Minneapolis, MN). Individual Luminex bead sets were coupled to cytokine-specific capture antibodies according to the manufacturer's recommendations. Conjugated beads were washed and are kept at 4°C until used. Biotinylated polyclonal antibodies were used at twice the concentrations recommended for a classical ELISA procedure. All of the assay procedures were performed in assay buffer made of phosphate-buffered saline (PBS) supplemented with 1% normal mouse serum, 1% normal goat serum, and 20 mM Tris-HCl (pH 7.4). The assays were run with 1,200 beads per set per well in a total volume of 50 μl. A total of 50 μl of each sample was added to the well, followed by incubation overnight at 4°C in a Millipore multiscreen plate. The liquid was then aspirated by using a Millipore vacuum manifold, and the plates were washed three times with 200 μl of assay buffer. The beads were then resuspended in 50 μl of solution containing biotinylated polyclonal antibodies for 1 h at room temperature. The plates were washed three times with PBS, the beads were resuspended in 50 μl of assay buffer, and 50 μl per well of a 16-μg/ml solution of streptavidin-PE (Molecular Probes, Eugene, OR) was added for 15 min, the plates were washed with PBS, and 150 μl of assay buffer was added to each well prior to reading in Bioplex. For each bead set, a minimum of 61 beads were collected.
In addition, semiquantitative evaluation of the production of 120 cytokines and chemokines was performed by using a protein array. Briefly, supernatants from HHV-7-infected tissue and matched uninfected control were incubated on arrayed antibody supports from RayBio human cytokine antibody array C series 1000 (RayBiotech, Inc., Atlanta, GA). After incubation with biotinylated antibodies and subsequently with horseradish peroxidase-conjugated streptavidin the signals were detected by using the chemiluminescence imaging system Fluorchem 9000 (Alpha Innotech Corp., San Leandro, CA).
CADA treatment.
Cyclotriazadisulfonamide (CADA) was resuspended in dimethyl sulfoxide at a concentration of 10 mg/ml; added to the tissue-bathing medium at concentrations of 0.25, 0.5, 1, 2, 4, or 8 μM; and left overnight prior to HIV-1 infection. The medium was changed every 3 days, and the drug was added with every medium change.
Statistical analysis.
Data obtained with tissue from one donor constituted the results of one experiment. To compare results obtained in different experiments, we normalized the data: for each experiment, we compared infected and control tissues obtained from an individual donor in replicates of 27 tissue blocks for each datum point and expressed the data as the percentage of controls. Statistical analysis performed on thus-normalized results included the calculation of the mean, the standard error of the mean (SEM), and P values by use of a two-way analysis of variance test or a paired Student t test. The significance level was set as P = 5 × 10−2, and the actual P values are indicated for each set of experiments. Statistical analysis of p24 ELISA data was performed with Deltasoft software (version 3.0; BioMetallics, Princeton, NJ), which combined data from three dilutions and calculated the interpolated weighed p24 concentration and the SEM. For chemokine detection with the Luminex platform, we performed data analysis of the median of the fluorescence intensity recorded for 61 beads of each bead set for the assays by using Bioplex Manager software (version 4.0; Bio-Rad), with a five-parameter regression algorithm.
RESULTS
Productive HHV-7 infection in human tonsillar tissue.
Human tonsillar tissue blocks were inoculated with HHV-7 AL strain by application of 5 μl of viral suspension (∼2 × 106 genome equivalents) onto each block. A productive infection was documented by measurement of the kinetics of accumulation of viral genomes in culture medium bathing 27 tonsillar blocks from each of nine donors using calibrated real-time PCR assay (Fig. 1A). Infection became evident at day 6 postinoculation and continued during the entire course of the experiment up to day 12. At day 12 postinfection, the average concentration of HHV-7 DNA in culture supernatant was (1.4 ± 0.4) × 108 genome equivalents per ml. Over this period, each block of tissue produced, on average, (6.6 ± 0.2) × 107 viral genome equivalents.
FIG. 1.

Replication of HHV-7 in human lymphoid tissue ex vivo. Tissue blocks (27 from each donor) were inoculated with HHV-7. The culture medium was changed every 3 days, and HHV-7 replication was measured by real-time PCR (A) or flow cytometry (B). (A) Kinetics of HHV-7 replication. Shown is the HHV-7 genome accumulation in culture medium during 3 days between medium changes. Each datum point is the mean ± the SEM for experiments with tissues from nine donors. (B) Bivariate density plots of lymphocytes stained for CD3 and HHV-7 pp85. Lymphocytes were isolated after 12 days in culture from HHV-7-infected (right panel) and matched uninfected tissue (left panel). A sample representative of experiments with tissues from 10 donors is displayed.
We also estimated infection by expression of a viral structural protein, the tegument phosphoprotein pp85, as evaluated by flow cytometry of cells mechanically isolated from infected tissue blocks and stained with anti-pp85 MAb in combination with various antibodies used for cell immunophenotyping. On average at day 12 postinoculation the fraction of HHV-7-positive cells among T (CD3+) lymphocytes was 20.8% ± 3.1% (n = 10), whereas the fraction of viral-antigen-positive cells among CD3− cells was 1.8% ± 0.4% (n = 10) (Fig. 1B). Thus, both real-time PCR and flow cytometry demonstrated productive HHV-7 infection in ex vivo-inoculated human lymphoid tissue. Since HHV-7 and HIV-1 (9) productively infect human lymphoid tissue ex vivo, it was possible to address the question of their interactions in this system.
HHV-7 differently affects the replication of R5 and X4 HIV-1 variants in coinfected lymphoid tissue.
