

Abstract
Rhinovirus (RV) is responsible for the majority of common colds and triggers exacerbations of asthma and chronic obstructive lung disease. We have shown that RV serotype 39 (RV39) infection activates phosphatidylinositol 3 (PI 3)-kinase and the serine threonine kinase Akt minutes after infection and that the activation of PI 3-kinase and Akt is required for maximal interleukin-8 (IL-8) expression. Here, we further examine the contributions of Src and PI 3-kinase activation to RV-induced Akt activation and IL-8 expression. Confocal fluorescent microscopy of 16HBE14o− human bronchial epithelial cells showed rapid (10-min) colocalization of RV39 with Src, p85α PI 3-kinase, p110β PI 3-kinase, Akt and Cit-Akt-PH, a fluorescent Akt pleckstrin homology domain which binds PI(3,4,5)P3. The chemical Src inhibitor PP2 {4-amino-5-(4-chlorophenyl)-7-(t-butyl)pyrazolo [3,4-d]pyrimidine} and the PI 3-kinase inhibitor LY294002 each inhibited Akt phosphorylation and the colocalization of RV39 with Akt. Digoxigenin-tagged RV coprecipitated with a Crosstide kinase likely to be Akt, and inhibition of Src blocked kinase activity. Digoxigenin-tagged RV39 colocalized with the lipid raft marker ceramide. In 16HBE14o− and primary mucociliary differentiated human bronchial epithelial cells, inhibition of Src kinase activity with the Src family chemical inhibitor PP2, dominant-negative Src (K297R), and Src small interfering RNA (siRNA) each inhibited RV39-induced IL-8 expression. siRNA against p110β PI 3-kinase also inhibited IL-8 expression. These data demonstrate that, in the context of RV infection, Src and p110β PI 3-kinase are upstream activators of Akt and the IL-8 promoter and that RV colocalizes with Src, PI 3-kinase, and Akt in lipid rafts.
Rhinovirus (RV) is a single-stranded RNA virus from the Picornaviridae family responsible for the majority of common colds. Viral infections trigger the majority of asthma exacerbations (17, 22), and RV accounts for 60% of virus-induced exacerbations (17). RV is also an important trigger of chronic obstructive pulmonary disease exacerbations (28, 32).
Numerous studies suggest a role for interleukin-8 (IL-8) in the pathogenesis of asthma and chronic obstructive pulmonary disease (COPD) exacerbations. IL-8, a CXC chemokine with the neutrophil attractant Glu-Leu-Arg (ELR) motif, and neutrophils are found in the nasal secretions and sputa of patients with RV-induced asthma exacerbations (8, 9, 12, 13, 26). Further, the number of neutrophils correlates with the level of IL-8 (9, 26). RV induces IL-8 expression in cultured airway epithelial cells (34, 38, 39). Increased neutrophil and IL-8 levels are a feature of asthma (23, 24) and COPD exacerbations (1, 10, 27). Together, these data suggest that RV may stimulate asthma and COPD exacerbations by inducing bronchial epithelial cell production of IL-8, leading to a neutrophilic inflammatory response.
We have recently shown that infection of human bronchial epithelial cells with RV serotype 39 (RV39) induces rapid phosphorylation of the p85 regulatory subunit of phosphatidylinositol 3 (PI 3)-kinase, as well as that of Akt, a downstream effector of PI 3-kinase (21). RV39 also colocalized with Cit-Akt-PH, a fluorescent fusion protein containing the pleckstrin homology domain of Akt, indicating that PI(3,4,5)P3 accumulates at the site of RV infection. Finally, inhibitions of PI 3-kinase activation with a chemical inhibitor and with dominant-negative p85α each inhibited RV39-induced IL-8 expression. However, the precise mechanism by which RV activates PI 3-kinase and the specific class IA PI 3-kinase catalytic subunit involved in RV-induced Akt phosphorylation were not determined.
Potential upstream activators of PI 3-kinase include the tyrosine kinase p60 Src and focal adhesion kinase, each of which regulates the remodeling of the actin cytoskeleton in response to cell adhesion and integrin clustering. Upon stimulation, Src translocates from the perinuclear region of the cell to peripheral sites of integrin clustering. Src binds to its substrates via its Src homology domains, which in turn interact with phosphotyrosine-containing or proline-rich sequences. Among its substrates are Src itself (autophosphorylation at tyrosine-416), focal adhesion kinase, and the p85 regulatory subunit of PI 3-kinase (15). p85 PI 3-kinase, in turn, may form heterodimers with one of three class IA PI 3-kinase catalytic subunits (p110α, p110β, and p110δ). p110α and p110β are ubiquitously expressed, whereas p110δ expression is largely restricted to cells of the immune system (4).
The human RVs include more than 100 serotypes, which are divided into two groups based on their cellular receptors. Intercellular adhesion molecule-1 (ICAM-1) is the airway epithelial cell receptor for major-subgroup RVs (e.g., RV14, -16, and -39). In endothelial cells, antibody-mediated clustering causes ICAM-1 to colocalize with Src in detergent-insoluble membrane domains, i.e., lipid rafts (35). ICAM-1 cross-linking increases phosphorylation and activation of Src in endothelial cells (7, 37). It has recently been shown that RVs infect human epithelial cells via ceramide-enriched membrane platforms (11). We therefore hypothesized that the Src-mediated activation of PI 3-kinase and its downstream effector Akt is a critical event in the transduction of RV signaling.
MATERIALS AND METHODS
Cell culture.
16HBE14o− human bronchial epithelial cells originating from bronchial epithelial tissue transfected with pSVori-, containing the origin-defective simian virus genome (5), were provided by Steven White (University of Chicago). Cells were grown in minimum essential medium supplemented with 10% fetal bovine serum and 2 mM of l-glutamine.
