

Abstract
The virion host shutoff (Vhs) protein (UL41) is a minor component of herpes simplex virus virions which, following penetration, accelerates turnover of host and viral mRNAs. Infected cells contain 58-kDa and 59.5-kDa forms of Vhs, which differ in the extent of phosphorylation, yet only a 58-kDa polypeptide is incorporated into virions. In pulse-chase experiments, the primary Vhs translation product comigrated in sodium dodecyl sulfate-polyacrylamide gel electrophoresis with the 58-kDa virion polypeptide, and could be chased to 59.5 kDa. While both 59.5-kDa and 58-kDa forms were found in nuclear and cytoplasmic fractions, the 59.5-kDa form was significantly enriched in the nucleus. Both forms were associated with intranuclear B and C capsids, yet only the 58-kDa polypeptide was found in enveloped cytoplasmic virions. A 58-kDa form, but not the 59.5-kDa form, was found in L particles, noninfectious particles that contain an envelope and tegument but no capsid. The data suggest that virions contain two populations of Vhs that are packaged by different pathways. In the first pathway, the primary translation product is processed to 59.5 kDa, is transported to the nucleus, binds intranuclear capsids, and is converted to 58 kDa at some stage prior to final envelopment. The second pathway does not involve the 59.5-kDa form or interactions between Vhs and capsids. Instead, the primary translation product is phosphorylated to the 58-kDa virion form and packaged through interactions with other tegument proteins in the cytoplasm or viral envelope proteins at the site of final envelopment.
Controls of the rate of mRNA decay play an important role in eukaryotic gene expression (3, 6-9, 47, 48, 55, 66). During lytic herpes simplex virus (HSV) infections, the half-lives of host and viral mRNAs are regulated by the HSV virion host shutoff (Vhs) protein (21, 34, 35, 52, 53, 56, 64, 73). Vhs is an endoribonuclease (12, 13, 16, 40, 41, 77, 87) that is a minor structural component of HSV virions (58, 68) and that, following release from infecting virions, accelerates the degradation of most cellular mRNAs in the cytoplasm (21, 64, 73). Following the onset of viral transcription, Vhs also accelerates the turnover of viral mRNAs (34, 35, 52, 53, 73), thereby facilitating the sequential expression of different classes of viral genes (56). Although partially purified Vhs shows some preference for A- or U-rich regions (77, 87), it is best viewed as a sequence-nonspecific endonuclease, since it cuts target RNAs at many sites (16, 77, 87). Mutations that inactivate Vhs stabilize most, if not all, viral mRNAs (34, 35, 52, 53, 73), as well as the vast majority of cellular mRNAs (21, 57, 64, 73), implying that Vhs degradation of most mRNAs is not dependent upon the recognition of a specific nucleotide sequence.
Although Vhs degrades many mRNAs, it is not totally nonselective, since in cells it targets mRNAs, as opposed to non-mRNAs (33, 52, 53, 63, 70, 73, 87), and cuts within mRNAs at preferred sites (12-14, 31, 76). For at least some mRNAs these preferred sites are in regions of translation initiation (12, 13, 31). The initial loading of Vhs onto mRNAs and targeting to regions of initiation may be due to its physical interaction with the cellular translation initiation factors eIF4H (11, 16, 19, 20), eIF4AII (20), and eIF4B (11). Of particular note, a point mutation in Vhs, which abrogates its binding to eIF4H (19), abolishes degradation of the vast majority of mRNAs that are translated by cap-dependent scanning (33-35, 52, 53, 57, 73), even though the protein retains RNase activity (41). Recently, it has been suggested that Vhs also may target cleavage sites within the 3′ untranslated regions of some mRNAs that contain AU-rich instability elements, perhaps through interactions with AU-rich instability element binding proteins (15, 75). Whether interactions with general translation factors also are required for the initial loading of Vhs onto these mRNAs remains to be determined.
The Vhs polypeptide is thought to be a component of the virion tegument (42, 58, 68). HSV is enveloped through a multistep process in which capsids acquire a primary envelope by budding through the inner nuclear membrane. This is followed by de-envelopment by fusion of the primary envelope with the outer nuclear membrane and reenvelopment of capsids by budding through membranes derived from the trans-Golgi network (5, 45, 67, 79). Nuclear and cytoplasmic sites have been proposed for the incorporation of different tegument proteins (62). Using immunogold electron microscopy, Naldinho-Souto et al. detected VP16, VP22, and VP13/14 in mature extracellular virions but only VP16 in primary enveloped virions, which had completed primary envelopment but not de-envelopment and reenvelopment (49). This suggests that VP22 and VP13/14 are acquired in the cytoplasm but that at least some molecules of VP16 are incorporated in the nucleus prior to or during primary envelopment. In contrast, VP22 and UL11 both associate with the cytoplasmic surfaces of trans-Golgi network-derived membranes, suggesting that they are incorporated into virions during final envelopment (1, 4, 38, 39). Recent data suggest that virions contain two populations of the UL17 protein (78). Some copies associate with capsids in the nucleus, while others are added through interactions with other tegument or glycoproteins in the cytoplasm or at cytoplasmic membranes. Of particular relevance to this study, Lee et al. found that infected cells contain a subpopulation of Vhs that is associated with lipid rafts (37), suggesting that at least some molecules of Vhs are incorporated into virions during final envelopment.
Earlier studies identified two forms of Vhs in infected cells; a phosphorylated 58-kDa form that comigrates on sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) with the virion polypeptide and a more highly phosphorylated 59.5-kDa form that, although present in infected cells, is missing from purified virions (58). The purpose of this study was threefold: (i) to investigate the role of the 59.5-kDa Vhs form in HSV infections, (ii) to begin to delineate the pathway(s) of Vhs packaging, and (iii) to determine whether both Vhs forms enter the packaging pathway and, if so, at which stage the 59.5-kDa form is converted to 58 kDa or lost from virions. The results suggest that virions contain two populations of Vhs: one that associates with capsids in the nucleus and another that is incorporated into virions in the cytoplasm or during final envelopment. The 59.5-kDa form is an intermediate in the nuclear pathway but plays no role in the cytoplasmic pathway.
MATERIALS AND METHODS
Cells and virus.
Procedures for the growth and maintenance of Vero and BHK-21 cells and for the growth of wild-type HSV type 1 (HSV-1), strain KOS, and Vhs mutants have been described previously (17, 53, 58). The mutant Vhs-ΔSma contains a deletion of 196 codons from the middle of the UL41 open reading frame (58). It encodes a Vhs polypeptide in which the first 147 amino acids of the wild-type protein are fused in frame to amino acids 344 through 489. The mutant polypeptide lacks mRNA-degradative activity and is not incorporated into virions. The mutant N237 contains a 26-bp polylinker with stop codons in all three reading frames inserted in the UL41 open reading frame (54). It encodes a Vhs polypeptide consisting of the first 237 amino acids of the wild-type protein followed by 6 amino acids encoded by the polylinker, and it lacks mRNA degradative activity (54).
Antibodies.
A polyclonal rabbit antiserum raised against a LacZ-Vhs fusion protein containing amino acids 239 through 489 of wild-type Vhs has been described previously (58). VP16-specific rabbit antisera were generously provided by Steve Triezenberg (22).
Plasmids.
pKOSamp contains the Vhs open reading frame from HSV-1 (KOS) cloned into pcDNA1.1amp (Invitrogen) downstream from a promoter for T7 RNA polymerase and the cytomegalovirus immediate-early promoter (17, 18).
Immunoprecipitations and Western blotting.
Infected cells were labeled with [35S]methionine or [32P]orthophosphate, and the Vhs polypeptides were analyzed by immunoprecipitation, SDS-PAGE, and autoradiography (58). Alternatively, Vhs polypeptides were analyzed by SDS-PAGE and Western blotting using an ECL Western blotting detection kit (Pierce) as previously described (58).
Phosphoamino acid analysis.
Procedures for phosphoamino acid analysis have been described previously (2). Vero cells were labeled with [32P]orthophosphate from 8 to 20 h after HSV-1 infection, at which time whole-cell lysates were prepared and the Vhs polypeptides immunoprecipitated. The 58-kDa and 59.5-kDa Vhs forms were resolved by preparative SDS-PAGE and transferred to Immobilon P membranes (Millipore Corp.). Strips containing the two forms were localized by autoradiography and excised from the membrane. The proteins were hydrolyzed by incubating the membrane strips under pressure in 6 N HCl at 110°C for 2 h. Phosphoamino acids were eluted with 30% (vol/vol) methanol and 0.1 N HCl, lyophilized, and resuspended, along with nonradioactive phosphoamino acid standards (Sigma), in pyridine acetate buffer (pH 3.5) (100:10:1,890 mixture of glacial acetic acid, pyridine, and water). Phosphoamino acids were resolved by one-dimensional (1-D) high-voltage electrophoresis on cellulose thin-layer plates (Merck), after which the plates were dried and stained with ninhydrin to visualize the standards. Radiolabeled phosphoamino acids were detected by autoradiography and identified by comparison to the stained standards.
Phosphatase treatment of Vhs.
Vero cells were labeled with [32P]orthophosphate or [35S]methionine from 3.5 to 19 h after infection. Whole-cell lysates were prepared and the Vhs polypeptides isolated by immunoprecipitation. The proteins were collected on protein A beads, after which the beads were washed five times with radioimmunoprecipitation assay buffer and three times with phosphatase buffer [40 mM piperazine-N,N′-bis(2-ethanesulfonic acid) (PIPES) (pH 6.0), 20 μg aprotinin per ml, 20 μg leupeptin per ml, and 1 mM dithiothreitol]. The beads were incubated in phosphatase buffer with or without 200 μg of potato acid phosphatase (Sigma) per ml at 37°C for 30 min (80), pelleted, resuspended in 200 μl of bead elution buffer (100 mM Tris[pH 7.0], 4% SDS, 20% glycerol, 5% β-mercaptoethanol) for every 50 μl (packed volume) of beads, and boiled for 10 min. The eluted Vhs polypeptides were analyzed by SDS-PAGE and autoradiography.
Pulse-chase analysis of the Vhs polypeptides.
Vero cells were infected with 20 PFU/cell of HSV-1 in Eagle's minimum essential medium (MEM) supplemented with 2% (vol/vol) calf serum. At 7.5 h after infection, the cells were washed two times and incubated for 30 min in MEM lacking methionine and serum. The cells were pulse-labeled for 15 min in MEM lacking cold methionine and serum but supplemented with 1 mCi of [35S]methionine per ml. The labeling medium was aspirated and the cells washed four times and incubated in chase medium consisting of MEM supplemented with 1 mM cold methionine and 1% calf serum. Whole-cell lysates were prepared after various chase intervals, and the Vhs polypeptides were analyzed by immunoprecipitation, SDS-PAGE, and autoradiography.
Cell fractionation.
