

Abstract
The severe acute respiratory syndrome (SARS), caused by a novel coronavirus (SARS-CoV), resulted in substantial morbidity, mortality, and economic losses during the 2003 epidemic. While SARS-CoV infection has not recurred to a significant extent since 2003, it still remains a potential threat. Understanding of SARS and development of therapeutic approaches have been hampered by the absence of an animal model that mimics the human disease and is reproducible. Here we show that transgenic mice that express the SARS-CoV receptor (human angiotensin-converting enzyme 2 [hACE2]) in airway and other epithelia develop a rapidly lethal infection after intranasal inoculation with a human strain of the virus. Infection begins in airway epithelia, with subsequent alveolar involvement and extrapulmonary virus spread to the brain. Infection results in macrophage and lymphocyte infiltration in the lungs and upregulation of proinflammatory cytokines and chemokines in both the lung and the brain. This model of lethal infection with SARS-CoV should be useful for studies of pathogenesis and for the development of antiviral therapies.
Severe acute respiratory syndrome (SARS) was first identified in Guangdong Province in China (28). Over the ensuing 9 months, more than 8,000 cases were identified throughout the world, with a ∼10% case fatality rate. A novel coronavirus, SARS coronavirus (SARS-CoV), was identified as the causative agent (6, 17, 29, 32). Initial investigations indicated that the virus spread to humans from infected exotic animals such as Himalayan palm civets (Paguma larvata) and Chinese ferret badgers (Melogale moschata) (12); more recent work has suggested that the natural reservoirs for the virus are wild bat populations in China (19, 24). Although SARS has not recurred in human populations to a significant extent since 2003, the potential severity of such a recurrence has spurred interest in developing an animal model for the human disease.
SARS-CoV infects and replicates in mice, ferrets, hamsters, and several species of nonhuman primates (cynomolgus and rhesus macaques, African green monkeys, and common marmosets) (reviewed in reference 37). However, none of these animals develop a clinical disease that is reproducible and equivalent in severity to that observed in SARS patients. A mouse model would be useful for answering many questions about SARS pathogenesis and for testing vaccine efficacy, in part because reagents for the study of the immune response are widely available. However, other than aged or immunocompromised (STAT1−/−) mice (37), these animals do not develop significant clinical disease, and lethality has not been demonstrated in any murine model of SARS. With the goal of developing a more robust murine model, we generated transgenic (Tg) mice in which expression of hACE2 (human angiotensin-converting enzyme 2, the primary host cell receptor for SARS-CoV [23]) was targeted to epithelial cells. While human ACE2 and murine ACE2 (mACE2) molecules are very homologous, mACE2 does not support SARS-CoV binding as efficiently as hACE2 (22). Here we show that the transgenic expression of hACE2 in epithelia converts a mild SARS-CoV infection into a rapidly fatal disease.
MATERIALS AND METHODS
Mice.
All animal studies were approved by the University of Iowa and the Veterans Administration Institutional Animal Care and Use committees. Mice transgenic for expression of hACE2 (K18-hACE2 mice) were generated as follows (see Fig. 1A). The hACE2 coding sequence was PCR amplified from IMAGE consortium clone ID 5243048 (ATCC, Manassas, VA) and cloned into the pCR2.1-TOPO vector (Invitrogen, Carlsbad, CA). The lacZ coding sequence in the previously described pK18mTElacZ-K18i6x7pA construct (16) (a kind gift from Jim Hu, Hospital for Sick Children, Toronto, Canada) was then replaced by the hACE2 coding sequence to create pK18-hACE2. 5′ of the hACE2 coding sequence, this plasmid contains 2.5 kb of upstream genomic sequence, the promoter, and the first intron (with a mutation in the 3′ splice acceptor site to reduce exon skipping) of the human cytokeratin 18 (K18) gene as well as a translational enhancer sequence from alfalfa mosaic virus. Downstream of the hACE2 coding sequence are exon 6, intron 6, exon 7, and the poly(A) signal of the human K18 gene. These elements were found to be necessary for high-level expression and epithelial cell specificity (4, 16). The purified 6.8-kb DNA fragment generated from an HpaI and XbaI double digest of pK18-hACE2 was used as the transgene for injection into pronuclei of fertilized (C57BL/6J × SJL/J)F2 mouse eggs to generate transgenic embryos. Mice used in this study were backcrossed two to three times onto a C57BL/6 background. Tail DNA was obtained from mice using an Extract-N-Amp tissue PCR kit (Sigma-Aldrich, St. Louis, MO). Mice transgenic for hACE2 expression were detected by PCR analysis using forward primer ACCTGGCTGAAAGACCAGAACAAG and reverse primer AATTAGCCACTCGCACATCC.
FIG. 1.

Generation and characterization of K18-hACE2 mice. (A) The hACE2 coding sequence (CDS) was cloned into a construct containing 5′ and 3′ genomic regions of the human K18 gene, which had previously been shown to be necessary for driving high-level epithelial-cell-specific expression. The K18 5′ genomic region consists of a 2.5-kb upstream genomic sequence, the promoter, and the first intron of the human K18 gene, while the K18 3′ region consists of exon 6, intron 6, exon 7, and ∼300 bp of the 3′ UTR of the human K18 gene, including the K18 poly(A) signal. Immediately upstream of the hACE2 start codon is a translational enhancer (TE) sequence from alfalfa mosaic virus. (B) hACE2 cDNA copy numbers in three transgenic founder lines determined by quantitative PCR, as described in Materials and Methods.
Determination of hACE2 copy number.
Genomic DNA from each founder line of transgenic mouse and from wild-type mice was isolated from the liver using DNAzol reagent (Invitrogen, Carlsbad, CA) according to the manufacturer's instructions. One nanogram of genomic DNA and three consecutive 1:1 dilutions were used as a template for TaqMan quantitative PCR. The primers and probe specific for hACE2 are as follows: forward primer, TCCTAACCAGCCCCCTGTT; reverse primer, TGACAATGCCAACCACTATCACT; probe, ATATGGCTGATTGTTTTTGGAGTTGTGATGGG. A single-copy reference gene, mouse PKD1, was also quantified using the same templates and on the same reaction plate as hACE2. The primer and probe for mouse PKD1 are as follows: forward primer, GGCTGCTGAGCGTCTGGTA; reverse primer, CCAGGTCCTGCGTGTCTGA; probe, ATCATTGAAGGTGGCTCATACCGGGTATG. To normalize for the DNA added, a rodent glyceraldehyde-3-phosphate dehydrogenase (GAPDH) primer and probe set (Applied Biosystems) was also used in each reaction. Each genomic DNA quantitative PCR was run in duplicate in 25-μl reaction mixtures using the ABI 9700HT thermocycler. Primer and probe efficiency and compatibility were validated by relative efficiency plots of threshold cycles (CT) from each DNA dilution for both the hACE2 and mouse PKD1 genes. To determine the hACE2 gene copy number in each transgenic mouse line, the 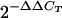 method was performed using the PKD1 gene as an endogenous calibrator (1). The
method was performed using the PKD1 gene as an endogenous calibrator (1). The 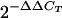 indicates the n-fold change in copy number of the hACE2 gene relative to the PKD1 gene, which is a single-copy gene in mice.
indicates the n-fold change in copy number of the hACE2 gene relative to the PKD1 gene, which is a single-copy gene in mice.