Human primary airway epithelial cells obtained from the tracheal trimmings of donor lungs at the time of double lung transplantation were cultured in collagen-coated plates with bronchial epithelial cell culture media (Cambrex, East Rutherford, NJ) as previously described (21, 30). First-passage cells were either grown under submerged conditions or, for selected experiments, differentiated to a mucociliary phenotype by seeding on collagen-coated transwells. After growth to confluence, cells were shifted to an air-liquid interface and maintained in a 1:1 mixture of bronchial epithelial cell culture medium and Dulbecco's modified Eagle medium for 3 weeks. The resulting epithelium was pseudostratified with ciliated cells interspersed among mucus-secreting cells.
Rhinovirus.
RV39 was obtained from American Type Culture Collection (Manassas, VA). Viral stocks were generated by infecting HeLa cells with RV until 80% of the cells were cytopathic. HeLa cells were serum deprived overnight, lysates were harvested, and cellular debris was pelleted by centrifugation (10,000 × g for 30 min at 4°C). RV was concentrated and partially purified by centrifugation with a 100,000-molecular-weight-cutoff Centricon filter (2,000 rpm at 4°C for 8 h; Millipore, Billerica, MA) (25). Similarly treated HeLa cell lysates from mock-infected cells served as controls (sham medium controls).
Transfection of cells and measurement of IL-8 promoter activity.
16HBE14o− cells grown in six-well plates were transiently transfected with the −162/+44 fragment of the human IL-8 promoter (3) and Renilla luciferase by use of Lipofectamine (Invitrogen, Carlsbad, CA). Selected cultures were cotransfected with cDNA encoding dominant-negative K296R/Y528F Src (20). Other cells were cotransfected with 100 nM of Src small interfering RNA (siRNA), p110β PI 3-kinase, or nontargeting RNA control (Dharmacon, Lafayette, CO). The following day, the cells were shifted to serum-free media and infected with RV at a multiplicity of infection (MOI) of 1.0 for 1 h. Inoculum was replaced with fresh serum-free medium and incubated at 33°C for 24 h, and the cells were harvested for analysis. Luciferase activity was measured with a luminometer. Promoter activity was normalized for transfection efficiency by dividing luciferase light units by Renilla luciferase light units. Results were reported as increases (n-fold) over values for the empty vector/untreated control.
Measurement of IL-8 production.
IL-8 production following RV39 infection was measured using Duoset enzyme-linked immunosorbent assay development kits purchased from R&D Systems (Minneapolis, MN). In selected cultures, cells were pretreated with dimethyl sulfoxide, PP2 {4-amino-5-(4-chlorophenyl)-7-(t-butyl)pyrazolo [3,4-d]pyrimidine} (16), or PP3 {4-amino-7-phenylpyrazol[3,4-d]pyrimidine } (36) for 1 h prior to viral infection. PP2 and PP3 were obtained from Calbiochem (San Diego, CA). 16HBE14o− cells were incubated with virus (MOI of 1.0) for 1 h, the virus-containing media removed, and fresh media with serum and the indicated concentrations of inhibitors added for a further 48 h before IL-8 release was assessed. Primary mucociliary differentiated cells were apically infected with virus (50% tissue culture infectivity dose [TCID50] of 5 × 106), and fresh medium and inhibitors added for a further 24 h before IL-8 release into the basolateral medium was assessed.
Western blot analysis of cell lysates and immunoprecipitates.
In selected experiments, 16HBE14o− cells were washed briefly in cold phosphate-buffered saline and incubated with homogenization buffer consisting of 50 mM Tris (pH 7.5), 100 mM NaCl, 50 mM NaF, 40 mM β-glycerophosphate, 2 mM EDTA, 200 μM Na3VO4, and 1% Triton X-100 containing complete protease inhibitors (Roche Diagnostics, Indianapolis, IN). Cells were homogenized by passing through a 28-gauge needle and centrifuged. Supernatants were subjected to sodium dodecyl sulfate-polyacrylamide gel electrophoresis, proteins transferred to nitrocellulose, and membranes probed with either rabbit C-terminal anti-Src recognizing amino acids 400 to 422 (Upstate Biotechnology, Charlottesville, VA), rabbit anti-phospho-Ser473, rabbit anti-Akt (both from Cell Signaling, Beverly, MA), or rabbit anti-p110β PI 3-kinase (Epitomics, Burlingame, CA). Bound antibody was detected by secondary antibody conjugated to horseradish peroxidase, and the signal detected by chemiluminescence. In other experiments, cell lysates were immunoprecipitated with mouse monoclonal 4G10 anti-phosphotyrosine (2 μg; Upstate Biotechnology). Immunoprecipitates were resolved by sodium dodecyl sulfate-polyacrylamide gel electrophoresis, transferred to nitrocellulose, and probed with either rabbit anti-phospho-Tyr416 Src (Cell Signaling) or mouse anti-Src (clone GD11 recognizing amino acids 82 to 169; Upstate Biotechnology). No bands were detected when isotype control antibodies were substituted for primary antibodies (not shown).
Immunofluorescent staining.
