

Abstract
Respiratory syncytial virus (RSV) is one of the most common viral pathogens causing severe lower respiratory tract infections in infants and young children. Infected host cells detect and respond to RNA viruses using different mechanisms in a cell-type-specific manner, including retinoic acid-inducible gene I (RIG-I)-dependent and Toll-like receptor (TLR)-dependent pathways. Because the relative contributions of these two pathways in the recognition of RSV infection are unknown, we examined their roles in this study. We found that RIG-I helicase binds RSV transcripts within 12 h of infection. Short interfering RNA (siRNA)-mediated RIG-I “knockdown” significantly inhibited early nuclear factor-κB (NF-κB) and interferon response factor 3 (IRF3) activation 9 h postinfection (p.i.). Consistent with this finding, RSV-induced beta interferon (IFN-β), interferon-inducible protein 10 (IP-10), chemokine ligand 5 (CCL-5), and IFN-stimulated gene 15 (ISG15) expression levels were decreased in RIG-I-silenced cells during the early phase of infection but not at later times (18 h p.i.). In contrast, siRNA-mediated TLR3 knockdown did not affect RSV-induced NF-κB binding but did inhibit IFN-β, IP-10, CCL-5, and ISG15 expression at late times of infection. Further studies revealed that TLR3 knockdown significantly reduced NF-κB/RelA transcription by its ability to block the activating phosphorylation of NF-κB/RelA at serine residue 276. We further found that TLR3 induction following RSV infection was regulated by RIG-I-dependent IFN-β secreted from infected airway epithelial cells and was mediated by both IFN response-stimulated element (ISRE) and signal transducer and activator of transcription (STAT) sites in its proximal promoter. Together these findings indicate distinct temporal roles of RIG-I and TLR3 in mediating RSV-induced innate immune responses, which are coupled to distinct pathways controlling NF-κB activation.
Respiratory syncytial virus (RSV) is the most frequent cause of bronchiolitis and pneumonia in young children requiring hospitalization worldwide (36). In the United States alone, lower respiratory tract infection with RSV is responsible for over 100,000 hospitalizations annually (47). RSV is a single-stranded RNA virus of the Paramyxoviridae family whose major site for productive replication is the epithelial cells in the airway mucosa (2, 18). Here, RSV replication induces cytokine and chemokine gene expression networks in a coordinated manner (6, 14, 43, 48, 56). One important cytokine network is the expression of type I interferons (IFNs), including alpha interferon (IFN-α), IFN-β, IFN-Ω, IFN-ɛ, and IFN-κ, which are responsible for producing an antiviral state and stimulating effector arms of cellular immunity (22, 42).
The detection of RNA virus by host cells occurs in a cell-type- and pathogen-type-specific manner. The sensors for viral infection primarily involve two kinds of receptors: the cytoplasmic pattern recognition receptors, including that for the retinoic acid-inducible gene I (RIG-I) and the pathogen-associated molecular pattern receptors known as the Toll-like receptors (TLRs) (20, 23, 37, 38). RIG-I is a highly inducible cytoplasmic RNA helicase that signals antiviral responses after binding double-stranded RNA (dsRNA), a pathway that has been implicated in antiviral responses to Sendai virus, vesicular stomatitis virus, and Newcastle disease virus, as well as the flaviviruses Japanese encephalitis virus, dengue virus 2, and hepatitis C virus (7, 24, 45). However, the role of RIG-I is cell-type dependent; for example, in fibroblast cells, RIG-I is the major sensor for viral infection, but in plasmacytoid dendritic cells, TLRs play a more important role (24, 53). The role of RIG-I in mediating epithelial cell response to RSV infection in airway epithelial cells has not been investigated. The TLRs are a family of membrane-bound pattern receptors expressed in a cell-type-specific manner that recognize distinct classes of virus- and bacterium-derived ligands (3, 4). In the case of RSV, TLRs 3 and 4 are thought to be major mediators of virus-inducible signaling. TLR3 binds dsRNA, the replication intermediate of RSV and other RNA viruses (10). In fact, others have recently shown that short interfering RNA (siRNA)-mediated TLR3 silencing results in the inhibition of RSV-induced chemokine ligand 5 (CCL-5) and interferon-inducible protein 10 (IP-10) expression in airway epithelial cells (32). In addition, although TLR4 binds lipopolysaccharide as its ligand, the RSV fusion glycoprotein binds TLR4 directly, mediating inducible-cytokine activation in monocytes (26). Among all TLRs, TLR3 is one of the most abundant isoforms expressed in airway epithelial cells (40). Moreover, two groups have recently reported that RSV infection induces TLR3 expression and cell-surface translocation through incompletely understood mechanisms (15, 33). Although these studies suggest that TLR3 may mediate epithelial responses to RSV infection, its antiviral role is controversial and the mechanism for its upregulation following RSV infection is unknown.
In this study, we examined the roles of RIG-I and TLR3 in RSV-induced gene expression in transformed human alveolar epithelial cells (A549). In the UV-cross-linking experiment, active RIG-I bound RSV transcripts within 12 h of RSV exposure. siRNA-mediated RIG-I silencing inhibited the activation of both NF-κB and IRF-3, as well as IFN-β, IP-10, CCL-5, and ISG15 expression, at the early phase of RSV infection (9 h p.i.). Surprisingly, siRNA-mediated TLR3 “knockdown” had little influence on the early response of RSV-induced genes but significantly inhibited their late expression. The role of TLR3 is not related to controlling RSV-induced NF-κB DNA binding, but rather is required to mediate the RSV-induced activating RelA at serine residue 276. We further found that the TLR3 expression was RSV inducible via a transcriptional mechanism mediated by two IFN-β-responsive ISRE and STAT binding sites in its proximal promoter. We further found that siRNA-mediated RIG-I silencing inhibited the upregulation of TLR3 and that the paracrine IFN-β secreted from infected cells was necessary and sufficient to induce the expression of TLR3. Together, these data indicate that TLR3 signaling in RSV infection is dependent on early RIG-I signaling and controls NF-κB/RelA subunit phosphorylation.
MATERIALS AND METHODS
Cell culture.
Human A549 pulmonary type II epithelial cells (American Type Culture Collection [ATCC]) were grown in F12K medium (Gibco) with 10% fetal bovine serum (FBS), penicillin (100 U/ml), and streptomycin (100 μg/ml) at 37°C in a 5% CO2 incubator. African green monkey kidney Vero cells (ATCC) were cultured in Eagle's minimum essential medium with 0.1 mM nonessential amino acids, 1.0 mM sodium pyruvate, and 10% FBS.
Virus preparation and infection.
The human RSV A2 strain was grown in HEp-2 cells and purified by centrifugation on discontinuous sucrose gradients (14). The viral titer of purified RSV pools was 8 to 9 log PFU/ml, determined by a methylcellulose plaque assay. Viral pools were aliquoted, quick-frozen on dry ice-ethanol, and stored at −70°C until they were used. For viral adsorption, cells were transferred into F12K medium containing 2% (vol/vol) FBS and RSV infected at a multiplicity of infection (MOI) of 1 for 18 h prior to harvest and assay.
siRNA-mediated gene silencing.
siRNA against human RIG-I (M-012511-00) and TLR3 (M-007745-00) and control siRNA (D-001206-13) were commercially obtained (Dharmacon Research, Inc., Lafayette, CO) and transfected at 100 nM into A549 cells by using a TransIT-siQuest transfection kit (Mirus Bio Corp., Madison, WI) according to the manufacturer's instructions. Forty-eight hours after transfection, cells were RSV infected for the times indicated in the figure legends. The efficiency of siRNA silencing was evaluated using reverse transcriptase PCR (RT-PCR).
Plasmid construction.