Cells were fractionated 12 h after infection into cytoplasmic, nuclear, and nuclear wash fractions as described previously (71). Briefly, the cells were scraped into ice-cold phosphate-buffered saline (PBS), pelleted, resuspended in 1 ml/2 × 106 cells of RSB (10 mM Tris [pH 7.5], 10 mM NaCl, 1.5 mM MgCl2), and allowed to swell on ice for 10 min. The cells were lysed by 10 strokes of a tight fitting Dounce homogenizer, after which 0.2 volume of 60% (wt/vol) sucrose in RSB was added. Centrifugation at 2,000 rpm for 5 min in an IEC SNII centrifuge produced a supernatant that was designated the cytoplasmic fraction and a crude nuclear pellet. Nuclei were resuspended in 1 ml/2 × 106 cells of RSB containing 10% sucrose, 1% (vol/vol) Triton X-100, 0.5% (vol/vol) sodium deoxycholate, and 0.5 mM phenylmethylsulfonyl fluoride and incubated on ice for 5 min. Centrifugation for 5 min in an IEC SNII centrifuge yielded a supernatant that was designated the nuclear wash fraction and a nuclear pellet. Proteins were precipitated from the cytoplasmic and nuclear wash fractions by the addition of 9 volumes of acetone. Precipitates were resuspended and boiled in SDS sample buffer (50 mM Tris[pH 7.0], 2% SDS, 10% glycerol, 5% β-mercaptoethanol), as was the nuclear pellet. Cytoplasmic, nuclear, and nuclear wash fractions were analyzed by SDS-PAGE and Western blotting.
Purification of virions and capsids. (i) Extracellular virions.
Virions from the extracellular medium were purified by sedimentation through gradients of 10 to 30% dextran T-10 prepared in MEM containing 0.5% (wt/vol) bovine serum albumin (BSA) (32, 58). Centrifugation was in a Beckman SW41 rotor for 1 h at 29,000 rpm. Fractions containing the virion peak were pooled and diluted with 2 volumes of MEM containing 0.5% (wt/vol) BSA, and the virions were pelleted by centrifugation at 40,000 rpm in a Beckman TLA-45 rotor. Virions were resuspended in a small volume of SDS sample buffer, boiled, and analyzed by SDS-PAGE and autoradiography or Western blotting.
(ii) Enveloped cytoplasmic virions.
Vero cells were harvested 18 h after infection by scraping into ice-cold PBS, pelleted, resuspended in 2 to 3 volumes of ice-cold 10 mM NaPO4 (pH 7.4), and incubated on ice for 10 min. The cells were disrupted by 10 strokes of a tight-fitting Dounce homogenizer and the nuclei pelleted (72). The resulting cytoplasmic supernatant was layered over a 10 to 30% gradient of dextran T-10, and enveloped virions were purified as described for extracellular virions.
(iii) B and C capsids.
B and C capsids were isolated from Vero cells labeled from 6 to 15 h after infection with both [35S]methionine and [3H]thymidine. Cells were scraped into ice-cold PBS; pelleted; resuspended in 1 ml/107 cells of 10 mM Tris (pH 7.5), 150 mM NaCl, and 2 mM MgCl2; and lysed by the addition of NP-40 to 1%. After 15 min on ice, the lysate was separated by low-speed centrifugation into a nucleus-containing pellet and a cytoplasmic supernatant (81). The supernatant was discarded. The nuclei were resuspended in a small volume of TNE (20 mM Tris [pH 7.5], 300 mM NaCl, 1 mM EDTA) supplemented with 1 μg/ml of aprotinin and were disrupted by three cycles of freezing and thawing, followed by brief sonication. Nuclear lysates were clarified by low-speed centrifugation and layered over gradients of 20 to 50% sucrose in TNE (10). Centrifugation was at 24,000 rpm for 100 min in a Beckman SW41 Ti rotor. Fractions of 500 μl were collected, and 10-μl aliquots were assayed for the incorporation of [35S]methionine and [3H]thymidine into trichloroacetic acid (TCA)-precipitable material. Fractions containing the B and C capsid peaks were identified by examining the profiles of TCA-precipitable radioactivity and diluted with 750 μl of TNE, and the capsids were pelleted by centrifugation in a Beckman TLA-45 rotor at 40,000 rpm for 40 min.
Washing capsids with GuHCl.
Capsids were washed with various concentrations of guanidine hydrochloride (GuHCl) as described by Newcombe and Brown (50). Intranuclear B capsids were purified by sedimentation through two consecutive 20 to 50% sucrose gradients, pelleted from the appropriate gradient fractions, and resuspended in a small volume of TNE. A solution of 6 M GuHCl in TNE was added dropwise with constant stirring until the desired GuHCl concentration had been reached. The mixtures were incubated for 30 min at 4°C with gentle rocking, and the extracted capsids were pelleted through a cushion of 25% sucrose in TNE by centrifugation for 1 h at 40,000 rpm in a Beckman TLA-45 rotor. The cushion was aspirated, and the capsids were resuspended in SDS sample buffer. Proteins were resolved by SDS-PAGE and transferred to Immobilon P membranes. Membranes were analyzed by autoradiography to detect labeled proteins and by Western blotting to detect Vhs.
Virion extractions.
Virions from the extracellular medium were purified by sedimentation through 10 to 30% gradients of dextran T-10, pelleted from the appropriate gradient fractions, and resuspended in a small volume of 10 mM Tris (pH 7.5). The virion suspensions were adjusted to one of the following conditions: (i) no additions, (ii) 1% Triton X-100, (iii) 1% NP-40, (iv) 0.5% Triton X-100 plus 0.5% sodium deoxycholate, or (v) 1% NP-40 plus 1 M NaCl. They were then incubated at room temperature for 1 h and centrifuged for 1 h at 36,000 rpm in a Beckman TLA-45 rotor. Pellet and supernatant fractions were analyzed by SDS-PAGE and the proteins transferred to Immobilon P membranes. The membranes were subjected to autoradiography to detect 35S-labeled proteins and to Western blotting to detect Vhs.
In some experiments virions were first extracted with 1% NP-40 plus 1 M NaCl as described above. The resulting capsids were then washed with various concentrations of GuHCl as described for washing intranuclear B capsids.
Virion and L-particle purification.
[35S]methionine-labeled virions and L particles were pelleted from the extracellular medium of infected BHK-21 cells, resuspended in a small volume of MEM, and sedimented through two successive 5 to 15% Ficoll gradients prepared in MEM (74, 78). Centrifugation was at 24,000 rpm for 100 min in a Beckman SW41 Ti rotor. Virions and L particles were pelleted from the appropriate gradient fractions and analyzed by SDS-PAGE, followed by autoradiography and Western blotting.
Indirect immunofluorescence.
Vero cells grown on coverslips were infected with 2.5 PFU/cell of wild-type HSV-1 or the Vhs mutant N237. Alternatively, cells were transfected with the Vhs expression plasmid pKOSamp (17, 18, 54). At 11.5 h after infection or 24 h after transfection, the cells were washed with PBS and fixed by incubation in 2% formaldehyde (in PBS) for 20 min at room temperature (26). The cells were washed three times with PBS, incubated for 10 min in PBS containing 50 mM ammonium chloride, and permeabilized by incubation in 0.1% Triton X-100 (in PBS) at room temperature for 4 min. Coverslips were washed three times with PBS containing 0.5% BSA and three times with wash buffer (PBS containing 1% goat serum and 0.2% Tween 20). Cells were incubated for 1 h in PBS containing a 1:80 dilution of rabbit antiserum raised against a LacZ-Vhs fusion protein containing Vhs amino acids 239 through 489 (58), washed three times with wash buffer, and incubated for 1 h in PBS containing a 1:3,200 dilution of fluorescein isothiocyanate-conjugated secondary antibody. The coverslips were mounted on glass slides and examined using a Zeiss LSM confocal microscope.
RESULTS
Kinetics of appearance of the 59.5-kDa form of Vhs and its dependence on de novo protein synthesis.
The 59.5-kDa form of Vhs was originally identified in cells 20 h after infection (58). To better define the kinetics of appearance of the 59.5-kDa and 58-kDa polypeptides, whole-cell extracts were prepared at various times after infection with 10 PFU/cell of HSV-1 and analyzed by Western blotting using a Vhs-specific antiserum (Fig. 1A). At this multiplicity of infection and exposure time, incoming copies of Vhs were not detected, as evidenced by the lack of a detectable signal at 3 h (Fig. 1A, lane a). Newly synthesized copies of the 58-kDa polypeptide were detected by 5 h after infection. The 59.5-kDa form was detected slightly later and, while always less abundant than the 58-kDa form, increased in abundance at late times. At higher multiplicities of infection and longer exposure times, the 59.5-kDa polypeptide could be detected as early as newly synthesized copies of the 58-kDa polypeptide (Fig. 1B), at 4 h after infection.
FIG. 1.

Appearance of the 59.5-kDa Vhs form requires de novo protein synthesis. (A) Time course of Vhs accumulation. Vero cells were infected with 20 PFU/cell of wild-type HSV-1 (KOS). Whole-cell lysates were prepared at the indicated times postinfection (p.i.) and analyzed by SDS-PAGE and Western blotting using antiserum raised against a UL41-LacZ fusion protein (58). The 58- and 59.5-kDa forms of Vhs are indicated to the right of lane f. (B) Vero cells were infected with 50 PFU/cell of wild-type HSV-1 in the absence of drugs (lanes b and e) or in the presence of 5 μg/ml of actinomycin D (ActD) (lanes c and f) or 50 μg/ml of cycloheximide (Cyclo) (lanes d and g). Whole-cell lysates were prepared at the indicated times and analyzed by SDS-PAGE and Western blotting using antiserum raised against a UL41-LacZ fusion protein. Lane a contains an aliquot of the infecting virions equivalent to the number used to infect the number of cells in each of the other samples. The 58- and 59.5-kDa forms of Vhs are indicated to the right of lane g.
Earlier studies showed that the 58-kDa and 59.5-kDa Vhs forms differ in the extent of phosphorylation and that, although present in infected cells, the 59.5-kDa form is not incorporated into virions (58). One can imagine that cellular or virion protein kinases might phosphorylate incoming copies of the 58-kDa polypeptide, converting them to 59.5 kDa. Alternatively, processing to 59.5 kDa might occur only for newly synthesized Vhs. To distinguish these possibilities, cells were infected with HSV-1 in the absence of drugs or in the presence of actinomycin D or cycloheximide to inhibit de novo transcription or translation, respectively. Whole-cell lysates were prepared and analyzed for Vhs by SDS-PAGE and Western blotting (Fig. 1B). These infections were performed with 50 PFU/cell to allow detection of incoming copies of Vhs. Only the 58-kDa polypeptide was detected at 2 h after infection in the presence or absence of the drugs (Fig. 1B, lanes b to d). However, both the 58-kDa and 59.5-kDa forms were detected at 4 h in the absence of drugs (Fig. 1B, lane e). In this case, the combined intensity of the 58-kDa and 59.5-kDa bands (Fig. 1B, lane e) was greater than that of the 58-kDa protein from incoming virions (Fig. 1B, lane a), indicating that de novo Vhs synthesis had occurred. In contrast, none of the 59.5-kDa form was detected at 4 h in cells infected in the presence of actinomycin D or cycloheximide (Fig. 1B, lanes f and g), indicating that there had been no detectable conversion of incoming Vhs to 59.5 kDa and that the appearance of the 59.5-kDa form required de novo protein synthesis. Since Vhs-mediated mRNA degradation occurs following infection in the presence of actinomycin D or cycloheximide (21, 64, 73), incoming copies of the 58-kDa virion polypeptide do not need to be converted to 59.5 kDa before they can induce mRNA decay.
