

Abstract
Tumor necrosis factor (TNF) is a multifunctional cytokine that has a role in induction and regulation of host innate and adaptive immune responses. The importance of TNF antiviral mechanisms is reflected by the diverse strategies adopted by different viruses, particularly members of the herpesvirus family, to block TNF responses. TNF binds and signals through two receptors, Tnfrsf1a (TNF receptor 1 [TNFR1], or p55) and Tnfrsf1b (TNFR2, or p75). We report here that herpes simplex virus 1 (HSV-1) infection of TNF−/− mice on the resistant C57BL/6 genetic background results in significantly increased susceptibility (P < 0.0001, log rank test) to fatal HSV encephalitis (HSE) and prolonged persistence of elevated levels of virus in neural tissues. In contrast, although virus titers in neural tissues of p55−/−N13 mice were elevated to levels comparable to what was found for the TNF−/− mice, the p55−/−N13 mice were as resistant as control C57BL/6 mice (P > 0.05). The incidence of fatal HSE was significantly increased by in vivo neutralization of TNF using soluble TNFR1 (sTNFR1) or depletion of macrophages in C57BL/6 mice (P = 0.0038 and P = 0.0071, respectively). Strikingly, in vivo neutralization of TNF in HSV-1-infected p55−/− p75−/− mice by use of three independent approaches (treatment with soluble p55 receptor, anti-TNF monoclonal antibody, or in vivo small interfering RNA against TNF) resulted in significantly increased mortality rates (P = 0.005), comparable in magnitude to those for C57BL/6 mice treated with sTNFR1 (P = 0.0018). Overall, these results indicate that while TNF is required for resistance to fatal HSE, both p55 and p75 receptors are dispensable. Precisely how TNF mediates protection against HSV-1 mortality in p55−/− p75−/− mice remains to be determined.
Early innate and subsequent adaptive immune responses to viral and bacterial pathogens are critically dependent on the tumor necrosis factor (TNF) superfamily of cytokines. These TNF superfamily cytokines act as effectors of host defense and regulate peripheral lymphoid tissue organogenesis and differentiation of natural killer cells and lymphoid cells (42, 46). TNF, a multifunctional cytokine produced primarily by activated macrophages (70), functions as a key regulator of leukocyte trafficking by affecting chemokine expression and stimulating antigen presentation, which it does by inducing dendritic cell maturation (26, 58). TNF exists in two forms, a precursor 26-kDa membrane-bound form (mTNF) and a 17-kDa soluble form (sTNF), both of which are bioactive (46, 71). TNF and the closely related ligand lymphotoxin-α (LT) bind as homotrimers to two receptors, TNF receptor 1 (TNFR1, or p55) and TNFR2 (p75), which are widely expressed on most cell types (71). Activation of p55 generally results in gene activation that leads to induction of inflammatory and cytotoxic responses, while activation of TNFR2 is associated with thymocyte proliferation and T-cell activation. In response to TNF binding, lipopolysaccharide (LPS) and several other stimuli in the extracellular domains of both TNFRs are released by proteolytic cleavage and these soluble TNFR (sTNFR) forms function as inhibitors of TNF signaling (1, 7, 18, 40). TNF has a role in several viral diseases of the central nervous system (CNS), including, for example, those caused by human immunodeficiency virus, feline immunodeficiency virus, herpes simplex virus (HSV), cytomegalovirus, Epstein-Barr virus, Sindbis virus, and Theiler's murine encephalomyelitis virus, with effects ranging from protective to toxic (31, 52).
Peripheral infection of mice with HSV involves local replication in epithelial tissues followed by rapid dissemination of virus via sensory axons for the corresponding ganglia and often the CNS (16). CNS infection in susceptible mouse strains can have effects ranging from mild to fatal encephalitis for virulent HSV strains. The early corneal infiltrate elicited by corneal infection is composed predominantly of neutrophils, and antibody-mediated depletion of neutrophils results in decreased clearance of virus and enhanced spread to the CNS (63, 66). Production of TNF and nitric oxide (NO), molecules with potent antiviral activities, may contribute to neutrophil-mediated clearance of HSV-1, whereas neutrophil production of interleukin-12 induces a CD4+ Th1-like response that mediates the development of herpes stromal keratitis, an immunopathologic disease (20, 30, 65). Activated macrophages are also present in the cornea early in infection and are responsible for the release of antiviral factors, like TNF, alpha interferon (IFN-α), and IFN-β. Synergism of TNF with IFN-α can induce IFN-β, resulting in potent suppression of HSV-1 infection both in vitro in cultured human fibroblasts and in vivo when expressed ectopically in the cornea (12, 57).
In contrast to what occurs in the cornea, macrophages rather than neutrophils dominate the early (day 3) inflammatory infiltrate in the trigeminal ganglion (Tg) after corneal inoculation of HSV (35, 59). Macrophages were shown to be the primary producers of TNF, interleukin-12, and inducible nitric oxide synthase, whereas γδ T cells produced IFN-γ in the ganglion, and both cell types were found in close proximity to infected neurons, suggesting a role in the control of HSV-1 replication (35). Accumulation of T cells, particularly CD8+ T cells, was delayed and occurred coincident with the clearance of HSV-1 antigen from the ganglion. We and others previously reported the unexpected observation that the inflammatory response persisted well into latency, with associated production of IFN-γ and TNF in close juxtaposition with infected neurons (11, 27, 41, 59). In one study, TNF was the major cytokine produced in the ganglion and the only cytokine detected on the CNS side of the dorsal root entry zone (60). These observations imply an important role for IFN-γ and TNF in the control of HSV-1 infection in neurons during acute and latent infection. Utilizing IFN-γ and IFN-γ receptor-null mutant mice, we demonstrated a role for IFN-γ in the control of in vivo-reactivated HSV-1, but the results did not support a role for IFN-γ in the control of acute infection (9, 10, 32); a role for IFN-γ in the control of HSV-1 latency has been confirmed and extended in recent studies (21).
Although TNF can potently inhibit HSV-1 in cultured cells, its in vivo role has not been clearly delineated (13, 24). Local TNF has been reported to both exacerbate herpes stromal keratitis and mediate protection from corneal scarring in ocular mouse models (25, 33). In a prior study, TNF pretreatment was shown to confer significant protection from lethal intraperitoneal HSV-1 challenge of resistant C57BL/6 mice by a mechanism independent of IFN production or natural killer cell activation (55). TNF and IFN-γ have also been shown to be important for macrophage activation and control of HSV and murine cytomegalovirus replication, independent of T and B cells (29). Further evidence that TNF signaling pathways are crucial for effective host immune defense against herpesviruses comes from recent reports that herpesviruses encode genes that target TNF-related cytokines and/or their associated receptors, as an immune evasion strategy (6, 37). Thus, HSV-1 exploits the herpesvirus entry mediator (HVEM, or HveA), a member of the TNFR superfamily, to enter lymphoid cells via glycoprotein Dbinding (38, 50). By antagonizing LIGHT, the lymphotoxin-related natural ligand for HVEM that is involved in T-cell activation, HSV-1 could potentially impede T-cell activation (14, 45, 64) and also prevent interaction with B- and T-lymphocyte attenuator, a known coinhibitory ligand for HVEM (17).
To better understand the role of TNF in the host immunity to HSV-1, we compared the outcome of infection in mice lacking TNFR1 (p55−/−) or both known receptors (p55−/− p75−/−) (53, 54) to that in mice deficient for TNF (36), all mice being on the resistant C57BL/6 background. Results from these studies showed that TNF signaling via p55 played a role in the control of HSV-1 replication in the eye, ganglion, and brain stem and also conferred protection against fatal HSV encephalitis (HSE). Surprisingly, neither p55 nor p75 was required for protection against fatal HSE, which implicates a novel TNF receptor in the mediation of the protective effects of TNF during HSV-1 infection.
MATERIALS AND METHODS
Mouse strains.