Infection with SARS-CoV.
The Urbani strain of SARS-CoV was obtained from W. Bellini and T. Ksiazek at the Centers for Disease Control, Atlanta, GA. The virus was propagated and titered on Vero E6 cells in a biosafety level 3 laboratory. The titer of virus used for all studies, as determined by a plaque assay, was 7.6 × 106 PFU/ml. Mice were lightly anesthetized with halothane and infected intranasally with the indicated dosage of SARS-CoV in 30 μl of Dulbecco's modified Eagle medium. Infected mice were examined and weighed daily. To obtain specimens for virus titers, animals were sacrificed and organs were aseptically removed into sterile phosphate-buffered saline. In some cases, blood was obtained via catheterization of the inferior vena cava. Tissues were homogenized using a manual homogenizer, and the 50% tissue culture infective dose (TCID50) was determined as described previously (36).
Whole-lung lavage.
Mice were euthanized with halothane, the chest was opened by midline incision, and lungs were lavaged in situ via PE-90 tubing inserted into the exposed trachea. Lungs were inflated with sterile saline to 25 cm H2O by adding 0.5 ml at a time (total lavage volume, approximately 4 ml). The recovered cells were pelleted, resuspended in 1 ml Hanks buffer, counted, and spun for 5 min onto a glass slide. Cells were stained with a Diff-Quik stain set using standard techniques.
Extraction of total RNA and quantitative reverse transcription-PCR (RT-PCR).
RNA was first extracted from mouse organs by using Trizol and then treated with RNase-free DNase I (Life Technologies, Gaithersburg, MD) for 30 min at 37°C. For each sample, 1 μg of total RNA was then used as a template for first-strand cDNA synthesis. The resulting cDNA was subjected to quantitative PCR amplification in the ABI 7900 sequence detection system to identify the hACE2 and rodent GAPDH amplicons and the mACE2 and rodent GAPDH amplicons in separate single reactions. Forward and reverse hACE2 and mACE2 primers and the fluorogenic TaqMan probe were designed using Primer Express software (Perkin-Elmer Applied Biosystems, Foster City, CA). Forward primers were 5′-TCCTAACCAGCCCCCTGTT-3′ for hACE2 and 5′-TCTGCCACCCCACAGCTT-3′ for mACE2; reverse primers were 5′-TGACAATGCCAACCACTATCACT-3′ for hACE2 and 5′-GGCTGTCAAGAAGTTGTCCATTG-3′ for mACE2; probes were 5′-ATATGGCTGATTGTTTTTGGAGTTGTGATGGG-3′for hACE2 and 5′-CACGGAGACTTCAGAATCAAGATC-5′for mACE2. The hACE2 and mACE2 probes are labeled with the fluorophore 6-carboxyfluorescein, and the rodent GAPDH probe is labeled with the fluorophore VIC. CT for hACE2 and mACE2 were normalized against CT for rodent GAPDH in each sample. A standard curve was generated using serial dilutions of hACE2 or mACE2 plasmids as the template in which 1 μg hACE2 plasmid DNA was determined to equal 1.2 × 1011 molecules and 1 μg mACE2 plasmid DNA was determined to equal 4.5 × 1011 molecules. The copy number for each sample was determined by using the formula extrapolated from this standard curve.
To measure levels of viral RNA, an aliquot of cDNA was subjected to PCR using a MyiQ single-color real-time PCR detection system (Bio-Rad, Hercules, CA) with iQ SYBR green supermix (Bio-Rad). The following primers were used: for the SARS-CoV nucleocapsid (N) gene, forward/leader primer 5′-ATATTAGGTTTTTACCCAGG-3′ and reverse primer 5′-CTTGCCCCATTGCGTCCTCC-3′; for human hypoxanthine phosphoribosyltransferase (HPRT), forward primer 5′-CCTCATGGACTGATTATGGAC-3′ and reverse primer 5′-CAGATTCAACTTGCGCTCATC-3′. Data were analyzed as previously described (30).
Histology and immunohistochemistry.
Organs were harvested from infected and uninfected mice and fixed in zinc formalin. For routine histology, sections were stained with hematoxylin and eosin. To detect viral antigen, sections were probed with a monoclonal antibody (MAb) to the SARS-CoV N protein (1:5,000; Zymed, San Francisco, CA) or a control immunoglobulin G2a MAb (E-Bioscience, San Diego, CA) followed by a biotinylated goat anti-mouse secondary antibody (1:200; Jackson Immunoresearch, West Grove, PA). Samples were developed by sequential incubation with a streptavidin-horseradish peroxidase conjugate (Jackson Immunoresearch) and diaminobenzidine (Sigma-Aldrich).
RNase protection assays for cytokines and chemokines.
Five micrograms of total RNA obtained from lungs or brains was analyzed by RNase protection assays using a custom set of probes purchased from BD PharMingen (San Diego, CA). After electrophoresis, gels were exposed to a phosphorimaging screen and analyzed using Bio-Rad Quantity One 4.4.0 software. Levels of RNA were normalized to those of a housekeeping gene (L32) in order to allow interanimal comparison of cytokine/chemokine mRNA levels.
Treatment with a neutralizing antibody.
In some cases, mice were treated intravenously with 25 mg of an anti-S neutralizing human MAb (MBL SARS-201; supplied by Donna Ambrosino, Massachusetts Biologic Laboratories) (10) per kg of body weight or a control humanized anti-respiratory syncytial virus MAb (palivizumab; Medimmune, Gaithersburg, MD) 1 day prior to infection.
RESULTS
Development and characterization of K18-hACE2 mice.
hACE2 is expressed in human airway and alveolar epithelia (13), and SARS-CoV infects primary airway epithelial cells in vitro (14, 34). Since studies with nonhuman primates have shown that SARS-CoV infection began in airway epithelia (25), we generated transgenic mice in which hACE2 expression was driven by the K18 promoter as described in Materials and Methods (Fig. 1A). The K18 promoter confers efficient transgene expression in airway epithelial cells (but not in alveolar epithelia), as well as in epithelia of other internal organs, including the liver, kidney, and gastrointestinal tract (4). We generated three founder lines that transmitted the hACE2 gene to their progeny. Levels of transgene DNA in founder lines ranged from 4 to 10 copies per genome as determined by quantitative PCR (Fig. 1B). We detected hACE2 mRNA in several tissues, including the lung, colon, liver, and kidney (Fig. 2A and C), whereas endogenous mACE2 was most abundantly expressed in the ileum (8). Notably, very low but measurable levels of ACE2 were detected in the brains of both non-Tg and K18-hACE2 mice (Fig. 2B and C). Using lung sections and an antibody that detects both hACE2 and mACE2 in immunofluorescence assays, we detected ACE2 in airway epithelia in both non-Tg and K18-hACE2 mice, with no obvious differences in distribution (data not shown).
FIG. 2.