For the TLR3-Luc reporter, 1.0 kb of the human TLR3 promoter was amplified from A549 cell genomic DNA by PCR using the forward primer 5′-TCAGAGGATCCGGCATGTTCTTAGGCAAACC-3′ and reverse primer 5′-TCAGAGATATCCTGTTGGATGACTGCTAGCC-3′. The PCR product was digested with BamHI/SmaI, gel purified, and ligated into the same sites in the pOLUC plasmid (5). Site-directed mutagenesis was conducted by rolling circle PCR (18 cycles) to mutate the ISRE1 site (forward primer, 5′-CCTCCCTAGGTTTCGCGCTCCTAATTTCTCAAA-3′; reverse primer, 5′-TTTGAGAAATTAGGAGCGCGAAACCTAGGGAGG-3′), the ISRE2 site (forward primer, 5′-AAGCTTTACTTTCACGATCGAGAGTGCCGTCT-3′; reverse primer, 5′-AGACGGCACTCTCGATCGTGAAAGTAAAGCTT-3′), and the STAT site (forward primer, 5′-TTTCTCCCTTTGCCCCCCTTGGAATGCACCAA-3′; reverse primer, 5′-TTGGTGCATTCCAAGGGGGGCAAAGGGAGAAA-3′). The pT1S vector was constructed by removing the cytomegalovirus (CMV) promoter from pCDNA6-V5-HisB (Invitrogen) and replacing it with a fragment containing the cytomegalovirus (CMV) promoter driving the tetracycline transactivator, followed by the simian virus 40 poly(A) signal and a tetracycline-responsive element (TRE). pT1S-RIG-I and pT1S-MDA5 were generated by inserting Flag-tagged RIG-I and Flag-tagged MDA5 into pT1S under the control of the TRE. Plasmids were purified by ion exchange chromatography (QIAGEN, Chatsworth, CA), and mutations were sequenced to verify authenticity. The PRDII luciferase plasmid was a kind gift from Michael Gale (12).
Helicase RNA UV cross-linking and immunoprecipitation.
The UV-cross-linking assay is modified from the assay described in reference 30. A549 cells were transfected with pT1S vector, pT1S-FLAG-RIG-I, or pT1S-FLAG-MDA5 for 48 h, and the transfectants were then infected with RSV (MOI = 1, 12 h). Cells were washed with prechilled phosphate-buffered saline (PBS) and UV irradiated for 5 min with an 8 W germicidal lamp at a 4-cm distance (GS gene linker; Bio-Rad). Cells were suspended in 500 μl of immunoprecipitation buffer (20 mM Tris-HCl, pH 7.4, 0.15 M NaCl, 5 mM EDTA, 4 μg/ml each leupeptin and pepstatin, and 1 mM phenylmethylsulfonyl fluoride), and lysed by sonication for 20 s. The lysates were treated with 20 U of RNase T1 and 10 μg of RNase A for 30 min at 37°C to remove unbound RNA. Immunoprecipitation was conducted with anti-FLAG M2 antibody (Ab) for 4 h at 4°C and with protein A-Sepharose for 1 h at 4°C. Total RNA was isolated from half of the immunoprecipitation using proteinase K, and the other half of the immunoprecipitation was fractionated by sodium dodecyl sulfate (SDS)-polyacrylamide gel electrophoresis for Western immunoblotting. Half of the RNA sample was subjected to 35 cycles of RT-PCR with RSV N protein-specific primers and visualized by agarose gel electrophoresis.
RT-PCR and quantitative real-time PCR (QRT-PCR).
Total RNA was extracted using acid guanidium phenol extraction (Tri Reagent; Sigma), and 1 μg of RNA was reverse transcribed using Moloney murine leukemia virus reverse transcriptase (New England Biolabs) in a 20-μl reaction mixture. One μl of cDNA product was diluted 1:2, and 2 μl was amplified in a 25-μl reaction mixture containing 12.5 μl of SYBR green supermix (Bio-Rad) and 0.4 μM each of forward and reverse gene-specific primers (Table 1), aliquoted into 96-well, 0.2-mm thin-wall PCR plates, and covered with optical-quality sealing tape. The plates were denatured for 90 s at 95°C and then subjected to 40 cycles of 15 s at 94°C, 60 s at 60°C, and 1 min at 72°C in an ABI 7000 thermocycler. After PCR was performed, PCR products were run on 2% agarose gels to assure a single amplification product. Duplicate cycle threshold (CT) values were analyzed by using the comparative CT (ΔΔCT) method (Applied Biosystems). The relative amount of target mRNA (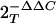 ) was obtained by normalizing to endogenous GAPDH (glyceraldehyde-3-phosphate dehydrogenase) reference and expressed relative to the amount from uninfected cells.
) was obtained by normalizing to endogenous GAPDH (glyceraldehyde-3-phosphate dehydrogenase) reference and expressed relative to the amount from uninfected cells.
TABLE 1.
Forward and reverse gene-specific primers
| Gene name | Primer sequence (5′-3′)
|
|
|---|---|---|
| Forward | Reverse | |
| RIG-I | ATTGCCACCTCAGTTGCTGAT | ACATACTCATAAAGGATGACAAGATTG |
| IFN-β | CAACTTGCTTGGATTCCTACAAAG | TGCCACAGGAGCTTCTGACA |
| CCL5 | CATCTGCCTCCCCATATTCCT | GCGGGCAATGTAGGCAAA |
| IP10 | ATTATTCCTGCAAGCCAATTTTG | TCACCCTTCTTTTTCATTGTAGCA |
| ISG15 | GGTGGACAAATGCGACGAA | TGCTGCGGCCCTTGTTAT |
| RSV N | AAGGGATTTTTGCAGGATTGTTT | TCCCCACCGTAACATCACTTG |
| GAPDH | GATCATCAGCAATGCCTCCT | TGTGGTCATGAGTCCTTCCA |
| TLR3 | GGTCCCAAGCCTTCAACGA | GGTGAAGGAGAGCTATCCACATTT |
Electrophoretic mobility shift assay (EMSA).
Nuclear extracts (NE) were prepared as described previously (17). A total of 5 μg NE was incubated in DNA binding buffer (5% glycerol, 12 mM HEPES, 80 mM NaCl, 5 mM dithiothreitol, 5 mM MgCl2, 0.5 mM EDTA) with 1.5 μg of poly(dA-dT) and 0.1 nM IRDye 700/IRDye 800-labeled ds oligonucleotide (Table 2) in a total volume of 10 μl. Complexes were fractionated by native polyacrylamide gels in 1× TBE buffer (89 mM Tris, 89 mM boric acid, 2 mM EDTA). For competition, unlabeled ds competitor was added at the time of the binding reaction. Gels were then scanned in an Odyssey infrared scanner (Odyssey system; Licor Biosciences, Lincoln, NE).
TABLE 2.
Sense and antisense EMSA probes
| Gene name | Strand | Primer sequence (5′-3′) |
|---|---|---|
| ISRE | Sense | GATCGGAAAGGGAAACCGAAACTGAAGCC |
| Antisense | GGCTTCAGTTTCGGTTTCCCTTTCCGATC | |
| ISREm | Sense | GATCGGGCGGGGGCGCCGGCGCTGAAGCC |
| Antisense | GGCTTCAGCGCCGGCGCCCCCGCCCGATC | |
| kB | Sense | GATGCCATTGGGGATTTCCTCTTTACTG |
| Antisense | CAGTAAAGAGGAAATCCCCAATGGCATC | |
| OCT | Sense | GATCCGAGCTTCACCTTATTTGCATAAGCGATTGA |
| Antisense | TCAATCGCTTATGCAAATAAGGTGAAGCTCGGATC |
Immunofluorescence microscopy.
A549 cells (105) plated on coverslips were mock or RSV infected (MOI = 1) for the times indicated in the legend to Fig. 3. The cells were fixed with 4% paraformaldehyde in PBS, pH 7.4. The cells were then incubated for 60 min at 37°C with anti-IRF-3 Ab or anti-NF-κB/RelA C20 Ab (Santa Cruz) diluted 1:200 in PBS-T (PBS, 0.1% Tween 20). Cells were washed three times in PBS-T and incubated with secondary fluorescein isothiocyanate-conjugated anti-rabbit Ab in PBS-T for 1 h at 22°C. Nuclei were visualized by staining for 15 min with SYTOX orange (Molecular Probes). Confocal microscopy was performed on a Zeiss LSM510 META system. Images were captured at a magnification of ×40. For each condition, 10 pictures were taken and the percentage of cells which showed the nuclear staining for IRF-3 or RelA was counted and expressed as the percentage of total cells examined.
FIG.3.


