

Abstract
Reverse transcription, an essential step in the life cycle of all retroelements, is a complex, multistep process whose regulation is not yet clearly understood. We have recently shown that reverse transcription in the pararetrovirus duck hepatitis B virus is associated with complete dephosphorylation of the viral core protein, which forms the nucleocapsid wherein reverse transcription takes place. Here we present a genetic study of the role of this dynamic nucleocapsid phosphorylation in regulating viral reverse transcription. Detailed analyses of the reverse transcription products synthesized within nucleocapsids composed of core phosphorylation site mutants revealed that alanine substitutions, mimicking the nonphosphorylated state, completely blocked reverse transcription at a very early stage. In contrast, aspartate substitutions, mimicking the phosphorylated state, allowed complete first-strand DNA synthesis but were severely defective in accumulating mature double-stranded DNA. The latter defect was due to a combination of mutant nucleocapsid instability during maturation and a block in mature second-strand DNA synthesis. Thus, the reversible phosphorylation of the nucleocapsids regulates the ordered progression of reverse transcription.
Reverse transcription, the synthesis of a double-stranded (DS) DNA copy of an RNA, is an essential step in the life cycle of all reverse-transcribing elements, including classical retroviruses, retrotransposons, and pararetroviruses, such as the hepadnaviruses. The family Hepadnaviridae, which includes the global human pathogen Hepatitis B virus (HBV) and closely related animal viruses, such as Duck hepatitis B virus (DHBV), is a group of small hepatotropic DNA viruses (7, 8). All hepadnaviruses replicate their DNA genomes via an RNA intermediate termed the pregenomic RNA (pgRNA) (31). Viral assembly begins with the packaging of the pgRNA, along with the virally encoded reverse transcriptase (RT), into an immature nucleocapsid (NC), followed by reverse transcription of the pgRNA within the NC into the characteristic, partially DS relaxed circular (RC) DNA genome. The DNA-containing mature NC is then enveloped and secreted extracellularly as a complete virion (10, 26, 31, 36).
The icosahedral NC consists of 180 or 240 copies of a single viral protein, the capsid or core protein (2, 6, 38). The N-terminal two-thirds of the core protein forms the capsid shell (1), while its basic, arginine-rich C-terminal domain is dispensable for capsid assembly but required for pgRNA packaging and reverse transcription (30, 39). Both the HBV and DHBV core proteins are known to be phosphorylated at the C-terminal domain in the host cells, which has been shown to modulate core protein functions, including pgRNA packaging and DNA synthesis (9, 18, 20, 22, 24, 28, 30, 40, 41).
Recently, we reported that the phosphorylation state of the DHBV NC undergoes a dynamic change during reverse transcription such that the phosphorylated, immature NCs become dephosphorylated as they mature (27). The immature NCs consist of core proteins that are phosphorylated at a total of six highly conserved sites, including four sites (T239, S245, S257, and S259) that were previously identified through metabolic labeling (41) and two additional sites (S230 and S232) that were recently identified by mass spectrometry (27). All of these phosphosites were found to be dephosphorylated in the mature NCs before envelopment and secretion (27, 28). These observations suggest that the sequential phosphorylation and dephosphorylation of core protein may regulate hepadnavirus reverse transcription and NC maturation. To test this hypothesis, we generated core protein mutants with substitutions that either block (S/T to A) or mimic (S/T to D) phosphorylation at these core phosphosites and analyzed their effects on the different steps in viral reverse transcription. Our results demonstrate that while core phosphorylation is required early during reverse transcription for the synthesis of the minus-strand DNA intermediate, the subsequent core dephosphorylation is required to facilitate the progression of reverse transcription to produce the mature DS RC DNA as well as to stabilize the mature RC DNA-containing NCs, thus strongly supporting the sequential phosphorylation and dephosphorylation model.
MATERIALS AND METHODS
Plasmids.