hACE2 expression in K18-hACE2 mice. (A) hACE2 transgene mRNA expression levels in the indicated mouse tissues. Quantitative RT-PCR was used to determine the relative abundances of the hACE2 transgene in the tissues given along the x axis, as described in Materials and Methods. Results are means ± standard errors for 3 to 6 mice per group. (B) hACE2 transgene mRNA expression in brains of K18-hACE2 mice. Results are means ± standard errors for 3 to 6 mice per group. Note the change in scale from panel A. (C) Expression of mouse ACE2 mRNA in lungs and brains of non-Tg and K18-hACE2 transgenic mice as determined by quantitative RT-PCR. Results are means ± standard errors for 3 to 6 mice per group.
SARS-CoV-infected K18-hACE2 mice develop severe clinical disease.
It was shown previously that intranasal inoculation of BALB/c or C57BL/6 mice with SARS-CoV resulted in minimal clinical disease, although C57BL/6 mice exhibited reduced weight gain after inoculation (9, 36). In agreement with these data, infection of non-Tg littermates resulted in no mortality or clinical disease, and mice gained weight over the course of the experiment (Fig. 3A and B). In marked contrast, K18-hACE2 mice inoculated intranasally with SARS-CoV began to lose weight by days 3 to 5 postinfection (p.i.). Concomitant with weight loss, mice became lethargic, with labored breathing. As shown in Fig. 3A, mice from all founder lines were dead by day 7 p.i., and nearly all mice from lines 1 and 2 were moribund by 4 days p.i. As noted above, mice from lines 1 and 2 contained the greatest number of hACE2 transgene copies. Since line 1 and line 2 mice exhibited nearly identical phenotypes, we used line 2 mice as representative of this more susceptible phenotype in the studies reported here.
FIG. 3.
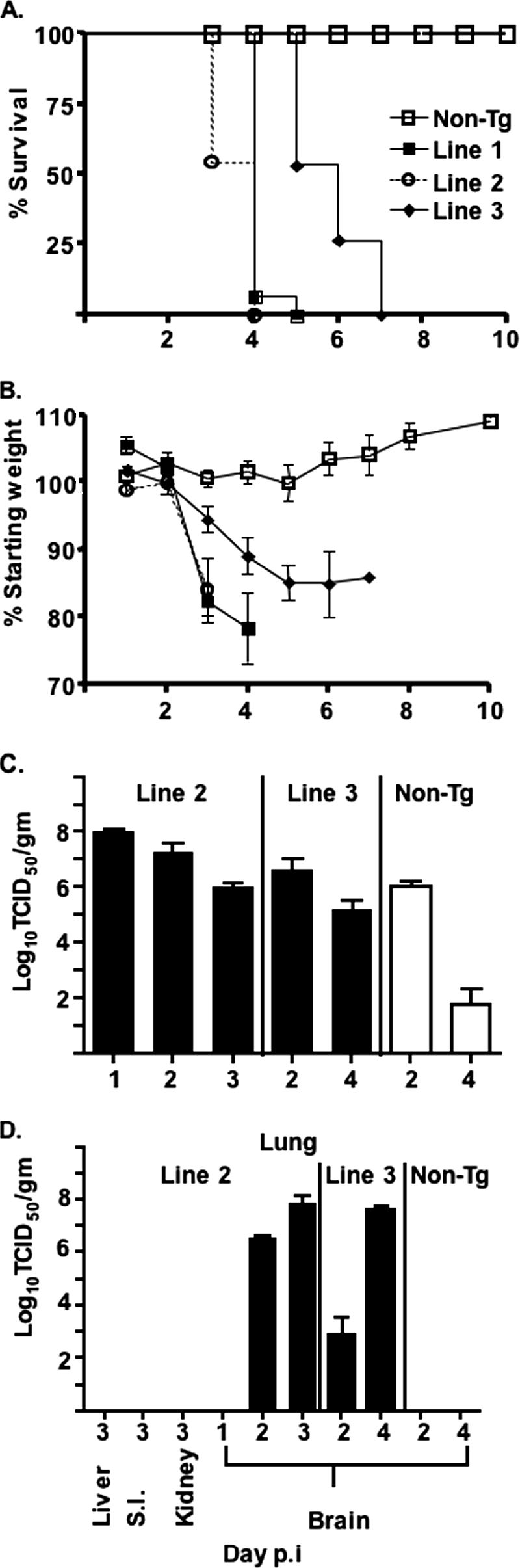
SARS-CoV causes lethal disease in K18-hACE2 mice. (A and B) K18-hACE2 mice (lines 1 [n = 15], 2 [n = 11], and 3 [n = 15]) and 15 non-Tg mice were infected intranasally with 2.3 × 104 PFU of SARS-CoV and were monitored daily for mortality (A) and weight (B). (C and D) Tissues were harvested from infected mice and assayed for infectious virus as described in Materials and Methods. Virus was detected only in the brains and lungs of K18-hACE2 mice and only in the lungs of non-Tg mice. Tissues from 3 to 6 mice were analyzed at each time point. Significantly more virus was detected in line 2 lungs at day 2 p.i. than in the lungs of non-Tg mice (P < 0.02). More virus was detected in line 3 lungs at day 4 p.i. (P < 0.0004), but not at day 2 p.i., than in the lungs of non-Tg mice.
Virus titers were 0.5 to 1 log unit higher in the lungs of K18-hACE2 mice than in those of their non-Tg littermates at day 2 p.i. and were higher in the lungs of K18-hACE2 mice that exhibited a more rapid disease course (line 2) than in those of mice surviving a few days longer (line 3) (Fig. 3C). While virus was partially cleared from the lungs of all mice by days 3 to 4 p.i., titers were 3 log units higher in K18-hACE2 mice than in non-Tg mice (line 3, day 4 p.i.; P < 0.0005). These results were confirmed by quantitative real-time RT-PCR, with higher levels of viral RNA present in the lung at 2 days p.i. than at 4 days p.i. (Fig. 4). Together, these data suggest that enhanced virus replication played a key role in the more severe disease observed in K18-hACE2 mice.
FIG. 4.

Quantitative RT-PCR for the N gene of SARS-CoV. RNA was prepared from the lungs and brains of K18-hACE2 (line 3) and non-Tg mice at days 2 and 4 p.i. RNA levels were detected by quantitative RT-PCR as described in Materials and Methods. Viral RNA levels parallel levels of infectious virus (Fig. 1). RNAs from six mice were analyzed in all groups, except that three brains from each group were analyzed at day 2. Significantly more viral RNA was detected in K18-hACE2 lungs than in non-Tg lungs at days 2 and 4. For K18-hACE2 lungs, significantly more viral RNA was detected at 2 days p.i. than at 4 days p.i. (P < 0.005).