RSV activates NF-κB and IRF-3 through the RIG-I pathway at the early phase of infection. (A) A549 cells were transfected with 100 nM of nonspecific siRNA as control (Con), RIG-I siRNA (RIG-I), or TLR3 siRNA (TLR3) for 48 h, followed by RSV infection for 9 or 18 h. NE from each siRNA treatment were prepared and assayed by EMSA. Shown are bound complexes on the IRDye 700-labeled κB oligonucleotides visualized by infrared scanning (left panel). A competition experiment was performed using the sample from the control siRNA-treated group that was infected with RSV for 18 h and incubated with 0, 0.5, or 1 nM unlabeled oligonucleotides (right panel). −, none. (B) IRF-3 binding at different times of RSV infection. EMSA was performed on NE using 0.1 nM IRDye 700-labeled ISRE binding site (left panel). A competition experiment with unlabeled probe and mutant probe was conducted (right panel). −, none. (C) OCT-1 binding. EMSA was performed using the same NE, binding 0.1 nM IRDye 800-labeled OCT-1 binding site. (D) A549 cells were transfected with either control (Con) or RIG-I siRNA (RIG-I) for 48 h and then RSV infected for 0, 9, or 18 h. The cells were fixed, incubated with rabbit anti-RelA Ab, and then stained with fluorescein isothiocyanate-conjugated anti-rabbit secondary Ab (top panels). The nuclei were stained with Sytox orange (middle panels). The slides were imaged using confocal microscopy, and colors were merged (bottom panels). Colocalization of RelA and nuclei is shown by light grey. White arrows indicate the cells which had RelA nuclear translocation. (E) A549 cells were treated as in (D), except that rabbit anti-IRF-3 Ab was used. Colocalization of IRF-3 and nuclei is shown by light grey and indicated by white arrows. (F) The percentages of cells with nuclei positive for RelA or IRF-3 at each time point and for each treatment were calculated based on five randomly photographed fields from two independent experiments. Asterisks indicate a significant difference between siRNA groups at the same time point of RSV infection (P < 0.05, Student's test). Error bars indicate standard deviations.
RSV-CM collection and IFN neutralization.
RSV-conditioned medium (RSV-CM) was prepared by infecting A549 monolayers with RSV (MOI = 1, 48-h incubation). The supernatant was collected, centrifuged at 3,000 × g, exposed to UV light to inactivate the live virus, quick-frozen, and stored at −70°C until used. For IFN neutralization, 20 μl of RSV-CM was mixed with either 15 μg of rabbit anti-human IFN-β Ab (Chemicon International) or 15 μg of rabbit immunoglobulin G (IgG) in a total volume of 2 ml of culture medium and incubated for 2 h at 37°C.
Northern blots.
Total RNA (30 μg) was fractionated by electrophoresis on a 1% agarose-formaldehyde gel and transferred to a nylon membrane (Zeta Probe GT; Bio-Rad). A TLR3 cDNA probe was made using asymmetric PCR. The membrane was hybridized with 2 × 106 cpm/ml of radiolabeled probe at 60°C overnight in 5% SDS hybridization buffer (55). The membrane was washed with 1× SSC (1× SSC is 0.15 M NaCl plus 0.015 M sodium citrate) with 0.1% SDS for 20 min at 60°C. Internal control hybridization was carried out with β-actin mRNA. The image was developed and quantified by exposing the membrane to a Molecular Dynamics phosphorimager cassette.
Western immunoblot.
Whole-cell extracts were prepared using modified radioimmunoprecipitation assay buffer (50 mM Tris-HCl [pH 7.4], 150 mM NaCl, 1 mM EDTA, 0.25% sodium deoxycholate, 1% Nonidet P-40, 1 mM phenylmethylsulfonyl fluoride, 1 mM NaF, 1 mM Na3VO4, and 1 μg each of aprotinin, leupeptin, and pepstatin/ml). One hundred micrograms protein was fractionated by 10% SDS-polyacrylamide gel electrophoresis and transferred to a polyvinylidene difluoride membrane by electroblotting. Membranes were blocked in 5% nonfat dry milk in Tris-buffered saline-0.1% Tween and probed with the primary Ab indicated in the figure legends. Membranes were washed and incubated with IRDye 700-conjugated anti-mouse Ab or IRDye 800-conjugated anti-rabbit Ab (Rockland, Inc.). Finally, the membranes were washed three times with PBS-T and scanned by infrared scanner. RelA C20 Ab (Santa Cruz), anti-phospho-276 RelA Ab (Cell Signaling), anti-phospho-536 RelA Ab (Cell Signaling), and Anti-FLAG M2 Ab (Sigma) were commercially obtained.
RESULTS
RSV infection activates NF-κB and IRF-3 in airway epithelial cells.
A549 cells are human epithelial cells that retain morphological features of type II alveolar cells, including surfactant secretion, and support productive RSV replication (27). Previous work has shown that RSV infection activates the nuclear factor-κB (NF-κB) and IRF-3 pathways (6, 16, 21), both central mediators of antiviral cytokine expression. To test their relative kinetics of activation, A549 cells were RSV infected (MOI = 1); NF-κB pathway activation was measured using an EMSA (Fig. 1A), and nuclear IRF-3 activation was measured by Western immunoblotting (Fig. 1B). Consistent with previous work, the NF-κB binding assay in the EMSA showed three distinct complexes (Fig. 1A) (14, 21). The less-mobile complex showed a time-dependent increase in response to RSV infection; previous work has shown that this species represents the heterodimer of the 65-kDa RelA-transactivating subunit and the 50-kDa NF-κB1 DNA binding subunit (14, 21). Conversely, the middle complex, representing homodimers of the 50-kDa NF-κB1 DNA subunit, was not affected by RSV treatment. The most-mobile band represents nonspecific DNA binding species. We noted that a significant increase in NF-κB binding was detected 6 h after RSV infection, which suggested the activation of NF-κB in response to RSV infection (Fig. 1A).
FIG. 1.

RSV activates NF-κB and IRF-3 in A549 cells. A549 cells were infected by the human RSV A2 strain (MOI = 1.0) for different times as indicated (in hours), and NE was prepared. (A) EMSA was performed on 5 μg NE using 0.1 nM IRDye 700-labeled DNA probe containing the κB element. The composition of the bound complexes is indicated. RelA · p50, RelA-p50 heterodimer; p50, p50 homodimer; N.S., nonspecific product. (B) Western immunoblotting was conducted on 50 μg NE. Top panel, the membrane was stained with anti-IRF-3 Ab; bottom panel, β-actin was stained as a loading control.
Similarly, IRF-3 pathway activation was rapid, with an increase of nuclear IRF-3 6 h after RSV infection, peaking after 12 h (Fig. 1B). The multiple bands detected represented different phosphorylation forms of IRF-3, characteristic of its activation mechanism (6, 41). Together, these data indicate that RSV activates both NF-κB and IRF-3 translocation rapidly, within 6 h after RSV infection.
The RIG-I pathway mediates the early phase of RSV-induced gene expression.
Although RSV-induced activation of the epithelial TLR3 pathway has been discovered recently, the role of the RIG-I pathway is unclear. RIG-I is a cytoplasmic helicase that activates signaling when it binds RNA. To investigate whether there is a physical interaction between RIG-I and RSV replication products, an RNA helicase UV-cross-linking experiment was performed. In this assay, eukaryotic expression vectors encoding nothing, FLAG-tagged RIG-I, or FLAG-tagged MDA5 were transfected into A549 cells. After 24 h, the transfectants were RSV infected for 12 h and RNA was UV cross-linked to cellular proteins. To determine whether RIG-I or MDA5 bound RSV transcripts, epitope-flagged protein was immunoprecipitated by anti-FLAG Ab, and the released RNA was assayed by RT-PCR. We found that a specific 69-bp band corresponding to the RSV N protein transcript was detected only in immunoprecipitates from RIG-I-transfected cells (Fig. 2A).
FIG. 2.