Phosphoamino acid analysis of Vhs.
The 58-kDa and 59.5-kDa Vhs forms differ in their extents of phosphorylation, with the 59.5-kDa form being more highly phosphorylated (58). It was therefore of interest to compare the types of amino acid on which they are phosphorylated. To this end, cells were labeled with [32P]orthophosphate from 4 to 18 h after HSV-1 infection. Both Vhs forms were immunoprecipitated, resolved by preparative SDS-PAGE, and subjected to phosphoamino acid analysis. Both the 58-kDa and 59.5-kDa forms were phosphorylated predominantly on serines (Fig. 2A). Thus, although the 58-kDa and 59.5-kDa forms evidently differ in the number of serines that are phosphorylated, there was no detectable difference in the types of amino acids that are phosphorylated.
FIG. 2.

Phosphoamino acid analysis of Vhs. (A) The 58-kDa and 59.5-kDa forms of Vhs are phosphorylated predominantly on serines. Vero cells were infected with 20 PFU/cell of wild-type HSV-1 and labeled from 4 to 18 h after infection by incubation in medium containing [32P]orthophosphate. After preparation of whole-cell lysates, the 58-kDa and 59.5-kDa forms of Vhs were immunoprecipitated and purified by preparative SDS-PAGE. Both forms were acid hydrolyzed and mixed with unlabeled phosphoamino acid standards, and the phosphoamino acids were analyzed by 1-D high-voltage electrophoresis on cellulose thin-layer plates. Unlabeled phosphoamino acid standards were localized by staining the plate with ninhydrin, while 32P-labeled amino acids were visualized by autoradiography. The locations of phosphoserine, phosphothreonine, and phosphotyrosine standards are indicated by the dashed circles. (B) The difference in electrophoretic mobility of the 58-kDa and 59-kDa Vhs forms is due primarily to differences in the extent of phosphorylation. Vero cells were infected with 20 PFU/cell of wild-type HSV-1 and labeled from 3.5 to 19 h after infection by incubation in medium containing either [35S]methionine (lanes c and d) or [32P]orthophosphate (lanes a and b). After preparation of whole-cell lysates, the Vhs polypeptides were immunoprecipitated and treated with potato acid phosphatase (lanes a and c) or left untreated (lanes b and d). The products were analyzed by SDS-PAGE and autoradiography. The locations of the 58-kDa and 59.5-kDa forms of the Vhs polypeptide are indicated to the right of lane d.
To determine whether the difference in phosphorylation is responsible for the different electrophoretic mobilities of the 58-kDa and 59.5-kDa Vhs forms, we examined whether phosphatase treatment would cause them to migrate as one band on SDS-PAGE (Fig. 2B). Cells were labeled with [35S]methionine or [32P]orthophosphate from 3.5 to 19 h after infection, and whole-cell lysates were prepared. The Vhs polypeptides were immunoprecipitated and either treated with acid phosphatase or left untreated, and the products were analyzed by SDS-PAGE and autoradiography. Phosphatase treatment removed most, if not all, of the 32P label from the Vhs polypeptides (Fig. 2B, compare lanes a and b). It also caused most of the 35S-labeled protein to migrate as a single band on SDS-PAGE with a mobility indistinguishable from that of the phosphorylated 58-kDa Vhs form (Fig. 2B, lanes b to d). The different mobilities of the 58-kDa and 59-kDa Vhs forms are, therefore, primarily due to differences in phosphorylation.
Pulse-chase analysis of the Vhs polypeptides.
To examine whether the 58-kDa and 59.5-kDa forms have a precursor-product relationship, Vero cells were pulse-labeled with [35S]methionine for 15 min, beginning 8 h after HSV-1 infection, after which the label was removed and chased by incubating the cells in medium containing an excess of cold methionine. Whole-cell lysates were prepared after various chase intervals, and the Vhs polypeptides were immunoprecipitated and analyzed by SDS-PAGE and autoradiography (Fig. 3). The first detectable Vhs translation product migrated with an apparent molecular mass of 58 kDa, identical to that of the phosphorylated virion polypeptide. The 59.5-kDa form was detectable after a chase of as short as 15 min, and it increased in abundance with increasing chase times until, after an 8-h chase, the ratio of the 59.5-kDa form to the 58-kDa form was almost 1.0. Thus, the 59.5-kDa form of Vhs is produced by posttranslational modification of a primary translation product that comigrates on SDS-PAGE with the 58-kDa virion polypeptide.
FIG. 3.

Pulse-chase analysis of the Vhs polypeptides. Vero cells were infected with 20 PFU/cell of wild-type HSV-1 (KOS) and pulse-labeled for 15 min with [35S]methionine beginning 8 h after infection. The label was removed, and the cells were washed and incubated for the indicated chase intervals in medium containing an excess of unlabeled methionine. Whole-cell lysates were prepared and the Vhs polypeptides immunoprecipitated and analyzed by SDS-PAGE and autoradiography. The upper panel shows an autoradiogram of the resulting gel. The locations of the 58-kDa and 59.5-kDa polypeptides are shown to the right of lane g. For each time point, the amounts of 58-kDa and 59.5-kDa polypeptides were quantified by densitometric scanning of the autoradiogram. The ratio of the amounts of 59.5-kDa and 58-kDa polypeptides (59.5 kDa/58 kDa) is plotted in the lower panel.
2-D gel analysis of Vhs.
Even after an 8-h chase, not all of the pulse-labeled Vhs had been chased to 59.5 kDa (Fig. 3). One possible explanation is that there is more than one possible fate for newly synthesized Vhs but only one involves processing to 59.5 kDa. This possibility is addressed later in this study. A second, not mutually exclusive, possibility is that the 58-kDa band contains more than one Vhs species, which are not resolved by 1-D SDS-PAGE. One of these might be the nonphosphorylated primary translation product, while another is the phosphorylated virion protein. In this case, the 58-kDa primary translation product would never be completely chased to 59.5 kDa because, after processing to 59.5 kDa, some copies of Vhs would be further processed to the phosphorylated virion protein, which comigrates with the primary translation product on SDS-PAGE. The data from Fig. 2 are consistent with this second possibility, since dephosphorylation of the phosphorylated 58-kDa polypeptide did not alter its electrophoretic mobility.
To investigate the second possibility, whole-cell lysates were prepared 24 h after HSV-1 infection and analyzed by 2-D gel electrophoresis involving isoelectric focusing in the first dimension and SDS-PAGE in the second (Fig. 4). One predominant 59.5-kDa form that exhibited a more acidic pI than the 58-kDa forms was detected. A smear of products was observed at 58 kDa and was composed predominantly of two concentrated spots with apparent pIs of 8.85 and 8.74. The results indicate that the 58-kDa Vhs band seen upon SDS-PAGE analysis of whole-cell lysates contains at least two forms of the Vhs polypeptide.
FIG. 4.

2-D gel electrophoresis of the Vhs polypeptides. Vero cells were harvested 24 h after infection with 10 PFU/cell of wild-type HSV-1, and solubilized in 2-D gel sample buffer. The lysate was analyzed by 2-D gel electrophoresis, involving isoelectric focusing in the first dimension and SDS-PAGE in the second. Following electrophoresis, the proteins were transferred to an Immobilon P membrane and detected by immunoblotting using rabbit antiserum raised against a UL41-lacZ fusion protein. The three predominant Vhs polypeptides are indicated by arrowheads. The positions of the 58-kDa and 59.5-kDa polypeptides are indicated to the left of the gel, while the pH values at different positions in the isoelectric focusing gel are indicated at the top.
The 59.5-kDa form of Vhs is enriched in the nucleus.
To examine whether the 58-kDa and 59.5-kDa Vhs forms differ with regard to subcellular localization, Vero cells were harvested 8 h after infection, disrupted by hypotonic lysis, and separated by low-speed centrifugation into a cytoplasmic fraction (supernatant) and a crude nuclear pellet. Nuclei were resuspended, washed in buffer containing ionic and nonionic detergents, and recentrifuged to yield the nuclear wash (supernatant) and nuclear (pellet) fractions. Analysis of the fractions by SDS-PAGE and Western blotting revealed two-thirds of the total Vhs protein in the cytoplasmic and nuclear wash fractions and one-third in the nuclear fraction (Fig. 5). Overall, cells contained 4 to 5 times more of the 58-kDa Vhs polypeptide than the 59.5-kDa polypeptide. However, while the ratios of the 59.5-kDa form to the 58-kDa form were 0.14 and 0.17 for the cytoplasmic and nuclear wash fractions, respectively, the ratio was 0.72 for the nuclear fraction. Thus, although each fraction contained both forms of the Vhs polypeptide, the 59.5-kDa form was enriched three- to fourfold in the nuclear fraction relative to the cytoplasm and nuclear wash fractions.
FIG. 5.

The 59.5-kDa form of Vhs is enriched in the nucleus. Vero cells were harvested 8 h after infection with 20 PFU/cell of wild-type HSV-1 and separated into nuclear, nuclear wash, and cytoplasmic fractions. Aliquots of fractions from equal numbers of cells were analyzed by SDS-PAGE and Western blotting to detect the 59.5-kDa and 58-kDa forms of Vhs. The relative amounts of the 59.5-kDa and 58-kDa polypeptides were quantified for each fraction by densitometric scanning of the film resulting from the Western blot, and the results are shown in the four lines beneath the gel. Line 1 shows the percentage of the total amount (59.5 kDa plus 58 kDa) of Vhs present in each fraction. Line 2 shows the ratio of the amounts of the 59.5-kDa and 58-kDa polypeptides in each fraction. Line 3 shows the percentage of the total amount of the 59.5-kDa polypeptide that was present in each fraction, and line 4 shows the percentage of the total amount of the 58-kDa polypeptide in each fraction.
Both 59.5-kDa and 58-kDa Vhs polypeptides bind intranuclear capsids.