Plasmid pCMV-DHBV/C− was derived from pCMV-DHBV (35, 37) by cleavage with NsiI (at nucleotide [nt] 2850), filling in, and religation, resulting in a frameshift mutation in the core gene after codon 67. pCMV-DHBV/C−/YMHA was derived from pCMV-DHBV/C− by the D513H and D514A double substitutions at the catalytic YMDD motif of the RT gene, which eliminates reverse transcription (4). Similarly, pCMV-DHBV/C−/RH− was derived from pCMV-DHBV/C− by the D715V substitution at codon 715 of the RT gene, destroying the RNase H activity (5). pCMV-DHBVΔXM is a derivative of pCMV-DHBV with a deletion of the DHBV sequence from nt 1217 to nt 2375 (35), such that it expresses a pgRNA with a large internal deletion, allowing the production of only the WT DHBV core protein and no other viral proteins. The core phosphosite mutations were introduced into pCMV-DHBVΔXM by PCR-mediated mutagenesis using mutagenic oligonucleotide primers, resulting in replacement of the phosphorylation site residue S or T by either D or A (Fig. 1). The coding sequences for the WT and certain mutant core proteins (including the SSDDDD mutant) were also cloned into pcDNA3 (Invitrogen) downstream of the cytomegalovirus (CMV) immediate-early promoter to make pcDNA-Dcore, which was used in the experiment shown in Fig. 4B. Mutations were confirmed by automated DNA sequencing. The single S-to-A substitution core mutant, the S245A mutant, was described before (40) and kindly provided by Jesse Summers.
FIG. 1.

Summary of DHBV core phosphorylation mutants. The sequences of the C-terminal 33 amino acids of the WT and various mutants are shown. The S or T residues at the six known phosphorylation sites (shown in bold) were changed to either A or D, as indicated. The mutants are designated on the left. The average levels in the various core mutants, compared to those in the WT (set to 100), of core protein expression (as measured by SDS-PAGE and Western blot analysis), pgRNA packaging (RNA pack) (as measured by the native agarose gel electrophoresis assay), SS DNA (extracted with micrococcal nuclease [MNase] digestion), RC DNA (extracted with or without exogenous nuclease digestion), and full-length plus-strand DNA [FL (+), extracted with or without nuclease digestion and measured using denatured core DNA], are indicated. ND, not determined; −, levels of DNA were too low to be quantified reliably.
FIG. 4.

Comparative analysis of viral core DNA isolated with or without nuclease digestion. (A) LMH cells were transfected as described in the legend to Fig. 2. Core DNA was isolated either following micrococcal nuclease (MNase) treatment of cytoplasmic lysates (lanes 1 to 7) or by the SDS-KCl precipitation method without nuclease treatment (lanes 8 to 14) (see Materials and Methods for details) and analyzed by Southern blot hybridization using a radiolabeled DHBV DNA probe. (B) LMH cells were transfected as described in the legend to Fig. 2, except that the complementing construct used was pcDNA-Dcore (rather than pCMVDHBVΔXM) expressing either the WT or the SSDDDD mutant core protein. RC, relaxed circular DNA; SS, full-length single-stranded DNA. The arrowhead in panel A indicates the internally deleted minus-strand DNA, as described in the legend to Fig. 2.
Transient transfection and analyses of core protein expression and DNA synthesis.
The chicken hepatoma cell line LMH (16) was transfected by the calcium phosphate method as previously described (14, 35). Transfected cells were harvested on day 5 posttransfection. Core protein expression levels in the cell lysates were analyzed by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and Western blotting as described previously (14, 35). Core DNA from NCs was isolated and analyzed by Southern blot analysis as described previously (14, 35), except that DNase I was replaced with micrococcal nuclease. Alternatively, to isolate core DNA without nuclease digestion, replicative viral DNA intermediates covalently bound to the RT protein were separated from other forms of viral DNA and cellular as well as transfected plasmid DNA by potassium precipitation of the protein-dodecyl sulfate complex by adopting a previously published procedure (32). Briefly, one 60-mm dish of transfected LMH cells was lysed with 0.5 ml of 0.2% NP-40 in TE (10 mM Tris, 1 mM EDTA, pH 7.5). To the cytoplasmic extract, 0.5 ml of 2% SDS was added and mixed well. Thereafter, 0.25 ml of 2.5 M KCl was added to precipitate the protein-dodecyl sulfate complex. To resuspend the pellet, 1 ml TE was added, and the sample was treated at 50°C for a few minutes, followed by vortexing. To the dispersed pellet, 0.25 ml of 2.5 M KCl was again added to precipitate the protein-dodecyl sulfate complex. The precipitate was then resuspended in 0.4 ml of TE with warming, as before, adjusted to 0.5 mg/ml proteinase K, and incubated at 50°C for 1 hour. Viral DNA was isolated by phenol extraction and ethanol precipitation. A genome-length, 32P-labeled DHBV DNA probe was routinely used to detect the viral DNA replicative intermediates by Southern blot analysis. Where indicated, 32P-labeled strand-specific riboprobes were used to detect either the minus-strand or plus-strand DNA.