TNF receptor p55 (Tnfrsf1a)-null mutant mice backcrossed 13 times to C57BL/6 (p55−/− N13) mice were obtained from Amgen Inc. (Thousand Oaks, CA). TNF double-receptor knockout (p55−/− p75−/−N5) mice, originally derived by Peschon et al. (53) by crossing a p75−/− N4 strain with a p55−/− strain produced with B6 embryonic stem (ES) cells, were obtained from Lyle Moldawer (University of Florida, Gainsville, FL) or The Jackson Laboratory (Bar Harbor, ME). TNF−/− mice (also produced using B6 ES cells) (15) were obtained from DNAX (Palo Alto, CA). C57BL/6 mice were obtained from The Jackson Laboratory (Bar Harbor, ME), and 129S6 mice were from Taconic (Germantown, NY).
Virus stocks and inoculation of mice.
Master stocks of HSV-1 strain 17+ composed only of cell-released virus were prepared in, and their titers determined on, mycoplasma-free CV-1 cell monolayers. Single-use aliquots of virus in Hanks balanced salt solution supplemented with 2% fetal bovine serum (FBS) were stored at −80°C. Mice were inoculated with HSV-1 by corneal scarification. The right corneas of the mice, deeply anesthetized by intraperitoneal injection of ketamine and xylazine, were gently scarified using a 27-gauge needle as follows: 10 vertical strokes, followed by an application of HSV in a volume of 4 μl of Hanks balanced salt solution, followed by another 10 horizontal strokes and gentle massaging of the eye with the eyelid to promote virus uptake. The same virus master stock was used for all experiments reported here. The City of Hope animal care committee approved all animal procedures.
Determination of NO levels in macrophage cultures.
Resident peritoneal exudate macrophages (PE-MP) were obtained by lavage with RPMI medium supplemented with 5% FBS. The cells were washed and plated in a 100-cm2 tissue culture dish in RPMI medium-10% FBS. The next day, the culture was washed, and the adherent cells were removed by scraping them in cell dissociation buffer and replated at a density of 2.5 × 105 cells per well in a 96-well plate. Macrophages were activated by treatment with IFN-γ/LPS, and 24 h later, NO levels in macrophage culture supernatants were determined as nitrite concentrations by use of the Greiss reagent and quantitated by comparison to a standard curve generated using sodium nitrate (62). Briefly, a 100-μl aliquot of medium from the macrophage cultures was mixed with an equal volume of Greiss reagent [1% sulfanilamide, 0.1% N-(1-napthyl) etheylenediamine dihydrochlororide, 2.1% phosphoric acid], and after 5 min at room temperature, the absorbance was read at 540 nm. The data presented are averages ± standard errors of the means (SEM) for duplicate cultures assayed in duplicate and are representative of the results from three to six experiments.
Antibody responses to HSV.
Blood was collected by cardiac puncture immediately following CO2 asphyxiation of mice, and serum samples were produced by allowing overnight clotting at 4°C. NaN3 (0.05%) was added to the serum samples, and the samples were stored at 4°C until enzyme-linked immunosorbent assay (ELISA) analysis. HSV-specific immunoglobulin G (IgG) production was determined by ELISA on serum samples obtained at >28 days postinfection (PI). Briefly, whole-HSV antigen in phosphate-buffered saline-NaN3 (PBSN) was adsorbed to high-protein-binding polystyrene ELISA plates (Corning, Corning, NY) at 4°C overnight and washed three times with PBSN-0.05% Tween 20 (PBST). The plates were blocked for 2 h with PBS SuperBlock (Pierce, Rockford, IL) and then incubated with serum samples for 4 h, followed by addition of 2 μg/ml horseradish peroxidase-goat anti-mouse IgG (Southern Biotech, Birmingham, AL) in PBST for a further 2 h. ELISAs were developed with one-step Turbo TMB solution (Pierce, Rockford, IL) and read on a THERMOmax microplate reader (Molecular Devices, Sunnyvale, CA).
siRNA down-regulation of TNF in RAW 264.7 macrophage cells.
Three small interfering RNAs (siRNAs) were designed to target different sites in TNF mRNA (Mark Belke, IDT). Procedures for siRNA down-regulation of TNF in RAW 264.7 cells were carried out according to our published detailed protocol (5). Briefly, RAW cells transfected with various concentrations of siRNA targeting TNF (siTNF) were incubated for approximately 18 h and then stimulated with 3 ng/ml LPS for 6 h, with brefeldin A added for the last 5 h, after which TNF was detected by intracellular staining and flow cytometry using conventional methods.
In vivo neutralization of TNF and depletion of macrophages by use of clodronate.
Mice were treated on days 0, 2, 4, 6, and 8 with 30 mg/kg PEGylated monomeric sTNFR1, which binds TNF but is not known to bind LT (22), 250 μg hamster anti-mouse TNF (clone 5B8), which does not bind LT (Hiko Kohno, Amgen, personal communication), or a total dose of 22 μg of a 27-mer siTNF (IDT, Coralville, IA). The siTNF was delivered as a complex with TransIT TKO (Mirus Bio, Madison, WI) to the peritoneal cavity as we have described previously (5), using six doses over 9 days (2 μg on day 0 and 4 μg each on days 1, 2, 4, 6, and 8). Following injection, the siRNA was distributed by massage throughout the peritoneum. The TNF antibody and sTNFR1 were administered to the peritoneum after dilution in PBS. Liposome-encapsulated clodronate (8.0 ml/kg) was also given intraperitoneally, and equal-volume PBS injections were used as the appropriate control per the manufacturer's recommendation (www.clodronateliposomes.org).
RESULTS
Lack of TNF increases HSE mortality in mice on the C57BL/6 background.
We have previously shown that B6 mice lacking either or both TNF receptors are as resistant to fatal HSE as are B6 control mice (43). However, a protective role for TNF is suggested by results showing that intraperitoneal administration of TNF can protect against fatal HSE (55). Hence, we compared HSV-1 infection in TNF−/− mice to that in C57BL/6 wild-type mice. TNF−/− mice were derived using B6 ES cells, which avoids the confounding effects that would result from replacement of the entire major histocompatibility complex with 129-derived DNA if 129 ES had been used. Mice were inoculated with a dose of HSV-1 previously determined to result in >85% mortality for susceptible 129S6 and BALB/c mice, compared to <15% mortality for resistant C57BL/6 mice (43). The survival rate for TNF-deficient mice (8/18) was significantly lower than that for either the p55−/− mice (31/40) or the control C57BL/6 mice (45/49) (P = 0.02 and P < 0.0002, respectively) (Fig. 1A). Mice that died of fatal HSE were necropsied, and HSV-1 titers were determined in the eyes, trigeminal ganglia, and brain stems. Compared to what was found for control C57BL/6 mice, HSV-1 titers were elevated in all target tissues of TNF−/− and p55−/− mice, with eyes showing the greatest difference (Fig. 1B). Thus, TNF appears to be important for the control of HSV-1 in the eye. Necropsy HSV-1 titers in resistant p55−/− and susceptible TNF−/− mice that died were not significantly different (Fig. 1A), which is contrary to the customary expectation that higher virus loads in target tissues of mice that succumb to HSV infection would allow distinction between susceptible and resistant strains. Since necropsy titers did not correlate with fatal HSE, we confirmed a role for TNF in the control of acute HSV-1 replication in the same three strains impaired for TNF signaling. Mice were inoculated with HSV-1 by corneal scarification, and the persistence of infectious virus in the eyes, trigeminal ganglia, and brain stems was determined at different times PI. Compared to what was found for B6 mice, HSV-1 persisted to a greater extent in target tissues, particularly in the inoculated ipsilateral eye, for both the TNF−/− and the p55−/− mice, with titers tending to be somewhat higher in the TNF−/− than in the p55−/− mice (Fig. 1C). Trigeminal ganglion and brain stem titers tended to be higher for TNF−/− mice than for control C57BL/6 and p55−/− mice; however, the trend was not statistically significant (P > 0.05) in paired one-tailed t tests comparing tissue titers over time between TNF−/− mice and either B6 or p55−/− mice.