RIG-I mediates early innate immune response to RSV infection. (A) RNA helicase UV cross-linking and immunoprecipitation assay. A549 cells were transfected with 2 μg pT1S vector as control (Con), pT1S-FLAG-RIG-I (RIG), or pT1S-FLAG-MDA5 (MDA) for 24 h. Thereafter, cells were infected with RSV for 12 h. UV cross-linking and immunoprecipitation experiments were conducted as described in Materials and Methods. RT-PCR yielded two bands in the sample corresponding to RIG-I immunoprecipitation. The upper band was confirmed as RSV N protein RNA by sequencing (top panel); the bottom band is a nonspecific product (N.S.). Molecular sizes in base pairs (bp) are shown on the left. The bottom panel shows a Western immunoblot of the immunoprecipitates, including RIG-I and MDA5. Specific bands are indicated by asterisks. Molecular sizes are shown on the left. M, marker. (B) A549 cells were transfected with 100 nM of nonspecific siRNA as control (Con), RIG-I siRNA (RIG-I), or TLR3 siRNA (TLR3) for 48 h. Cells were infected by RSV for 0, 9, or 18 h, and total RNA was extracted. QRT-PCR was performed to determine changes in RIG-I (top panel) or TLR3 (bottom panel) expression levels as indicated. *, P is <0.01 compared to control siRNA (Student's t test). (C and D) A549 cells were transfected with control (Con), RIG-I siRNA (RIG-I), or TLR3 siRNA (TLR3) for 48 h and then RSV infected for 0, 9, or 18 h. CCL-5, IP-10, IFN-β, and ISG15 expression levels were determined by QRT-PCR; shown are the changes (n-fold) relative to unstimulated cells transfected with control siRNA (0 h). *, P is <0.05 relative to the corresponding group at the same time point (Student's t test). (C) Results of QRT-PCR of cells transfected with RIG-I siRNA and (D) cells transfected with TLR3 siRNA. Error bars indicate standard deviations.
To further understand the roles of RIG-I and TLR3 in RSV-induced gene expression, A549 cells were transfected with RIG-I, TLR3, or control siRNA and subsequently RSV infected for 0, 9, or 18 h, and QRT-PCR was used to measure the antiviral gene expression. RSV induced RIG-I expression by 30-fold within 9 h of infection, which, significantly, fell to 20-fold after 18 h (Fig. 2B). Relative to control siRNA-transfected cells, RIG-I siRNA-transfected cells showed significant inhibition of RIG-I expression at 9 h and less inhibition after 18 h of RSV infection (Fig. 2B). Similarly, RSV induced TLR3 expression 22- and 25-fold at 9 and 18 h, respectively. TLR3 knockdown significantly reduced RSV-induced TLR3 expression at both times. These data indicate that RSV induced both RIG and TLR3 expression and that siRNA knockdown was effective.
We next examined the effect of RIG-I knockdown on RSV-induced IFN-β, IP-10, ISG15, and CCL-5 expression (Fig. 2C). RSV strongly induced IFN-β, IP-10, and ISG15 production, at 100-fold, 280-fold, and 75-fold, respectively, after 9 h of infection. By contrast, CCL-5 expression peaked at 2,000-fold 18 h after RSV infection. In the RIG-I knockdown mutants, basal expression levels of IFN-β and the IFN-responsive IP-10 and ISG15 genes were all increased relative to those of the control siRNA transfectants but were not significantly induced by RSV infection 9 h later (Fig. 2C). For CCL-5, basal expression was not detectable and, in the RIG-I knockdown, its expression was reduced at 9 h as well. In addition, we noted that after 18 h of RSV infection, levels of IP-10, ISG15, and CCL-5 were enhanced significantly in RIG-I-silenced cells, whereas there was no significant difference in the level of expression of IFN-β between RIG-I-silenced cells and control cells.
RSV-induced expression of the same genes was then examined after TLR3 knockdown (Fig. 2D). In the TLR3 knockdown mutants, the basal expression levels of all investigated genes were increased, and after 9 h of RSV infection, the gene expression levels of IP-10 and CCL-5 were not affected. In contrast to the effects of RIG-I knockdown, expression levels of both IFN-β and ISG15 were increased, rather than inhibited, 9 h after infection. In this group of genes, expression levels were inhibited 18 h after RSV infection (Fig. 2D). These results suggest that the RIG-I pathway is involved in the early response of host cells to RSV infection. At later phases of RSV infection, other RIG-I-independent pathways are activated that mediate downstream gene expression, including that of TLR3.
The RIG-I pathway mediates early NF-κB/RelA and IRF-3 activation in response to RSV infection.
To determine whether RSV-induced DNA binding of NF-κB or IRF-3 was affected after RIG-I or TLR3 expression was silenced, DNA binding activity was measured in NE by EMSA using either double-stranded NF-κB or ISRE sites. NF-κB-specific DNA binding activity was abolished in RIG-I-silenced cells at 9 h or 18 h after RSV infection compared to that in controls, whereas only a slight decrease in binding occurred in cells after TLR3 silencing (Fig. 3A, left panel). Conversely, siRNA-mediated RIG-I silencing inhibited specific RSV-inducible IRF-3 binding to its cognate ISRE element 9 h after RSV infection, but a significant increase of IRF-3 binding was observed later (18 h after infection; Fig. 3B). Similar to its lack of effect on RSV-induced NF-κB DNA binding, siRNA-mediated TLR3 silencing had little detectable effect on inducible IRF-3 binding at any time point (Fig. 3B). An EMSA experiment using the oligonucleotide containing the OCT1 element was conducted, which confirmed equivalent nuclear protein preparation for each extract (Fig. 3C). Together, these data indicated that the RIG-I, but not the TLR3 signal, was required for RSV-induced NF-κB and IRF-3 DNA binding.
To determine whether RIG-I was required for cytoplasmic-nuclear translocation, we next examined the subcellular distributions of NF-κB and IRF-3 using confocal microscopy in RSV-infected cells transfected with control siRNA or RIG-I siRNA. In control siRNA-transfected cells, 32% of cells showed nuclear RelA accumulation 9 h after RSV infection; this number increased to 63% 18 h after infection. By contrast, in RIG-I-silenced cells, nuclear RelA translocation was significantly inhibited, with only 13% of cells after 9 h and 22% of cells after 18 h showing nuclear RelA signals (Fig. 3D and F). In the case of IRF-3 activation, in control siRNA-transfected cells, 21% (9 h) and 38% of cells (18 h) showed IRF-3 nuclear translocation after RSV infection, whereas in RIG-I-silenced cells, 8% and 29% of cells showed nuclear IRF-3 accumulation 9 and 18 h after infection, respectively (Fig. 3E and F). These data indicated that RIG-I expression was necessary for nuclear translocation of both RelA and IRF-3 at early times after RSV infection (9 h). In addition, we noted that there was no statistically significant difference between the RIG-I siRNA group and control groups for IRF-3 nuclear translocation 18 h after RSV infection, and this later increase in IRF-3 binding was consistent with the preserved RSV-induced IRF-3 DNA binding detected by EMSA (Fig. 3B), suggesting that, in the absence of RIG-I, other redundant pathways may mediate IRF-3 activation later during the infection.
The TLR3 pathway regulates the phosphorylation of NF-κB/RelA at serine 276.
Our findings suggested that the TLR3 pathway was not required for inducing NF-κB/RelA or IRF-3 DNA binding activity in response to RSV infection (Fig. 3A and B), yet it significantly inhibited the expression of genes which we have found to be NF-κB dependent, including the CCL-5 and IP-10 genes (48). To examine whether TLR3 signaling affected NF-κB/RelA transcriptional activity, we tested RSV-inducible NF-κB activity using a luciferase reporter plasmid containing the IFN-β PRDII domain in control or TLR3 siRNA-transfected cells. Reporter gene expression levels increased four- and eightfold after 9 and 18 h of RSV infection in the cells transfected with control siRNA, whereas NF-κB-dependent reporter gene expression was significantly inhibited in TLR3-silenced cells (Fig. 4A). This suggested that the TLR3 pathway controls NF-κB transcriptional activation.
FIG. 4.