To study the interactions between HHV-7 and HIV-1, we coinfected tissue blocks with HHV-7 and one of two biologically different HIV-1 variants: a prototypic X4 variant, LAI.04, and a prototypic R5 variant, SF162. We compared the replication of each of these viruses in coinfected tissue blocks with its replication in matched singly infected tissues (Fig. 2). As shown earlier (8), both X4 and R5 HIV-1 variants readily replicate in ex vivo-infected tissues (Fig. 2A and B). Replication became evident at day 6 postinoculation and increased thereafter to reach levels that varied among different donors between 1 and 160 ng of p24 per ml of culture medium. Despite this variability, within tissue from an individual donor X4LAI.04 and R5SF162 HIV-1 variants replicated approximately to the same level: the cumulative production of p24 in culture medium was, on average, 63.4 ± 22.4 ng and 33.5 ± 9.8 ng for R5SF162 and X4LAI.04, respectively (P = 1.8 × 10−1, n = 8). As shown in Fig. 2, coinfection with HHV-7 differentially affected the replication of R5 and X4 HIV-1 variants. Replication of the R5 strain HIV-1SF162 was consistently suppressed by, on average, 71.9 ± 6.1% (P = 2 × 10−2, n = 8) relative to that in matched tissues infected by HIV-1SF162 alone (Fig. 2C). This inhibition typically became evident between days 6 and 9 and reached its maximum level at day 12 postinfection, coinciding with HHV-7 highest level of replication (Fig. 2B). Consistent with the suppression of HIV-1 replication revealed by p24 measurements, there was a decrease in the average frequency of HIV-1-productively infected (p24+) T cells determined by flow cytometry from 1.5 ± 0.4% in tissues infected with HIV-1SF162 alone to 0.63 ± 0.4 in matched tissues coinfected with HHV-7 (P = 5 × 10−3, n = 4).
FIG. 2.

R5SF162 and X4LAI.04 replication in HHV-7-coinfected human lymphoid tissues. Sets of matched tissue blocks (27 blocks for each experimental condition from each donor) were infected with one of the HIV-1 variants alone or together with HHV-7. HIV replication was evaluated by measurement of the p24 in culture medium. (A and B) Average kinetics of X4LAI.04 (A) and R5SF162 (B) replication in singly HIV-infected and HHV-7-coinfected tissues (mean ± the SEM, n = 8). The data are expressed as the percentage of the maximum p24 value in singly HIV-infected tissues to account for the variation in absolute replication levels in tissues from different donors. (C) Total production of R5SF162 and X4LAI.04 in HHV-7-coinfected tissues. Presented are the means ± the SEM for the total HIV-1 production over 12 days postinoculation for tissues from eight donors expressed as a percentage of the total HIV-1 production in matched singly infected tissues. An asterisk denotes a significant difference (P < 5 × 10−2). (D) Correlation between HHV-7 replication and HIV-1 inhibition in coinfected tissues. The data for HHV-7 replication are expressed as a log percentage of the maximum for each tissue; R5SF162 HIV-1 inhibition is expressed as a percentage of the decrease in p24 relative to p24 production in matched singly HIV-infected tissue. The datum points from experiments with tissues from eight donors (days 6, 9, and 12 postinoculation) are plotted.
In contrast, the replication of X4 HIV-1LAI.04 was only marginally inhibited (by 31.4% ± 12.2% relative to matched tissues infected with HIV-1LAI.04 alone; P = 6 × 10−2, n = 8) (Fig. 2C). Accordingly, the average frequency of p24+ T cells, as detected by flow cytometry, in HHV-7 coinfected tissues was not different from that in singly HIV-1LAI.04-infected tissue (5.1% ± 2.5% and 5.1% ± 2.5%, P = 6.8 × 10−1, n = 4). However, when we inoculated HIV-1 in one tissue 3 days later than HHV-7, the replication of HIV-1 LAI.04 was suppressed by 56.6% compared to singly HIV-1-infected tissue, whereas in the matched tissue coinfected simultaneously, HIV-1LAI.04 replication was suppressed by 31.6%.
HHV-7 replication is differentially affected by coinfection with R5 and X4 HIV-1 variants.
To interpret the differential effects of HHV-7 on R5 and X4 HIV-1 strains, we evaluated the level of HHV-7 replication in tissues coinfected with R5SF162 or X4LAV.04 using a calibrated real-time PCR assay. In tissues coinfected with R5 HIV-1, the level of HHV-7 replication was not statistically different from that in tissues infected by HHV-7 alone (replication at day 12 postinoculation compared to singly HHV-7-infected tissues was 109.0% ± 19.0%; P = 4.9 × 10−1, n = 8) (Fig. 3). In contrast, the replication of HHV-7 was significantly suppressed in tissues coinfected with the X4 HIV-1 variant (relative replication at day 12 postinoculation of 42.2% ± 16.8%; P = 4 × 10−2, n = 8). In the tissue in which HHV-7 was inoculated 3 days prior to X4 HIV-1 LAI.04 it replicated similarly to singly HHV-7-infected tissue until day 9 and then was inhibited, although it still replicated 4.7 times more efficiently than in matched tissue infected with both viruses simultaneously.
FIG. 3.