FIG. 1.

(A) HSV-1 induced mortality in C57BL/6, p55−/−, and TNF−/− mice. Mice were inoculated with 3,200 PFU HSV-1 and monitored for mortality and symptoms of encephalitis necessitating euthanasia. Shown are the cumulative survival data from four experiments using 18 to 28 mice from the C57BL/6 (black circles), p55−/− (gray circles), and TNF−/− (gray squares) strains. Values for the TNF−/− mice are significantly different from those for the C57BL/6 mice (P < 0.0001), whereas those for the p55−/− N13 mice are not (P > 0.05). (B) HSV-1 titers in necropsy tissues from C57BL/6, p55−/−, and TNF−/− mice. Tissues from dead mice were collected shortly after death, and virus titers were determined. Titers in the indicated tissues are shown for C57BL/6 (black bars), p55−/− (gray bars), and TNF−/− (white bars) mice. Animals were inoculated with 3,200 PFU HSV-1 and monitored for mortality; mice with pronounced symptoms of encephalitis were euthanized. Combined data from two experiments resulting in 4 to 11 deaths per strain are shown as average HSV-1 titers ± SEM. (C) Persistence of HSV-1 in C57BL/6, p55−/−, and TNF−/− mice after corneal inoculation. The amounts of infectious HSV recovered from the infected right eye (R. Eye), the right trigeminal ganglion (R. Tg), the brain stem (BS), the left trigeminal ganglion (L. Tg), and the left eye (L. Eye) are shown. HSV-1 titers were determined by a plaque assay for tissues collected at the indicated time points. Combined data for five experiments using three to five mice per strain are shown, and the data are presented as average HSV titers ± SEM.
NO production by peritoneal macrophages.
Macrophages and neutrophils produce NO, which has been shown to block HSV-1 replication in vitro and in vivo (3, 35, 44). As TNF is involved in induction of NO (51), we determined whether the deficiency in TNF signaling in p55−/− and TNF−/− mice impaired TNF and NO production in macrophages. Compared to that in PE-MP from control C57BL/6 mice, NO production in PE-MP from p55−/− and TNF−/− mice was significantly reduced in response to in vitro activation with IFN-γ and LPS (Fig. 2A). Although HSV-1 infection synergized with IFN-γ for induction of NO in control and p55−/− mice, overall it reduced the levels of NO produced by B6 PE-MP compared to those produced by uninfected PE-MP; the same trend was evident for p55−/− and TNF−/− macrophages, although the effects were smaller (Fig. 2B). Additionally, TNF production in p55−/− PE-MP was reduced compared to that in B6 PE-MP, and interestingly, HSV-1 infection failed to augment TNF production in PE-MP activated with IFN-γ (Fig. 2C). Thus, deficiencies in TNF signaling result in reduced NO production in PE-MP, and this could contribute to the greater persistence of HSV-1 in p55−/− and TNF−/− mice (Fig. 1C).
FIG. 2.

Nitric oxide and TNF production by peritoneal exudate macrophages from C57BL/6, p55−/−, and TNF−/− mice. (A) LPS-induced NO production from PE-MP of C57BL/6 (black bars), p55−/− (gray bars), and TNF−/− (white bars) mice in the absence of HSV-1 infection during in vitro culture. (B) LPS-induced NO production from PE-MP in the presence of infectious HSV-1 during in vitro culture. (C) TNF production elicited by LPS stimulation in the absence (black bars) or presence (hatched bars) of infectious HSV-1 in PE-MP from C57BL/6 (black bars) and p55−/− (gray bars) mice. Nonstimulated culture supernatants contained no TNF. All PE-MP cultures were pretreated overnight with IFN-γ before use in culture assays. Representative data from three experiments using pooled cells from five to seven mice are shown. (not done), insufficient peritoneal exudate cells recovered.
HSE resistance in wild-type C57BL/6 mice is dependent on TNF.
To further investigate the discrepancy wherein TNF−/− mice are susceptible while neither TNF receptor appeared to be involved in protection against fatal HSE, we tested the effect of treating C57BL/6 mice with an sTNFR1 preparation capable of neutralizing TNF in vivo during HSV-1 infection. C57BL/6 mice treated with sTNFR1 during the course of acute infection showed dose-dependent increases (R2 = 0.964) in mortality (Fig. 3A). Compared to what was found for untreated mice, mortality was increased approximately threefold (P < 0.01) after intraperitoneal administration of sTNFR1 at 30 mg/kg body weight. Although injection of sTNFR1 at 10 mg/kg increased mortality, the difference did not reach statistical significance with the number of mice tested. Because macrophages are the major producers of TNF (70), we anticipated that their depletion would increase susceptibility to fatal HSE. B6 mice injected intraperitoneally with the liposome-encapsulated macrophage toxin clodronate (Cl2-MDP) to ablate macrophages in vivo (69) showed a threefold increase in mortality (P < 0.005) (Fig. 3B), which is comparable to results obtained with sTNFR1 treatment (Fig. 3A).
FIG. 3.
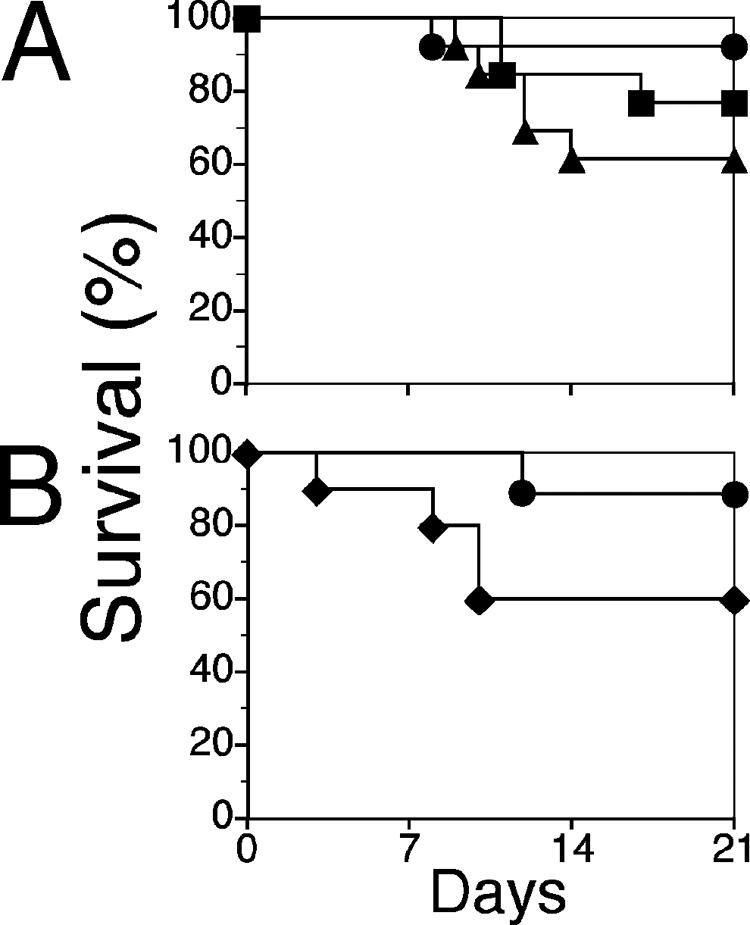
In vivo TNF and macrophage depletion increases mortality in C57BL/6 mice. (A) C57BL/6 mice were inoculated with 3,200 PFU HSV-1, given sTNFR1 on days 0, 2, 4, 6, 8, and 10, and monitored daily for mortality; mice with overt symptoms of encephalitis were euthanized. Results for mice treated with 10 mg/kg or 30 mg/kg sTNFR1 are indicated by squares or triangles, respectively, and results for untreated mice are shown by circles. (B) C57BL/6 (black diamonds) mice were inoculated with 3,200 PFU HSV-1, and macrophages were depleted by intraperitoneal administration of liposome-encapsulated clodronate on days 0, 2, 4, 6, 8, and 10 PI. Untreated control mice were given saline (black circles). Mice were monitored for mortality, and animals with pronounced symptoms of encephalitis were euthanized. Combined data from five experiments using 10 to 25 mice per strain are shown.