To visualize RV39 internalization into 16HBE14o− and submerged primary airway epithelial cells, cells were plated on collagen-coated slides (Becton Dickinson Labware, Bedford, MA) and infected with virus at an MOI of between 10 and 100, or an equal volume of cell lysate from uninfected HeLa cells, for 10 min at 33°C. The cells were then washed extensively and fixed in 1% paraformaldehyde. Cells were permeabilized with 1% (vol/vol) Triton X-100 in phosphate-buffered saline, blocked, and incubated with either guinea pig anti-RV39 (American Type Culture Collection), rabbit anti-phospho-Tyr416 Src, mouse anti-Src (monoclonal GD11), rabbit anti-phospho-Ser473 Akt, rabbit anti-Akt, mouse or sheep anti-digoxigenin (Roche Diagnostics), rabbit anti-p85α PI 3-kinase, rabbit anti-p110β PI 3-kinase, or mouse anti-ceramide immunoglobulin M (Sigma Chemical, St. Louis, MO). After being incubated with the appropriate Alexa-Fluor-conjugated secondary antibody (Molecular Probes, Portland, OR), cells were mounted with ProLong Antifade reagent (Molecular Probes) and visualized by confocal fluorescent microscopy with a Zeiss LSM 510 confocal microscope mounted on a Zeiss Axiovert 100 M inverted microscope. Some experiments were performed with 16HBE14o− cells stably transfected with Cit-Akt-PH, a cDNA encoding a fusion protein of Citrogen and the PI(3,4,5)P3-binding Akt pleckstrin homology domain (21). Minimal staining was detected when isotype control antibodies were substituted for primary antibodies (not shown).
RV39 labeling.
In selected experiments, virus was labeled for immunoprecipitation and fluorescent microscopy using an N-hydroxysuccinimide derivative of digoxigenin, an amine-reactive form of this epitope tag (Roche Diagnostics). In a modification of described methods previously used to label RV with Alexa-Fluor dye (21), 0.4 ml of 0.1 M NaHCO3 was added to a 0.1-ml aliquot of purified, concentrated virus to raise the pH to 8.5. The virus was then incubated in the dark for 1 h with 250 μg of probe. This virus maintained infectivity, with no reduction in TCID50 compared with unlabeled virus. The incorporation of digoxigenin into labeled virus or sham protein was quantified on an fmole digoxigenin-per-μg protein basis by methods described by the manufacturer.
Crosstide kinase assay.
After serum deprivation for 24 h, cells were incubated with digoxigenin-labeled sham protein or digoxigenin-labeled RV39 at an MOI of 1.0 for 10 min. Cell homogenates were immunoprecipitated with mouse anti-digoxigenin antibody (Roche Diagnostics) and precipitates incubated with Crosstide (Cell Signaling) and [γ-32P]ATP. Crosstide is a glycogen synthase kinase α/β fusion protein sequence (GRPRTSSFAEG) which is a substrate for Akt (6). Samples were processed for autoradiography and immunoblotting using rabbit anti-phospho-Tyr416 Src, mouse anti-Src (clone GD11), rabbit anti-phospho-Ser473, or rabbit anti-Akt.
Reverse transcriptase PCR (RT-PCR).
cDNA was generated using 0.2 to 0.5 μg of RNA using Superscript reverse transcriptase (Invitrogen) and specific antisense first-strand primers for PI 3-kinase class IAα (5′CT TTT CAG TTC AAT GCA TGC), IAβ (5′TTA AGA TCT GTA GTC TTT CC), or IBγ (5′TTA GGC TGA ATG TTT CTC TGG) cDNA. Taq DNA polymerase (Invitrogen) and 2.5 μM of specific primer were used.
To detect class I PI 3-kinases, we used 0.4 μM of each primer and 10 ng of cDNA template in a reaction volume of 100 μl for a total of 30 cycles. Specific PI 3-kinase primer sequences were as follows. For IAα (accession no. NM_006218), forward primer 5′ TGGGATGTATTTGAAGCACC 3′ and nested reverse primer 5′ TTTCGCACCACCTCAATAAG 3′ was used to produce a 474-bp product. For IAβ (accession no. NM_006219), forward primer 5′ GTTGCGCTTGATGGATTTACT 3′ and nested reverse primer 5′ TCACAACACTGGCGGAACC 3′ were expected to produce a 506-bp product. Finally, for IBγ (accession no. NM_002649), forward primer 5′ ATGCTGCACGACTTTACCC 3′ and nested reverse primer 5′ TGGGGCTTGGGGGTCTTCTG 3′ were expected to produce a 490-bp product.
Data analysis.
All experiments were performed a minimum of three times. Statistical significance was assessed by repeated-measures analysis of variance (ANOVA). Differences identified by ANOVA were pinpointed by the Student Newman-Keuls multiple range text.
RESULTS
Src is activated by RV infection and required for RV39-induced Akt activation.
We have recently shown that the infection of human bronchial epithelial cells with RV39 induces the rapid phosphorylation of the p85 regulatory subunit of PI 3-kinase (21). To determine whether Src is involved in RV-induced PI 3-kinase activation, we examined the localization of RV39, Src, and PI 3-kinase in RV39-infected 16HBE14o− human bronchial epithelial cells by confocal fluorescence microscopy. We found a close association of RV, Src, and p85 PI 3-kinase (Fig. 1, upper panel). There was also colocalization of RV, Src, and stably expressed Cit-Akt-PH, a marker of PI(3,4,5)P3-containing membranes (middle panel). Finally, RV and Cit-Akt-PH colocalized with p110β, a catalytic subunit of PI 3-kinase (lower panel).
FIG. 1.

RV39 increases the association of Src with PI 3-kinase containing membranes. 16HBE14o− cells were infected with sham HeLa cell lysates or RV39 at an MOI of 100 for 10 min. (Upper panel) Confocal fluorescent microscopy was initially performed with guinea pig anti-RV39 (blue channel in all panels), mouse anti-Src (clone GD-11 [red]), and rabbit p85α (green). The colocalization of RV, Src, and p85α in the merged image appears white. (Middle panel) Colocalization of RV39, Src (using rabbit anti-Src specific for the C terminus [red]), and stably expressed Cit-Akt-PH, a marker of PI(3,4,5)P3-containing membranes (green), appears white in the merged image. (Lower panel) RV and p110β, a catalytic subunit of PI 3-kinase (red), colocalize with Cit-Akt-PH (green), appearing white in the merged image.