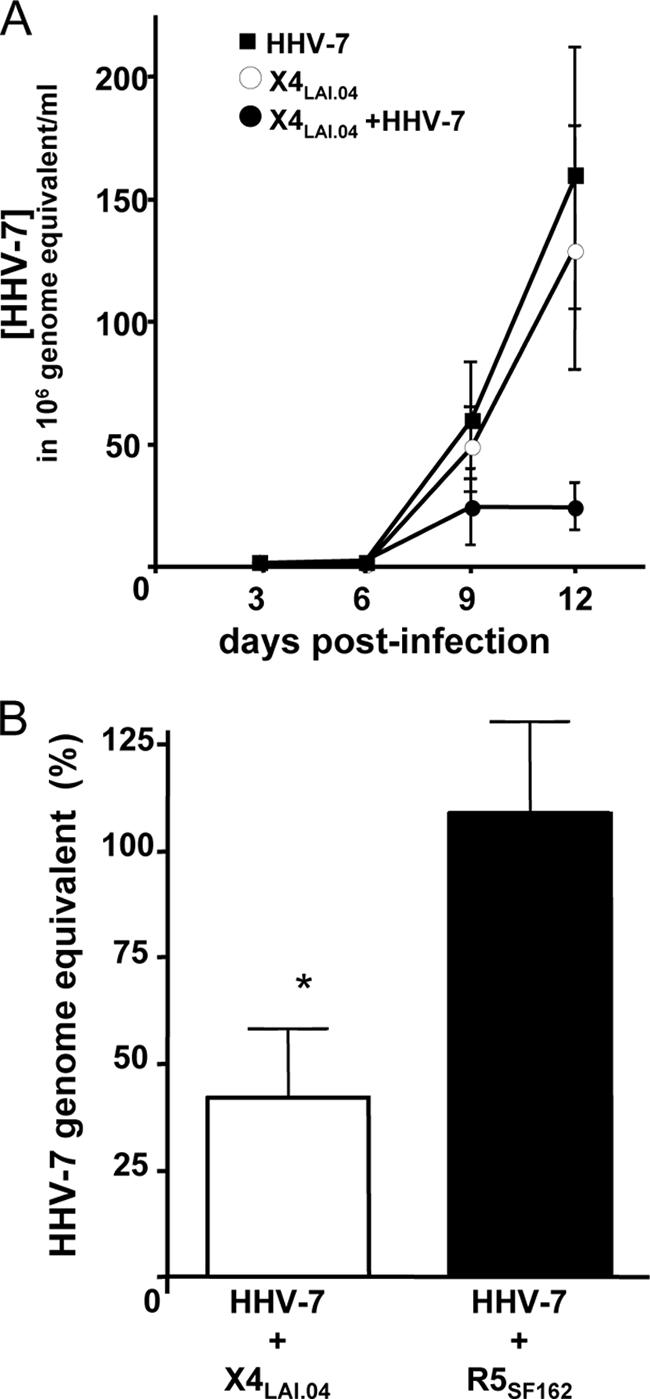
HHV-7 replication in human lymphoid tissues coinfected with R5SF162 or X4LAI.04. Sets of matched tissue blocks (27 blocks for each experimental condition from each donor) were infected with HHV-7 alone or together with one of the HIV-1 variants. HHV-7 replication was evaluated by measuring the number of HHV-7 genome equivalents accumulated in the culture medium. (A) Typical (n = 8) kinetics of HHV-7 replication in singly infected and HIV coinfected tissues. Shown are the means ± the SEM of the accumulation of HHV-7 genomes in culture medium during 3 days between medium changes. (B) Relative replication of HHV-7 in tissues coinfected with R5SF162 and X4LAI.04 Presented are the means ± the SEM of the production of HHV-7 genomes in culture medium over 12 days postinoculation for tissues from eight donors expressed as a percentage of the HHV-7 genome production in matched singly infected tissues. An asterisk denotes a significant difference (P < 5 × 10−2).
Consistent with the suppression of HHV-7 replication revealed by real-time PCR, flow cytometry showed a significant reduction in the number of cells productively infected with HHV-7 in tissues coinfected with X4 HIV-1 (the average fraction of HHV-7-positive cells at day 12 postinfection was reduced by 66.6% ± 14.1% relative to that in matched tissues singly infected with HHV-7; P = 9 × 10−3, n = 5). In contrast, the fraction of cells productively infected with HHV-7 in tissues coinfected with R5 HIV-1 was not significantly reduced (by 35.0% ± 15.7% relative to matched tissues singly infected with HHV-7; P = 9 × 10−2, n = 5).
Lack of upregulation of CC chemokines in HHV-7-infected lymphoid tissues.
Since interaction of HIV-1 with several microbes, including HHV-6, was shown to be mediated by upregulation of CC chemokines (11, 12, 24), we compared cytokine profiles in uninfected and HHV-7-infected human lymphoid tissues ex vivo. We measured the release of RANTES, MIP-1α, and MIP-1β, as well as of 16 additional cytokines (IL-1α, IL-1β, IL-2, IL-4, IL-6, IL-7, IL-8, IL-12, IL-15, IL-16, MIG, granulocyte-macrophage colony-stimulating factor, IP-10, SDF-1β, tumor necrosis factor alpha, and gamma interferon) by uninfected tissues, singly infected tissues, and tissues coinfected with HHV-7 and either R5 or X4 HIV-1 between days 3 and 12 postinfection. For these tissues we compared the total production throughout the culture, as well as at each time interval. No significant change of the cytokine/chemokine profile was observed in HHV-7-infected tissues compared to matched uninfected controls or in tissues coinfected with HHV-7 and either R5 or X4 HIV-1 compared to those infected with HIV-1 only (P ≥ 1.4 × 10−1, n = 9). Total IL-16 production was slightly decreased in HHV-7-infected tissues: by 10.5% ± 0.5% in singly HHV-7-infected tissues relative to matched uninfected controls and by 8.4% ± 0.7% in the case of HHV-7-R5 HIV-1 coinfection relative to matched tissues infected by R5 HIV-1 only. The entire decrease in IL-16 production in singly HHV-7-infected or R5 HIV-1 coinfected tissues occurred between days 6 and 9 (23.7% ± 9% and 13% ± 4%, respectively, P≤ 4 × 10−2, n = 9) and, in contrast to the drop in the total IL-16 production, was statistically significant. In X4 HIV-1-coinfected tissue the production of IL-16 did not change.
As shown earlier (16), R5 HIV-1 infection ex vivo did not change the cytokine profile in human lymphoid tissue, whereas X4 HIV-1 significantly upregulated the production of the CCR5-binding chemokines, MIP-1α, MIP-1β, and RANTES, both in singly X4 HIV-1-infected tissues and in X4 HIV-1-HHV-7 coinfected tissues (data not shown).
To investigate whether other cytokines not included in our multiplex panel were changed as a result of HHV-7 infection, we used a proteomics chip that semiquantitatively evaluates 120 cytokines. This assay did not reveal any change in cytokine profiles in HHV-7-infected tissues relative to a matched infected control (data not shown). Thus, HHV-7 infection does not seem to change the cytokine profile significantly. Likewise, HHV-7 coinfection of HIV-infected tissue failed to change the cytokine profile relative to tissues infected with HIV-1 alone.
HHV-7 downregulates the expression of CD4 on T lymphocytes.
To further investigate the molecular mechanisms of HIV-1 interactions with HHV-7, we characterized cells targeted by HHV-7 in lymphoid tissue (Fig. 4). In HHV-7-infected tissues from 10 donors, the vast majority of viral antigen-positive cells were CD3+ (on average, 89.8% ± 2.0% at day 12 postinfection). Among these cells 60.4% ± 3.7% expressed neither CD4 nor CD8, whereas 32.1% ± 3.9% were CD4+ CD8−, 3.8% ± 0.6% were CD4− CD8+, and 3.4% ± 0.5% CD4+ CD8+ (Fig. 4). Thus, the majority of HHV-7-infected cells were CD3+ lymphocytes of the double-negative (CD4− CD8−) phenotype which in uninfected tissue accounts for only 10.9% ± 1.4% of the CD3+ cell population since most CD3+ cells are either CD4+ (72.0% ± 2.3%, n = 9) or CD8+ (12.4% ± 1.4%, n = 9).