Humoral immune responses in mice deficient in TNF or macrophages.
Protective immunity to HSV-1 is thought to depend primarily on antigen-specific cellular Th1 responses as well as antibody responses, both processes involving regulation by TNF that reflects on the efficiency of antigen processing by the host. Total HSV-specific IgG levels were determined by ELISA in pooled sera from two or three mice sacrificed at >28 days after infection with HSV-1. HSV-1-specific IgG levels in p55−/− and TNF−/− mice were reduced relative to those in control C57BL/6 mice, as shown in Fig. 4A, implicating TNF signaling in the regulation of antibody production. Similar defects in primary antibody responses were noted for TNF−/− and p55−/− mice challenged with Leishmania sp. strains or immunized with a schistosome vaccine (61, 72). However, neutralizing TNF or depleting macrophages by treatment with sTNFR1 or the macrophage toxin clodronate, respectively, dramatically increased HSV-specific IgG levels in wild-type C57BL/6 mice relative to those in control PBS-treated mice (Fig. 4B). While these results reveal a role for TNF in the regulation of HSV-1-specific IgG production, they do not support a protective role for HSV-specific antibody responses against fatal HSE because mortality was also increased in mice whose macrophages were ablated or mice treated with sTNFR1 (Fig. 3).
FIG. 4.
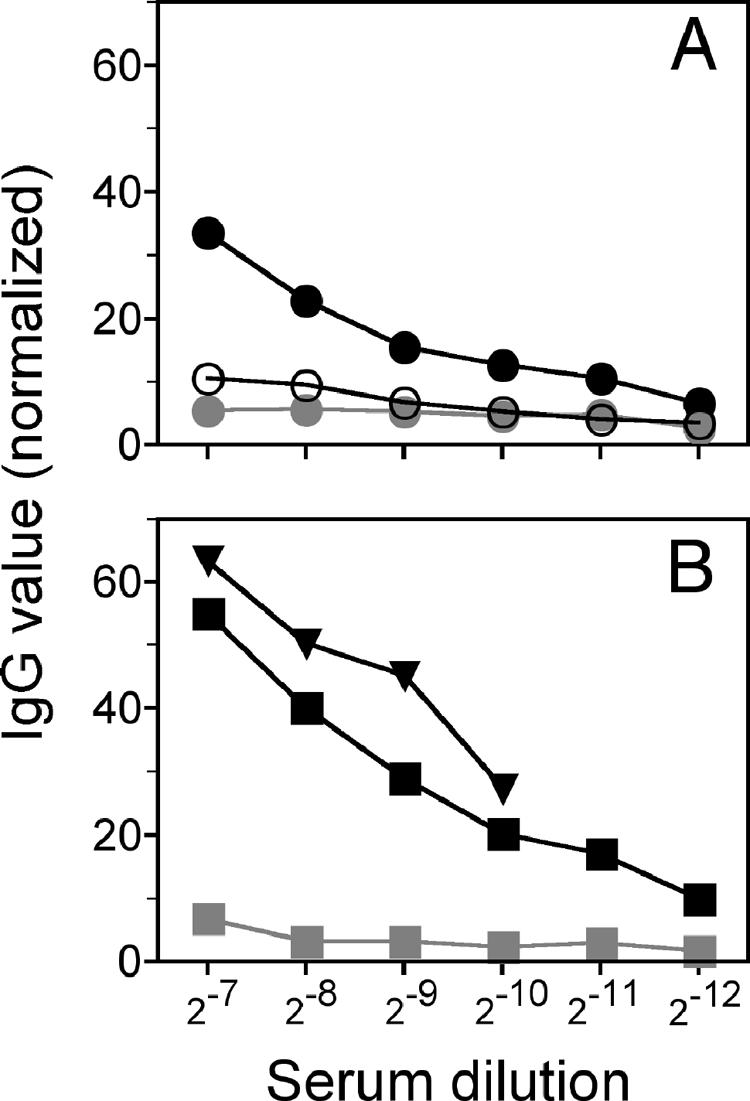
HSV-specific IgG production in C57BL/6, p55−/−, and TNF−/− mice. (A) Relative amounts of anti-HSV-1 IgG in sera from C57BL/6 (black circles), p55−/− (gray circles), and TNF−/− (white circles) mice sacrificed at >28 days PI are shown. (B) Relative anti-HSV IgG levels in sera after treatment with either 30 mg/kg sTNFR1 in C57BL/6 (black squares) or clodronate liposomes to deplete macrophages (black triangles); as controls, mice were treated with PBS (gray squares). Animals were inoculated with 3,200 PFU HSV-1, and serum samples were collected from mice at >28 days PI. Absorbance values (450 to 570 nm; TMB horseradish peroxidase substrate; Pierce) were normalized to those observed for 1:10 dilutions of day 28 HSV-positive serum samples (100%) and HSV-negative serum samples (0%). Ranges shown are 1:128 to 1:4,096 dilutions of the respective serum samples.
Depletion of TNF increases HSE mortality equally in wild-type B6 and p55−/− p75−/− mice.
The discrepant mortality of TNF−/− mice and TNF receptor-null mutant mice in response to HSV-1 infection raised the possibility that TNF-mediated protection against fatal HSE was independent of either TNFR1 or TNFR2. Therefore, we tested the prediction that resistance of TNFR double mutant mice would be sensitive to in vivo TNF depletion. From our previous studies, we knew that cumulative mortality for C57BL/6 mice was indistinguishable from that for p55−/− p75−/− mice; hence, we tested whether in vivo neutralization of TNF in these mice would increase their susceptibility. Indeed, relative to what was found for untreated mice, administration of sTNFR1 increased mortality to the same extent in C57BL/6 and p55−/−p75−/− double-knockout mice (P = 0.0018) (Fig. 5A). This result reinforces the conclusion that TNF-mediated protection against HSV-1-induced mortality is independent of signaling via the known TNF receptors, p55 and p75. Although the monomeric sTNFR1 preparation used does not bind LT when tested in vitro, there is a remote possibility that in vivo it might bind LT in addition to TNF, both of which are natural ligands for p55 that have been implicated in the mediation of resistance to HSV-1 (8, 24, 39). Consequently, we also tested an anti-TNF monoclonal antibody (MAb) that does not bind LT and demonstrated that mortality due to HSE was increased to an extent similar to that observed with sTNFR1 treatment (Fig. 5D). Another remote possibility that we considered is that reverse signaling through mTNF might be elicited by either sTNFR1- or TNF-neutralizing antibodies(23, 34, 49). To mitigate these potential confounding effects, we developed a procedure utilizing siRNA for efficient down-regulation of TNF in vivo (5) as a highly specific alternative approach for demonstrating TNF-mediated protection in HSV-1-infected p55−/− p75−/− mice. The siRNA targeting TNF was designed to react specifically with TNF but not LT. We evaluated three independent siRNAs targeting different sites in the TNF mRNA. The RAW 264.7 macrophage cell line was transfected with siTNF or irrelevant siRNA (siIRR) and treated or not treated with LPS for 6 hours to induce TNF production, which was measured by intracellular staining and flow cytometry analysis. siTNF site 1 (siTNF-S1) was highly effective and reduced TNF protein levels virtually to background levels obtained with siIRR-transfected RAW 264.7 cells (Fig. 5B). siTNF-S2 was much less efficient, whereas siTNF-S3 activity was intermediate between siTNF-S1 and siTNF-S2 activities (Fig. 5C); therefore, siTNF-S1 was used for subsequent in vivo experiments with p55−/− p75−/− mice. Compared to treatment with siIRR, treatments with sTNFR1, anti-TNF MAb, and siTNF-S1 resulted in significantly increased mortality for HSV-1-infected p55−/− p75−/− mice (P = 0.005) (Fig. 5D). Mortality rates for control siIRR-treated or untreated p55−/− p75−/− mice were not different; therefore, the siIRR-treated mice served as a control for the anti-TNF MAb-treated mice as well. Infection and treatment of mice with isotype control IgG to serve as a separate control could not be justified, since we and others have previously shown that treatment with normal IgG has no effect on the outcome of HSV infection (9, 67). Procedures for in vivo neutralization of TNF are summarized in Table 1, and Fig. 6 illustrates how these different TNF antagonists interfere with TNF signaling.