Compared to sham-infected cells, 16HBE14o− cells infected with RV39 also demonstrated colocalization of RV39 and phospho-Tyr416 Src (Fig. 2, upper panel). Colocalization was blocked when cells were preincubated with the Src family kinase inhibitor PP2 but not with its less potent analogue PP3. A similar pattern of colocalization was observed with RV and phospho-Ser473 Akt (Fig. 2, lower panel). Again, colocalization was demonstrated by preincubation with PP2 but not with PP3. The PI 3-kinase inhibitor LY294002 blocked virus internalization and colocalization of RV39 and phospho-Akt but did not block virus binding.
FIG. 2.

RV39 internalization and colocalization with phospho-Tyr416 Src (upper panel) and phospho-Ser473Akt (lower panel) are blocked by chemical Src inhibitors. 16HBE14o− cells were incubated with sham or RV39 at an MOI of 100. RV39 (shown in green) induced Tyr416 phosphorylation of Src (red, upper panel) and colocalized with Src (yellow). The high-affinity Src inhibitor PP2 blocked pTyr416 Src colocalization of RV but did not block virus binding. The low-affinity inhibitor PP3 did not block the association of RV and Src. RV39 also induced Ser473 phosphorylation of Akt (red, lower panel) and colocalized with an Akt fraction (yellow). PP2 appeared to block Ser473 Akt phosphorylation and the colocalization of RV and Akt. The low-affinity inhibitor PP3 did not block the association of RV and Akt. LY294002 also appeared to block virus internalization and colocalization with phospho-Ser473Akt.
Infection of 16HBE14o− cells with RV39 (MOI of 1.0 for 10 min at 33°C) was also sufficient to increase Tyr416 phosphorylation of a 60-kDa Src family kinase (Fig. 3A and B). Immunoblots of cell extracts with the C-terminal anti-Src antibody showed the presence of Src. We were unable to detect Fyn, another Src family kinase with a molecular mass of approximately 60 kDa (not shown). Src phosphorylation was efficiently blocked by PP2 but not by PP3. RV39 infection also induced phosphorylation of Ser473 Akt (Fig. 3C). Akt phosphorylation was blocked by PP2 as well as the PI 3-kinase inhibitor LY294002.
FIG. 3.

RV39 increases phosphorylation of Ser473Akt in a Src- and PI 3-kinase-dependent manner. (A) 16HBE14o− cells without or with inhibitor (1 h at 37°C) were treated with sham or RV39 infection (MOI of 1, 10 min at 33°C). Detergent-soluble proteins (500 μg) were immunoprecipitated with mouse monoclonal 4G10 anti-phosphotyrosine (2 μg). Immunoprecipitates were immunoblotted for phospho-Tyr416 (p-Tyr416) Src (top panel) or total Src. (B) Group mean data for three experiments (means ± standard errors of the means; asterisk indicates a value different from that for RV alone; P < 0.05 by ANOVA). (C) Whole-cell lysates were immunoblotted for phospho-Ser473 Akt (20 μg protein/lane). The PP2 Src family kinase inhibitor blocked phosphorylation of both Src and Akt more effectively than PP3 did. These results are representative of three experiments.
Digoxigenin-labeled RV39 associates with an active complex of signaling molecules, including Src and Akt.
To better study such RV39-protein complexes, an immunoprecipitable RV39 was developed using a chemically modified form of the virus similar to that used in our previous work (21). An N-hydroxysuccinimide derivative of digoxigenin was used to tag the capsid surface, thereby providing a specific epitope for immunochemistry. The chemically modified form of RV39 retained infectivity identical to that of unmodified virus. We reasoned that immunoprecipitation with anti-digoxigenin would obtain a Triton-insoluble complex of RV39 bound to its receptor and associated signaling molecules.
16HBE14o− cells were infected with digoxigenin-labeled RV39 (MOI of 1.0) for 10 min. Cells were lysed in 1% Triton X-100 and centrifuged (10,000 × g), and the supernatant was collected for immunoprecipitation with anti-digoxigenin. Immunoprecipitates were incubated with [γ-32P]ATP and Crosstide, a glycogen synthase kinase α/β fusion protein sequence which is a substrate for Akt (6). Cells exposed to RV39 demonstrated a Crosstide kinase activity in vitro (Fig. 4). This kinase activity was not found when cells were exposed to an equimolar (based on digoxigenin) amount of labeled sham protein. Serine phosphorylation of Crosstide was confirmed by immunoblotting (not shown), and phosphorylation was blocked by PP2 but not by PP3. These data suggest that RV infection activates Akt in a Src-dependent manner.
FIG. 4.
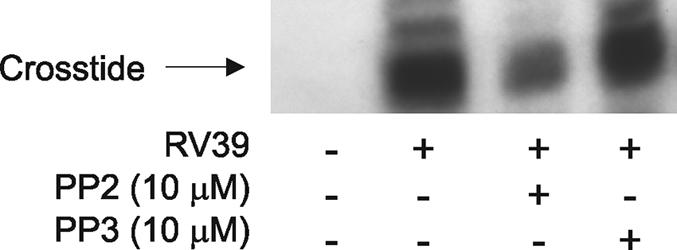
Activated Akt coimmunoprecipitates with labeled RV39 after infection, and its activity is inhibited by a chemical Src inhibitor. Cells were treated with 3 nM digoxigenin-labeled sham or digoxigenin-labeled RV39 (MOI of 1 for 10 min). Selected cultures were pretreated with PP2 or PP3 (10 μM). Cell lysates were immunoprecipitated for digoxigenin. Immunoprecipitates were incubated with [γ-32P]ATP and Crosstide. Digoxigenin immunoprecipitates contained Crosstide kinase activity, which was reduced by PP2. PP2 also reduced the abundance of phosphorylated Akt in the immunoprecipitates. This experiment was repeated twice.