FIG. 4.

Phenotype of HHV-7-infected cells in human lymphoid tissues. Tissue blocks (27 blocks for each condition from each donor) were inoculated with HHV-7 and cultured for 12 days. Cells isolated from these blocks and from matched uninfected blocks were stained for lymphocytic markers and analyzed with flow cytometry. (A) Distribution of CD4 and CD8 in uninfected tissues and in matched HHV-7-infected tissue. Bars: □, T cells in uninfected tissue; ▪, uninfected T cells in HHV-7-infected tissue; ▤, HHV-7-infected T cells in infected tissue. Each bar represents the mean ± the SEM for tissues from nine donors. Asterisks denote significant differences: *, P < 10−2; and **, P < 10−3. (B) Bivariate density plot of T (CD3+) cells isolated from HHV-7-infected tissue on day 12 postinfection and stained for lymphocytic markers. Panels: left, distribution of T cells in uninfected tissue; middle, distribution of T cells in HHV-7-infected tissues; right, distribution of HHV-7-infected T cells (red dots) among T cells (gray) in HHV-7-infected tissues. A typical example of experiments with tissues from 10 donors is presented.
To investigate whether HHV-7-producing cells expressed CD4 at an earlier time point of infection, in one experiment we immunophenotyped HHV-7-infected cells both at day 6 and at day 12 postinfection. At day 6 postinfection 58.1% of HHV-7-infected T lymphocytes expressed CD4, whereas CD4− CD8− T lymphocytes constituted less than 41.9%. At day 12 the amount of CD4+ T cells in HHV-7-infected fraction dropped to 18.9%, while the amount of CD4− CD8− cells in this fraction increased to 81.1%. Thus, it seems that, in agreement with the data for isolated peripheral blood mononuclear cells (19), CD4+ T cells are the predominant target cells for HHV-7 infection in lymphoid tissue, but HHV-7 dramatically downregulates CD4 in the course of the infection. To confirm this hypothesis, we compared CD4 expression on CD3+ CD4+ pp85+ cells in infected tissues with that on CD4+ T cells in matched uninfected controls by measuring the distribution of the fluorescence intensity for CD4 (Fig. 5A and B). On average, on CD4+pp85+ T cells the median fluorescence intensity was reduced by 40.3% ± 7.1% compared to that of CD4+ T cells in matched control (P = 9 × 10−4, n = 11). To evaluate the overall decrease in CD4 expression on HHV-7-infected T cells, we gated on CD8− T cells since this subset comprises T cells with different levels of CD4 expression. On HHV-7-infected cells of this phenotype (CD3+ CD8− pp85+) the expression of CD4 was reduced by 86.3% ± 3.3% (P = 10−10, n = 10) compared to CD3+ CD8− cells in a matched uninfected control (Fig. 5B and C).
FIG. 5.


CD4 expression in T lymphocytes isolated from HHV-7-infected human lymphoid tissues. Cells from matched sets of uninfected or HHV-7-infected tissue blocks (27 blocks for each experimental condition from each donor) were stained for lymphocytic markers and for HHV-7 pp85 and then analyzed by flow cytometry. (A) Distribution of HHV-7+ CD3+ CD8− and HHV-7− CD3+ CD8− cells isolated from uninfected tissues (left panel) and matched HHV-7-infected tissues (right panel) according to their CD4 fluorescence intensity. A typical bivariate dot plot for tissues from 11 donors is shown. (B) Distribution of CD4 fluorescence intensity in HHV-7+ (red histogram) and HHV-7− (green histogram) cells from HHV-7-infected tissue blocks and in cells from matched uninfected control tissue (blue histogram). Presented are data for the same tissue as in panel A. A typical distribution for tissues from 11 donors is shown. (C) Cumulative frequency distribution of CD4 fluorescence intensities. The percentage of infected tissue HHV-7+ (red) and HHV-7− (green) cells with the CD4 fluorescence below the median fluorescence of a matched uninfected control (blue) are indicated. Presented are data for the same tissue as in panels A and B. Typical for data from 11 donors are shown. (D) Downregulation of CD4 in HHV-7-infected and HHV-7-uninfected CD8− T cells isolated from HHV-7-infected tissues and matched uninfected controls. Downregulation of CD4 was defined as the difference (percent) between the ratio of the numbers of cells with the CD4 fluorescence higher and lower than the median fluorescence for uninfected control tissue (by definition equals one) and a similar ratio for matched infected tissues. That is, DCD4 = (Flhctrl/Fllctrl − Flhinf/Fllinf) × 100 = (1 − Flhinf/Fllinf) × 100, where DCD4 is the downregulation of CD4 on the cell surface, Fllinf and Flhinf are the numbers of cells in infected tissues, with fluorescence lower and higher (respectively) than the median in an uninfected control, and Flhctrl and Fllctrl are similar parameters for a matched uninfected control. By definition, the median the ratio of Flhcntrl/Fllcntrl is equal to 1. Presented are means ± the SEM for tissues from 11 donors.
Strikingly, HHV-7 infection decreased CD4 expression also on the surface of uninfected bystander T cells (Fig. 4A and Fig. 5C and D). We observed, on average, a reduction of CD4 expression on CD3+ CD8− pp85− cells by 58.2% ± 5% relative to matched uninfected control tissue (P = 4 × 10−7, n = 10). Overall, the expression of CD4 in both HHV-7-infected and in bystander T cells considered together was decreased by 65.9% ± 4.8% (P = 8 × 10−8, n = 11) (Fig. 5D). Furthermore, there was a significant correlation between the number of HHV-7-infected cells and the degree of CD4 downregulation in both infected (pp85+) T cells (R = 0.64, P = 3 × 10−2, n = 11) and uninfected (pp85−) bystander T cells (R = 0.78, P = 4 × 10−3, n = 11).