FIG. 5.

In vivo TNF depletion increases mortality in both C57BL/6 and p55−/− p75−/− mice. (A) C57BL/6 (squares) and p55−/− p75−/− (circles) mice inoculated with 3200 PFU HSV 17+ were untreated (black symbols) or treated with sTNFR (blue symbols) on days 0, 2, 4, 6, 8, and 10 PI and monitored for mortality; animals with pronounced symptoms of encephalitis were euthanized. Combined survival data from six experiments using totals of 69 to 121 mice per strain are shown. (B) Histogram showing TNF down-regulation in LPS-stimulated RAW 267.4 cells treated with 25 nM siTNF site 1 (red line) compared to that in siIRR-treated, LPS-stimulated (solid black line) or nonstimulated (dashed black line) RAW cells. (C) Dose response for down-regulation of TNF by three siRNAs targeting different sites in TNF mRNA; data are normalized to values for RAW cells treated with LPS plus siIRR as a control. (D) In vivo neutralization of TNF in p55−/− p75−/− mice. Mice were treated with sTNFR1, 27-mer siTNF (22 μg in six doses over 9 days), anti-TNF MAb, or siIRR as a control and monitored for mortality; animals with pronounced symptoms of encephalitis were euthanized. Combined data from three experiments using 10 to 34 mice per group are shown.
TABLE 1.
Effects and complications from in vivo TNF depletion methodsa
| Treatment | Target(s) | Complication in data interpretation |
|---|---|---|
| sTNFR1 | TNF, LT? | Reverse signaling via mTNF, LT binding? |
| Anti-TNF MAb | TNF | Reverse signaling via mTNF |
| siTNF | TNF | TLR3 activation at high concnb |
| Clodronate | Macrophages | Depletion of nonmacrophage cells |
Abbreviations: sTNFR1, soluble monomeric mouse p55; mTNF, membrane-bound TNF (26-kDa form); anti-TNF MAb, TNF antibody that does not bind LT; siTNF, siRNA targeting TNF; TLR3, toll-like receptor 3, specific for dsRNA. A question mark indicates uncertainty about whether the monomeric sTNFR1 binds LT in vivo.
The optimized amount of siTNF used in this study did not cause nonspecific activation capable of overcoming TNF down-regulation by the siRNA (not shown).
FIG. 6.

Diagram illustrating potential interactions of TNF antagonists with relevant TNF superfamily members. sTNFR1 and anti-TNF MAb can bind soluble or membrane-bound TNF. PEGylated monomeric sTNFR1, which was used in several of the studies reported here, does not bind LT, and the anti-TNF MAb binds both sTNF and mTNF but not LT. Thus, the only potential side effect in using sTNFR1 and anti-TNF MAb for in vivo neutralization of TNF is reverse signaling via mTNF. In contrast, siTNF specifically down-regulates TNF but does not interact with other TNF superfamily member ligands or receptors.
DISCUSSION
We presented here data that demonstrate an important role for TNF in resistance to mortality following ocular inoculation of HSV-1. Prolonged persistence and increased titers for HSV-1 in the eyes, trigeminal ganglia, and brain stems of TNF−/− and p55−/− mutant mice compared to what was found for wild-type C57BL/6 mice reveal a role for TNF in the control of replication (Fig. 1). A protective role for NO produced via induction of inducible nitric oxide synthase has been demonstrated in several models of HSV-1 infection (2, 44); hence, we suspect that the suboptimal NO production observed for p55−/− and TNF−/− macrophages contributes to the impaired control of HSV-1 in these mice (Fig. 2). These data and other reports of early induction of TNF expression in tissues targeted by HSV-1 are consistent with a protective role for TNF in HSV-1 infection (11, 27, 28, 41, 60). Additionally, intraperitoneal injection of TNF 4 h before or 8 h after intraperitoneal HSV-1 inoculation of C57BL/6 mice significantly extended their survival rate compared to that for untreated C57BL/6 mice (55). Hence, we anticipated and indeed observed significantly higher mortality rates (P < 0.0002) for TNF-null mutant mice (10/18, 56%) than for wild-type C57BL/6 mice (4/49, 8%). Similar mortality rates were reported in previous studies comparing the survival rates of C57BL/6 TNF−/− and control C57BL/6 mice challenged with HSV-1 by the corneal route (47, 48).
Most important effects of TNF, including antiviral activity, are generally ascribed to signaling via p55 rather than p75, which interacts preferentially with mTNF (4, 71, 73). Finding that p55−/− mice were as resistant to HSV-1 ocular challenge as control C57BL/6 mice (P > 0.05) (Fig. 1A) suggested that TNF signaling via p75 exerted anti-HSV effects. The antiviral effects of TNF on two poxviruses, vaccinia virus and ectromelia virus, were shown to depend on both p55 and p75 TNF receptors (56). However, we have reported that C57BL/6 and p55−/− p75−/− mortality rates were indistinguishable, ranging from 13% to 15% (P > 0.05) in HSV-1-infected mice (43). These results imply that while TNF is required for protection against fatal HSV-1 infection, both p55 and p75 receptors are dispensable. Strong support for this conclusion is provided by the nearly identical increases in mortality resulting from treatment of HSV-1-infected C57BL/6 and p55−/− p75−/− mice with sTNFR1 or anti-TNF MAb (Fig. 5A), both of which are known to neutralize TNF but not LT. A comparable increase in mortality was observed for C57BL/6 mice depleted of macrophages by intraperitoneal injection of liposomes encapsulating a macrophage toxin that is widely used for this purpose (68). This result implicates macrophage-produced TNF in protective antiviral responses to HSV-1, consistent with results from studies by others (29, 35, 51). Although TNF contributes to the control of HSV-1 replication, the mechanisms by which TNF protects against fatal HSE are uncertain since HSV-1 titers in CNS tissues were comparable in susceptible TNF−/− and resistant p55−/− mice (Fig. 1B and C).
To mitigate possible confounding effects of reverse signaling through mTNF by sTNFR1 and anti-TNF MAb (34, 49), we utilized siRNA to down-regulate TNF in vivo in HSV-1-infected p55−/− p75−/− mice and observed an increase in mortality comparable to that obtained with either sTNFR1 or anti-TNF MAb treatment (Fig. 5B). It is important to note that siTNF specifically targets TNF and has no cross-reactivity with other TNF family member ligands or receptors, as illustrated in Fig. 6. siTNF specifically down-regulated TNF production, as demonstrated by the dose-dependent down-regulation of TNF using three independent target sites in TNF mRNA and by siRNA targeting an irrelevant transcript having no effect (Fig. 5B and C). Additionally, by in vivo titration, we determined an siTNF dose that was highly effective in down-regulating TNF at the protein level while avoiding nonspecific innate immune responses (5, 19). The most reasonable interpretation of these results is that TNF-mediated resistance to fatal HSV-1 infection in mice on the C57BL/6 genetic background is independent of either of the known TNF receptors, p55 and p75. The mechanism by which TNF protects against fatal HSE in p55−/− p75−/− mice remains speculative in the absence of formal proof for the existence of a novel TNFR. The fact that three mechanistically different approaches, namely, treatment with sTNFR1, anti-TNF MAb, and siTNF, increased mortality to the same extent for HSV-1-infected C57BL/6 and p55−/− p75−/− mice is compelling evidence that only TNF neutralization was involved and argues against reverse signaling via mTNF for sTNFR1 and anti-TNF MAb or neutralization of other TNF ligands. In a related study, the existence of a third, unknown receptor was invoked to explain the observed resistance of p55−/− p75−/− mice compared to that of TNF−/− mice on the C57BL/6 background to a rapidly fatal leishmaniasis (72). The possibility of developmental defects in secondary lymphoid organs of C57BL/6 TNF−/− mice influencing the course of disease was excluded in this study by using reciprocal bone marrow chimeras.