The TLR3 pathway mediates the phosphorylation of NF-κB/RelA at serine 276. (A) A549 cells were transfected with control siRNA (Con) or TLR3 siRNA, and a luciferase reporter plasmid containing the PRDII domain was cotransfected for 48 h. Cells were infected with RSV for 0, 9, or 18 h before cell lysis was performed. Shown is normalized luciferase activity expressed as change (n-fold) relative to that of uninfected cells. *, P < 0.05 (Student's t test). Error bars indicate standard deviations. (B) A549 cells were transfected with control siRNA (Con) or TLR3 siRNA (TLR3) and then RSV infected for 0, 9, or 18 h. Western immunobloting was performed to detect changes in phospho-Ser276 RelA (top panel), phospho-Ser536 RelA (middle panel), and RelA (bottom panel) levels using 100 μg of whole-cell extract.
NF-κB is known to be a nuclear phosphoprotein with activating sites at serine residues 276 (50, 58) and 536 (8, 35). We therefore investigated whether RSV induced RelA phosphorylation and, if so, whether it was inhibited by TLR3 silencing. In control siRNA-transfected cells, RelA phosphorylation on serine 276 and 536 sites increased 9 and 18 h after RSV infection. By contrast, in TLR3 siRNA transfectants, serine 276 phosphorylation was significantly inhibited 18 h after RSV infection (Fig. 4B). Together, these data suggested that the activation of the TLR3 pathway in airway epithelial cells controls the phosphorylation of RelA at serine 276 as its mechanism for regulating RSV-induced NF-κB-dependent gene expression at the late phase of infection.
The RIG-I pathway mediates RSV-induced TLR3 upregulation by increasing paracrine IFN-β secretion.
Previous studies (15, 31, 32) and ours (Fig. 2B) have shown that TLR3 expression is induced by RSV infection. To further investigate the interaction between the RIG-I pathway and the TLR3 pathway, we next explored the effect of RIG-I knockdown on TLR3 transcription and expression. QRT-PCR was conducted to measure endogenous TLR3 mRNA levels after RIG-I expression was silenced. In the control siRNA group, the TLR3 mRNA levels were increased 6- and 18-fold after 9 or 18 h of RSV infection. siRNA-mediated RIG-I silencing abolished the RSV-induced TLR3 induction. These data suggested that the activation of the epithelial RIG-I pathway is required for RSV-induced TLR3 upregulation. To initially localize the regulatory regions in the TLR3 gene, a computational analysis of the human TLR3 promoter was conducted using position weight matrices (TRANSFAC) (51). In this analysis, we predicted two interferon response elements (ISRE1 and ISRE2) and one STAT site. A 1.0-kb fragment of the human TLR3 promoter containing these regulatory regions was cloned and inserted into a luciferase reporter plasmid, generating hTLR3/LUC. To determine their relative contributions, each ISRE and STAT site was individually mutated to non-DNA binding sequences in the context of the 1-kb hTLR3/LUC (Fig. 5B). The wild-type hTLR3/LUC and its respective site mutants were then transfected into A549 cells and luciferase reporter activity was measured in the absence or presence of RSV infection. We found that hTLR3/LUC was induced sixfold by RSV relative to activity in the uninfected control (Fig. 5C). In addition, mutation of the ISRE1 site did not affect RSV-induced reporter gene expression, but mutation of either the ISRE2 or STAT site significantly decreased RSV-induced reporter gene activity (Fig. 5C). Because RSV is known to induce IFN-β secretion by airway epithelial cells (22, 42, 43) and others have reported that type I IFNs enhance TLR3 expression (49), we next investigated whether RSV-induced TLR3 expression is controlled in a paracrine manner by IFN-β secretion. CM from RSV-infected A549 cells (24 h after infection) was collected, and naïve A549 cells were incubated with 2.5% (vol/vol) of UV-inactivated RSV-CM (UV-RSV-CM). Six and twelve hours after exposure, RNA was extracted and Northern blotting was conducted to measure TLR3 expression. We found that UV-RSV-CM induced TLR3 expression in naïve A549 cells, indicating that paracrine activators of TLR3 expression were present in infected A549 cell culture supernatants. To determine whether the paracrine mediator in the RSV-CM was IFN-β, two other plates were treated with UV-RSV-CM neutralized with either rabbit IgG or neutralizing anti-IFN-β Ab. The UV-RSV-CM induction of TLR3 was significantly and selectively inhibited in the medium after IFN-β was neutralized (Fig. 5D). These data suggested that IFN-β acts in a paracrine manner to up-regulate TLR3 expression. To further establish that paracrine IFN-β secretion is necessary for RSV-induced TLR3 expression, RSV-induced TLR3 expression was measured in Vero cells, cells deficient in IFN-β expression but capable of productive RSV replication (44, 54). Although RSV infection increased TLR3 expression in A549 cells, it did not induce TLR3 in Vero cells. Importantly, adding UV-RSV-CM to Vero cells induced TLR3 gene expression (Fig. 5E). These data indicate that IFN-β is necessary and sufficient for TLR3 upregulation.
FIG. 5.

RSV-induced TLR3 expression depends on RIG-I-induced IFN-β secreted from infected cells. (A) A549 cells were transfected with control siRNA (Con) and RIG-I siRNA for 48 h and RSV infected for 0, 9, or 18 h. QRT-PCR was performed using TLR3 probe. #, P is <0.01; *, P is <0.05 relative to control siRNA at the same time point. Error bars indicate standard deviations. The results shown here are representative of two independent experiments. (B) Noncontiguous genomic sequence of hTLR3 promoter. The location relative to the major transcription start site is shown at left. Underlines, two predicted ISRE sites (ISRE1 and ISRE2) and one STAT site; bold font, site-directed mutagenesis of each individual regulatory element was performed by rolling-circle PCR. (C) A549 cells were transfected with either wild-type hTLR3/LUC reporter gene or different site mutants. Twenty-four hours later, cells were RSV infected and normalized luciferase activity was measured 12 h later. WT, wild type; *, P is <0.001 relative to wild-type hTLR3/LUC activity at 12 h. Error bars indicate standard deviations. (D) Naïve A549 cells were treated with 20% (vol/vol) UV-RSV-CM taken from RSV-infected cells for the indicated times (in hours). Prior to its addition to A549 cells, UV-RSV-CM was preincubated with PBS, rabbit IgG (IgG), or neutralizing anti-IFN-β Ab for 2 h. An autoradiogram from Northern blot hybridization is shown. Top panel, hybridization using radiolabeled TLR3 cDNA; bottom panel, hybridization with β-actin as an internal control. α, anti. (E) IFN-β-deficient Vero cells were infected with RSV for 12 h or were treated with 20% (vol/vol) UV-RSV-CM for 12 h. The conditioned medium was collected from A549 cells 24 h after RSV infection. Top panel, 20 μg total RNA was isolated and Northern blot hybridization conducted using TLR3 cDNA probe; bottom panel, β-actin hybridization. −, absent; +, present.
DISCUSSION
RSV is the major etiologic agent of epidemic wheezing and bronchiolitis in children, leading causes of hospitalization in children (36). In natural infections, airway epithelial cells are the primary sites for RSV invasion and these represent the cell type where productive replication takes place (2). Previous studies have shown that host cells primarily use two different classes of “sensors” for viral detection. One is a group of pattern recognition receptors localized in the cytoplasm that includes the DExD/H box-containing RNA helicases, RIG-I, and the melanoma differentiation-associated gene 5 (MDA5), and the second is a group of membrane-bound pathogen-associated molecular pattern receptors known as the TLRs (20). In this study, we found that the expression of both RIG-I and TLR3 is rapidly induced by RSV infection in alveolar-like A549 cells. Which of these two mechanisms is used by airway epithelial cells to detect RSV infection and their interrelationships are not understood.
In this study, we are the first to demonstrate that RIG-I mediates RSV-induced early signaling events leading to the activation of NF-κB and IRF-3, two key transcription factors controlling inflammatory cytokine and chemokine expression in airway epithelial cells (20). We have shown that RIG-I, but not the related MDA5 molecule, specifically binds RSV RNA. In addition, siRNA-mediated RIG-I knockdown significantly inhibits the activation of NF-κB and IRF-3, especially at the early phase of RSV infection (9 h p.i.). The mechanisms by which RIG-I couples to transcription factor activation are partially understood. After RNA virus infection, transcription and replication of the virus yields RNA intermediates which are bound by the helicase domain of RIG-I. This event activates the two amino-terminal caspase-recruiting domains (CARD), which in turn, are required for binding another CARD-containing molecule, known as mitochondrion antiviral signaling (MAVS, also known as IPS-1/VISA/Cardif) (25, 29, 39, 52). Although we have not investigated its role here, MAVS has been identified as the only downstream adaptor for RIG-I. The CARD-mediated association between RIG-I and MAVS then leads to the activation of NF-κB and IRF-3. IRF-3 activation appears to be mediated by activation of the atypical IKKs, TBK1/IKKι/ɛ, that phosphorylate IRF-3, resulting in its dimerization and nuclear translocation.