In addition to downregulating CD4, HHV-7 also exerts cytopathic effects during productive infection. We did not observe a cytopathic effect of HHV-7 on CD8+ T cells since there was no significant difference in the numbers of CD8+ T cells between HHV-7-infected and matched uninfected tissues (the mean difference between the two sets of tissues was 1.0% ± 0.6% [n = 3]). To assess the cytopathic effect of HHV-7 on CD4+ T cells and to evaluate the relative contributions of HHV-7-induced CD4 downregulation and cell death to the decrease in the number of CD4+ T cells in infected lymphoid tissue, we enumerated CD3+ CD4+ and CD3+ CD8− cells in HHV-7-infected tissues and in matched control tissues. Since almost all CD3+ cells in uninfected tissues are either CD4+ or CD8+, we assumed that the death of a CD3+ CD4+ cell in HHV-7-infected tissues should be reflected by the loss of a CD3+ CD8− cell. Conversely, if a CD3+ CD4+ cell ceased to express CD4 it would still be counted in our analysis as a CD3+ CD8− cell. Enumeration of the CD3+ CD4+ and CD3+ CD8− cells revealed a significant difference between the decrease in the number of cells in these two subsets (41.9% ± 7.5% versus 21.2% ± 5.7%, P = 8 × 10−5, n = 10). Thus, overall, HHV-7 infection resulted in the death of ca. 21% of CD4+ T cells, complete downregulation of CD4 in another 20% of the CD4+ T cells, and partial downregulation in the rest of the CD4+ T cells.
A similar pattern of CD4 downregulation was observed in tissues coinfected with HHV-7 and R5 HIV-1: complete downregulation of CD4 in 29.2% ± 12% of the CD4+ T cells and downregulation of CD4 expression on the rest of CD4+ T cells by 49.7% ± 10.2% (P = 4 × 10−2, n = 6). In contrast, compared to single HIV-1 infection, CD4 expression on X4 HIV-coinfected tissues was completely downregulated on only 5.5% ± 5.7% of the CD4+ T cells and downregulated by 17.1% ± 6.1% on the rest of the CD4+ T cells (n = 7, P = 4 × 10−2).
Further analysis of T-cell subsets revealed neither a preferential loss of CCR5+ or CXCR4+ T cells nor a downregulation of CCR5, whereas CXCR4 was slightly downregulated in HHV-7-infected tissues (data not shown).
Finally, we determined the naive (CD45RA+ CD62L+) and non-naive (CD45RA− CD62L−, CD45RA− CD62L+, and CD45RA+ CD62L−) status of HHV-7-infected cells. HHV-7 demonstrated a strong preference for infection of non-naive over naive T cells, whereas in control tissues the ratio between non-naive and naive T cells was, on average, 3.7 ± 1.4 (n = 3); in infected tissues among HHV-7-infected cells this ratio was 14.0 ± 6.9 (n = 3). After correction for the relative abundance of naive versus non-naive T cells in matched uninfected control tissues the incidence of HHV-7 infection among non-naive T cells was calculated to be 0.1 ± 0.05 (n = 3), whereas in naive cells it was 0.03 ± 0.02 (n = 3). Thus, the probability of a non-naive T-cell becoming productively infected by HHV-7 in the context of human lymphoid tissue was ∼3-fold higher than that of a naive T cell.
Downregulation of CD4 is sufficient to suppress HIV-1 replication.
To test whether the inhibition of R5 HIV-1 replication by HHV-7 can be ascribed to the downregulation of CD4, we mimicked the effects of HHV-7 by inducing CD4 downregulation by nonvirologic means. We used the synthetic macrocycle CADA, which specifically downregulates CD4 surface expression by interfering with the posttranslational processing of CD4 (22). After culturing the lymphoid tissue for 12 days in the presence of CADA, we observed a dose-dependent downregulation of CD4 with a 50% effective concentration of 1 μM. At high a concentration (8 μM) CD4 was almost completely downregulated (by 98.4% compared to the matched untreated control). In HHV-7-infected tissues, CADA at this concentration almost completely prevented HHV-7 infection: the number of HHV-7+ T cells at day 12 postinfection decreased from 19.0% to 0.6%. Treatment with CADA at 1 and 2 μM better mimicked the impact of HHV-7 on the expression of CD4, reducing its expression by 53.1% ± 4.3% and 85.7% ± 1.3%, respectively, compared to that in matched untreated controls (n = 3) (Fig. 6A).
FIG. 6.

CD4 expression and HIV replication in CADA-treated human lymphoid tissues. Matched sets of tissue (27 blocks for each experimental condition from each of two donors) were incubated with CADA (1 or 2 μM) for 12 days or used as untreated controls. Some of these sets were infected with R5SF162 and X4LAI.04. (A) Cumulative frequency distribution of CD4 fluorescence intensities in T cells isolated from untreated (blue histogram) tissues and from tissues incubated with CADA at 1 μM (green histogram) and 2 μM (red histogram). The percentages of T cells with the CD4 fluorescence below the median fluorescence of matched uninfected control (blue) are indicated. The distributions for the tissues from three donors were similar, and only one is presented here. (B) Relative replication of R5SF162 and X4LAI.04 in tissues incubated with 1 or 2 μM CADA expressed as a percentage of p24 production in matched untreated tissues infected with HIV-1. Shown are means ± the SEM of the cumulative production of HIV over 12 days of infection.
Based on these results, we evaluated the effect of CADA at 1 or 2 μM on the replication of X4 and R5 HIV-1. At concentrations of 1 and 2 μM, CADA suppressed the replication of HIV-1SF162 by 69.0% ± 1.8% (n = 2) and 73.7% ± 2.4% (n = 2), respectively, relative to that in matched untreated controls (Fig. 6B). CADA was also efficient, although to a lesser extent, in suppressing the replication of X4LAI.04: replication was decreased by 37.3% ± 13.4% (n = 2) and 48.2% ± 0.3% (n = 2) for 1 and 2 μM concentrations of CADA, respectively, compared to the untreated control. Thus, the downregulation of CD4 expression in human lymphoid tissue by pharmacological methods resulted in suppression of HIV-1 replication similar to that caused by HHV-7.