Colocalization of RV, Src, and Akt with ceramide, a constituent of lipid rafts.
It has recently been shown that RVs infect human epithelial cells via ceramide-enriched membrane platforms (11). While nonionic detergents like Triton X-100 are sufficient to dissociate many membrane-associated complexes, initial experiments to biochemically enrich virus-associated rafts by sucrose density flotation met with unsatisfactory results because the virus tends to sediment in sucrose density gradients. We therefore colocalized RV and relevant signaling intermediates with ceramide. In this set of experiments, digoxigenin-labeled RV (MOI of 10) was used. Composite projection views show colocalization of RV39, ceramide, phospho-Tyr416 Src, and phospho-Ser473 Akt (Fig. 5A). The association of RV39 with Src and Akt in ceramide-enriched lipid domains strongly suggests the formation of raft protein aggregates.
FIG. 5.

Digoxigenin-labeled RV39 colocalizes with phosphorylated Src and Akt in ceramide-containing lipid rafts. (A) RV39 immunoreactivity (blue) colocalized with ceramide (red), phospho-Tyr416 Src (green, left panel), and phospho-Ser473 Akt (green, right panel) in a white merged image. The colocalization of both Src and Akt with ceramide in punctate structures at the cell surface suggests that these protein kinases are associated with raft complexes where bound RV is aggregated. (B) Digoxigenin-labeled RV39 also colocalized with Src in primary mucociliary differentiated airway epithelial cells. The apical surface of primary airway epithelial cells grown at an air-liquid interface were incubated with sham or digoxigenin-labeled RV39 at an MOI of 100. Digoxigenin-labeled RV39 (shown in green) localized to the airway epithelial cell surface. Src was visualized using the clone GD11 antibody (shown in red). RV39 appeared to colocalize with Src just under the plasma membrane (see z-axis section). The high-affinity Src inhibitor PP2 appeared to block the colocalization of RV and Src. The low-affinity inhibitor PP3 did not block the association of RV and Src.
Digoxigenin-labeled RV39 colocalizes with Src in primary airway epithelial cells.
We tested whether the colocalization of digoxigenin-labeled RV39 and Src occurs in primary human airway epithelial cells. Compared to sham-infected cells, cells infected with RV39 showed colocalization of RV and Src, as recognized by the GD11 antibody (Fig. 5B). A z-axis section more clearly demonstrates the three-dimensional arrangement of RV and Src, with colocalization occurring just below the cell surface. Colocalization of RV39 and Src was blocked by PP2 but not by PP3.
Src kinase activity is required for the IL-8 response to RV39.
We have recently shown that infection of human bronchial epithelial cells with RV39 induces rapid activation of PI 3-kinase and phosphorylation of the PI 3-kinase p85α regulatory subunit (21). Since Src is associated with focal adhesion complexes and phosphorylates p85 (15), we tested the dependence of IL-8 production induced by RV39 on Src kinase activation. Src siRNA specifically reduced Src protein expression (Fig. 6A). In 16HBE14o− human bronchial epithelial cells, expression of the dominant-negative K296R/Y528F Src or Src siRNA or treatment with the Src family-specific kinase inhibitor PP2 (0.1 to 1 μM) significantly inhibited RV39-induced transcription from the IL-8 promoter (Fig. 6B). PP2 also attenuated IL-8 release from primary mucociliary differentiated airway epithelial cells infected with RV39 (Fig. 6C). PP3 (1 μM), an inactive analogue of PP2, did not significantly inhibit IL-8 release. Together, these findings suggest that Src is required for RV-stimulated IL-8 expression.
FIG. 6.

Inhibition of Src kinase activity blocks the IL-8 response to RV39. (A) Immunoblots showing knockdown of Src and p110β PI 3-kinase with specific siRNAs. (B) 16HBE14o− cells were transiently transfected with an IL-8 luciferase reporter and either 100 ng empty vector, dominant-negative K297R Src (dnSrc), 100 nM nontargeting RNA (nt siRNA), or Src siRNA. Dominant-negative Src inhibited RV39-induced IL-8 promoter activity. Selected cells were also treated either with dimethyl sulfoxide carrier, with the indicated concentration of Src family tyrosine kinase inhibitor PP2, or with its less potent and specific analog PP3. Cells were infected with RV39 (MOI of 1 for 1 h), incubated for 24 h, and harvested for assessment of luciferase activity. (C) Primary mucociliary differentiated human airway epithelial cells were infected with 5 × 106 TCID50/ml RV39 for 1 h in the presence of either PP2 or PP3 and incubated for an additional 48 h. IL-8 protein abundance was measured by enzyme-linked immunosorbent assay for 48 h. Protein abundance was inhibited by PP2 but not by a comparable concentration of PP3. For each panel, results from three experiments are shown, data shown are means ± standard errors of the means, and asterisks indicate P values of <0.05 versus those for RV39, RV39 plus empty vector, or RV39 plus nontargeting siRNA by ANOVA.
p110β PI 3-kinase is required for RV39-induced IL-8 expression.
Using RT-PCR, we confirmed that p110β is present in airway epithelial cells (Fig. 7A). Transient transfection of 16HBE14o− human bronchial epithelial cells with siRNA against p110β significantly inhibited p110β protein expression (Fig. 7A) and RV39-induced transcription from the IL-8 promoter (Fig. 7B), demonstrating that p110β is required for the response.
FIG. 7.

p110β PI 3-kinase is required for RV39-induced IL-8 expression. (A) RT-PCR showing presence of p110β (IAα) but not p110α (IAβ) or p110γ (IAγ), in airway epithelial cells. (B) Transient transfection of 16HBE14o− human bronchial epithelial cells with p110β siRNA, but not with nontargeting (nt) siRNA, inhibited RV39-induced transcription from the IL-8 promoter (n = 3; *, P < 0.05 by ANOVA).