Although the K18 promoter is active in the epithelia of multiple organs, virus was not detected in the liver, kidney, or small intestine (ileum) at either 2 or 4 days p.i. (Fig. 3D). We also analyzed the brain for evidence of SARS-CoV, since the virus has been detected in patient brains in some studies (5, 11, 42) (Fig. 3D). Virus was never detected in the brains of non-Tg mice at days 2 to 4 p.i. In line 2 mice, virus was not detected in the brain at day 1 p.i. but was present at 2 days p.i. and was present at very high levels by 3 days p.i. Virus was also detected at low levels on day 2 and at high levels on day 4 p.i. in the brains of line 3 mice, even though levels of hACE2 mRNA in the brains of these mice were barely above background (Fig. 2B). Levels of viral RNA in the brain also increased dramatically from day 2 to day 4 p.i. (Fig. 4). Of note, SARS-CoV infects the brains of C57BL/6 mice at later times (9 days) p.i. (9), showing that the central nervous system (CNS) is a secondary site of infection even in non-Tg mice.
Mice were inoculated intranasally with 2.3 × 104 PFU in these initial experiments. In subsequent experiments, we showed that virus was lethal at even lower dosages, since 3/3 and 5/6 mice (line 2) died after infection with 2.3 × 103 PFU and 2.3 × 102 PFU, respectively. Thus, the 50% lethal dose of SARS-CoV for K18-hACE2 mice was less than 230 PFU after intranasal inoculation. SARS-CoV was not transmitted from moribund mice to uninfected K18-hACE2 mice (n = 4) housed in the same cages. This was not surprising, however, since mice do not cough or sneeze, and virus was not detected in the gastrointestinal tract or kidney.
Inflammatory changes and viral antigen in the lungs and brains of K18-hACE2 mice infected with SARS-CoV.
To better understand the pulmonary lesions associated with the virulent phenotype of K18-hACE2 mice, we performed histologic analysis of the lungs. At day 2 p.i., both nontransgenic and K18-hACE2 mice showed evidence of perivascular and peribronchiolar inflammation (Fig. 5C and D). We observed more-widespread inflammatory cell infiltrates, increased inflammatory cell margination through vessels, more epithelial cell sloughing, and more signs of lung injury in infected K18-hACE2 mice (Fig. 5D) compared to their nontransgenic littermates (Fig. 5C). Staining for viral antigen revealed similar localization of SARS-CoV in the airway epithelia of the two groups of mice (Fig. 5E and F). By day 4 p.i., nontransgenic mice showed near-complete resolution of the pulmonary findings, with minimal evidence of inflammatory changes (Fig. 5I). In contrast, K18-hACE2 mice showed continued perivascular and peribronchial inflammation, hemorrhage, and congestion of alveolar septa (Fig. 5J). Staining for viral antigen was negative for both infected K18-hACE2 and non-Tg mice at day 4 p.i. (data not shown). These findings for the K18-hACE2 mouse share some features with the pulmonary lesions described for SARS patients, including modest mixed inflammatory cell infiltrates (11, 39), the detection of virus in conducting airway epithelia (11), alveolar septal thickening (39), and epithelial shedding and proliferation (7, 27). We saw no evidence of diffuse alveolar damage or acute respiratory distress syndrome, but it should be noted that patients with such findings commonly received assisted ventilation and supplemental oxygen, which complicate postmortem pulmonary findings.
FIG. 5.

Pulmonary disease in SARS-CoV-infected K18-hACE2 and non-Tg mice. (A through J and L) K18-hACE2 and non-Tg mice were either left uninfected (A and B) or infected with 2.3 × 104 PFU of SARS-CoV. Lungs were fixed in zinc formalin and stained with hematoxylin and eosin. Non-Tg mice showed mild perivascular and peribronchiolar inflammation in response to SARS-CoV 2 (C) and 4 (I) days following infection. K18-hACE2 mice demonstrated more-extensive disease 2 days following infection, characterized by epithelial sloughing (D, arrowheads) and more-extensive areas of mixed inflammatory cell infiltrates within and around airways, blood vessels, and the alveolar parenchyma. At day 2 p.i., viral antigen was localized to conducting airway epithelia in non-Tg (E) and K18-hACE2 (F) mice. Cells recovered from BAL specimens of infected K18-hACE2 mice (H) included macrophages with more vacuoles, consistent with activation, as well as enhanced neutrophilia and lymphocytosis compared to non-Tg mice (G). By 4 days p.i., inflammation in infected non-Tg lungs was resolving (I), while perivascular and peribronchiolar infiltrates and hemorrhage (arrowhead) were detected in K18-hACE2 mice (J). In some animals, bronchioles were completely occluded by neutrophils with marked intra-alveolar edema and without vasculitis (L), consistent with aspiration. BAL specimens were obtained from uninfected and infected K18-hACE2 and non-Tg mice and results pooled for 3 and 4 days p.i. Bars, 50 μm. (K) BAL analysis. Means (standard errors) are shown. K18-hACE2 Tg mice exhibited increased numbers of lymphocytic and neutrophilic cells in BAL specimens compared to non-Tg mice. n ≥ 6 for all conditions except for naïve non-Tg mice (n = 3). *, P < 0.05 for comparison to naïve mice. Mac, macrophages; L, lymphocytes; PMN, neutrophils.
In addition to the findings described above, patchy, intense neutrophilic infiltrates were noted in the lungs of some K18-hACE2 mice (Fig. 5L). These lesions obstructed the bronchioles with degenerate neutrophil aggregates and were associated with foci of necrotizing bronchopneumonia and alveolar flooding with seroproteinaceous fluid. In some areas, the neutrophilic inflammation was centered on foreign material (identical to esophageal contents), consistent with aspiration pneumonia (data not shown). We suspect that these aspiration events are neurogenic in nature, a consequence of pharyngeal and laryngeal dysfunction that may occur secondary to the spread of the virus to the CNS. Aspiration pneumonia has also been noted in mouse models of influenza infection (33) and occasionally for patients with SARS (26).
To characterize the inflammatory cell infiltrates observed in infected lungs, we obtained bronchoalveolar lavage (BAL) specimens from K18-hACE2 and non-Tg mice as described in Materials and Methods (Fig. 5G, H, and K). Total cell numbers were increased in lavage fluid from infected K18-hACE2 and non-Tg mice, and, as with SARS-CoV-infected patients (7, 26, 27), large numbers of macrophages were recovered in BAL specimens from infected mice. Macrophages from infected K18-hACE2 mice were larger than non-Tg macrophages and contained more vacuoles and cell debris in their cytoplasm, consistent with activation (Fig. 5G and H). Of note, we detected greater numbers of lymphocytes in BAL samples obtained from K18-hACE2 mice than in those from non-Tg mice. Low levels of neutrophils were also present in infected K18-hACE2 mice; neutrophils were not detected in infected human lungs, but no tissue samples were obtained prior to 5 day p.i. in any published report.
In agreement with the high levels of virus assayed in the brains of infected K18-hACE2 mice at day 4 p.i., we also detected viral antigen in large numbers of neurons throughout the cerebrum, thalamus, and brainstem, with relative sparing of the olfactory bulb and cerebellum (Fig. 6A and C). Although cytokeratin 18 is an epithelial cell protein, K18-based expression of a LacZ reporter in cortical and brainstem neurons has been reported previously (4). Infection of the CNS was accompanied by relatively minimal meningeal and perivascular infiltration (Fig. 6B), suggesting that mice died prior to a substantial cellular host immune response in the brain. No virus antigen was detected in the brains of non-Tg mice at day 4 p.i. (Fig. 6E) or in those of any mice at day 2 p.i.