By contrast, the mechanism for NF-κB activation is not fully understood. Our previous work has shown that RSV controls NF-κB nuclear translocation by its effects on IκBα proteolysis (21); interpreted together with our studies demonstrating that RIG-I is required for NF-κB nuclear translocation, these data suggest that RIG-I is upstream of the canonical NF-κB pathway. We note that a recent publication showed that TNF receptor-associated factor 3 (TRAF3) was involved in the activation of the IKK complex through the RIG-I-MAVS pathway (34). In this regard, it will be of interest to examine whether TRAF3 is involved in RSV-induced NF-κB activation. Finally, our immunohistochemistry experiments indicate that the NF-κB translocation response, as well as the IRF-3 response, occurs only in a subpopulation (∼30%) of RSV-infected cells. These findings are consistent with other recent studies, which used an even higher multiplicity of infection (43). These findings indicate that there is significant heterogeneity in the RSV-induced antiviral cell-signaling response.
The activation of both RIG-I-dependent and RIG-I-independent pathways by West Nile virus has also been reported recently (13). Using RIG-I-null embryonic fibroblasts, it was found that West Nile virus activated IRF-3 through the RIG-I pathway at the early phase of viral infection. However, at late times of infection, West Nile virus was still able to activate IRF-3 through a RIG-I-independent pathway whose mechanism is unknown (13). These data are consistent with our findings (Fig. 3B). We suspect that the activation of the RIG-I-independent pathway may have a higher threshold, signaling only under conditions of higher levels of viral replication. It will be of interest to identify the mediators of this RIG-I-independent pathway; these could include protein kinase R, TLR, or other cytoplasmic RNA helicases not yet identified.
In our data and the work published by others (32), TLR3 pathway inhibition affects the expression of some antiviral genes, such as CCL-5 and IP-10. However, the exact mechanism underlying this observation was not known previously. We show here that the TLR3 pathway does not contribute to the DNA binding activity of NF-κB and IRF-3 following RSV infection (Fig. 3A and B). Rather, it regulates the transcription of these genes by modulating the phosphorylation of RelA at serine residue 276 (Fig. 4B). Serine 276 phosphorylation is controlled by several protein kinases, including the catalytic subunit of PKA and the mitogen- and stress-activated protein kinase-1 (50, 58). Serine 276 phosphorylation is required to induce intermolecular interaction between RelA and the p300 coactivator, thereby resulting in transcriptional activation (57). It is not presently known which kinase mediates RelA serine 276 phosphorylation in response to RSV, and we will investigate this in future studies.
The results of our study indicate that the TLR3 pathway functions only later during the evolution of RSV infection. The subcellular localization of TLR3 in uninfected cells may determine its kinetics and role in antiviral response. In particular, some studies have indicated that TLR3 is localized, at least partially, in an endosomal compartment in unstimulated cells (9, 28). Because RSV is a paramyxovirus which enters the cell directly by pH-independent fusion with the plasma membrane (19), TLR3 would likely encounter its dsRNA ligand only later in the viral life cycle. The implication here is that other viruses that enter via the endosomal pathway may be able to activate the TLR3 pathway as a primary event. Although TLR3 is partially endosomal in unstimulated cells, in response to stimulation, newly synthesized TLR3 distributes to the plasma membrane (15). The translocation of TLR3 might allow RSV-infected cells to respond to extracellular dsRNA. Alternatively, dsRNA released during late RSV infection might be taken up by cells and transported to the endosomal compartments where it can be recognized by TLR3. This hypothesis will require further study.
Recently, two groups have reported the interaction between RSV and TLR3 (15, 31, 32). The identical phenomenon that they observed was that the expression of TLR3 was induced by RSV infection. However, the mechanism of this induction is not clearly understood. In this study, we found that IFN-β secreted from RSV-infected epithelial cells is necessary and sufficient to activate TLR3 expression. Importantly, induction of TLR3 is not directly the result of cytoplasmic RSV replication, because TLR3 expression was not induced by RSV infection in Vero cells. Vero cells are capable of high levels of RSV replication (44, 54), but are deficient in IFN-β expression (11). The induction of TLR3 in response to RSV infection was absent in Vero cells, but when Vero cells were treated with RSV-conditioned media, rich in IFN-β, TLR3 was induced. These observations suggest that TLR3 activation is a secondary paracrine response mediated by local IFN-β secreted by RSV-infected epithelial cells. The induction of TLR3 expression in epithelial cells by measles virus and type I interferon has also been reported recently (46, 49). Like RSV, measles virus is a negative-sense single-stranded paramyxovirus. Measles virus infection increased the expression of TLR3 through a transcriptional mechanism involving the ISRE2 binding sites in the hTLR3 promoter (46). Consistent with this finding, we also found that the ISRE2 site was essential for TLR3 induction in response to the IFN-β present in UV-RSV-CM. However, we found that a proximal STAT site is also required for hTLR3 promoter expression. Here, IFN-β binds to its IFNAR1 receptor, inducing the activation of receptor-associated Jak/Tyk tyrosine kinases and phosphorylation of receptor-associated STATs. This process induces the formation of interferon-stimulated gene factor 3, including STAT1 and STAT2 and IRF-9 (1). Because the hTLR3 promoter contains a functionally important ISRE and a STAT site, it is uniquely poised to integrate signals from the RSV-IRF and IFN-STAT pathways into enhanced transcriptional activation. Since IFN-β played an essential role for inducing TLR3 expression in response to RSV infection, it was reasonable to find that RIG-I was involved in this process. siRNA-mediated RIG-I silencing abolished the endogenous TLR3 induction in response to RSV. This result suggests that RIG-I is a primary sensor for RSV detection in airway epithelial cells and that TLR3 expression is secondary to RIG-I-signaling action.
In summary, we found that the RIG-I pathway mediates the early response of airway epithelial cells to RSV infection, which initiates the innate immune response. The TLR3 pathway only affects the late-time gene expression, which regulates the phosphorylation of RelA at serine 276. TLR3 expression is induced by RSV in a paracrine manner that depends on RIG-I-induced IFN-β secretion.
Acknowledgments
This project was supported by NIAID grant R01 AI40218 (to A.R.B.) and, in part, by PO1 AI062885 (to A.R.B.). Core laboratory support was funded by NIEHS grant P30 ES06676 (to J. Halpert, UTMB).
Footnotes
Published ahead of print on 15 November 2006.