The observation that 33% of the total Vhs protein is found in the nuclear fraction and that this fraction is enriched for the 59.5-kDa polypeptide was surprising because the known function of Vhs is to accelerate mRNA degradation in the cytoplasm. However, Vhs is also a structural component of virions. This raised the possibility that nuclear copies of the protein are a packaging intermediate, in the process of being incorporated into progeny virions, perhaps by binding intranuclear capsids prior to primary envelopment. To investigate this possibility, Vero cells were labeled with [35S]methionine and [3H]thymidine from 4 to 18 h after infection. Nuclear lysates were prepared and analyzed by sedimentation through gradients of 20 to 50% sucrose to determine whether either or both forms of Vhs were associated with intranuclear capsids (Fig. 6). Aliquots of each gradient fraction were analyzed to determine the amounts of 35S and 3H label incorporated into TCA-precipitable material (Fig. 6A) and by SDS-PAGE and Western blotting to detect the Vhs polypeptides (Fig. 6B). This analysis revealed two peaks of labeled proteins centered around fractions 11 and 14 (Fig. 6A). The peak centered at fraction 14 coincided with a peak of labeled DNA and was, therefore, tentatively identified as containing C capsids. The peak centered at fraction 11 did not coincide with a peak of labeled DNA and was, therefore, tentatively identified as the peak containing B and, perhaps, some A capsids. Examination of the Western blot revealed that much of both Vhs polypeptides cosedimented with low-molecular-mass material in the upper third of the gradient (Fig. 6B). However, a significant amount of both 58-kDa and 59.5-kDa polypeptides cosedimented with the putative B and C capsids.
FIG. 6.

Both 59.5-kDa and 58-kDa Vhs polypeptides bind intranuclear B and C capsids. Vero cells were infected with 20 PFU/cell of wild-type HSV-1 and incubated from 4 to 18 h after infection in MEM containing [35S]methionine and [3H]thymidine to label protein and DNA, respectively. A nuclear lysate was prepared and analyzed by sedimentation through a gradient of 20 to 50% sucrose prepared in TNE. Aliquots of each gradient fraction were analyzed to determine the amounts of [35S]methionine and [3H]thymidine incorporated into TCA-precipitable material (A) and by SDS-PAGE and Western blotting to detect the 59.5-kDa and 58-kDa Vhs polypeptides (B). The peak centered at fraction 11 was presumed to be B capsids, and that at fraction 14 was presumed to be C capsids. The putative B capsids were pelleted from fraction 11 of the first gradient, resuspended in TNE, and resedimented through a second 20 to 50% sucrose gradients. An aliquot from each fraction of the second gradient was analyzed by SDS-PAGE, and the resolved proteins were blotted onto an Immobilon P membrane. The membrane was subjected to autoradiography (C) to detect all 35S-labeled proteins and probed with Vhs-specific antiserum (D) to allow immunologic detection of the Vhs polypeptides. The locations of major capsid proteins are shown by dots to the right of lane 12 of panel C and labeled to the left of lane 1 of panel C. The 59.5-kDa and 58-kDa Vhs polypeptides are indicated by dots to the right of lane 12 of panel D and labeled to the left of lane 1 of panel D. In this and other figures, the smallest of the major capsid proteins, VP26, was run off the end of the gel. This was necessary in order to run the gel long enough to resolve the 58-kDa and 59.5-kDa forms of Vhs.
To verify that the peak centered at fraction 11 actually contained B capsids and that the sedimentation of Vhs in this fraction was due to its association with the capsids, the putative B capsids were pelleted from fraction 11 of the first gradient, resuspended, and recentrifuged through a second gradient of 20 to 50% sucrose. The proteins in each fraction of the second gradient were resolved by SDS-PAGE and transferred to an Immobilon P membrane. The membrane was analyzed by autoradiography to detect all 35S-labeled proteins (Fig. 6C) and probed with Vhs-specific antiserum to detect the Vhs polypeptides (Fig. 6D). On the second gradient, both Vhs polypeptides sedimented in a peak spanning fractions 7 through 12, which coincided with the peak of 35S-labeled capsid proteins. However, in contrast to the first gradient, little Vhs or labeled capsid protein was observed in the upper third of the gradient or in other fractions outside of the peak.
To better compare the Vhs polypeptides associated with B and C capsids, 35S-labeled B and C capsids were both purified by sedimentation through two consecutive sucrose gradients and the peak fractions analyzed on adjacent lanes of an SDS-polyacrylamide gel, followed by autoradiography to detect 35S-labeled proteins (Fig. 7A) and by immunoblotting to detect Vhs (Fig. 7B). The profiles of labeled proteins were similar for B and C capsids, with the exception that B capsids contained the scaffolding protein VP22a, which was missing from C capsids (Fig. 7A). This agreed with previous studies of intranuclear B and C capsids (23, 24, 28), verifying the tentative identification of the peaks. Of most importance to the current study, both 58-kDa and 59.5-kDa forms of Vhs were associated with B and C capsids purified over two consecutive sucrose gradients (Fig. 7B). For both types of capsid, several times more of the 58-kDa polypeptide than of the 59.5-kDa form was observed. In addition, similar amounts of the 58-kDa form were observed in B and C capsids, as was also the case for the 59.5-kDa polypeptide. While the strongest bands on this Western blot corresponded to the two Vhs polypeptides, faint bands were also observed at the positions of VP5 and other capsid proteins (Fig. 7A). These bands were not observed when the Western analysis was performed on unlabeled capsids (data not shown). Since the last step in the immunoblotting procedure was detection of the chemiluminescent signal by exposing the membrane to film, these bands were judged to result from autoradiographic detection of the 35S-labeled capsid proteins rather than from cross-reaction of the UL41-specific antibody with the capsid proteins.
FIG. 7.

Both 59.5-kDa and 58-kDa Vhs polypeptides are found associated with intranuclear B and C capsids, but VP16 is not. [35S]methionine-labeled B and C capsids were purified from nuclear lysates of infected Vero cells by sedimentation through two sequential gradients of 20 to 50% sucrose as described for B capsids in the legend to Fig. 6. Proteins in the peak fractions from the second B and C capsid gradients were resolved by SDS-PAGE and blotted onto an Immobilon P membrane. The membrane was subjected to autoradiography (A) to detect all 35S-labeled proteins and probed with Vhs-specific antiserum (B) to allow immunologic detection of the Vhs polypeptides. The locations of major capsid proteins are shown to the left of panel A, and those of the 59.5-kDa and 58-kDa Vhs polypeptides are shown to the right of panel B. The proteins in B and C capsids were compared by Western blotting to those in extracellular virions purified as described in the text. Following SDS-PAGE, the proteins were blotted onto an Immobilon P membrane. The membrane was probed using UL41-specific antiserum to detect the Vhs polypeptides (C), after which it was stripped and reprobed using VP16-specific antiserum to detect the VP16 polypeptide (D).
B and C capsids contain associated Vhs but not VP16.
A potential problem with these experiments would occur if the capsid preparations were contaminated with enveloped virions from the cytoplasm. These would provide a source of Vhs protein that might be mistaken for Vhs that was associated with intranuclear capsids. Therefore, as an additional control, B and C capsids were examined by SDS-PAGE and Western blotting for the presence of both Vhs and VP16. Although it is a major component of the virion tegument, VP16 has been reported to not be associated with intranuclear B and C capsids (82). An observation that it is absent from B and C capsid preparations that contain Vhs would be an important control that those preparations are not contaminated with enveloped virions. It was also of interest to look for VP16 because Vhs and VP16 can be coimmunoprecipitated from infected cells (58, 65, 69), and newly synthesized VP16 appears to play an important role in controlling the activity of newly synthesized Vhs (36).
As expected, both forms of the Vhs polypeptide were detected in preparations of B and C capsids (Fig. 7C), while purified virions contained only the 58-kDa protein (Fig. 7C). However, in accord with earlier results, no VP16 was detected in the B and C capsid preparations, although it was easily detected in purified virions (Fig. 7D). Thus, Vhs and VP16 differ in that both forms of Vhs are associated with intranuclear capsids, while VP16 is not. It follows that the B and C capsid preparations were not contaminated with detectable amounts of enveloped virions. Furthermore, the interaction between Vhs and VP16 must occur elsewhere within the cell than on B and C capsids.
Limited removal of Vhs from B capsids by washing with GuHCl.
To investigate the strength of the interaction between Vhs and capsids, we examined whether the 58-kDa or 59.5-kDa form could be released from B capsids by various washing protocols. In preliminary experiments, no detectable Vhs could be released by washing B capsids with a variety of nonionic and ionic detergents or at a range of salt concentrations (data not shown). These treatments included washing the capsids for an hour with 1% NP-40, with 1% NP-40 plus 1 M NaCl, with 0.5% Triton X-100 plus 0.5% sodium deoxycholate, or with 1 M urea.
Next, [35S]methionine-labeled B capsids were purified by sedimentation through two successive sucrose gradients, washed with buffers containing increasing concentrations of GuHCl, and then pelleted through a sucrose cushion. Polypeptides that remained associated with pelleted capsids were resolved by SDS-PAGE, transferred to an Immobilon P membrane, and analyzed by autoradiography to visualize the major capsid proteins or by probing the membrane with UL41-specific antiserum to detect Vhs (Fig. 8). In accord with previous results (50), washing B capsids with increasing concentrations of GuHCl led to the progressive removal of VP22a, while almost all VP5 and VP23 remained associated with the capsid structures at concentrations of up to 2 M (Fig. 8). The associations of the 58-kDa and 59.5-kDa forms of Vhs with capsids showed intermediate sensitivities to washing with GuHCl. Both were released from capsids more efficiently than VP5 and VP23 but less readily than VP22a. It is noteworthy that while a significant amount of each Vhs form could be released by washing, 40 to 50% of each polypeptide remained associated with capsids at GuHCl concentrations of as high as 2 M, indicating a strong association with B capsids.
FIG. 8.

Limited removal of the Vhs polypeptides from B capsids by washing with GuHCl. [35S]methionine-labeled B capsids were purified from nuclear lysates from infected Vero cells by sedimentation through two sequential gradients of 20 to 50% sucrose prepared in TNE. B capsids were pelleted from the appropriate fractions of the second gradient, resuspended, and washed for 30 min in TNE containing 0, 0.2 M, 0.5 M, 1 M, or 2 M GuHCl. The capsids were then pelleted through a cushion of 25% sucrose in TNE. Polypeptides that remained associated with the pelleted capsids were resolved by SDS-PAGE, transferred to an Immobilon P membrane, and analyzed by autoradiography (A) to visualize the major capsid proteins or by probing the membrane using Vhs-specific antiserum (C) for immunologic detection of the Vhs polypeptides. For comparison, lane f of panel A contains a sample of C capsids. The profile of proteins in C capsids was similar to that in B capsids, except that C capsids lack VP22a. In this and other figures, the smallest of the major capsid proteins, VP26, was run off the end of the gel. This was necessary in order to run the gel long enough to resolve the 58-kDa and 59.5-kDa forms of Vhs. The relative amounts of capsid proteins and Vhs polypeptides remaining bound to washed and unwashed capsids were determined by densitometric scanning of the autoradiogram in panel A or the Western blot in panel C and are plotted in panel B.
Virions contain two populations of Vhs as defined by extraction with detergent and salt.