FIG. 6.

SARS-CoV infects the brains of K18-hACE2 but not non-Tg mice. Brains were harvested from infected K18-hACE2 (A to C) and non-Tg (D and E) mice and stained with hematoxylin and eosin (B and D) or for virus antigen (A, C, and E). (A) Virus (brown) is detected in large numbers of cells in the cerebrum (C), thalamus, and brainstem but not in the olfactory bulb (OB) or cerebellum (Ce). Brainstem (B) and cerebellum (Ce) tissues are shown in panels B to E. (B and D) Little inflammation is present in the brains of infected non-Tg or K18-hACE2 mice. (C) Extensive infection of neurons is detected in the brainstem but not in the adjacent cerebellum. (E) No antigen labeling is detected in the brains of non-Tg mice. Bar, 100 μm.
Upregulation of proinflammatory cytokines and chemokines in SARS-CoV-infected K18-hACE2 mice.
Elevated levels of several cytokines and chemokines, including interleukin-1 (IL-1), IL-6, IL-12, CXCL8, CXCL10, and CCL2, were detected in the serum of SARS patients and may have contributed to clinical disease (15, 38, 40, 41, 43). Similarly, levels of several proinflammatory cytokine and chemokine mRNAs, including gamma interferon (IFN-γ), CXCL9, CXCL10, CCL2, and CCL7, were elevated in the lungs of K18-hACE2 mice and, to a lesser extent, in those of non-Tg mice at 2 days p.i.; in parallel with virus levels, levels of these cytokine and chemokine mRNAs were greatly diminished by 4 days p.i. (Fig. 7A and B). Conversely, no cytokine or chemokine mRNAs were elevated in the brain at day 2 p.i., but several, most notably IL-6, IFN-γ, CCL2, and CCL12, were detected at high levels in the infected K18-hACE2 CNS at day 4 p.i. (Fig. 7C). Remarkably, no IFN-α/β mRNA was detected in infected lungs, and only low levels of IFN-β mRNA were detected in the brain, consistent with the observation that SARS-CoV does not induce type 1 IFN in fibroblasts, macrophages, or dendritic cells (3, 20, 35).
FIG. 7.

Detection of proinflammatory cytokine and chemokine mRNAs in the lungs and brains of infected K18-hACE2 and non-Tg mice. Infected K18-hACE2 (line 3) and non-Tg mice were sacrificed at day 2 p.i. (6 mice each) (A) and day 4 p.i. (6 mice each) (B and C). RNAs were prepared from lungs (A and B) and brains (C) and assayed for cytokine and chemokine mRNA levels by using an RNase protection assay as described in Materials and Methods. Data are shown as levels of RNA normalized to the level of a housekeeping gene (L32). (A and B) There were significant differences (P < 0.05) in pulmonary mRNA levels of CCL7, CCL12, CXCL10, and IL-12p35 between K18-hACE2 and non-Tg mice at day 2 (A). Differences in levels of tumor necrosis factor alpha and IL-6 were nearly significant (P < 0.06). At day 4 p.i., there was a significant difference in IL-1β levels between K18-hACE2 and non-Tg mice (B). There was a statistically significant decrease (P < 0.05) in the levels of all cytokines and chemokines when infected K18-hACE2 mice at days 2 and 4 p.i. were compared, except for CXCL10 (P = 0.06) and IL-1β (P = 0.47). IL-2, IL-4, IL-10, IL-12p40, IFN-β, IFN-α, CCL3, and CXCL2 (MIP-2) were not detected in lungs. (C) Levels of all cytokine and chemokine mRNAs in infected K18-hACE2 and non-Tg brains were indistinguishable from those of naïve brains at day 2 p.i. (data not shown). By day 4 p.i., all cytokine and chemokine mRNA levels were statistically higher in K18-hACE2 mice than in non-Tg mice (P < 0.02). IL-2, IL-4, IL-10, IFN-α, CCL3, and CXCL2 were not detected in brains. Stippled bars, naive mice; open bars, non-Tg mice; solid bars, K18-hACE2 mice.
Pretreatment of K18-hACE2 mice with a human anti-SARS-CoV MAb prevents clinical disease.
To determine whether K18-hACE2 mice will be useful for evaluating anti-SARS-CoV therapy, animals were treated, as proof of principle, with a human MAb that binds to the hACE2 receptor binding domain of the SARS-CoV surface glycoprotein (MAb 201). MAb 201 has previously been shown to diminish virus replication and the severity of pathological changes in SARS-CoV-infected mice and hamsters (10, 31). Intravenous administration of MAb 201 (25 mg/kg of body weight), but not of a control antibody, to K18-hACE2 mice 1 day prior to SARS-CoV infection completely prevented death (Fig. 8), clinical disease, and weight loss (data not shown).
FIG. 8.
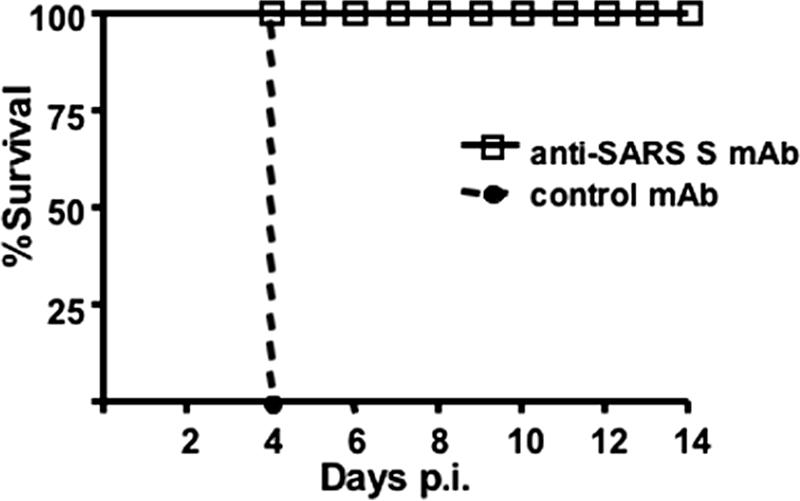
Treatment with an anti-SARS-CoV neutralizing antibody protects K18-hACE2 mice against clinical disease. K18-hACE2 mice (line 2) received 25 mg of MAb 201 (9 mice) or a control antibody (7 mice)/kg 1 day prior to infection with 2.3 × 104 PFU of SARS-CoV. Mice were monitored for survival and weight loss. All infected K18-hACE2 mice that received MAb 201 survived and exhibited no weight loss (data not shown).