Agarose gel analysis of native NCs (RNA packaging assay).
Native agarose gel electrophoresis of intact NCs for analyzing pgRNA packaging was carried out by using micrococcal nuclease-digested cell lysates, as previously reported (35, 40). Viral RNA was detected using a 32P-labeled DHBV plus-strand-specific riboprobe against the 5′ end of the pgRNA, and the same membranes were subsequently probed with an anti-DHBV core polyclonal antibody (15) to detect the core protein.
RESULTS
Experimental approach.
To test the role of core phosphorylation and dephosphorylation in NC maturation, a DHBV core expression construct, pCMV-DHBVΔXM, was used to express the WT or mutant core proteins. S/T-to-A or -D substitutions were made to mimic the unphosphorylated or phosphorylated state, respectively (Fig. 1) (see Materials and Methods for details). The effects of these core substitutions on NC maturation were then tested in a transcomplementation assay, whereby the indicated core expression construct was cotransfected into permissive hepatoma cells, together with a DHBV genomic construct, pCMV-DHBV/C−, that would be replication competent except for a frameshift mutation in the core gene eliminating core protein expression. Core protein expression and viral DNA synthesis were then monitored by standard Western and Southern blot analyses. To test specifically the effects of the core substitutions on pgRNA packaging, we used the genomic construct pCMV-DHBV/C−/YMHA, which harbors an additional mutation in the RT active site that eliminates any viral DNA synthesis but has no effect on pgRNA packaging. Therefore, the plus-strand viral nucleic acid detected in NCs by the native agarose gel packaging assay was derived exclusively from the packaged pgRNA (not from plus-strand DNA). Since the phosphorylation motif (S-K/R) at the recently identified S230 and S232 sites (27) was different from the S/T-P motif at the previously reported T239, S245, S257, and S259 sites (41), we considered it plausible that the regulation of phosphorylation at these two different groups of sites, and their effects on NC maturation, would be distinct. Therefore, we decided to mutagenize these two groups of phosphosites separately as well as in combination.
Phosphorylation at S230 and S232 was required for viral DNA synthesis.
To test the role of the recently identified S230 and S232 phosphosites on NC maturation, four mutants with single substitutions, namely, S230A (ASTSSS), S232A (SATSSS), S230D (DSTSSS), and S232D (SDTSSS), as well as two mutants with double substitutions, namely, S230/232A (AATSSS) and S230/232D (DDTSSS), were constructed (Fig. 1). SDS-PAGE and Western blot analysis showed that these core mutants were expressed in the cells at WT-like levels (Fig. 1 and 2B). Furthermore, they all displayed a migration heterogeneity similar to that of the WT, which was shown to be dependent on phosphorylation at the four downstream S/T-P sites (41). These results thus suggested that the mutations at positions 230 and 232 did not affect core protein expression or phosphorylation at the S/T-P sites. They also indicated that phosphorylation at S230 and S232 had no appreciable effect on the migration of core protein by SDS-PAGE. Most of these mutations had no or only a slight effect (ca. twofold reduction) on pgRNA packaging, but the AATSSS mutant decreased pgRNA packaging approximately fourfold (Fig. 1 and 3).
FIG. 2.

Effects of core substitutions on protein expression and DNA synthesis. LMH cells were cotransfected with pCMV-DHBV/C−, which expresses a core-defective DHBV genome, together with the indicated core (WT or mutant) expression plasmid. (A) Replicative viral DNA intermediates were isolated (with nuclease digestion [see Materials and Methods for details]) from the cytoplasmic NCs and analyzed by Southern hybridization using a radiolabeled DHBV DNA probe. (B) Core protein expression was analyzed by SDS-PAGE and Western blotting with a core-specific antibody. RC, relaxed circular DNA; SS, full-length single-stranded DNA. The arrowhead indicates the minus-strand DNA (ca. 2.0 kb) reverse transcribed from an internally deleted pgRNA. See the text for details. The marker DNA sizes (kb) are indicated.
FIG. 3.