Earlier studies have shown that several tegument proteins can be released from virions by extraction with 1% NP-40 plus 1 M NaCl or with 0.5% Triton X-100 plus 0.5% sodium deoxycholate (88). In addition, Zelus et al. showed that a Vhs-dependent RNase activity could be released from virions by disruption of the envelope with NP-40 followed by extraction of the capsid-tegument structure with buffer containing 400 mM potassium acetate, although they did not directly monitor release of the Vhs polypeptide (87). Our observation that 40% of Vhs remained associated with B capsids, even after extraction with 2 M guanidine hydrochloride, was therefore somewhat surprising, especially in light of earlier studies suggesting that Vhs is a component of the tegument. To examine this apparent discrepancy, we investigated whether Vhs could be released from virions by using a variety of extraction procedures.
35S-labeled wild-type or Vhs-ΔSma virions were purified from the extracellular media of infected cultures by centrifugation through gradients of 10% to 30% dextran T-10 (Fig. 9A, lanes b and c), as were 32P-labeled wild-type virions (Fig. 9A, lane a). Comparison of the profiles of wild-type and Vhs-ΔSma virions aided in the visualization of the wild-type Vhs polypeptide, since the Vhs protein of Vhs-ΔSma is not incorporated into virions (58). Comparison of the protein profiles of 35S-labeled and 32P-labeled wild-type virions illustrates the previous finding (58) that the virion copies of Vhs are phosphorylated (Fig. 9A, lanes a and c). 35S-labeled virions were then extracted with buffer containing one of the following: (i) 1% NP-40, (ii) 1% Triton X-100, (iii) 1% NP-40 plus 1 M NaCl, or (iv) 0.5% Triton X-100 plus 0.5% sodium deoxycholate. After extraction, the virion preparations were separated by centrifugation into a capsid-containing pellet and a supernatant containing the solubilized proteins. These fractions were analyzed by SDS-PAGE, followed by Western blotting and/or autoradiography (Fig. 9). Vhs and other tegument proteins fell into two categories with regard to the buffer conditions required to initiate their extraction from virions. In accord with previous results (88), extraction with 1% NP-40 or 1% Triton X-100 solubilized most of the envelope glycoproteins, as well as some of the tegument proteins VP7/8 and VP16 (Fig. 9). However, these extraction conditions did not release any detectable Vhs from the capsids or detectable amounts of the other tegument proteins, VP13/14 and VP22. In contrast, extraction of virions with 1% NP-40 plus 1 M NaCl or with 0.5% Triton X-100 plus 0.5% sodium deoxycholate released some of the VP13/14 and VP22 and approximately half of the Vhs protein into the soluble supernatant fraction.
FIG. 9.

Extraction of virions with detergents, salt, and GuHCl. (A) [32P]orthophosphate-labeled wild-type (WT) virions (lane a) and [35S]methionine-labeled wild-type (lanes c to e) and Vhs-ΔSma (lane b) virions were extracted with 1% Triton X-100 (Tx) and separated into pellet (P) (lane d) and supernatant (S) (lane e) fractions as described in the text or were left untreated (lanes a to c). The various virions and fractions were analyzed by SDS-PAGE and autoradiography. Various virion polypeptides are labeled between lanes c and d. The wild-type Vhs polypeptide is indicated by arrowheads to the right of lane c and left of lane d. The Vhs polypeptide is missing from Vhs-ΔSma virions. (B) [35S]methionine-labeled wild-type virions were extracted for 1 h with the following buffers as described in the text: (i) 10 mM Tris (pH 7.5) (control), (ii) 1% NP-40, (iii) 0.5% Triton X-100 plus 0.5% sodium deoxycholate (Doc), and (iv) 1% NP-40 plus 1 M NaCl. The extracted virions were separated by centrifugation into a capsid-containing pellet and a supernatant. Proteins in the various fractions were resolved by SDS-PAGE, transferred to an Immobilon P membrane, and visualized by autoradiography to detect all virion proteins. (C) The same membrane was then probed using UL41-specific antiserum to detect the Vhs polypeptide. (D) Wild-type virions were extracted with 1% NP-40 plus 1 M NaCl, after which the capsids were pelleted. The capsids, retaining approximately 50% of the Vhs protein, were washed further with TNE containing 0 M, 0.5 M, 1 M, or 2 M GuHCl. The washed capsids were pelleted through a cushion of 25% sucrose, resuspended, and analyzed by SDS-PAGE and Western blotting to detect the Vhs polypeptide. The amount of Vhs remaining bound to capsids washed with various concentrations of GuHCl was quantified by densitometric scanning and expressed as a percentage of the amount bound to TNE-washed capsids.
This raised the possibility that virions contain two populations of the Vhs polypeptide: one population that can be released relatively easily by extraction with nonionic detergents plus salt or sodium deoxycholate and a second population that is bound to capsids more tightly. To investigate this possibility, virions were extracted with 1% NP-40 plus 1 M NaCl and then separated by centrifugation into a capsid-containing pellet and a soluble supernatant. The pellet, which contained approximately half of the initial amount of Vhs, was resuspended and then washed with increasing concentrations of GuHCl to see if this released any of the remaining Vhs protein. The GuHCl washes had a similar effect upon the remaining Vhs as they had upon the Vhs polypeptides that were associated with intranuclear capsids (Fig. 9). Although some of the Vhs was removed, 60% to 70% remained associated with the capsids even after washing with 2 M GuHCl. The results support a model in which virions contain two populations of Vhs, one of which is strongly associated with the capsid, just as intranuclear capsids contain tightly bound copies of the Vhs protein.
Only the 58-kDa form of Vhs is found in enveloped cytoplasmic virions.
Although the 58-kDa and 59.5-kDa forms of Vhs are both associated with intranuclear B and C capsids, only the 58-kDa form is found in extracellular virions (58). This implies that the 59.5-kDa form is converted to 58 kDa or lost from the virus particle at some stage between binding to intranuclear capsids and the release of mature virions from infected cells. To better define the step at which this conversion or loss takes place, we analyzed whether both the 58-kDa and 59.5-kDa forms, or only the 58-kDa polypeptide, are found in enveloped virions isolated from the cytoplasm. The Western blot in Fig. 10 clearly shows that, as is the case for extracellular virions, enveloped cytoplasmic virions contain only the 58-kDa form of Vhs.
FIG. 10.

Enveloped cytoplasmic virions contain only the 58-kDa form of Vhs. Enveloped cytoplasmic virions (lane 3), and intranuclear B (lane 1) and C (lane 2) capsids were purified as described in the text and analyzed by SDS-PAGE and Western blotting using Vhs-specific antiserum. The locations of the 59.5-kDa and 58-kDa forms of Vhs are indicated to the left of lane 1.
L particles contain a 58-kDa form but not the 59.5-kDa form of Vhs.
The preceding experiments indicate that intranuclear capsids and virions both contain a population of Vhs that is strongly associated with capsids. While this suggests that some copies of Vhs are incorporated into virions through a pathway involving binding to intranuclear capsids, other data suggest that some copies of Vhs are added to assembling virions in the cytoplasm through interactions with other tegument proteins or the tails of envelope glycoproteins. In particular, McLauchlan et al. have reported that L particles exhibit virion host shutoff activity, although they did not assay for the Vhs protein directly (42). Since L particles are noninfectious enveloped particles that contain tegument and glycoproteins, but no capsids, the incorporation of Vhs into L particles would not be due to Vhs-capsid protein interactions.
To investigate this more fully, [35S]methionine-labeled virions and L particles were isolated from the extracellular media of cultures of infected BHK-21 cells and analyzed by SDS-PAGE and Western blotting to detect the Vhs polypeptides or by autoradiography to detect the other viral proteins (Fig. 11). BHK-21 cells were used because, in our hands, they produce much larger amounts of L particles than Vero cells (data not shown). As expected, L particles contained large amounts of the major tegument proteins VP16 and VP7/8 but little if any of the major capsid protein, VP5 (Fig. 11, lane 3). Virions and L particles both contained readily detectable amounts of the Vhs protein (Fig. 11, lanes 2 and 4). However, as for virions, L cells contained only a 58-kDa form of Vhs.
FIG. 11.

L particles contain a 58-kDa, but not the 59.5-kDa, form of Vhs. [35S]methionine-labeled virions (lanes 1 and 2) and L particles (lanes 3 and 4) were purified from the extracellular medium from cultures of BHK-21 cells infected with wild-type HSV-1. Proteins were resolved by SDS-PAGE and transferred to an Immobilon P membrane, after which the membrane was subjected to autoradiography (A) (lanes 1 and 3) to detect all labeled viral proteins or probed using a UL41-specific antiserum (W) (lanes 2 and 4) for immunologic detection of the Vhs polypeptides.
Subcellular localization of Vhs by indirect immunofluorescence.
The experiments described above suggest that Vhs is present in both the nucleus and cytoplasm of infected cells. To investigate this, the intracellular distribution of Vhs was examined in infected as well as transfected cells, by indirect immunofluorescence using antiserum raised against a LacZ-Vhs fusion protein containing Vhs amino acids 239 through 489 (58). By 11.5 h after infection with wild-type virus, most cells showed Vhs staining at many sites throughout the cytoplasm, including intense staining at some perinuclear sites (Fig. 12A). In addition, a particularly striking feature of infected cells was a thin, intense band of Vhs staining around the nuclear rim. Some cells also showed a less intense, diffuse staining throughout their nuclei. Whether this was due to the presence of Vhs in the nucleoplasm or to visualization of nuclear rim staining at the nuclear membrane overlaying some nuclei in this optical section is uncertain. Either possibility indicates a nuclear presence for some copies of Vhs. The specificity of the staining for Vhs was indicated by the virtual absence of staining in cells infected with the Vhs mutant N237 (Fig. 12B). N237 is a viable nonsense mutant encoding a truncated Vhs polypeptide containing amino acids 1 through 237 of the wild-type protein and which therefore lacks the portion of Vhs that reacts with the antiserum. Cells transfected with a plasmid expressing wild-type Vhs exhibited Vhs staining throughout much of the cytoplasm (Fig. 12C), some of which was concentrated in punctuate spots. This is consistent with earlier observations that transfection with a wild-type Vhs allele induces the decay of cytoplasmic mRNAs (29, 54). However, transfected cells lacked the intense nuclear rim staining characteristic of infected cells, suggesting that the nuclear rim localization requires the expression of additional viral proteins beside Vhs.
FIG. 12.