DISCUSSION
We have recently shown that infection of human airway epithelial cells with RV39 induces rapid activation of PI 3-kinase and phosphorylation of Akt. (21). PI 3-kinase activation was accompanied by phosphorylation of the PI 3-kinase p85 regulatory subunit. RV39 also colocalized with Cit-Akt-PH, a fluorescent fusion protein containing the pleckstrin homology domain of Akt, indicating that 3-phosphorylated PI accumulates at the site of RV infection. We now show that, following the engagement of ICAM-1 by RV39, Src functions as an upstream activator of Akt and that RV colocalizes with Src, PI 3-kinase p110β, and Akt in lipid rafts. These data are consistent with previous work with endothelial cells showing that antibody cross-linking of ICAM-1 causes colocalization with Src in lipid rafts (35) as well as Src phosphorylation and activation (7, 37).
Src/PI 3-kinase signaling has been noted to be activated in the context of viral infection twice previously. Engagement of the B lymphocyte Epstein-Barr virus receptor activates PI 3-kinase via Src (2). Expression of human herpesvirus 8 envelope glycoprotein gB induces Src/PI 3-kinase signaling in human foreskin fibroblasts (33). We now extend this mode of entry to RV-infected human bronchial epithelial cells. The data shown here demonstrate that activation of a Src/p110β PI 3-kinase/Akt pathway occurs coincident with the binding of RV into ceramide-enriched lipid domains as a prelude to internalization.
Expression of dominant-negative K296R/Y528F Src and Src siRNA, as well as pretreatment with a Src family kinase chemical inhibitor, each significantly attenuated RV39-induced IL-8 expression, demonstrating that Src is required for maximal RV-induced IL-8 expression. However, IL-8 expression was not completely eliminated, suggesting that other pathways are also required. Consistent with this, we have found that Toll-like receptor-3, a receptor for double-stranded RNA, is also required for RV-induced chemokine expression (31), as it is for respiratory syncytial virus and influenza virus, two other positive-strand RNA viruses (14, 29).
Confocal micrographs showed that while RV39 infection induces Tyr416 phosphorylation of Src, only a fraction of phosphoprotein colocalizes with RV. Lipid-lipid interactions involving amino-terminal acyl groups on Src family kinases, for example, palmitoylation, are the primary mechanism for membrane localization, particularly localization to membrane microdomains or lipid rafts (18). The exclusion of a Src fraction from rafts implies that it may not encounter substrates needed for localization to lipid rafts and that this Src fraction may be performing functions distinct from those of raft-localized Src. Similarly, Ser473 Akt phosphorylation was not confined to areas of colocalization with RV, suggesting diffusion of the class IA PI 3-kinase product PI(3,4,5)P3 outside the raft area. These data are consistent with our previous data indicating membrane and cytoplasmic localization of the pleckstrin homology domain of Akt following RV infection (21).
As in a previous study (11), we did not biochemically enrich RV-associated rafts by sucrose density flotation, as the virus tended to sediment in sucrose density gradients. Interestingly, rafts not associated with RV contained inactive/closed Tyr527-phosphorylated Src (19) (not shown). However, the association of RV, Src, Akt, and ceramide, which brings about the coalescence of microscopic rafts into large-membrane macrodomains, strongly suggests that RV initiates the formation of Src- and Akt-containing membrane platforms. Src may therefore represent a cellular target for intervention against RV-associated respiratory disease.
Acknowledgments
These studies were supported by National Institutes of Health grants HL56399, HL81420, and HL82550 and by the Cystic Fibrosis Foundation.
We thank Allan Brasier and Steven White for providing needed reagents.
Footnotes
Published ahead of print on 22 November 2006.
REFERENCES
- 1.Aaron, S. D., J. B. Angel, M. Lunau, K. Wright, C. Fex, N. Le Saux, and R. E. Dales. 2001. Granulocyte inflammatory markers and airway infection during acute exacerbation of chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 163:349-355. [DOI] [PubMed] [Google Scholar]
- 2.Barel, M., M. Balbo, M. Le Romancer, and R. Frade. 2003. Activation of Epstein-Barr virus/C3d receptor (gp140, CR2, CD21) on human cell surface triggers pp60src and Akt-GSK3 activities upstream and downstream to PI 3-kinase, respectively. Eur. J. Immunol. 33:2557-2566. [DOI] [PubMed] [Google Scholar]
- 3.Casola, A., R. P. Garofalo, M. Jamaluddin, S. Vlahopoulos, and A. R. Brasier. 2000. Requirement of a novel upstream response element in respiratory syncytial virus-induced IL-8 gene expression. J. Immunol. 164:5944-5951. [DOI] [PubMed] [Google Scholar]