DISCUSSION
HHV-7 and HHV-6 are two closely related human herpesviruses with a high degree of genetic homology. Both viruses were isolated from cultured human T lymphocytes and efficiently replicate in CD4+ T lymphocytes in vitro (7, 20). As shown here for HHV-7 and earlier for HHV-6 (12), both viruses suppress the replication of R5 HIV-1 in coinfected human lymphoid tissues, without significant effects on X4 HIV-1. In spite of striking similarities in the pattern of HHV-6- and HHV-7-mediated modulation of HIV-1 infection, we found that the molecular mechanisms of HIV-1 suppression caused by these two closely related herpesviruses are dramatically different. Whereas HHV-6 suppression of R5 HIV-1 is mediated primarily by the upregulation of RANTES (12), HHV-7 suppresses R5 HIV-1 by downregulation of the major HIV receptor, CD4.
To study the effects of HHV-7 on HIV-1 in coinfected tissues, we first investigated whether human lymphoid tissue can support productive AL HHV-7 infection ex vivo. We consider this virus a typical HHV-7 since no significant strain variation in terms of cellular tropism or other biological features has been noticed thus far both in our laboratory and in the literature. We found that blocks of human lymphoid tissue inoculated with HHV-7 become productively infected without exogenous activation. This was evidenced by a steep increase in the release of HHV-7 DNA into the culture medium bathing the inoculated tissue, as measured by real-time PCR, and by an increase in the number of HHV-7 antigen-positive lymphocytes, as estimated by flow cytometry. In 12 days of infection an inoculated tissue block produced on average more than 50 million copies of HHV-7 DNA.
Flow cytometry revealed that almost all of the infected cells were T lymphocytes. At the end of the experiment 20% of these lymphocytes, mostly of non-naive (central and effector memory) phenotype, were HHV-7 infected. Only a few of these infected cells expressed CD8. This is in agreement with HHV-7 usage of CD4 as its major cellular receptor (19). Also, the critical role of CD4 in HHV-7 infection was evident in experiments with CADA, a synthetic macrocycle that specifically downregulates CD4 in a dose-dependent manner by interfering with the CD4 translation machinery (22). A high dose of CADA almost completely suppressed CD4 expression on tissue T cells, and this downregulation was accompanied by a dramatic resistance to HHV-7 infection.
In spite of the known CD4+ T-cell tropism of HHV-7 (19), most HHV-7-infected tissue T lymphocytes expressed neither CD4 nor CD8. Did these cells express CD4 and modulate its expression as a result of HHV-7 infection? To address this question, we enumerated lymphocytes in different subsets and found that, whereas the number of CD4+ T cells dropped by more than 40% in the course of HHV-7 infection, only half of this drop could be accounted by cell death. Thus, HHV-7 infection almost completely abolished CD4 expression in 20% of T lymphocytes, mostly in those that were productively infected. The complete loss of CD4 expression on infected T lymphocytes was directly demonstrated by the measurement of CD4 fluorescence intensity on these cells. The drop in CD4 expression gradually developed over the course of HHV-7 infection and was more evident at day 12 than at day 6 postinfection. Moreover, measurement of the CD4 fluorescence showed that in HHV-7-infected T cells, which still expressed CD4, the level of expression was significantly diminished. In addition to HHV-7+ T cells, HHV-7− T cells also showed a significant downregulation of CD4. The latter may be either affected by their productively infected neighbors (e.g., through the secretion of soluble factors) or by HHV-7 virions binding to the CD4 molecule without causing infection. Whichever the mechanism of CD4 downregulation in both productively infected and bystander cells, HHV-7-infected tissues are dramatically depleted of CD4+ T cells, and those that are left express CD4 at much lower levels than in uninfected tissue.
In summary, we identified three mechanisms whereby HHV-7 infection resulted in a generalized decrease in the number of potential HIV targets, reducing the susceptibility of these tissues to HIV-l infection: mild depletion of CD4+ T cells, severe downregulation of CD4 on the surface of productively infected T cells, and downregulation of CD4 on uninfected cells. These effects should decrease the replication of both R5 and X4 HIV-1. However, only R5 HIV-1 replication was affected in our experiments, suggesting that different mechanisms operate in HHV-7-R5 HIV-1- and HHV-7-X4 HIV-1-coinfected tissues. Therefore, these two cases of coinfection will be discussed separately below.
In the case of HHV-7-R5 HIV-coinfected tissues, HHV-7 replication was not affected, whereas that of R5 HIV-1 was dramatically suppressed. The severity of this suppression temporally correlated with HHV-7 replication. Does HHV-7-induced downregulation of CD4 contribute to the observed suppression of R5 HIV-1?
To address this question, we mimicked the effects of HHV-7 by downregulating CD4 in an HHV-7-independent way. We chose the concentration of CADA that downregulates CD4 in tissue lymphocytes to approximately the same extent as HHV-7 infection. We inoculated CADA-treated tissue blocks with R5 HIV-1 and observed suppression of HIV replication comparable to that in HHV-7-coinfected tissues. In conclusion, downregulation of CD4 by HHV-7 alone seems to be sufficient to account for R5 HIV-1 suppression in human lymphoid tissues coinfected with both viruses.
Additional mechanisms may contribute to R5 HIV-1 suppression by HHV-7 as well. A similar suppression of R5 HIV-1 variant in coinfected tissues was earlier reported for HHV-6 (12), measles virus (11), and GB virus C (24), and in all of these cases it was explained by the upregulation of CCR5-binding chemokines. In contrast, in the case of HHV-7, the production of none of these chemokines nor of 16 additional cytokines except IL-16 was altered either in singly infected tissues or in tissues coinfected with HIV-1 compared to singly infected tissues. The production of IL-16 was decreased between days 6 and 9 when HHV-7 replication expanded rapidly. This decrease may be related to the downregulation of CD4 since IL-16 is a ligand for this receptor.
However, since we did not monitor cytokines continuously but measured them in samples collected every 3 days, it is conceivable that some transient changes in cytokine release may occur within the 3-day intervals. Thus, it seems that the mechanism by which HHV-7 suppresses R5 HIV-1 replication is different from that enacted by other microbes.