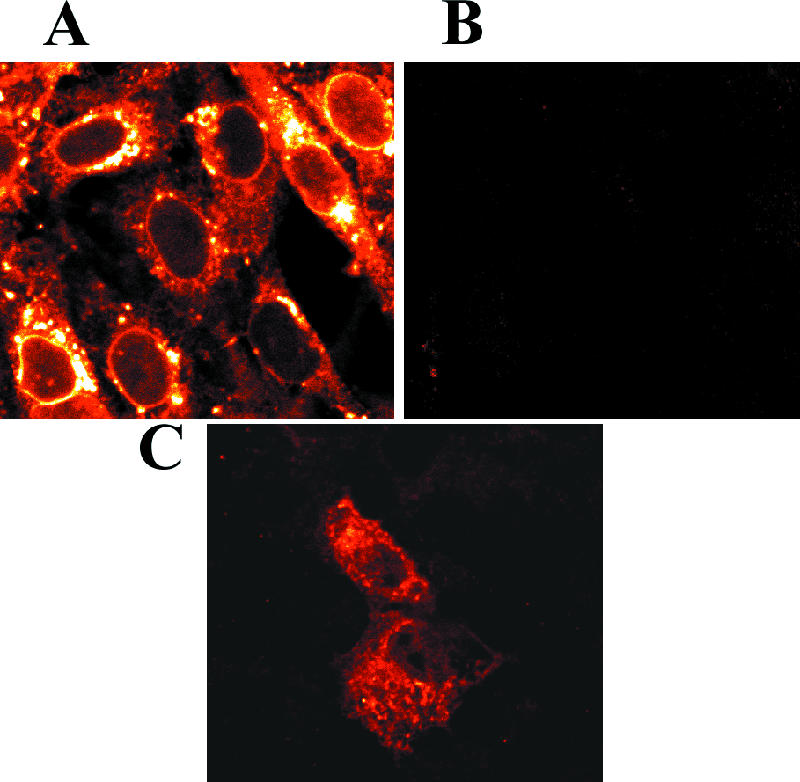
Indirect immunofluorescence localization of Vhs in infected and transfected cells. Vero cells were infected with 2.5 PFU/cell of wild-type HSV-1 (A) or the Vhs mutant N237 (B) or were transfected with the wild-type Vhs expression plasmid pKOSamp (C). At 11.5 h after infection or 24 h after transfection, the cells were fixed and stained for indirect immunofluorescence using rabbit antiserum raised against a LacZ-Vhs fusion protein containing amino acids 239 through 489 of wild-type Vhs and were examined using a Zeiss LSM confocal microscope.
DISCUSSION
These studies were initiated to investigate the pathway(s) by which Vhs is packaged into virions and the role of the 59.5-kDa Vhs form during HSV-1 infections. Previous studies showed that infected cells contain two forms of Vhs: a phosphorylated 58-kDa polypeptide that is incorporated into virions and a more highly phosphorylated 59.5-kDa protein that, while present in infected cells, is not incorporated into virions (58). The principal contribution of the present experiments is to demonstrate the association of both the 59.5-kDa and 58-kDa Vhs polypeptides with intranuclear B and C capsids. The implication is that at least some copies of Vhs are incorporated into virions by binding capsids in the nucleus prior to primary envelopment. The association of the 59.5-kDa polypeptide with intranuclear capsids also suggests that it may be an intermediate in this Vhs packaging pathway.
Several observations indicate that the association of Vhs with B and C capsids is valid and is not due to artifactual sticking of Vhs to capsids or to contamination of the B and C capsid preparations with enveloped cytoplasmic virions, enveloped primary virions, or cytoplasmic capsids resulting from the de-envelopment of primary virions. First, the overall protein profiles of the capsids were similar to those previously published for B and C capsids (23, 24, 28, 50, 51) and showed no obvious contamination with tegument or envelope proteins. Second, the B and C capsid preparations lacked detectable VP16, a major tegument protein that is easily detected within enveloped cytoplasmic and extracellular virions (27, 58, 62, 69, 72). VP16 has also been reported to be a component of enveloped primary virions (49) and to be associated with nonenveloped cytoplasmic capsids (46). However, it has been reported to not be associated with intranuclear B and C capsids (82). Thus, our observation that VP16 was missing from B and C capsid preparations that contained Vhs indicates that they were not contaminated with appreciable amounts of enveloped cytoplasmic or primary virions or nonenveloped cytoplasmic capsids. Third, the presence of the 59.5-kDa Vhs polypeptide on intranuclear capsids cannot be explained by contamination of the capsid preparations with enveloped cytoplasmic virions, because purified cytoplasmic virions lack the 59.5-kDa form of Vhs. Fourth, washing B capsids with increasing concentrations of GuHCl led to only the gradual and incomplete removal of Vhs from the capsids. The 59.5-kDa and 58-kDa Vhs polypeptides were removed with approximately equal efficiencies, more completely than some integral capsid proteins (50) but not as readily as others, indicating that the association of both Vhs forms with B capsids is strong and probably not due to fortuitous sticking. Finally, indirect immunofluorescence using anti-Vhs antibody indicates that a portion of Vhs is localized to a thin band around the nuclear rim, reminiscent of the UL31 and UL34 polypeptides, which modify the nuclear lamina and play a role in primary envelopment (59, 60). Whether this localization reflects an accumulation of Vhs just inside the inner nuclear membrane or in primary virions between the two nuclear membranes is uncertain. However, in either case, the results are consistent with the idea that some molecules of Vhs bind intranuclear capsids prior to primary envelopment at the inner nuclear membrane.
2-D gel electrophoresis resolved three forms of the Vhs polypeptide, two with an apparent molecular mass of 58 kDa and one 59.5-kDa form. This is consistent with the observation that the primary Vhs translation product and the phosphorylated virion form of the protein comigrate at 58 kDa on SDS-PAGE. It also provides a potential explanation for why the primary translation product never appeared to be completely chased to 59.5 kDa when the products of a pulse-chase experiment were analyzed by SDS-PAGE, since some of the 59.5-kDa packaging intermediate would be converted to the 58-kDa virion form, which is indistinguishable from the primary translation product on SDS-PAGE. While the difference between the apparent molecular masses of the 59.5-kDa and 58-kDa forms is due primarily to a difference in phosphorylation, it is not clear that the less acidic of the two major 58-kDa forms observed on 2-D gels is the primary Vhs translation product or that the two 58-kDa forms differ only in the extent of phosphorylation. The primary translation product may be short-lived and not very abundant and may fall in the smear of minor 58-kDa products seen in the Western blot of the 2-D gel. In addition, preliminary data indicate that one or more Vhs forms are modified by the addition of O-linked N-acetylglucosamine (M. Patterson and G. S. Read, unpublished data), a modification that is observed on many cytoplasmic and nuclear proteins (83-86). Clearly, further experiments are required to fully characterize the covalent modifications of the various forms of Vhs.
In pulse-chase experiments, processing of Vhs to 59.5 kDa could be detected within 15 min of translation, suggesting that it occurs in the cytoplasm. However, while some of the 59.5-kDa polypeptide was found in the cytoplasm, it was significantly enriched in the nucleus, suggesting that processing to 59.5 kDa correlates with transport to or retention in the nucleus. This, together with the data on the association of Vhs with B and C capsids, suggests a pathway for Vhs packaging in which some of the 58-kDa primary translation product is processed to 59.5 kDa, transported to the nucleus, binds intranuclear capsids, and is converted to the phosphorylated 58-kDa virion form at some point prior to or coincident with final envelopment.
While this is an attractive model in outline, several important questions remain. First, even though the 59.5-kDa polypeptide is enriched in the nucleus, there is no direct evidence that prior processing to 59.5 kDa is actually required for nuclear import. A significant amount of 58-kDa Vhs is found in the nucleus, and it is possible that some of the primary translation product is processed directly to the phosphorylated 58-kDa virion form, enters the nucleus, and binds B and C capsids without having to pass through the 59.5-kDa form of the protein. Within the nucleus, at least some B and C capsids contain tightly bound copies of the 59.5-kDa and 58-kDa polypeptides. Given the strength of this association, it is attractive to postulate that the Vhs polypeptides remain bound to capsids throughout the subsequent steps of primary envelopment, de-envelopment, and final envelopment at the trans-Golgi. If this is the case, bound copies of the 59.5-kDa protein must be subsequently converted to the 58-kDa virion form while associated with capsids. Alternatively, one cannot exclude the possibility that bound copies of Vhs are lost from intranuclear capsids and then replaced by new Vhs molecules at a later stage of virion maturation. However, if this is the case, it is unclear what would cause the dissociation of strongly bound copies of Vhs from the capsids. In addition, although the populations of intranuclear B and C capsids contain bound copies of the 59.5-kDa and 58-kDa polypeptides, it is unclear whether individual B or C capsids contain both forms of Vhs or just the 59.5-kDa or 58-kDa polypeptides. Clearly, many details of this pathway remain to be elucidated.
While the above data strongly support a packaging pathway involving the binding of Vhs to intranuclear capsids, other experiments suggest a second packaging pathway, similar to that proposed for a number of other tegument proteins, in which Vhs is added to virions through interactions with other tegument proteins in the cytoplasm following primary envelopment and de-envelopment of capsids or through interactions with the cytoplasmic domains of viral envelope proteins at the surface of the trans-Golgi prior to final envelopment (4, 25, 30, 37, 39, 43-45, 49, 67). The data supporting such a second pathway are severalfold. First, a subpopulation of Vhs has been reported to be associated with lipid rafts in HSV-infected cells (37). Second, as is confirmed in this study, Vhs is present in L particles, enveloped structures that are released from some infected cells and contain tegument and envelope proteins but not capsids. While the precise relevance of L particles to HSV assembly is not entirely clear, the presence of Vhs in L particles indicates that it is capable of interactions with other tegument or envelope proteins in the absence of capsids, giving rise to enveloped tegument-like structures. Previous studies showed that L particles possess virion host shutoff activity in that they can induce mRNA decay in L-particle-infected cells (42, 61). However, they did not demonstrate the presence of the Vhs protein. The contribution of the present studies is to show that L particles contain a 58-kDa Vhs polypeptide but lack the 59.5-kDa form of the protein. The implication is that the 59.5-kDa Vhs polypeptide is involved in the intranuclear pathway of Vhs incorporation but not the cytoplasmic pathway. Taken together, the data suggest a model in which virions contain two populations of Vhs that are incorporated through different pathways. The first involves binding of Vhs to intranuclear capsids prior to primary envelopment and includes the 59.5-kDa polypeptide as a packaging intermediate. The second does not involve the 59.5-kDa form or interactions between Vhs and capsids. Instead, the primary translation product is phosphorylated to the virion form and incorporated into virions in the cytoplasm, presumably through interactions with other tegument proteins or the tails of viral glycoproteins at the site of final envelopment.
This model of two Vhs populations that are distinguished by their packaging pathways is consistent with the observation that virions contain two populations of Vhs as defined by their ease of extraction with nonionic detergents and salt. Thus, approximately half of the Vhs can be released from virions by washing with 1% NP-40 and 1 M NaCl. The remaining Vhs molecules are tightly associated with capsids, as evidenced by the fact that they are removed only inefficiently and incompletely by further washing with 2 M GuHCl; in this respect they are similar to Vhs molecules bound to intranuclear B and C capsids. Whether the population of Vhs molecules that are tightly bound to capsids within virions corresponds to the population that initially interacted with capsids within the nucleus remains to be determined.