DISCUSSION
We show here that transgenic expression of hACE2 behind an epithelial cell-specific promoter, with no other modifications of the virus or murine host, is sufficient to convert a mild infection with SARS-CoV to a lethal infection. This finding contrasts markedly with the findings of a study of mice infected with another strain of human coronavirus, HCoV-229E, in which only immunodeficient (STAT1−/−) mice transgenically expressing the human receptor developed mild disease, and even this required the use of a mouse-adapted strain of the virus (18). Our results are consistent with the notion that SARS-CoV, unlike HCoV-229E, has the ability to “jump” species, requiring adaptation to the host ACE2 receptor for enhanced virus replication. Since we could not detect ACE2 expression in novel sites in the lungs of K18-hACE2 mice, we favor the conclusion that hACE2 facilitates more efficient viral entry into, and replication in, the airways, resulting in a prolonged infection and a greater virus burden (Fig. 3C).
Like findings for infected nonhuman primates (25), our results suggest that SARS-CoV infection of K18-hACE2 mice began in airway epithelial cells. Viral antigen is detected primarily in pneumocytes and alveolar macrophages in human autopsy material (11, 26). However, this observation may reflect the difficulty of obtaining specimens from patients with SARS at early times; virus may be cleared from infected airways by the time that samples are obtained. Enhanced virus replication in airway cells results in increased inflammatory cell infiltration, more epithelial cell sloughing and proliferation (consistent with repair), and augmented cytokine and chemokine production in the lungs of K18-hACE2 mice compared to non-Tg mice (Fig. 5D and J and 7A and B).
One consequence of elevated cytokine and chemokine production is increased blood-brain barrier permeability, which likely facilitated widespread entry of SARS-CoV into the K18-hACE2 CNS. In agreement with this hypothesis, we could detect low levels of virus in the blood of some mice (titers, <500 TCID50/ml; two, two, and none of three mice were positive at days 1, 2, and 3 p.i., respectively). Neurotropic coronaviruses generally enter the CNS via olfactory neurons, with subsequent transneuronal spread to other sites within the brain (2). However, it is unlikely that SARS-CoV spread to the brain via the olfactory system, because we detected no virus in the olfactory bulb (Fig. 6A). Further, virus was detected throughout the brain, without preferential infection of sites transneuronally connected to the olfactory bulb.
Our results show that the CNS is an important target for SARS-CoV in K18-hACE2 mice, even in line 3 mice, which express very low levels of hACE2 in the brain (Fig. 2C). While the CNS is not considered a major site of infection in infected humans, SARS-CoV has been detected in the brains of infected patients (5, 11, 42). Also, some SARS survivors have neurological/psychological sequelae that are not well understood (21, 42); our results support the notion that direct virus infection contributes to the CNS dysfunction that is observed in these patients. Disease in the K18-hACE2 CNS may also be partially immunopathological, as evidenced by the high levels of proinflammatory cytokines and chemokines detected in infected brains (Fig. 7C). While patients with SARS generally die from pulmonary failure, it is likely that infection of the CNS is a major factor contributing to the fatal outcome observed for SARS-CoV-infected K18-hACE2 mice.
We anticipate that these mice will be useful for studies of pathogenesis, especially for examining the role of proinflammatory chemokines and cytokines in pulmonary and CNS disease and the basis of viremia and extrapulmonary spread. Furthermore, the lack of cellular infiltration into the CNS is unexpected, given the levels of SARS-CoV (Fig. 3D, 4, and 6), raising the possibility that the virus specifically inhibits inflammatory cell migration into this organ. In addition to their utility in studies of SARS pathogenesis, K18-hACE2 mice will also be very useful in vaccine and other therapeutic studies, especially those directed against human strains of SARS-CoV. We have shown that treatment with a fully human anti-SARS-CoV neutralizing MAb 1 day prior to infection prevented clinical disease. These results suggest that treatment of exposed persons with this antibody could be completely protective, even against severe disease.
Acknowledgments
We thank Michael Welsh, John Harty, Beverly Davidson, Noah Butler, and Tony Fischer for careful review of the manuscript. We thank Jim Hu for providing the K18 plasmid construct and Donna Ambrosino for supplying the anti-SARS-CoV MAb 201. We thank Jian Shao and Jan Janssen for technical advice and assistance.
We acknowledge the support of the NIH (PO1 AI060699-02). We also acknowledge the support of the Cell Morphology Core, partially supported by the Center for Gene Therapy for Cystic Fibrosis (NIH P30 DK-54759) and the Cystic Fibrosis Foundation, and that of the Transgenic Mouse Facility, supported in part by the College of Medicine and the Center for Gene Therapy for Cystic Fibrosis.
Footnotes
Published ahead of print on 1 November 2006.
REFERENCES
- 1.Ballester, M., A. Castello, E. Ibanez, A. Sanchez, and J. M. Folch. 2004. Real-time quantitative PCR-based system for determining transgene copy number in transgenic animals. BioTechniques 37:610-613. [DOI] [PubMed] [Google Scholar]
- 2.Barnett, E., M. Cassell, and S. Perlman. 1993. Two neurotropic viruses, herpes simplex virus type I and mouse hepatitis virus, spread along different neural pathways from the main olfactory bulb. Neuroscience 57:1007-1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cheung, C. Y., L. L. Poon, I. H. Ng, W. Luk, S. F. Sia, M. H. Wu, K. H. Chan, K. Y. Yuen, S. Gordon, Y. Guan, and J. S. Peiris. 2005. Cytokine responses in severe acute respiratory syndrome coronavirus-infected macrophages in vitro: possible relevance to pathogenesis. J. Virol. 79:7819-7826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chow, Y. H., H. O'Brodovich, J. Plumb, Y. Wen, K. J. Sohn, Z. Lu, F. Zhang, G. L. Lukacs, A. K. Tanswell, C. C. Hui, M. Buchwald, and J. Hu. 1997. Development of an epithelium-specific expression cassette with human DNA regulatory elements for transgene expression in lung airways. Proc. Natl. Acad. Sci. USA 94:14695-14700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ding, Y., L. He, Q. Zhang, Z. Huang, X. Che, J. Hou, H. Wang, H. Shen, L. Qiu, Z. Li, J. Geng, J. Cai, H. Han, X. Li, W. Kang, D. Weng, P. Liang, and S. Jiang. 2004. Organ distribution of severe acute respiratory syndrome (SARS) associated coronavirus (SARS-CoV) in SARS patients: implications for pathogenesis and virus transmission pathways. J. Pathol. 203:622-630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Drosten, C., S. Gunther, W. Preiser, S. van der Werf, H. R. Brodt, S. Becker, H. Rabenau, M. Panning, L. Kolesnikova, R. A. Fouchier, A. Berger, A. M. Burguiere, J. Cinatl, M. Eickmann, N. Escriou, K. Grywna, S. Kramme, J. C. Manuguerra, S. Muller, V. Rickerts, M. Sturmer, S. Vieth, H. D. Klenk, A. D. Osterhaus, H. Schmitz, and H. W. Doerr. 2003. Identification of a novel coronavirus in patients with severe acute respiratory syndrome. N. Engl. J. Med. 348:1967-1976. [DOI] [PubMed] [Google Scholar]