Although HHV-7-mediated downregulation of CD4 expression in both HHV-7-infected and bystander cells emerged as the major mechanism of R5 HIV-1 suppression in coinfected tissue, coinfection with HHV-7 and X4 HIV-1 presents a very different scenario. In contrast to R5 HIV-1-HHV-7 coinfection, in tissues inoculated with HHV-7 and X4 HIV-1 it was the replication of HHV-7 that was severely suppressed, whereas the replication of HIV-1 was lower but not statistically different from that in singly HIV-infected tissue. Since in X4 HIV-HHV-7-coinfected tissue the replication of HHV-7, although reduced, was not inhibited completely, this suppression of X4 HIV-1 may be attributed to the residual HHV-7 replication in these tissues. In agreement with this, when X4 HIV-1 was inoculated after HHV-7 infection was already established, X4 HIV-1 replication was suppressed more and HHV-7 was suppressed less than in matched tissue infected with both viruses simultaneously. This finding supports our hypothesis regarding the HHV-7 replication that triggers CD4 downregulation as the major factor for HIV-1 suppression.
Moreover, the downregulation of CD4 may have less impact on X4 than on R5 HIV-1 since X4 HIV-1 was also less sensitive to CADA-triggered downregulation of CD4 than R5 HIV-1. The mechanisms for the apparently different requirement for CD4 for X4 and R5 HIV-1 are currently unknown and are beyond the scope of the present study.
Thus, whereas the inhibition of both R5 and X4 HIV-1 by replicating HHV-7 seems to be mediated by CD4 downregulation, the mechanisms of HHV-7 suppression in X4 HIV-1-coinfected tissues are not yet understood. We speculate that the difference in the effects of X4 and R5 HIV-1 on HHV-7 is due to the different target cell specificities for HIV-1 of these phenotypes. As we have demonstrated, HHV-7 predominantly infects non-naive CD4+ T cells. These are the cells that express the highest levels of CCR5 (10) and thus are the predominant natural targets for R5 HIV-1 infection. Thus, HHV-7 and R5 HIV-1 largely have the same pool of target cells on which they share the same receptor, and HHV-7 outcompetes R5 HIV-1 in infecting this pool. In contrast, X4 HIV-1 has a broader pool of target cells, which includes both non-naive T cells and naive resting T cells (14). Therefore, in the case of X4 HIV-1 coinfection, there is a target pool of non-naive CD4+ T cells that both viruses share and a large pool of naive CD4+ T cells that X4 HIV-1 can infect without competing with HHV-7. The latter pool may produce X4 HIV-1 virions without the interfering effect of HHV-7, which in turn would outcompete HHV-7 in the infection of non-naive CD4+ T cells. Furthermore, X4 HIV-1 infection depletes up to 90% of CD4+ T cells in this system, in particular of the non-naive phenotype (14), thus depleting almost all of the potential HHV-7 target cells and therefore progressively suppressing HHV-7 infection. This mechanism is in agreement with the recent observation that the HHV-7 load in HIV-coinfected individuals correlates with the CD4+ T-cell count (3).
In summary, ex vivo coinfection of human lymphoid tissues with HHV-7 and either R5 or X4 HIV-1 results in different outcomes. The former results in an almost complete suppression of R5 HIV-1, whereas HHV-7 replication is not affected; the latter results in a severe suppression of HHV-7, whereas the replication of X4 HIV-1 was suppressed slightly.
The model system described here is the first that permits the study of HHV-7-induced pathogenesis in the context of human lymphoid tissue under controlled experimental conditions and the comparison with tissue pathogenesis of other lymphotropic viruses. In particular, this system emphasized the striking difference in the pathogenesis of HHV-7 and the closely related HHV-6. HHV-7 has a strong CD4 tropism restricting its target cells to CD4+ T cells, whereas HHV-6 uses CD46 and infects a broader pool of T and non-T cells, regardless of CD4 expression (15). Both viruses infect predominantly non-naive tissue T cells and on these cells downregulate surface molecules essential for T-cell activation: HHV-7 downregulates CD4, whereas HHV-6 downregulates CD3 (15). This may be part of a Roseolovirus strategy to interfere with the T-cell activation and to preserve the herpesvirus-infected cells in a quiescent state favoring long-term persistent infection and/or viral replication.
The molecular mechanism of HIV-1 suppression by HHV-7 described here is novel and different from the earlier reported mechanisms of interactions of other microbes with HIV, all of which were mediated by the upregulation of chemokines. The model for studying the interactions of these viruses in the context of human lymphoid tissue provides an opportunity to investigate mechanisms of this interference under physiologically relevant conditions. The results in ex vivo tissues suggest that HHV-7 may interfere with HIV-1 in lymphoid tissue in vivo, thus affecting disease progression, in particular facilitating a switch of dominance from R5 to X4 HIV-1, which in turn may affect HHV-7 replication. Knowledge of the mechanisms of the interactions of HIV-1 with HHV-7, as well as with other pathogens that modulate HIV-1 replication, may provide new insights into HIV pathogenesis and lead to the development of new anti-HIV therapeutic strategies.
Footnotes
Published ahead of print on 25 October 2006.