We show here that resistance in wild-type (p55+/+) and p55−/− N13 mice is strictly dependent on TNF signaling, as it is impaired by in vivo neutralization of TNF. TNF thus plays a pivotal role in resistance to HSV, which is genetically very complex, involving multiple interacting loci (unpublished data). We previously reported that the C57BL/6 allele of the herpes resistance locus, Hrl, linked to p55 on mouse chromosome 6 confers resistance to HSV-1 and HSV-2 (43) in mice lacking p55 (p55−/− N13). Resistance in p55−/− N13 mice is also abrogated by in vivo neutralization of TNF (unpublished observation), which indicates a general requirement for TNF in the resistance of mice on the B6 background to HSV-1 infection.
Acknowledgments
We thank Lyle Moldawer for generously supplying mice and Harry Openshaw for many useful discussions and critical comments. Cl2-MDP (or clodronate) was a gift from Roche Diagnostics GmbH, Mannheim, Germany, sTNFR1 and anti-TNF MAb were gifts from Amgen, siRNA targeting TNF was a gift from Integrated DNA Technologies, and TransIT TKO reagent was a gift from Mirus Bio Corporation.
This work was supported by NIH grant EY13814.
Footnotes
Published ahead of print on 15 November 2006.
REFERENCES
- 1.Aderka, D., P. Sorkine, S. Abu-Abid, D. Lev, A. Setton, A. P. Cope, D. Wallach, and J. Klausner. 1998. Shedding kinetics of soluble tumor necrosis factor (TNF) receptors after systemic TNF leaking during isolated limb perfusion. Relevance to the pathophysiology of septic shock. J. Clin. Investig. 101:650-659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Adler, H., J. L. Beland, N. C. Del-Pan, L. Kobzik, J. P. Brewer, T. R. Martin, and I. J. Rimm. 1997. Suppression of herpes simplex virus type 1 (HSV-1)-induced pneumonia in mice by inhibition of inducible nitric oxide synthase (iNOS, NOS2). J. Exp. Med. 185:1533-1540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Adler, H., J. L. Beland, N. C. Del-Pan, L. Kobzik, R. A. Sobel, and I. J. Rimm. 1999. In the absence of T cells, natural killer cells protect from mortality due to HSV-1 encephalitis. J. Neuroimmunol. 93:208-213. [DOI] [PubMed] [Google Scholar]
- 4.Aggarwal, B. B., and K. Natarajan. 1996. Tumor necrosis factors: developments during the last decade. Eur. Cytokine Netw. 7:93-124. [PubMed] [Google Scholar]
- 5.Amarzguioui, M., P. Lundberg, E. Cantin, J. Hagstrom, M. A. Behlke, and J. J. Rossi. 2006. Rational design and in vitro and in vivo delivery of Dicer substrate siRNA. Nat. Protoc. 1:508-517. [DOI] [PubMed] [Google Scholar]
- 6.Benedict, C. A., and C. F. Ware. 2001. Virus targeting of the tumor necrosis factor superfamily. Virology 289:1-5. [DOI] [PubMed] [Google Scholar]
- 7.Bjornberg, F., M. Lantz, I. Olsson, and U. Gullberg. 1994. Mechanisms involved in the processing of the p55 and the p75 tumor necrosis factor (TNF) receptors to soluble receptor forms. Lymphokine Cytokine Res. 13:203-211. [PubMed] [Google Scholar]
- 8.Bossen, C., K. Ingold, A. Tardivel, J. L. Bodmer, O. Gaide, S. Hertig, C. Ambrose, J. Tschopp, and P. Schneider. 2006. Interactions of tumor necrosis factor (TNF) and TNF receptor family members in the mouse and human. J. Biol. Chem. 281:13964-13971. [DOI] [PubMed] [Google Scholar]
- 9.Cantin, E., B. Tanamachi, and H. Openshaw. 1999. Role for gamma interferon in control of herpes simplex virus type 1 reactivation. J. Virol. 73:3418-3423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cantin, E., B. Tanamachi, H. Openshaw, J. Mann, and K. Clarke. 1999. Gamma interferon (IFN-γ) receptor null-mutant mice are more susceptible to herpes simplex virus type 1 infection than IFN-γ ligand null-mutant mice. J. Virol. 73:5196-5200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cantin, E. M., D. R. Hinton, J. Chen, and H. Openshaw. 1995. Gamma interferon expression during acute and latent nervous system infection by herpes simplex virus type 1. J. Virol. 69:4898-4905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Carr, D. J. J., and S. Noisakran. 2002. The antiviral efficacy of the murine alpha-1 interferon transgene against ocular herpes simplex virus type 1 requires the presence of CD4+, α/β T-cell receptor-positive T lymphocytes with the capacity to produce gamma interferon. J. Virol. 76:9398-9406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chen, S. H., J. E. Oakes, and R. N. Lausch. 1994. Synergistic anti-herpes effect of TNF-alpha and IFN-gamma in human corneal epithelial cells compared with that in corneal fibroblasts. Antivir. Res. 25:201-213. [DOI] [PubMed] [Google Scholar]
- 14.Cheung, T. C., I. R. Humphreys, K. G. Potter, P. S. Norris, H. M. Shumway, B. R. Tran, G. Patterson, R. Jean-Jacques, M. Yoon, P. G. Spear, K. M. Murphy, N. S. Lurain, C. A. Benedict, and C. F. Ware. 2005. Evolutionarily divergent herpesviruses modulate T cell activation by targeting the herpesvirus entry mediator cosignaling pathway. Proc. Natl. Acad. Sci. USA 102:13218-13223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cook, M. C., H. Korner, D. S. Riminton, F. A. Lemckert, J. Hasbold, M. Amesbury, P. D. Hodgkin, J. G. Cyster, J. D. Sedgwick, and A. Basten. 1998. Generation of splenic follicular structure and B cell movement in tumor necrosis factor-deficient mice. J. Exp. Med. 188:1503-1510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Corey, L., and P. G. Spear. 1986. Infections with herpes simplex viruses (1). N. Engl. J. Med. 314:686-691. [DOI] [PubMed] [Google Scholar]
- 17.Croft, M. 2005. The evolving crosstalk between co-stimulatory and co-inhibitory receptors: HVEM-BTLA. Trends Immunol. 26:292-294. [DOI] [PubMed] [Google Scholar]
- 18.Crowe, P. D., T. L. VanArsdale, R. G. Goodwin, and C. F. Ware. 1993. Specific induction of 80-kDa tumor necrosis factor receptor shedding in T lymphocytes involves the cytoplasmic domain and phosphorylation. J. Immunol. 151:6882-6890. [PubMed] [Google Scholar]
- 19.Cullen, B. R. 2006. Enhancing and confirming the specificity of RNAi experiments. Nat. Methods 3:677-681. [DOI] [PubMed] [Google Scholar]
- 20.Daheshia, M., S. Kanangat, and B. T. Rouse. 1998. Production of key molecules by ocular neutrophils early after herpetic infection of the cornea. Exp. Eye Res. 67:619-624. [DOI] [PubMed] [Google Scholar]
- 21.Decman, V., M. L. Freeman, P. R. Kinchington, and R. L. Hendricks. 2005. Immune control of HSV-1 latency. Viral Immunol. 18:466-473. [DOI] [PubMed] [Google Scholar]