- 7.Franks, T. J., P. Y. Chong, P. Chui, J. R. Galvin, R. M. Lourens, A. H. Reid, E. Selbs, C. P. McEvoy, C. D. Hayden, J. Fukuoka, J. K. Taubenberger, and W. D. Travis. 2003. Lung pathology of severe acute respiratory syndrome (SARS): a study of 8 autopsy cases from Singapore. Hum. Pathol. 34:743-748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gembardt, F., A. Sterner-Kock, H. Imboden, M. Spalteholz, F. Reibitz, H. P. Schultheiss, W. E. Siems, and T. Walther. 2005. Organ-specific distribution of ACE2 mRNA and correlating peptidase activity in rodents. Peptides 26:1270-1277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Glass, W. G., K. Subbarao, B. Murphy, and P. M. Murphy. 2004. Mechanisms of host defense following severe acute respiratory syndrome-coronavirus (SARS-CoV) pulmonary infection of mice. J. Immunol. 173:4030-4039. [DOI] [PubMed] [Google Scholar]
- 10.Greenough, T. C., G. J. Babcock, A. Roberts, H. J. Hernandez, W. D. Thomas, Jr., J. A. Coccia, R. F. Graziano, M. Srinivasan, I. Lowy, R. W. Finberg, K. Subbarao, L. Vogel, M. Somasundaran, K. Luzuriaga, J. L. Sullivan, and D. M. Ambrosino. 2005. Development and characterization of a severe acute respiratory syndrome-associated coronavirus-neutralizing human monoclonal antibody that provides effective immunoprophylaxis in mice. J. Infect. Dis. 191:507-514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gu, J., E. Gong, B. Zhang, J. Zheng, Z. Gao, Y. Zhong, W. Zou, J. Zhan, S. Wang, Z. Xie, H. Zhuang, B. Wu, H. Zhong, H. Shao, W. Fang, D. Gao, F. Pei, X. Li, Z. He, D. Xu, X. Shi, V. M. Anderson, and A. S. Leong. 2005. Multiple organ infection and the pathogenesis of SARS. J. Exp. Med. 202:415-424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Guan, Y., B. J. Zheng, Y. Q. He, X. L. Liu, Z. X. Zhuang, C. L. Cheung, S. W. Luo, P. H. Li, L. J. Zhang, Y. J. Guan, K. M. Butt, K. L. Wong, K. W. Chan, W. Lim, K. F. Shortridge, K. Y. Yuen, J. S. Peiris, and L. L. Poon. 2003. Isolation and characterization of viruses related to the SARS coronavirus from animals in southern China. Science 302:276-278. [DOI] [PubMed] [Google Scholar]
- 13.Hamming, I., W. Timens, M. L. Bulthuis, A. T. Lely, G. J. Navis, and H. van Goor. 2004. Tissue distribution of ACE2 protein, the functional receptor for SARS coronavirus. A first step in understanding SARS pathogenesis. J. Pathol. 203:631-637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jia, H. P., D. C. Look, L. Shi, M. Hickey, L. Pewe, J. Netland, M. Farzan, C. Wohlford-Lenane, S. Perlman, and P. B. McCray, Jr. 2005. ACE2 receptor expression and severe acute respiratory syndrome coronavirus infection depend on differentiation of human airway epithelia. J. Virol. 79:14614-14621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jiang, Y., J. Xu, C. Zhou, Z. Wu, S. Zhong, J. Liu, W. Luo, T. Chen, Q. Qin, and P. Deng. 2005. Characterization of cytokine/chemokine profiles of severe acute respiratory syndrome. Am. J. Respir. Crit. Care Med. 171:850-857. [DOI] [PubMed] [Google Scholar]
- 16.Koehler, D. R., Y. H. Chow, J. Plumb, Y. Wen, B. Rafii, R. Belcastro, M. Haardt, G. L. Lukacs, M. Post, A. K. Tanswell, and J. Hu. 2000. A human epithelium-specific vector optimized in rat pneumocytes for lung gene therapy. Pediatr. Res. 48:184-190. [DOI] [PubMed] [Google Scholar]
- 17.Ksiazek, T. G., D. Erdman, C. S. Goldsmith, S. R. Zaki, T. Peret, S. Emery, S. Tong, C. Urbani, J. A. Comer, W. Lim, P. E. Rollin, S. F. Dowell, A. E. Ling, C. D. Humphrey, W. J. Shieh, J. Guarner, C. D. Paddock, P. Rota, B. Fields, J. DeRisi, J. Y. Yang, N. Cox, J. M. Hughes, J. W. LeDuc, W. J. Bellini, and L. J. Anderson. 2003. A novel coronavirus associated with severe acute respiratory syndrome. N. Engl. J. Med. 348:1953-1966. [DOI] [PubMed] [Google Scholar]
- 18.Lassnig, C., C. M. Sanchez, M. Egerbacher, I. Walter, S. Majer, T. Kolbe, P. Pallares, L. Enjuanes, and M. Muller. 2005. Development of a transgenic mouse model susceptible to human coronavirus 229E. Proc. Natl. Acad. Sci. USA 102:8275-8280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lau, S. K., P. C. Woo, K. S. Li, Y. Huang, H. W. Tsoi, B. H. Wong, S. S. Wong, S. Y. Leung, K. H. Chan, and K. Y. Yuen. 2005. Severe acute respiratory syndrome coronavirus-like virus in Chinese horseshoe bats. Proc. Natl. Acad. Sci. USA 102:14040-14045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Law, H. K., C. Y. Cheung, H. Y. Ng, S. F. Sia, Y. O. Chan, W. Luk, J. M. Nicholls, J. S. Peiris, and Y. L. Lau. 2005. Chemokine upregulation in SARS coronavirus infected human monocyte derived dendritic cells. Blood 106:2366-2376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lee, D. T., Y. K. Wing, H. C. Leung, J. J. Sung, Y. K. Ng, G. C. Yiu, R. Y. Chen, and H. F. Chiu. 2004. Factors associated with psychosis among patients with severe acute respiratory syndrome: a case-control study. Clin. Infect. Dis. 39:1247-1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Li, W., T. C. Greenough, M. J. Moore, N. Vasilieva, M. Somasundaran, J. L. Sullivan, M. Farzan, and H. Choe. 2004. Efficient replication of severe acute respiratory syndrome coronavirus in mouse cells is limited by murine angiotensin-converting enzyme 2. J. Virol. 78:11429-11433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li, W., M. J. Moore, N. Vasilieva, J. Sui, S. K. Wong, M. A. Berne, M. Somasundaran, J. L. Sullivan, K. Luzuriaga, T. C. Greenough, H. Choe, and M. Farzan. 2003. Angiotensin-converting enzyme 2 is a functional receptor for the SARS coronavirus. Nature 426:450-454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Li, W., Z. Shi, M. Yu, W. Ren, C. Smith, J. H. Epstein, H. Wang, G. Crameri, Z. Hu, H. Zhang, J. Zhang, J. McEachern, H. Field, P. Daszak, B. T. Eaton, S. Zhang, and L. F. Wang. 2005. Bats are natural reservoirs of SARS-like coronaviruses. Science 310:676-679. [DOI] [PubMed] [Google Scholar]