REFERENCES
- 1.Ablashi, D. V., Z. N. Berneman, B. Kramarsky, Y. Asano, S. Choudhury, and G. R. Pearson. 1994. Human herpesvirus-7 (HHV-7). In Vivo 8:549-554. [PubMed] [Google Scholar]
- 2.Black, J. B., and P. E. Pellett. 1999. Human herpesvirus 7. Rev. Med. Virol. 9:245-262. [DOI] [PubMed] [Google Scholar]
- 3.Boutolleau, D., O. Bonduelle, A. Sabard, L. Devers, H. Agut, and A. Gautheret-Dejean. 2005. Detection of human herpesvirus 7 DNA in peripheral blood reflects mainly CD4+ cell count in patients infected with HIV. J. Med. Virol. 76:223-228. [DOI] [PubMed] [Google Scholar]
- 4.Broccolo, F., S. Bossolasco, A. M. Careddu, G. Tambussi, A. Lazzarin, and P. Cinque. 2002. Detection of DNA of lymphotropic herpesviruses in plasma of human immunodeficiency virus-infected patients: frequency and clinical significance. Clin. Diagn. Lab. Immunol. 9:1222-1228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Broccolo, F., G. Locatelli, L. Sarmati, S. Piergiovanni, F. Veglia, M. Andreoni, S. Butto, B. Ensoli, P. Lusso, and M. S. Malnati. 2002. Calibrated real-time PCR assay for quantitation of human herpesvirus 8 DNA in biological fluids. J. Clin. Microbiol. 40:4652-4658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fabio, G., S. N. Knight, I. M. Kidd, S. M. Noibi, M. A. Johnson, V. C. Emery, P. D. Griffiths, and D. A. Clark. 1997. Prospective study of human herpesvirus 6, human herpesvirus 7, and cytomegalovirus infections in human immunodeficiency virus-positive patients. J. Clin. Microbiol. 35:2657-2659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Frenkel, N., E. C. Schirmer, L. S. Wyatt, G. Katsafanas, E. Roffman, R. M. Danovich, and C. H. June. 1990. Isolation of a new herpesvirus from human CD4+ T cells. Proc. Natl. Acad. Sci. USA 87:748-752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Glushakova, S., B. Baibakov, L. B. Margolis, and J. Zimmerberg. 1995. Infection of human tonsil histocultures: a model for HIV pathogenesis. Nat. Med. 1:1320-1322. [DOI] [PubMed] [Google Scholar]
- 9.Glushakova, S., B. Baibakov, J. Zimmerberg, and L. B. Margolis. 1997. Experimental HIV infection of human lymphoid tissue: correlation of CD4+ T-cell depletion and virus syncytium-inducing/non-syncytium-inducing phenotype in histocultures inoculated with laboratory strains and patient isolates of HIV type 1. AIDS Res. Hum. Retrovir. 13:461-471. [DOI] [PubMed] [Google Scholar]
- 10.Gondois-Rey, F., J. C. Grivel, A. Biancotto, M. Pion, R. Vigne, L. B. Margolis, and I. Hirsch. 2002. Segregation of R5 and X4 HIV-1 variants to memory T-cell subsets differentially expressing CD62L in ex vivo infected human lymphoid tissue. AIDS 16:1245-1249. [DOI] [PubMed] [Google Scholar]
- 11.Grivel, J. C., M. Garcia, W. J. Moss, and L. B. Margolis. 2005. Inhibition of HIV-1 replication in human lymphoid tissues ex vivo by measles virus. J. Infect. Dis. 192:71-78. [DOI] [PubMed] [Google Scholar]
- 12.Grivel, J. C., Y. Ito, G. Faga, F. Santoro, F. Shaheen, M. S. Malnati, W. Fitzgerald, P. Lusso, and L. Margolis. 2001. Suppression of CCR5- but not CXCR4-tropic HIV-1 in lymphoid tissue by human herpesvirus 6. Nat. Med. 7:1232-1235. [DOI] [PubMed] [Google Scholar]
- 13.Grivel, J. C., N. Malkevitch, and L. Margolis. 2000. Human immunodeficiency virus type 1 induces apoptosis in CD4+ but not in CD8+ T cells in ex vivo-infected human lymphoid tissue. J. Virol. 74:8077-8084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Grivel, J. C., and L. B. Margolis. 1999. CCR5- and CXCR4-tropic HIV-1 are equally cytopathic for their T-cell targets in human lymphoid tissue. Nat. Med. 5:344-346. [DOI] [PubMed] [Google Scholar]
- 15.Grivel, J. C., F. Santoro, S. Chen, G. Faga, M. S. Malnati, Y. Ito, L. Margolis, and P. Lusso. 2003. Pathogenic effects of human herpesvirus 6 in human lymphoid tissue ex vivo. J. Virol. 77:8280-8289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ito, Y., J. C. Grivel, S. Chen, Y. Kiselyeva, P. Reichelderfer, and L. Margolis. 2004. CXCR4-tropic HIV-1 suppresses replication of CCR5-tropic HIV-1 in human lymphoid tissue by selective induction of CC-chemokines. J. Infect. Dis. 189:506-514. [DOI] [PubMed] [Google Scholar]
- 17.Kempf, W., B. Muller, R. Maurer, V. Adams, and G. Campadelli Fiume. 2000. Increased expression of human herpesvirus 7 in lymphoid organs of AIDS patients. J. Clin. Virol. 16:193-201. [DOI] [PubMed] [Google Scholar]
- 18.Kidd, I. M., D. A. Clark, C. A. Sabin, D. Andrew, A. F. Hassan-Walker, P. Sweny, P. D. Griffiths, and V. C. Emery. 2000. Prospective study of human betaherpesviruses after renal transplantation: association of human herpesvirus 7 and cytomegalovirus coinfection with cytomegalovirus disease and increased rejection. Transplantation 69:2400-2404. [DOI] [PubMed] [Google Scholar]
- 19.Lusso, P., P. Secchiero, R. W. Crowley, A. Garzino-Demo, Z. N. Berneman, and R. C. Gallo. 1994. CD4 is a critical component of the receptor for human herpesvirus 7: interference with human immunodeficiency virus. Proc. Natl. Acad. Sci. USA 91:3872-3876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Salahuddin, S. Z., D. V. Ablashi, P. D. Markham, S. F. Josephs, S. Sturzenegger, M. Kaplan, G. Halligan, P. Biberfeld, F. Wong-Staal, B. Kramarsky, et al. 1986. Isolation of a new virus, HBLV, in patients with lymphoproliferative disorders. Science 234:596-601. [DOI] [PubMed] [Google Scholar]
- 21.Secchiero, P., Z. N. Berneman, R. C. Gallo, and P. Lusso. 1994. Biological and molecular characteristics of human herpesvirus 7: in vitro growth optimization and development of a syncytia inhibition test. Virology 202:506-512. [DOI] [PubMed] [Google Scholar]
- 22.Vermeire, K., and D. Schols. 2005. Cyclotriazadisulfonamides: promising new CD4-targeted anti-HIV drugs. J. Antimicrob. Chemother. 56:270-272. [DOI] [PubMed] [Google Scholar]
- 23.Ward, K. N. 2005. The natural history and laboratory diagnosis of human herpesvirus-6 and -7 infections in the immunocompetent. J. Clin. Virol. 32:183-193. [DOI] [PubMed] [Google Scholar]
- 24.Xiang, J., S. L. George, S. Wunschmann, Q. Chang, D. Klinzman, and J. T. Stapleton. 2004. Inhibition of HIV-1 replication by GB virus C infection through increases in RANTES, MIP-1α, MIP-1β, and SDF-1. Lancet 363:2040-2046. [DOI] [PubMed] [Google Scholar]