REFERENCES
- 1.Aaronson, D. S., and C. M. Horvath. 2002. A road map for those who don't know JAK-STAT. Science 296:1653-1655. [DOI] [PubMed] [Google Scholar]
- 2.Aherne, W., T. Bird, S. D. Court, P. S. Gardner, and J. McQuillin. 1970. Pathological changes in virus infections of the lower respiratory tract in children. J. Clin. Pathol. 23:7-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Akira, S., and K. Takeda. 2004. Toll-like receptor signalling. Nat. Rev. Immunol. 4:499-511. [DOI] [PubMed] [Google Scholar]
- 4.Beutler, B. 2004. Inferences, questions and possibilities in Toll-like receptor signalling. Nature 430:257-263. [DOI] [PubMed] [Google Scholar]
- 5.Brasier, A. R., J. E. Tate, and J. F. Habener. 1989. Optimized use of the firefly luciferase assay as a reporter gene in mammalian cell lines. BioTechniques 7:1116-1122. [PubMed] [Google Scholar]
- 6.Casola, A., R. P. Garofalo, H. Haeberle, T. F. Elliott, R. Lin, M. Jamaluddin, and A. R. Brasier. 2001. Multiple cis regulatory elements control RANTES promoter activity in alveolar epithelial cells infected with respiratory syncytial virus. J. Virol. 75:6428-6439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chang, T. H., C. L. Liao, and Y. L. Lin. 2006. Flavivirus induces interferon-beta gene expression through a pathway involving RIG-I-dependent IRF-3 and PI3K-dependent NF-kappaB activation. Microbes Infect. 8:157-171. [DOI] [PubMed] [Google Scholar]
- 8.Chen, L. F., S. A. Williams, Y. Mu, H. Nakano, J. M. Duerr, L. Buckbinder, and W. C. Greene. 2005. NF-κB RelA phosphorylation regulates RelA acetylation. Mol. Cell. Biol. 25:7966-7975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.de Bouteiller, O., E. Merck, U. A. Hasan, S. Hubac, B. Benguigui, G. Trinchieri, E. E. Bates, and C. Caux. 2005. Recognition of double-stranded RNA by human toll-like receptor 3 and downstream receptor signaling requires multimerization and an acidic pH. J. Biol. Chem. 280:38133-38145. [DOI] [PubMed] [Google Scholar]
- 10.Doyle, S., S. Vaidya, R. O'Connell, H. Dadgostar, P. Dempsey, T. Wu, G. Rao, R. Sun, M. Haberland, R. Modlin, and G. Cheng. 2002. IRF3 mediates a TLR3/TLR4-specific antiviral gene program. Immunity 17:251-263. [DOI] [PubMed] [Google Scholar]
- 11.Emeny, J. M., and M. J. Morgan. 1979. Regulation of the interferon system: evidence that Vero cells have a genetic defect in interferon production. J. Gen. Virol. 43:247-252. [DOI] [PubMed] [Google Scholar]
- 12.Foy, E., K. Li, C. Wang, R. Sumpter, Jr., M. Ikeda, S. M. Lemon, and M. Gale, Jr. 2003. Regulation of interferon regulatory factor-3 by the hepatitis C virus serine protease. Science 300:1145-1148. [DOI] [PubMed] [Google Scholar]
- 13.Fredericksen, B. L., and M. Gale, Jr. 2006. West Nile virus evades activation of interferon regulatory factor 3 through RIG-I-dependent and -independent pathways without antagonizing host defense signaling. J. Virol. 80:2913-2923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Garofalo, R., M. Sabry, M. Jamaluddin, R. K. Yu, A. Casola, P. L. Ogra, and A. R. Brasier. 1996. Transcriptional activation of the interleukin-8 gene by respiratory syncytial virus infection in alveolar epithelial cells: nuclear translocation of the RelA transcription factor as a mechanism producing airway mucosal inflammation. J. Virol. 70:8773-8781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Groskreutz, D. J., M. M. Monick, L. S. Powers, T. O. Yarovinsky, D. C. Look, and G. W. Hunninghake. 2006. Respiratory syncytial virus induces TLR3 protein and protein kinase R, leading to increased double-stranded RNA responsiveness in airway epithelial cells. J. Immunol. 176:1733-1740. [DOI] [PubMed] [Google Scholar]
- 16.Haeberle, H. A., R. Takizawa, A. Casola, A. R. Brasier, H. J. Dieterich, R. N. Van, Z. Gatalica, and R. P. Garofalo. 2002. Respiratory syncytial virus-induced activation of nuclear factor-kappaB in the lung involves alveolar macrophages and toll-like receptor 4-dependent pathways. J. Infect. Dis. 186:1199-1206. [DOI] [PubMed] [Google Scholar]
- 17.Hai, T., M. L. Yeung, T. G. Wood, Y. Wei, S. Yamaoka, Z. Gatalica, K. T. Jeang, and A. R. Brasier. 2006. An alternative splice product of IκB kinase (IKKγ), IKKγ-Δ, differentially mediates cytokine and human T-cell leukemia virus type 1 Tax-induced NF-κB activation. J. Virol. 80:4227-4241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hall, C. B. 2001. Respiratory syncytial virus and parainfluenza virus. N. Engl. J. Med. 344:1917-1928. [DOI] [PubMed] [Google Scholar]
- 19.Harris, J., and D. Werling. 2003. Binding and entry of respiratory syncytial virus into host cells and initiation of the innate immune response. Cell. Microbiol. 5:671-680. [DOI] [PubMed] [Google Scholar]
- 20.Hiscott, J., R. Lin, P. Nakhaei, and S. Paz. 2006. MasterCARD: a priceless link to innate immunity. Trends Mol. Med. 12:53-56. [DOI] [PubMed] [Google Scholar]
- 21.Jamaluddin, M., A. Casola, R. P. Garofalo, Y. Han, T. Elliott, P. L. Ogra, and A. R. Brasier. 1998. The major component of IκBα proteolysis occurs independently of the proteasome pathway in respiratory syncytial virus-infected pulmonary epithelial cells. J. Virol. 72:4849-4857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jamaluddin, M., S. Wang, R. P. Garofalo, T. Elliott, A. Casola, S. Baron, and A. R. Brasier. 2001. IFN-beta mediates coordinate expression of antigen-processing genes in RSV-infected pulmonary epithelial cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 280:L248-L257. [DOI] [PubMed] [Google Scholar]
- 23.Johnson, C. L., and M. Gale, Jr. 2006. CARD games between virus and host get a new player. Trends Immunol. 27:1-4. [DOI] [PubMed] [Google Scholar]
- 24.Kato, H., S. Sato, M. Yoneyama, M. Yamamoto, S. Uematsu, K. Matsui, T. Tsujimura, K. Takeda, T. Fujita, O. Takeuchi, and S. Akira. 2005. Cell type-specific involvement of RIG-I in antiviral response. Immunity 23:19-28. [DOI] [PubMed] [Google Scholar]
- 25.Kawai, T., K. Takahashi, S. Sato, C. Coban, H. Kumar, H. Kato, K. J. Ishii, O. Takeuchi, and S. Akira. 2005. IPS-1, an adaptor triggering RIG-I- and Mda5-mediated type I interferon induction. Nat. Immunol. 6:981-988. [DOI] [PubMed] [Google Scholar]
- 26.Kurt-Jones, E. A., L. Popova, L. Kwinn, L. M. Haynes, L. P. Jones, R. A. Tripp, E. E. Walsh, M. W. Freeman, D. T. Golenbock, L. J. Anderson, and R. W. Finberg. 2000. Pattern recognition receptors TLR4 and CD14 mediate response to respiratory syncytial virus. Nat. Immunol. 1:398-401. [DOI] [PubMed] [Google Scholar]
- 27.Lieber, M., B. Smith, A. Szakal, W. Nelson-Rees, and G. Todaro. 1976. A continuous tumor-cell line from a human lung carcinoma with properties of type II alveolar epithelial cells. Int. J. Cancer. 17:62-70. [DOI] [PubMed] [Google Scholar]
- 28.Matsumoto, M., K. Funami, M. Tanabe, H. Oshiumi, M. Shingai, Y. Seto, A. Yamamoto, and T. Seya. 2003. Subcellular localization of Toll-like receptor 3 in human dendritic cells. J. Immunol. 171:3154-3162. [DOI] [PubMed] [Google Scholar]
- 29.Meylan, E., J. Curran, K. Hofmann, D. Moradpour, M. Binder, R. Bartenschlager, and J. Tschopp. 2005. Cardif is an adaptor protein in the RIG-I antiviral pathway and is targeted by hepatitis C virus. Nature 437:1167-1172. [DOI] [PubMed] [Google Scholar]