While these studies provide evidence for two pathways of Vhs packaging, the relative importance of the two pathways is unclear; specifically, it is unclear what fraction of the Vhs molecules in a virion are incorporated via each pathway. In these experiments, L particles contained about half as much Vhs per molecule of VP16 as did extracellular virions (Fig. 11). However, one should not take this as a direct measure of the fraction of Vhs that is incorporated into virions by the cytoplasmic pathway, because the precise relevance of L particles to the assembly of infectious virions is unclear. Comparison of autoradiograms of 35S-labeled virions and capsids and Western blots of the same virion and capsid preparations allows one to estimate that, on average, intranuclear capsids contain about 20% as much Vhs per molecule of the major capsid protein VP5 as do extracellular virions (data not shown). However, this may underestimate the fraction of Vhs that is packaged via the intranuclear pathway. Some molecules of Vhs may have dissociated from B and C capsids during purification. In addition, it is uncertain whether the B and C capsid preparations were uniform populations in which every capsid contained Vhs molecules or whether they were mixtures containing some capsids that had already bound Vhs and some capsids that had not. The latter possibility would have an obvious effect on attempts to estimate the number of Vhs molecules per capsid. Finally, it is uncertain what would be the advantage to the virus of having two pathways for Vhs packaging and whether the existence of two pathways implies an additional function for Vhs beyond its known role in mRNA decay. These are among the questions currently under investigation.
Acknowledgments
This work was supported by Public Health Service grant RO1 AI-21501 from the National Institute of Allergy and Infectious Diseases and by a grant from the University of Missouri Research Board.
We thank Steve Triezenberg for providing the VP16 expression plasmid and VP16-specific antiserum.
Footnotes
Published ahead of print on 8 November 2006.
REFERENCES
- 1.Bowzard, J. B., R. J. Visalli, C. B. Wilson, J. S. Loomis, E. M. Callahan, R. J. Courtney, and J. W. Wills. 2000. Membrane targeting properties of a herpesvirus tegument protein-retrovirus Gag chimera. J. Virol. 74:8692-8699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Boyle, W. J., P. Van Der Geer, and T. Hunter. 1991. Phosphopeptide mapping and phosphoamino acid analysis by two-dimensional separation on thin-layer cellulose plates, p. 110-149. In T. Hunter and B. M. Sefton (ed.), Protein phosphorylation, part B. Academic Press, Inc., San Diego, CA. [DOI] [PubMed]
- 3.Brengues, M., D. Teixeira, and R. Parker. 2005. Movement of eukaryotic mRNAs between polysomes and cytoplasmic processing bodies. Science 310:486-489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Brignati, M. J., J. S. Loomis, J. W. Wills, and R. J. Courtney. 2003. Membrane association of VP22, a herpes simplex virus type 1 tegument protein. J. Virol. 77:4888-4898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Browne, H., S. Bell, T. Minson, and D. W. Wilson. 1996. An endoplasmic reticulum-retained herpes simplex virus glycoprotein H is absent from secreted virions: evidence for reenvelopment during egress. J. Virol. 70:4311-4316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cao, D., and R. Parker. 2001. Computational modeling of eukaryotic mRNA turnover. RNA 7:1192-1212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cao, D., and R. Parker. 2003. Computational modeling and experimental analysis of nonsense-mediated decay in yeast. Cell 113:533-545. [DOI] [PubMed] [Google Scholar]
- 8.Coller, J., and R. Parker. 2004. Eukaryotic mRNA decapping. Annu. Rev. Biochem. 73:861-890. [DOI] [PubMed] [Google Scholar]
- 9.Decker, C. J., and R. Parker. 2002. mRNA decay enzymes: decappers conserved between yeast and mammals. Proc. Natl. Acad. Sci. USA 99:12512-12514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Desai, P., N. A. DeLuca, J. C. Glorioso, and S. Person. 1993. Mutations in herpes simplex virus type 1 genes encoding VP5 and VP23 abrogate capsid formation and cleavage of replicated DNA. J. Virol. 67:1357-1364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Doepker, R. C., W. L. Hsu, H. A. Saffran, and J. R. Smiley. 2004. Herpes simplex virus virion host shutoff protein is stimulated by translation initiation factors eIF4B and eIF4H. J. Virol. 78:4684-4699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Elgadi, M. M., C. E. Hayes, and J. R. Smiley. 1999. The herpes simplex virus Vhs protein induces endoribonucleolytic cleavage of target RNAs in cell extracts. J. Virol. 73:7153-7164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Elgadi, M. M., and J. R. Smiley. 1999. Picornavirus internal ribosome entry site elements target RNA cleavage events induced by the herpes simplex virus virion host shutoff protein. J. Virol. 73:9222-9231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Esclatine, A., B. Taddeo, L. Evans, and B. Roizman. 2004. The herpes simplex virus 1 UL41 gene-dependent destabilization of cellular RNAs is selective and may be sequence-specific. Proc. Natl. Acad. Sci. USA 101:3603-3608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Esclatine, A., B. Taddeo, and B. Roizman. 2004. Herpes simplex virus 1 induces cytoplasmic accumulation of TIA-1/TIAR and both synthesis and cytoplasmic accumulation of tristetraprolin, two cellular proteins that bind and destabilize AU-rich RNAs. J. Virol. 78:8582-8592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Everly, D. N., Jr., P. Feng, I. S. Mian, and G. S. Read. 2002. mRNA degradation by the virion host shutoff (Vhs) protein of herpes simplex virus: genetic and biochemical evidence that Vhs is a nuclease. J. Virol. 76:8560-8571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Everly, D. N., Jr., and G. S. Read. 1997. Mutational analysis of the virion host shutoff gene (UL41) of herpes simplex virus (HSV): characterization of HSV type 1 (HSV-1)/HSV-2 chimeras. J. Virol. 71:7157-7166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Everly, D. N., Jr., and G. S. Read. 1999. Site-directed mutagenesis of the virion host shutoff gene (UL41) of herpes simplex virus (HSV): analysis of functional differences between HSV type 1 (HSV-1) and HSV-2 alleles. J. Virol. 73:9117-9129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Feng, P., D. N. Everly, Jr., and G. S. Read. 2001. mRNA decay during herpesvirus infections: interaction between a putative viral nuclease and a cellular translation factor. J. Virol. 75:10272-10280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Feng, P., D. N. Everly, Jr., and G. S. Read. 2005. mRNA decay during herpes simplex virus (HSV) infections: protein-protein interactions involving the HSV virion host shutoff protein and translation factors eIF4H and eIF4A. J. Virol. 79:9651-9664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fenwick, M. L., and M. M. McMenamin. 1984. Early virion-associated suppression of cellular protein synthesis by herpes simplex virus is accompanied by inactivation of mRNA. J. Gen. Virol. 65:1225-1228. [DOI] [PubMed] [Google Scholar]
- 22.Friedman, A. D., S. J. Triezenberg, and S. L. McKnight. 1988. Expression of a truncated viral trans-activator selectively impedes lytic infection by its cognate virus. Nature 335:452-454. [DOI] [PubMed] [Google Scholar]
- 23.Gibson, W., and B. Roizman. 1972. Proteins specified by herpes simplex virus. 8. Characterization and composition of multiple capsid forms of subtypes 1 and 2. J. Virol. 10:1044-1052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gibson, W., and B. Roizman. 1974. Proteins specified by herpes simplex virus. Staining and radiolabeling properties of B capsid and virion proteins in polyacrylamide gels. J. Virol. 13:155-165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gross, S. T., C. A. Harley, and D. W. Wilson. 2003. The cytoplasmic tail of herpes simplex virus glycoprotein H binds to the tegument protein VP16 in vitro and in vivo. Virology 317:1-12. [DOI] [PubMed] [Google Scholar]
- 26.Harlow, E., and D. Lane. 1999. Using antibodies: a laboratory manual. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- 27.Heine, J. W., R. W. Honess, E. Cassai, and B. Roizman. 1974. Proteins specified by herpes simplex virus. XII. The virion polypeptides of type 1 strains. J. Virol. 14:640-651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Homa, F. L., and J. C. Brown. 1997. Capsid assembly and DNA packaging in herpes simplex virus. Rev. Med. Virol. 7:107-122. [DOI] [PubMed] [Google Scholar]
- 29.Jones, F. E., C. A. Smibert, and J. R. Smiley. 1995. Mutational analysis of the herpes simplex virus virion host shutoff protein: evidence that Vhs functions in the absence of other viral proteins. J. Virol. 69:4863-4871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kamen, D. E., S. T. Gross, M. E. Girvin, and D. W. Wilson. 2005. Structural basis for the physiological temperature dependence of the association of VP16 with the cytoplasmic tail of herpes simplex virus glycoprotein H. J. Virol. 79:6134-6141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Karr, B. M., and G. S. Read. 1999. The virion host shutoff function of herpes simplex virus degrades the 5′ end of a target mRNA before the 3′ end. Virology 264:195-204. [DOI] [PubMed] [Google Scholar]
- 32.Kousoulas, K. G., D. J. Bzik, N. A. DeLuca, and S. Person. 1983. The effect of ammonium chloride and tunicamycin on the glycoprotein content and infectivity of herpes simplex virus type 1. Virology 125:468-474. [DOI] [PubMed] [Google Scholar]
- 33.Krikorian, C. R., and G. S. Read. 1991. In vitro mRNA degradation system to study the virion host shutoff function of herpes simplex virus. J. Virol. 65:112-122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kwong, A. D., and N. Frenkel. 1987. Herpes simplex virus-infected cells contain a function(s) that destabilizes both host and viral mRNAs. Proc. Natl. Acad. Sci. USA 84:1926-1930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kwong, A. D., J. A. Kruper, and N. Frenkel. 1988. Herpes simplex virus virion host shutoff function. J. Virol. 62:912-921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lam, Q., C. A. Smibert, K. E. Koop, C. Lavery, J. P. Capone, S. P. Weinheimer, and J. R. Smiley. 1996. Herpes simplex virus VP16 rescues viral mRNA from destruction by the virion host shutoff function. EMBO J. 15:2575-2581. [PMC free article] [PubMed] [Google Scholar]
- 37.Lee, G. E., G. A. Church, and D. W. Wilson. 2003. A subpopulation of tegument protein Vhs localizes to detergent-insoluble lipid rafts in herpes simplex virus-infected cells. J. Virol. 77:2038-2045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Loomis, J. S., J. B. Bowzard, R. J. Courtney, and J. W. Wills. 2001. Intracellular trafficking of the UL11 tegument protein of herpes simplex virus type 1. J. Virol. 75:12209-12219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Loomis, J. S., R. J. Courtney, and J. W. Wills. 2003. Binding partners for the UL11 tegument protein of herpes simplex virus type 1. J. Virol. 77:11417-11424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lu, P., F. E. Jones, H. A. Saffran, and J. R. Smiley. 2001. Herpes simplex virus virion host shutoff protein requires a mammalian factor for efficient in vitro endoribonuclease activity. J. Virol. 75:1172-1185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lu, P., H. A. Saffran, and J. R. Smiley. 2001. The Vhs1 mutant form of herpes simplex virus virion host shutoff protein retains significant internal ribosome entry site-directed RNA cleavage activity. J. Virol. 75:1072-1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.McLauchlan, J., C. Addison, M. C. Craigie, and F. J. Rixon. 1992. Noninfectious L-particles supply functions which can facilitate infection by HSV-1. Virology 190:682-688. [DOI] [PubMed] [Google Scholar]
- 43.Mettenleiter, T. C. 2002. Herpesvirus assembly and egress. J. Virol. 76:1537-1547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mettenleiter, T. C. 2004. Budding events in herpesvirus morphogenesis. Virus Res. 106:167-180. [DOI] [PubMed] [Google Scholar]
- 45.Mettenleiter, T. C., T. Minson, and P. Wild. 2006. Egress of alphaherpesviruses. J. Virol. 80:1610-1612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Miranda-Saksena, M., R. A. Boadle, P. Armati, and A. L. Cunningham. 2002. In rat dorsal root ganglion neurons, herpes simplex virus type 1 tegument forms in the cytoplasm of the cell body. J. Virol. 76:9934-9951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mitchell, P., and D. Tollervey. 2000. mRNA stability in eukaryotes. Curr. Opin. Genet. Dev. 10:193-198. [DOI] [PubMed] [Google Scholar]
- 48.Mitchell, P., and D. Tollervey. 2001. mRNA turnover. Curr. Opin. Cell Biol. 13:320-325. [DOI] [PubMed] [Google Scholar]
- 49.Naldinho-Souto, R., H. Browne, and T. Minson. 2006. Herpes simplex virus tegument protein VP16 is a component of primary enveloped virions. J. Virol. 80:2582-2584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Newcomb, W. W., and J. C. Brown. 1991. Structure of the herpes simplex virus capsid: effects of extraction with guanidine hydrochloride and partial reconstitution of extracted capsids. J. Virol. 65:613-620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Newcomb, W. W., F. L. Homa, D. R. Thomsen, Z. Ye, and J. C. Brown. 1994. Cell-free assembly of the herpes simplex virus capsid. J. Virol. 68:6059-6063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Oroskar, A. A., and G. S. Read. 1987. A mutant of herpes simplex virus type 1 exhibits increased stability of immediate-early (alpha) mRNAs. J. Virol. 61:604-606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Oroskar, A. A., and G. S. Read. 1989. Control of mRNA stability by the virion host shutoff function of herpes simplex virus. J. Virol. 63:1897-1906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Pak, A. S., D. N. Everly, K. Knight, and G. S. Read. 1995. The virion host shutoff protein of herpes simplex virus inhibits reporter gene expression in the absence of other viral gene products. Virology 211:491-506. [DOI] [PubMed] [Google Scholar]
- 55.Parker, R., and H. Song. 2004. The enzymes and control of eukaryotic mRNA turnover. Nat. Struct. Mol. Biol. 11:121-127. [DOI] [PubMed] [Google Scholar]
- 56.Read, G. S. 1997. Control of mRNA stability during herpes simplex virus infections, p. 311-321. In J. B. Harford and D. R. Morris (ed.), mRNA metabolism and post-transcriptional gene regulation. Wiley-Liss, Inc., New York, NY.