- 4.Chantry, D., A. Vojtek, A. Kashishian, D. A. Holtzman, C. Wood, P. W. Gray, J. A. Cooper, and M. F. Hoekstra. 1997. p110delta, a novel phosphatidylinositol 3-kinase catalytic subunit that associates with p85 and is expressed predominantly in leukocytes. J. Biol. Chem. 272:19236-19241. [DOI] [PubMed] [Google Scholar]
- 5.Cozens, A. L., M. J. Yezzi, K. Kunzelmann, T. Ohrui, L. Chin, K. Eng, W. E. Finkbeiner, J. H. Widdicombe, and D. C. Gruenert. 1994. CFTR expression and chloride secretion in polarized immortal human bronchial epithelial cells. Am. J. Respir. Cell Mol. Biol. 10:38-47. [DOI] [PubMed] [Google Scholar]
- 6.Cross, D. A., D. R. Alessi, P. Cohen, M. Andjelkovich, and B. A. Hemmings. 1995. Inhibition of glycogen synthase kinase-3 by insulin mediated by protein kinase B. Nature 378:785-789. [DOI] [PubMed] [Google Scholar]
- 7.Etienne-Manneville, S., J.-B. Manneville, P. Adamson, B. Wilbourn, J. Greenwood, and P.-O. Couraud. 2000. ICAM-1-coupled cytoskeletal rearrangements and transendothelial lymphocyte migration involve intracellular calcium signaling in brain endothelial cell lines. J. Immunol. 165:3375-3383. [DOI] [PubMed] [Google Scholar]
- 8.Fleming, H., F. Little, D. Schnurr, P. Avila, H. Wong, J. Liu, S. Yagi, and H. Boushey. 1999. Rhinovirus-16 colds in healthy and in asthmatic subjects: similar changes in upper and lower airways. Am. J. Respir. Crit. Care Med. 160:100-108. [DOI] [PubMed] [Google Scholar]
- 9.Gern, J. E., R. Vrtis, K. A. Grindle, C. Swenson, and W. W. Busse. 2000. Relationship of upper and lower airway cytokines to outcome of experimental rhinovirus infection. Am. J. Respir. Crit. Care Med. 162:2226-2231. [DOI] [PubMed] [Google Scholar]
- 10.Gompertz, S., C. O'Brien, D. L. Bayley, S. L. Hill, and R. A. Stockley. 2001. Changes in bronchial inflammation during acute exacerbations of chronic bronchitis. Eur. Respir. J. 17:1112-1119. [DOI] [PubMed] [Google Scholar]
- 11.Grassme, H., A. Riehle, B. Wilker, and E. Gulbins. 2005. Rhinoviruses infect human epithelial cells via ceramide-enriched membrane platforms. J. Biol. Chem. 280:26256-26262. [DOI] [PubMed] [Google Scholar]
- 12.Grunberg, K., H. H. Smits, M. C. Timmers, E. P. A. De Klerk, R. J. E. M. Dolhain, E. C. Dick, P. S. Hiemstra, and P. J. Sterk. 1997. Experimental rhinovirus 16 infection. Effects on cell differentials and soluble markers in sputum in asthmatic subjects. Am. J. Respir. Crit. Care Med. 156:609-616. [DOI] [PubMed] [Google Scholar]
- 13.Grunberg, K., M. C. Timmers, H. H. Smits, E. P. de Klerk, E. C. Dick, W. J. Spaan, P. S. Hiemstra, and P. J. Sterk. 1997. Effect of experimental rhinovirus 16 colds on airway hyperresponsiveness to histamine and interleukin-8 in nasal lavage in asthmatic subjects in vivo. Clin. Exp. Allergy 27:36-45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Guillot, L., R. Le Goffic, S. Bloch, N. Escriou, S. Akira, M. Chignard, and M. Si-Tahar. 2005. Involvement of Toll-like receptor 3 in the immune response of lung epithelial cells to double-stranded RNA and influenza A virus. J. Biol. Chem. 280:5571-5580. [DOI] [PubMed] [Google Scholar]
- 15.Haefner, B., R. Baxter, V. J. Fincham, C. P. Downes, and M. C. Frame. 1995. Cooperation of Src homology domains in the regulated binding of phosphatidylinositol 3-kinase. J. Biol. Chem. 270:7937-7943. [DOI] [PubMed] [Google Scholar]
- 16.Hanke, J. H., J. P. Gardner, R. L. Dow, P. S. Changelian, W. H. Brissette, E. J. Weringer, B. A. Pollok, and P. A. Connelly. 1996. Discovery of a novel, potent, and src family-selective tyrosine kinase inhibitor. J. Biol. Chem. 271:695-701. [DOI] [PubMed] [Google Scholar]
- 17.Johnston, S. L., P. K. Pattemore, G. Sanderson, S. Smith, F. Lampe, L. Josephs, P. Symington, S. O'Toole, S. H. Myint, D. A. Tyrrell, and S. T. Holgate. 1995. Community study of role of viral infections in exacerbations of asthma in 9-11 year old children. Br. Med. J. 310:1225-1229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liang, X., A. Nazarian, H. Erdjument-Bromage, W. Bornmann, P. Tempst, and M. D. Resh. 2001. Heterogeneous fatty acylation of src family kinases with polyunsaturated fatty acids regulates raft localization and signal transduction. J. Biol. Chem. 276:30987-30994. [DOI] [PubMed] [Google Scholar]
- 19.MacAuley, A., M. Okada, S. Nada, H. Nakagawa, and J. A. Cooper. 1993. Phosphorylation of Src mutants at Tyr 527 in fibroblasts does not correlate with in vitro phosphorylation by CSK. Oncogene 8:117-124. [PubMed] [Google Scholar]
- 20.Mukhopadhyay, D., L. Tsiokas, X. M. Zhou, D. Foster, J. S. Brugge, and V. P. Sukhatme. 1995. Hypoxic induction of human vascular endothelial growth factor expression through c-Src activation. Nature 375:577-581. [DOI] [PubMed] [Google Scholar]
- 21.Newcomb, D. C., U. Sajjan, S. Nanua, Y. Jia, A. M. Goldsmith, J. K. Bentley, and M. B. Hershenson. 2005. Phosphatidylinositol 3-kinase is required for rhinovirus-induced airway epithelial cell interleukin-8 expression. J. Biol. Chem. 280:36952-36961. [DOI] [PubMed] [Google Scholar]