- 25.McAuliffe, J., L. Vogel, A. Roberts, G. Fahle, S. Fischer, W. J. Shieh, E. Butler, S. Zaki, M. St Claire, B. Murphy, and K. Subbarao. 2004. Replication of SARS coronavirus administered into the respiratory tract of African green, rhesus and cynomolgus monkeys. Virology 330:8-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nicholls, J. M., J. Butany, L. L. Poon, K. H. Chan, S. L. Beh, S. Poutanen, J. S. Peiris, and M. Wong. 2006. Time course and cellular localization of SARS-CoV nucleoprotein and RNA in lungs from fatal cases of SARS. PLoS Med. 3:e27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nicholls, J. M., L. L. Poon, K. C. Lee, W. F. Ng, S. T. Lai, C. Y. Leung, C. M. Chu, P. K. Hui, K. L. Mak, W. Lim, K. W. Yan, K. H. Chan, N. C. Tsang, Y. Guan, K. Y. Yuen, and J. S. Peiris. 2003. Lung pathology of fatal severe acute respiratory syndrome. Lancet 361:1773-1778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Peiris, J. S., Y. Guan, and K. Y. Yuen. 2004. Severe acute respiratory syndrome. Nat. Med. 10:S88-S97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Peiris, J. S., S. T. Lai, L. L. Poon, Y. Guan, L. Y. Yam, W. Lim, J. Nicholls, W. K. Yee, W. W. Yan, M. T. Cheung, V. C. Cheng, K. H. Chan, D. N. Tsang, R. W. Yung, T. K. Ng, and K. Y. Yuen. 2003. Coronavirus as a possible cause of severe acute respiratory syndrome. Lancet 361:1319-1325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pewe, L., H. Zhou, J. Netland, C. Tangadu, H. Olivares, L. Shi, D. Look, T. M. Gallagher, and S. Perlman. 2005. A severe acute respiratory syndrome-associated coronavirus-specific protein enhances virulence of an attenuated murine coronavirus. J. Virol. 79:11335-11342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Roberts, A., W. D. Thomas, J. Guarner, E. W. Lamirande, G. J. Babcock, T. C. Greenough, L. Vogel, N. Hayes, J. L. Sullivan, S. Zaki, K. Subbarao, and D. M. Ambrosino. 2006. Therapy with a severe acute respiratory syndrome-associated coronavirus-neutralizing human monoclonal antibody reduces disease severity and viral burden in golden Syrian hamsters. J. Infect. Dis. 193:685-692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rota, P. A., M. S. Oberste, S. S. Monroe, W. A. Nix, R. Campagnoli, J. P. Icenogle, S. Penaranda, B. Bankamp, K. Maher, M. H. Chen, S. Tong, A. Tamin, L. Lowe, M. Frace, J. L. DeRisi, Q. Chen, D. Wang, D. D. Erdman, T. C. Peret, C. Burns, T. G. Ksiazek, P. E. Rollin, A. Sanchez, S. Liffick, B. Holloway, J. Limor, K. McCaustland, M. Olsen-Rasmussen, R. Fouchier, S. Gunther, A. D. Osterhaus, C. Drosten, M. A. Pallansch, L. J. Anderson, and W. J. Bellini. 2003. Characterization of a novel coronavirus associated with severe acute respiratory syndrome. Science 300:1394-1399. [DOI] [PubMed] [Google Scholar]
- 33.Shinya, K., A. Suto, M. Kawakami, H. Sakamoto, T. Umemura, Y. Kawaoka, N. Kasai, and T. Ito. 2005. Neurovirulence of H7N7 influenza A virus: brain stem encephalitis accompanied with aspiration pneumonia in mice. Arch. Virol. 150:1653-1660. [DOI] [PubMed] [Google Scholar]
- 34.Sims, A. C., R. S. Baric, B. Yount, S. E. Burkett, P. L. Collins, and R. J. Pickles. 2005. Severe acute respiratory syndrome coronavirus infection of human ciliated airway epithelia: role of ciliated cells in viral spread in the conducting airways of the lungs. J. Virol. 79:15511-15524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Spiegel, M., A. Pichlmair, L. Martinez-Sobrido, J. Cros, A. Garcia-Sastre, O. Haller, and F. Weber. 2005. Inhibition of beta interferon induction by severe acute respiratory syndrome coronavirus suggests a two-step model for activation of interferon regulatory factor 3. J. Virol. 79:2079-2086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Subbarao, K., J. McAuliffe, L. Vogel, G. Fahle, S. Fischer, K. Tatti, M. Packard, W. J. Shieh, S. Zaki, and B. Murphy. 2004. Prior infection and passive transfer of neutralizing antibody prevent replication of severe acute respiratory syndrome coronavirus in the respiratory tract of mice. J. Virol. 78:3572-3577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Subbarao, K., and A. Roberts. 2006. Is there an ideal animal model for SARS? Trends Microbiol. 14:299-303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tang, N. L., P. K. Chan, C. K. Wong, K. F. To, A. K. Wu, Y. M. Sung, D. S. Hui, J. J. Sung, and C. W. Lam. 2005. Early enhanced expression of interferon-inducible protein-10 (CXCL-10) and other chemokines predicts adverse outcome in severe acute respiratory syndrome. Clin. Chem. 51:2333-2340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tsang, K. W., P. L. Ho, G. C. Ooi, W. K. Yee, T. Wang, M. Chan-Yeung, W. K. Lam, W. H. Seto, L. Y. Yam, T. M. Cheung, P. C. Wong, B. Lam, M. S. Ip, J. Chan, K. Y. Yuen, and K. N. Lai. 2003. A cluster of cases of severe acute respiratory syndrome in Hong Kong. N. Engl. J. Med. 348:1977-1985. [DOI] [PubMed] [Google Scholar]
- 40.Wang, W. K., S. Y. Chen, I. J. Liu, C. L. Kao, H. L. Chen, B. L. Chiang, J. T. Wang, W. H. Sheng, P. R. Hsueh, C. F. Yang, P. C. Yang, and S. C. Chang. 2004. Temporal relationship of viral load, ribavirin, interleukin (IL)-6, IL-8, and clinical progression in patients with severe acute respiratory syndrome. Clin. Infect. Dis. 39:1071-1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wong, C. K., C. W. Lam, A. K. Wu, W. K. Ip, N. L. Lee, I. H. Chan, L. C. Lit, D. S. Hui, M. H. Chan, S. S. Chung, and J. J. Sung. 2004. Plasma inflammatory cytokines and chemokines in severe acute respiratory syndrome. Clin. Exp. Immunol. 136:95-103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Xu, J., S. Zhong, J. Liu, L. Li, Y. Li, X. Wu, Z. Li, P. Deng, J. Zhang, N. Zhong, Y. Ding, and Y. Jiang. 2005. Detection of severe acute respiratory syndrome coronavirus in the brain: potential role of the chemokine Mig in pathogenesis. Clin. Infect. Dis. 41:1089-1096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhang, Y., J. Li, Y. Zhan, L. Wu, X. Yu, W. Zhang, L. Ye, S. Xu, R. Sun, Y. Wang, and J. Lou. 2004. Analysis of serum cytokines in patients with severe acute respiratory syndrome. Infect. Immun. 72:4410-4415. [DOI] [PMC free article] [PubMed] [Google Scholar]