- 22.Edwards, C. K., III. 1999. PEGylated recombinant human soluble tumour necrosis factor receptor type I (r-Hu-sTNF-RI): novel high affinity TNF receptor designed for chronic inflammatory diseases. Ann. Rheum. Dis. 58(Suppl. 1):173-181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Eissner, G., S. Kirchner, H. Lindner, W. Kolch, P. Janosch, M. Grell, P. Scheurich, R. Andreesen, and E. Holler. 2000. Reverse signaling through transmembrane TNF confers resistance to lipopolysaccharide in human monocytes and macrophages. J. Immunol. 164:6193-6198. [DOI] [PubMed] [Google Scholar]
- 24.Feduchi, E., M. A. Alonso, and L. Carrasco. 1989. Human gamma interferon and tumor necrosis factor exert a synergistic blockade on the replication of herpes simplex virus. J. Virol. 63:1354-1359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ghiasi, H., S. L. Wechsler, R. Kaiwar, A. B. Nesburn, and F. M. Hofman. 1995. Local expression of tumor necrosis factor alpha and interleukin-2 correlates with protection against corneal scarring after ocular challenge of vaccinated mice with herpes simplex virus type 1. J. Virol. 69:334-340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Green, M. R., R. Treisman, and T. Maniatis. 1983. Transcriptional activation of cloned human beta-globin genes by viral immediate-early gene products. Cell 35:137-148. [DOI] [PubMed] [Google Scholar]
- 27.Halford, W. P., B. M. Gebhardt, and D. J. Carr. 1996. Persistent cytokine expression in trigeminal ganglion latently infected with herpes simplex virus type 1. J. Immunol. 157:3542-3549. [PubMed] [Google Scholar]
- 28.He, J., H. Ichimura, T. Iida, M. Minami, K. Kobayashi, M. Kita, C. Sotozono, Y. I. Tagawa, Y. Iwakura, and J. Imanishi. 1999. Kinetics of cytokine production in the cornea and trigeminal ganglion of C57BL/6 mice after corneal HSV-1 infection. J. Interferon Cytokine Res. 19:609-615. [DOI] [PubMed] [Google Scholar]
- 29.Heise, M. T., and H. W. Virgin IV. 1995. The T-cell-independent role of gamma interferon and tumor necrosis factor alpha in macrophage activation during murine cytomegalovirus and herpes simplex virus infections. J. Virol. 69:904-909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hendricks, R. L. 1999. Immunopathogenesis of viral ocular infections. Chem. Immunol. 73:120-136. [DOI] [PubMed] [Google Scholar]
- 31.Herbein, G., and W. A. O'Brien. 2000. Tumor necrosis factor (TNF)-alpha and TNF receptors in viral pathogenesis. Proc. Soc. Exp. Biol. Med. 223:241-257. [DOI] [PubMed] [Google Scholar]
- 32.Holterman, A. X., K. Rogers, K. Edelmann, D. M. Koelle, L. Corey, and C. B. Wilson. 1999. An important role for major histocompatibility complex class I-restricted T cells, and a limited role for gamma interferon, in protection of mice against lethal herpes simplex virus infection. J. Virol. 73:2058-2063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Keadle, T. L., N. Usui, K. A. Laycock, J. K. Miller, J. S. Pepose, and P. M. Stuart. 2000. IL-1 and TNF-alpha are important factors in the pathogenesis of murine recurrent herpetic stromal keratitis. Investig. Ophthalmol. Vis. Sci. 41:96-102. [PubMed] [Google Scholar]
- 34.Kirchner, S., S. Boldt, W. Kolch, S. Haffner, S. Kazak, P. Janosch, E. Holler, R. Andreesen, and G. Eissner. 2004. LPS resistance in monocytic cells caused by reverse signaling through transmembrane TNF (mTNF) is mediated by the MAPK/ERK pathway. J. Leukoc. Biol. 75:324-331. [DOI] [PubMed] [Google Scholar]
- 35.Kodukula, P., T. Liu, N. V. Rooijen, M. J. Jager, and R. L. Hendricks. 1999. Macrophage control of herpes simplex virus type 1 replication in the peripheral nervous system. J. Immunol. 162:2895-2905. [PubMed] [Google Scholar]
- 36.Korner, H., M. Cook, D. S. Riminton, F. A. Lemckert, R. M. Hoek, B. Ledermann, F. Kontgen, B. Fazekas de St. Groth, and J. D. Sedgwick. 1997. Distinct roles for lymphotoxin-alpha and tumor necrosis factor in organogenesis and spatial organization of lymphoid tissue. Eur. J. Immunol. 27:2600-2609. [DOI] [PubMed] [Google Scholar]
- 37.Krajcsi, P., and W. S. Wold. 1998. Inhibition of tumor necrosis factor and interferon triggered responses by DNA viruses. Semin. Cell Dev. Biol. 9:351-358. [DOI] [PubMed] [Google Scholar]
- 38.Krummenacher, C., A. V. Nicola, J. C. Whitbeck, H. Lou, W. Hou, J. D. Lambris, R. J. Geraghty, P. G. Spear, G. H. Cohen, and R. J. Eisenberg. 1998. Herpes simplex virus glycoprotein D can bind to poliovirus receptor-related protein 1 or herpesvirus entry mediator, two structurally unrelated mediators of virus entry. J. Virol. 72:7064-7074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kumaraguru, U., I. A. Davis, S. Deshpande, S. S. Tevethia, and B. T. Rouse. 2001. Lymphotoxin alpha-/- mice develop functionally impaired CD8+ T cell responses and fail to contain virus infection of the central nervous system. J. Immunol. 166:1066-1074. [DOI] [PubMed] [Google Scholar]
- 40.Lantz, M., F. Bjornberg, I. Olsson, and J. Richter. 1994. Adherence of neutrophils induces release of soluble tumor necrosis factor receptor forms. J. Immunol. 152:1362-1369. [PubMed] [Google Scholar]
- 41.Liu, T., Q. Tang, and R. L. Hendricks. 1996. Inflammatory infiltration of the trigeminal ganglion after herpes simplex virus type 1 corneal infection. J. Virol. 70:264-271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Locksley, R. M., N. Killeen, and M. J. Lenardo. 2001. The TNF and TNF receptor superfamilies: integrating mammalian biology. Cell 104:487-501. [DOI] [PubMed] [Google Scholar]
- 43.Lundberg, P., P. Welander, H. Openshaw, C. Nalbandian, C. Edwards, L. Moldawer, and E. Cantin. 2003. A locus on mouse chromosome 6 that determines resistance to herpes simplex virus also influences reactivation, while an unlinked locus augments resistance of female mice. J. Virol. 77:11661-11673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.MacLean, A., X. Q. Wei, F. P. Huang, U. A. Al-Alem, W. L. Chan, and F. Y. Liew. 1998. Mice lacking inducible nitric-oxide synthase are more susceptible to herpes simplex virus infection despite enhanced Th1 cell responses. J. Gen. Virol. 79:825-830. [DOI] [PubMed] [Google Scholar]
- 45.Mauri, D. N., R. Ebner, R. I. Montgomery, K. D. Kochel, T. C. Cheung, G. L. Yu, S. Ruben, M. Murphy, R. J. Eisenberg, G. H. Cohen, P. G. Spear, and C. F. Ware. 1998. LIGHT, a new member of the TNF superfamily, and lymphotoxin alpha are ligands for herpesvirus entry mediator. Immunity 8:21-30. [DOI] [PubMed] [Google Scholar]
- 46.McDermott, M. F. 2001. TNF and TNFR biology in health and disease. Cell. Mol. Biol. (Noisy-le-Grand) 47:619-635. [PubMed] [Google Scholar]
- 47.Minagawa, H., K. Hashimoto, and Y. Yanagi. 2004. Absence of tumour necrosis factor facilitates primary and recurrent herpes simplex virus-1 infections. J. Gen. Virol. 85:343-347. [DOI] [PubMed] [Google Scholar]