- 30.Nanbo, A., K. Inoue, K. Adachi-Takasawa, and K. Takada. 2002. Epstein-Barr virus RNA confers resistance to interferon-alpha-induced apoptosis in Burkitt's lymphoma. EMBO J. 21:954-965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ritter, M., D. Mennerich, A. Weith, and P. Seither. 2005. Characterization of Toll-like receptors in primary lung epithelial cells: strong impact of the TLR3 ligand poly(I:C) on the regulation of Toll-like receptors, adaptor proteins and inflammatory response. J. Inflamm. 2:16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rudd, B. D., E. Burstein, C. S. Duckett, X. Li, and N. W. Lukacs. 2005. Differential role for TLR3 in respiratory syncytial virus-induced chemokine expression. J. Virol. 79:3350-3357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rudd, B. D., J. J. Smit, R. A. Flavell, L. Alexopoulou, M. A. Schaller, A. Gruber, A. A. Berlin, and N. W. Lukacs. 2006. Deletion of TLR3 alters the pulmonary immune environment and mucus production during respiratory syncytial virus infection. J. Immunol. 176:1937-1942. [DOI] [PubMed] [Google Scholar]
- 34.Saha, S. K., E. M. Pietras, J. Q. He, J. R. Kang, S. Y. Liu, G. Oganesyan, A. Shahangian, B. Zarnegar, T. L. Shiba, Y. Wang, and G. Cheng. 2006. Regulation of antiviral responses by a direct and specific interaction between TRAF3 and Cardif. EMBO J. 25:3257-3263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sasaki, C. Y., T. J. Barberi, P. Ghosh, and D. L. Longo. 2005. Phosphorylation of RelA/p65 on serine 536 defines an IκBα-independent NF-κB pathway. J. Biol. Chem. 280:34538-34547. [DOI] [PubMed] [Google Scholar]
- 36.Schmidt, A. C., T. R. Johnson, P. J. Openshaw, T. J. Braciale, A. R. Falsey, L. J. Anderson, G. W. Wertz, J. R. Groothuis, G. A. Prince, J. A. Melero, and B. S. Graham. 2004. Respiratory syncytial virus and other pneumoviruses: a review of the international symposium—RSV 2003. Virus Res. 106:1-13. [DOI] [PubMed] [Google Scholar]
- 37.Schroder, M., and A. G. Bowie. 2005. TLR3 in antiviral immunity: key player or bystander? Trends Immunol. 26:462-468. [DOI] [PubMed] [Google Scholar]
- 38.Sen, G. C., and S. N. Sarkar. 2005. Hitching RIG to action. Nat. Immunol. 6:1074-1076. [DOI] [PubMed] [Google Scholar]
- 39.Seth, R. B., L. Sun, C. K. Ea, and Z. J. Chen. 2005. Identification and characterization of MAVS, a mitochondrial antiviral signaling protein that activates NF-kappaB and IRF 3. Cell 122:669-682. [DOI] [PubMed] [Google Scholar]
- 40.Sha, Q., A. Q. Truong-Tran, J. R. Plitt, L. A. Beck, and R. P. Schleimer. 2004. Activation of airway epithelial cells by Toll-like receptor agonists. Am. J. Respir. Cell Mol. Biol. 31:358-364. [DOI] [PubMed] [Google Scholar]
- 41.Sharma, S., B. R. tenOever, N. Grandvaux, G. P. Zhou, R. Lin, and J. Hiscott. 2003. Triggering the interferon antiviral response through an IKK-related pathway. Science 300:1148-1151. [DOI] [PubMed] [Google Scholar]
- 42.Spann, K. M., K.-C. Tran, B. Chi, R. L. Rabin, and P. L. Collins. 2004. Suppression of the induction of alpha, beta, and lambda interferons by the NS1 and NS2 proteins of human respiratory syncytial virus in human epithelial cells and macrophages. J. Virol. 78:4363-4369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Spann, K. M., K. C. Tran, and P. L. Collins. 2005. Effects of nonstructural proteins NS1 and NS2 of human respiratory syncytial virus on interferon regulatory factor 3, NF-κB, and proinflammatory cytokines. J. Virol. 79:5353-5362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sugrue, R. J., C. Brown, G. Brown, J. Aitken, and H. W. M. Rixon 2001. Furin cleavage of the respiratory syncytial virus fusion protein is not a requirement for its transport to the surface of virus-infected cells. J. Gen. Virol. 82:1375-1386. [DOI] [PubMed] [Google Scholar]
- 45.Sumpter, R., Jr., Y. M. Loo, E. Foy, K. Li, M. Yoneyama, T. Fujita, S. M. Lemon, and M. Gale, Jr. 2005. Regulating intracellular antiviral defense and permissiveness to hepatitis C virus RNA replication through a cellular RNA helicase, RIG-I. J. Virol. 79:2689-2699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tanabe, M., M. Kurita-Taniguchi, K. Takeuchi, M. Takeda, M. Ayata, H. Ogura, M. Matsumoto, and T. Seya. 2003. Mechanism of up-regulation of human Toll-like receptor 3 secondary to infection of measles virus-attenuated strains. Biochem. Biophys. Res. Commun. 311:39-48. [DOI] [PubMed] [Google Scholar]
- 47.Thompson, W. W., D. K. Shay, E. Weintraub, L. Brammer, N. Cox, L. J. Anderson, and K. Fukuda. 2003. Mortality associated with influenza and respiratory syncytial virus in the United States. JAMA 289:179-186. [DOI] [PubMed] [Google Scholar]
- 48.Tian, B., Y. Zhang, B. A. Luxon, R. P. Garofalo, A. Casola, M. Sinha, and A. R. Brasier. 2002. Identification of NF-κB-dependent gene networks in respiratory syncytial virus-infected cells. J. Virol. 76:6800-6814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tissari, J., J. Siren, S. Meri, I. Julkunen, and S. Matikainen. 2005. IFN-alpha enhances TLR3-mediated antiviral cytokine expression in human endothelial and epithelial cells by up-regulating TLR3 expression. J. Immunol. 174:4289-4294. [DOI] [PubMed] [Google Scholar]
- 50.Vermeulen, L., G. De Wilde, P. Van Damme, W. Vanden Berghe, and G. Haegeman. 2003. Transcriptional activation of the NF-kappaB p65 subunit by mitogen- and stress-activated protein kinase-1 (MSK1). EMBO J. 22:1313-1324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wingender, E., P. Dietze, H. Karas, and R. Knuppel. 1996. TRANSFAC: a database on transcription factors and their DNA binding sites. Nucleic Acids Res. 24:238-241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Xu, L. G., Y. Y. Wang, K. J. Han, L. Y. Li, Z. Zhai, and H. B. Shu. 2005. VISA is an adapter protein required for virus-triggered IFN-beta signaling. Mol. Cell 19:727-740. [DOI] [PubMed] [Google Scholar]
- 53.Yoneyama, M., M. Kikuchi, T. Natsukawa, N. Shinobu, T. Imaizumi, M. Miyagishi, K. Taira, S. Akira, and T. Fujita. 2004. The RNA helicase RIG-I has an essential function in double-stranded RNA-induced innate antiviral responses. Nat. Immunol. 5:730-737. [DOI] [PubMed] [Google Scholar]
- 54.Zhang, W., H. Yang, X. Kong, S. Mohapatra, H. San Juan-Vergara, G. Hellermann, S. Behera, R. Singam, R. F. Lockey, and S. S. Mohapatra. 2005. Inhibition of respiratory syncytial virus infection with intranasal siRNA nanoparticles targeting the viral NS1 gene. Nat. Med. 11:56-62. [DOI] [PubMed] [Google Scholar]
- 55.Zhang, Y., M. Jamaluddin, S. Wang, B. Tian, R. P. Garofalo, A. Casola, and A. R. Brasier. 2003. Ribavirin treatment up-regulates antiviral gene expression via the interferon-stimulated response element in respiratory syncytial virus-infected epithelial cells. J. Virol. 77:5933-5947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zhang, Y., B. A. Luxon, A. Casola, R. P. Garofalo, M. Jamaluddin, and A. R. Brasier. 2001. Expression of respiratory syncytial virus-induced chemokine gene networks in lower airway epithelial cells revealed by cDNA microarrays. J. Virol. 75:9044-9058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zhong, H., M. J. May, E. Jimi, and S. Ghosh. 2002. The phosphorylation status of nuclear NF-kappa B determines its association with CBP/p300 or HDAC-1. Mol. Cell 9:625-636. [DOI] [PubMed] [Google Scholar]
- 58.Zhong, H., R. E. Voll, and S. Ghosh. 1998. Phosphorylation of NF-kappa B p65 by PKA stimulates transcriptional activity by promoting a novel bivalent interaction with the coactivator CBP/p300. Mol. Cell 1:661-671. [DOI] [PubMed] [Google Scholar]