- 22.Nicholson, K. G., J. Kent, and D. C. Ireland. 1993. Respiratory viruses and exacerbations of asthma in adults. Br. Med. J. 307:982-986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Norzila, M. Z., K. Fakes, R. L. Henry, J. Simpson, and P. G. Gibson. 2000. Interleukin-8 secretion and neutrophil recruitment accompanies induced sputum eosinophil activation in children with acute asthma. Am. J. Respir. Crit. Care Med. 161:769-774. [DOI] [PubMed] [Google Scholar]
- 24.Ordonez, C. L., T. E. Shaughnessy, M. A. Matthay, and J. V. Fahy. 2000. Increased neutrophil numbers and IL-8 levels in airway secretions in acute severe asthma. Clinical and biologic significance. Am. J. Respir. Crit. Care Med. 161:1185-1190. [DOI] [PubMed] [Google Scholar]
- 25.Papi, A., and S. L. Johnston. 1999. Rhinovirus infection induces expression of its own receptor intercellular adhesion molecule 1 (ICAM-1) via increased NF-kB-mediated transcription. J. Biol. Chem. 274:9707-9720. [DOI] [PubMed] [Google Scholar]
- 26.Pizzichini, M. M., E. Pizzichini, A. Efthimiadis, A. J. Chauhan, S. L. Johnston, P. Hussack, J. Mahony, J. Dolovich, and F. E. Hargreave. 1998. Asthma and natural colds. Inflammatory indices in induced sputum: a feasibility study. Am. J. Respir. Crit. Care Med. 158:1178-1184. [DOI] [PubMed] [Google Scholar]
- 27.Qiu, Y., J. Zhu, V. Bandi, R. L. Atmar, K. Hattotuwa, K. K. Guntupalli, and P. K. Jeffery. 2003. Biopsy neutrophilia, neutrophil chemokine and receptor gene expression in severe exacerbations of chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 168:968-975. [DOI] [PubMed] [Google Scholar]
- 28.Rohde, G., A. Wiethege, I. Borg, M. Kauth, T. T. Bauer, A. Gillissen, A. Bufe, and G. Schultze-Werninghaus. 2003. Respiratory viruses in exacerbations of chronic obstructive pulmonary disease requiring hospitalisation: a case-control study. Thorax 58:37-42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rudd, B. D., E. Burstein, C. S. Duckett, X. Li, and N. W. Lukacs. 2005. Differential role for TLR3 in respiratory syncytial virus-induced chemokine expression. J. Virol. 79:3350-3357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sajjan, U., S. Keshavjee, and J. Forstner. 2004. Responses of well-differentiated airway epithelial cell cultures from healthy donors and patients with cystic fibrosis to Burkholderia cenocepacia infection. Infect. Immun. 72:4188-4199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sajjan, U. S., Y. Jia, D. C. Newcomb, J. K. Bentley, N. W. Lukacs, J. J. LiPuma, and M. B. Hershenson. 2006. H. influenzae potentiates airway epithelial cell responses to rhinovirus by increasing ICAM-1 and TLR3 expression. FASEB J. 20:2121-2123. [DOI] [PubMed] [Google Scholar]
- 32.Seemungal, T., R. Harper-Owen, A. Bhowmik, I. Moric, G. Sanderson, S. Message, P. Maccallum, T. W. Meade, D. J. Jeffries, S. L. Johnston, and J. A. Wedzicha. 2001. Respiratory viruses, symptoms, and inflammatory markers in acute exacerbations and stable chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 164:1618-1623. [DOI] [PubMed] [Google Scholar]
- 33.Sharma-Walia, N., P. P. Naranatt, H. H. Krishnan, L. Zeng, and B. Chandran. 2004. Kaposi's sarcoma-associated herpesvirus/human herpesvirus 8 envelope glycoprotein gB induces the integrin-dependent focal adhesion kinase-Src-phosphatidylinositol 3-kinase-Rho GTPase signal pathways and cytoskeletal rearrangements. J. Virol. 78:4207-4223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Subauste, M. C., D. B. Jacoby, S. M. Richards, and D. Proud. 1995. Infection of a human respiratory epithelial cell line with rhinovirus. Induction of cytokine release and modulation of susceptibility to infection by cytokine exposure. J. Clin. Investig. 96:549-557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tilghman, R. W., and R. L. Hoover. 2002. E-selectin and ICAM-1 are incorporated into detergent-insoluble membrane domains following clustering in endothelial cells. FEBS Lett. 525:83-87. [DOI] [PubMed] [Google Scholar]
- 36.Traxler, P., G. Bold, J. Frei, M. Lang, N. Lydon, H. Mett, E. Buchdunger, T. Meyer, M. Mueller, and P. Furet. 1997. Use of a pharmacophore model for the design of EGF-R tyrosine kinase inhibitors: 4-(phenylamino)pyrazolo[3,4-d]pyrimidines. J. Med. Chem. 40:3601-3616. [DOI] [PubMed] [Google Scholar]
- 37.Wang, Q., G. R. Pfeiffer II, and W. A. Gaarde. 2003. Activation of SRC tyrosine kinases in response to ICAM-1 ligation in pulmonary microvascular endothelial cells. J. Biol. Chem. 278:47731-47743. [DOI] [PubMed] [Google Scholar]
- 38.Yamaya, M., K. Sekizawa, T. Suzuki, N. Yamada, M. Furukawa, S. Ishizuka, K. Nakayama, M. Terajima, Y. Numazaki, and H. Sasaki. 1999. Infection of human respiratory submucosal glands with rhinovirus: effects on cytokine and ICAM-1 production. Am. J. Physiol. Lung Cell. Mol. Physiol. 277:L362-L371. [DOI] [PubMed] [Google Scholar]
- 39.Zhu, Z., W. Tang, J. M. Gwaltney, Y. Wu, and J. A. Elias. 1997. Rhinovirus stimulation of interleukin-8 in vivo and in vitro: role of NF-kappa B. Am. J. Physiol. Lung Cell. Mol. Physiol. 273:L814-L824. [DOI] [PubMed] [Google Scholar]