- 48.Minami, M., M. Kita, X. Q. Yan, T. Yamamoto, T. Iida, K. Sekikawa, Y. Iwakura, and J. Imanishi. 2002. Role of IFN-gamma and tumor necrosis factor-alpha in herpes simplex virus type 1 infection. J. Interferon Cytokine Res. 22:671-676. [DOI] [PubMed] [Google Scholar]
- 49.Mitoma, H., T. Horiuchi, N. Hatta, H. Tsukamoto, S. Harashima, Y. Kikuchi, J. Otsuka, S. Okamura, S. Fujita, and M. Harada. 2005. Infliximab induces potent anti-inflammatory responses by outside-to-inside signals through transmembrane TNF-alpha. Gastroenterology 128:376-392. [DOI] [PubMed] [Google Scholar]
- 50.Montgomery, R. I., M. S. Warner, B. J. Lum, and P. G. Spear. 1996. Herpes simplex virus-1 entry into cells mediated by a novel member of the TNF/NGF receptor family. Cell 87:427-436. [DOI] [PubMed] [Google Scholar]
- 51.Paludan, S. R., L. Malmgaard, S. Ellermann-Eriksen, L. Bosca, and S. C. Mogensen. 2001. Interferon (IFN)-gamma and herpes simplex virus/tumor necrosis factor-alpha synergistically induce nitric oxide synthase 2 in macrophages through cooperative action of nuclear factor-kappa B and IFN regulatory factor-1. Eur. Cytokine Netw. 12:297-308. [PubMed] [Google Scholar]
- 52.Perry, S. W., S. Dewhurst, M. J. Bellizzi, and H. A. Gelbard. 2002. Tumor necrosis factor-alpha in normal and diseased brain: conflicting effects via intraneuronal receptor crosstalk? J. Neurovirol. 8:611-624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Peschon, J. J., D. S. Torrance, K. L. Stocking, M. B. Glaccum, C. Otten, C. R. Willis, K. Charrier, P. J. Morrissey, C. B. Ware, and K. M. Mohler. 1998. TNF receptor-deficient mice reveal divergent roles for p55 and p75 in several models of inflammation. J. Immunol. 160:943-952. [PubMed] [Google Scholar]
- 54.Pfeffer, K., T. Matsuyama, T. M. Kundig, A. Wakeham, K. Kishihara, A. Shahinian, K. Wiegmann, P. S. Ohashi, M. Kronke, and T. W. Mak. 1993. Mice deficient for the 55 kd tumor necrosis factor receptor are resistant to endotoxic shock, yet succumb to L. monocytogenes infection. Cell 73:457-467. [DOI] [PubMed] [Google Scholar]
- 55.Rossol-Voth, R., S. Rossol, K. H. Schutt, S. Corridori, W. de Cian, and D. Falke. 1991. In vivo protective effect of tumour necrosis factor alpha against experimental infection with herpes simplex virus type 1. J. Gen. Virol. 72:143-147. [DOI] [PubMed] [Google Scholar]
- 56.Ruby, J., H. Bluethmann, and J. J. Peschon. 1997. Antiviral activity of tumor necrosis factor (TNF) is mediated via p55 and p75 TNF receptors. J. Exp. Med. 186:1591-1596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sainz, B., Jr., and W. P. Halford. 2002. Alpha/beta interferon and gamma interferon synergize to inhibit the replication of herpes simplex virus type 1. J. Virol. 76:11541-11550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Sedgwick, J. D., D. S. Riminton, J. G. Cyster, and H. Korner. 2000. Tumor necrosis factor: a master-regulator of leukocyte movement. Immunol. Today 21:110-113. [DOI] [PubMed] [Google Scholar]
- 59.Shimeld, C., J. L. Whiteland, S. M. Nicholls, E. Grinfeld, D. L. Easty, H. Gao, and T. J. Hill. 1995. Immune cell infiltration and persistence in the mouse trigeminal ganglion after infection of the cornea with herpes simplex virus type 1. J. Neuroimmunol. 61:7-16. [DOI] [PubMed] [Google Scholar]
- 60.Shimeld, C., J. L. Whiteland, N. A. Williams, D. L. Easty, and T. J. Hill. 1997. Cytokine production in the nervous system of mice during acute and latent infection with herpes simplex virus type 1. J. Gen. Virol. 78:3317-3325. [DOI] [PubMed] [Google Scholar]
- 61.Street, M., P. S. Coulson, C. Sadler, L. J. Warnock, D. McLaughlin, H. Bluethmann, and R. A. Wilson. 1999. TNF is essential for the cell-mediated protective immunity induced by the radiation-attenuated schistosome vaccine. J. Immunol. 163:4489-4494. [PubMed] [Google Scholar]
- 62.Stuehr, D. J., S. S. Gross, I. Sakuma, R. Levi, and C. F. Nathan. 1989. Activated murine macrophages secrete a metabolite of arginine with the bioactivity of endothelium-derived relaxing factor and the chemical reactivity of nitric oxide. J. Exp. Med. 169:1011-1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Stumpf, T. H., R. Case, C. Shimeld, D. L. Easty, and T. J. Hill. 2002. Primary herpes simplex virus type 1 infection of the eye triggers similar immune responses in the cornea and the skin of the eyelids. J. Gen. Virol. 83:1579-1590. [DOI] [PubMed] [Google Scholar]
- 64.Tamada, K., K. Shimozaki, A. I. Chapoval, Y. Zhai, J. Su, S. F. Chen, S. L. Hsieh, S. Nagata, J. Ni, and L. Chen. 2000. LIGHT, a TNF-like molecule, costimulates T cell proliferation and is required for dendritic cell-mediated allogeneic T cell response. J. Immunol. 164:4105-4110. [DOI] [PubMed] [Google Scholar]
- 65.Thomas, J., S. Gangappa, S. Kanangat, and B. T. Rouse. 1997. On the essential involvement of neutrophils in the immunopathologic disease: herpetic stromal keratitis. J. Immunol. 158:1383-1391. [PubMed] [Google Scholar]
- 66.Tumpey, T. M., S. H. Chen, J. E. Oakes, and R. N. Lausch. 1996. Neutrophil-mediated suppression of virus replication after herpes simplex virus type 1 infection of the murine cornea. J. Virol. 70:898-904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Tumpey, T. M., H. Cheng, X. T. Yan, J. E. Oakes, and R. N. Lausch. 1998. Chemokine synthesis in the HSV-1-infected cornea and its suppression by interleukin-10. J. Leukoc. Biol. 63:486-492. [DOI] [PubMed] [Google Scholar]
- 68.van Rooijen, N., J. Bakker, and A. Sanders. 1997. Transient suppression of macrophage functions by liposome-encapsulated drugs. Trends Biotechnol. 15:178-185. [DOI] [PubMed] [Google Scholar]
- 69.van Rooijen, N., A. Sanders, and T. K. van den Berg. 1996. Apoptosis of macrophages induced by liposome-mediated intracellular delivery of clodronate and propamidine. J. Immunol. Methods 193:93-99. [DOI] [PubMed] [Google Scholar]
- 70.Vassalli, P. 1992. The pathophysiology of tumor necrosis factors. Annu. Rev. Immunol. 10:411-452. [DOI] [PubMed] [Google Scholar]
- 71.Wallach, D., E. E. Varfolomeev, N. L. Malinin, Y. V. Goltsev, A. V. Kovalenko, and M. P. Boldin. 1999. Tumor necrosis factor receptor and Fas signaling mechanisms. Annu. Rev. Immunol. 17:331-367. [DOI] [PubMed] [Google Scholar]
- 72.Wilhelm, P., U. Ritter, S. Labbow, N. Donhauser, M. Rollinghoff, C. Bogdan, and H. Korner. 2001. Rapidly fatal leishmaniasis in resistant C57BL/6 mice lacking TNF. J. Immunol. 166:4012-4019. [DOI] [PubMed] [Google Scholar]
- 73.Wong, G. H., L. A. Tartaglia, M. S. Lee, and D. V. Goeddel. 1992. Antiviral activity of tumor necrosis factor is signaled through the 55-kDa type I TNF receptor [corrected]. J. Immunol. 149:3350-3353. [PubMed] [Google Scholar]