- 57.Read, G. S., and N. Frenkel. 1983. Herpes simplex virus mutants defective in the virion-associated shutoff of host polypeptide synthesis and exhibiting abnormal synthesis of alpha (immediate-early) viral polypeptides. J. Virol. 46:498-512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Read, G. S., B. M. Karr, and K. Knight. 1993. Isolation of a herpes simplex virus type 1 mutant with a deletion in the virion host shutoff gene and identification of multiple forms of the Vhs (UL41) polypeptide. J. Virol. 67:7149-7160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Reynolds, A. E., B. J. Ryckman, J. D. Baines, Y. Zhou, L. Liang, and R. J. Roller. 2001. U(L)31 and U(L)34 proteins of herpes simplex virus type 1 form a complex that accumulates at the nuclear rim and is required for envelopment of nucleocapsids. J. Virol. 75:8803-8817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Reynolds, A. E., E. G. Wills, R. J. Roller, B. J. Ryckman, and J. D. Baines. 2002. Ultrastructural localization of the herpes simplex virus type 1 UL31, UL34, and US3 proteins suggests specific roles in primary envelopment and egress of nucleocapsids. J. Virol. 76:8939-8952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Rixon, F. J., C. Addison, and J. McLauchlan. 1992. Assembly of enveloped tegument structures (L particles) can occur independently of virion maturation in herpes simplex virus type 1-infected cells. J Gen. Virol. 73:277-284. [DOI] [PubMed] [Google Scholar]
- 62.Roizman, B., and D. M. Knipe. 2001. Herpes simplex viruses and their replication, p. 2399-2459. In D. M. Knipe, P. M. Howley, D. E. Griffin, R. A. Lamb, M. A. Martin, B. Roizman, and S. E. Straus (ed.), Fields virology, 4th ed. Lippincott Williams & Williams, Philadelphia, PA.
- 63.Sacks, W. R., and P. A. Schaffer. 1987. Deletion mutants in the gene encoding the herpes simplex virus type 1 immediate-early protein ICP0 exhibit impaired growth in cell culture. J. Virol. 61:829-839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Schek, N., and S. L. Bachenheimer. 1985. Degradation of cellular mRNAs induced by a virion-associated factor during herpes simplex virus infection of Vero cells. J. Virol. 55:601-610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Schmelter, J., J. Knez, J. R. Smiley, and J. P. Capone. 1996. Identification and characterization of a small modular domain in the herpes simplex virus host shutoff protein sufficient for interaction with VP16. J. Virol. 70:2124-2131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Sheth, U., and R. Parker. 2003. Decapping and decay of messenger RNA occur in cytoplasmic processing bodies. Science 300:805-808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Skepper, J. N., A. Whiteley, H. Browne, and A. Minson. 2001. Herpes simplex virus nucleocapsids mature to progeny virions by an envelopment→deenvelopment→reenvelopment pathway. J. Virol. 75:5697-5702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Smibert, C. A., D. C. Johnson, and J. R. Smiley. 1992. Identification and characterization of the virion-induced host shutoff product of herpes simplex virus gene UL41. J. Gen. Virol. 73:467-470. [DOI] [PubMed] [Google Scholar]
- 69.Smibert, C. A., B. Popova, P. Xiao, J. P. Capone, and J. R. Smiley. 1994. Herpes simplex virus VP16 forms a complex with the virion host shutoff protein Vhs. J. Virol. 68:2339-2346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Sorenson, C. M., P. A. Hart, and J. Ross. 1991. Analysis of herpes simplex virus-induced mRNA destabilizing activity using an in vitro mRNA decay system. Nucleic Acids Res. 19:4459-4465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Spang, A. E., P. J. Godowski, and D. M. Knipe. 1983. Characterization of herpes simplex virus 2 temperature-sensitive mutants whose lesions map in or near the coding sequences for the major DNA-binding protein. J. Virol. 45:332-342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Spear, P. G., and B. Roizman. 1972. Proteins specified by herpes simplex virus. V. Purification and structural proteins of the herpesvirion. J. Virol. 9:143-159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Strom, T., and N. Frenkel. 1987. Effects of herpes simplex virus on mRNA stability. J. Virol. 61:2198-2207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Szilagyi, J. F., and C. Cunningham. 1991. Identification and characterization of a novel non-infectious herpes simplex virus-related particle. J. Gen. Virol. 72:661-668. [DOI] [PubMed] [Google Scholar]
- 75.Taddeo, B., A. Esclatine, and B. Roizman. 2004. Post-transcriptional processing of cellular RNAs in herpes simplex virus-infected cells. Biochem. Soc. Trans. 32:697-701. [DOI] [PubMed] [Google Scholar]
- 76.Taddeo, B., A. Esclatine, W. Zhang, and B. Roizman. 2003. The stress-inducible immediate-early responsive gene IEX-1 is activated in cells infected with herpes simplex virus 1, but several viral mechanisms, including 3′ degradation of its RNA, preclude expression of the gene. J. Virol. 77:6178-6187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Taddeo, B., W. Zhang, and B. Roizman. 2006. The U(L)41 protein of herpes simplex virus 1 degrades RNA by endonucleolytic cleavage in absence of other cellular or viral proteins. Proc. Natl. Acad. Sci. USA 103:2827-2832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Thurlow, J. K., F. J. Rixon, M. Murphy, P. Targett-Adams, M. Hughes, and V. G. Preston. 2005. The herpes simplex virus type 1 DNA-packaging protein UL17 is a virion protein that is present in both the capsid and the tegument compartments. J. Virol. 79:150-158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Whiteley, A., B. Bruun, T. Minson, and H. Browne. 1999. Effects of targeting herpes simplex virus type 1 gD to the endoplasmic reticulum and trans-Golgi network. J. Virol. 73:9515-9520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Yaciuk, P., and E. Moran. 1991. Analysis with specific polyclonal antiserum indicates that the E1A-associated 300-kDa product is a stable nuclear phosphoprotein that undergoes cell cycle phase-specific modification. Mol. Cell. Biol. 11:5389-5397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Yao, F., and R. J. Courtney. 1989. A major transcriptional regulatory protein (ICP4) of herpes simplex virus type 1 is associated with purified virions. J. Virol. 63:3338-3344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Yao, F., and R. J. Courtney. 1992. Association of ICP0 but not ICP27 with purified virions of herpes simplex virus type 1. J. Virol. 66:2709-2716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Zachara, N. E., W. D. Cheung, and G. W. Hart. 2004. Nucleocytoplasmic glycosylation, O-GlcNAc: identification and site mapping. Methods Mol. Biol. 284:175-194. [DOI] [PubMed] [Google Scholar]
- 84.Zachara, N. E., and G. W. Hart. 2004. O-GlcNAc, a sensor of cellular state: the role of nucleocytoplasmic glycosylation in modulating cellular function in response to nutrition and stress. Biochim. Biophys. Acta 1673:13-28. [DOI] [PubMed] [Google Scholar]
- 85.Zachara, N. E., and G. W. Hart. 2006. Cell signaling, the essential role of O-GlcNAc! Biochim. Biophys. Acta 1761:599-617. [DOI] [PubMed] [Google Scholar]
- 86.Zachara, N. E., N. O'Donnell, W. D. Cheung, J. J. Mercer, J. D. Marth, and G. W. Hart. 2004. Dynamic O-GlcNAc modification of nucleocytoplasmic proteins in response to stress. A survival response of mammalian cells. J. Biol. Chem. 279:30133-30142. [DOI] [PubMed] [Google Scholar]
- 87.Zelus, B. D., R. S. Stewart, and J. Ross. 1996. The virion host shutoff protein of herpes simplex virus type 1: messenger ribonucleolytic activity in vitro. J. Virol. 70:2411-2419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Zhang, Y., and J. L. McKnight. 1993. Herpes simplex virus type 1 UL46 and UL47 deletion mutants lack VP11 and VP12 or VP13 and VP14, respectively, and exhibit altered viral thymidine kinase expression. J. Virol. 67:1482-1492. [DOI] [PMC free article] [PubMed] [Google Scholar]