Effects of core mutations on pgRNA packaging. LMH cells were cotransfected as described in the legend to Fig. 2, except that the core-defective DHBV genome also harbored a mutant polymerase defective in DNA synthesis but competent for pgRNA packaging (DHBV/C−/YMHA). Cytoplasmic capsids were analyzed by native agarose gel electrophoresis and transferred to a nitrocellulose membrane. A radiolabeled plus-strand-specific riboprobe was used to detect the packaged pgRNA (top), and an anticore antibody was used to detect the capsids on the same membrane (bottom).
Southern blot analysis of viral DNA replicative intermediates showed that the single or double D substitutions (DSTSSS, SDTSSS, and DDTSSS) did not significantly affect viral DNA synthesis, whereas the single A substitution at S232 (ASTSSS) moderately (four- to fivefold) decreased viral DNA synthesis (Fig. 1 and 2A, lanes 3 to 7). Strikingly, the double A substitution (AATSSS) drastically inhibited viral DNA synthesis (Fig. 1 and 2A, lane 8). These results thus indicated that phosphorylation of at least one of the two S-K/R sites (S230 and S232) was required for efficient viral DNA synthesis but that dephosphorylation at these two sites was dispensable, at least when the other downstream core phosphosites remained intact.
Phosphorylation and subsequent dephosphorylation at T239, S245, S257, and S259 were required for minus- and plus-strand DNA synthesis, respectively.
A previous study examining the effects of single A or D substitutions at the four S/T-P phosphorylation sites showed that the S259A and S245A substitutions decreased minus-strand and mature RC DNA synthesis, respectively, but that none of the single D substitutions affected NC maturation (40). In light of the dramatic dephosphorylation at all of the core phosphosites in association with NC maturation (27) and the synergistic effect of the double A substitutions at S230 and S232, as described above, we decided to construct quadruple A or D substitutions at all four S/T-P sites (SSDDDD and SSAAAA) (Fig. 1). Neither of these sets of substitutions significantly affected core protein expression (Fig. 1 and 2B). The quadruple A substitution mutant displayed a single band by SDS-PAGE, comigrating with the fastest-migrating species of the WT core, as expected from the loss of phosphorylation at the S/T-P sites. Interestingly, the quadruple D substitution mutant also migrated as a fast single band, similar to the SSAAAA mutant. Both of these quadruple substitution mutants only modestly affected pgRNA packaging (ca. twofold reduction) (Fig. 1 and 3).
In contrast, Southern blot analysis of viral DNA replicative intermediates showed a dramatic effect of these quadruple substitutions on viral DNA synthesis. Consistent with a requirement of phosphorylation at these sites for viral DNA synthesis, the SSAAAA mutant showed very little (barely detectable) DNA synthesis (Fig. 1 and 2A, lane 12). Most interestingly, the SSDDDD mutant was able to synthesize single-stranded (SS) (minus-strand) DNA at close to (ca. 90%) WT levels but showed little (1.7% of the WT level) mature RC DNA (Fig. 1 and 2A, lane 11).
Previous studies have shown that certain core mutations, including the single A substitution at S245 (S245A), preferentially destabilize mature NCs so that the mature RC DNA becomes degraded by nuclease during core DNA extraction, resulting in an apparent phenotype similar to that of our D substitution mutants (17, 19). To test if the quadruple D substitution showed a similar effect, we analyzed viral DNA intermediates synthesized by this mutant, with or without nuclease digestion during viral DNA extraction. The results showed that without nuclease digestion, higher levels of mature RC DNA were indeed detectable in both the quadruple D (Fig. 4, lane 10 versus lane 3) and S245A (data not shown) mutants, but its levels were still reduced compared to that of the WT (17.6%) (Fig. 1). Thus, the almost complete lack of mature RC DNA in these mutants likely resulted from a combined effect of decreased RC DNA synthesis and instability of the mature NCs. On the other hand, the SSDDDD mutant still contained some immature DS DNAs migrating above the full-length minus strand, either with or without nuclease digestion during DNA extraction (Fig. 2A, lane 11, and 4A, lanes 3 and 10).
To analyze further the lengths of the viral minus-strand and plus-strand DNAs individually, we performed Southern blot analyses of heat-denatured viral DNA replicative intermediates, extracted with or without exogenous nuclease digestion, using strand-specific riboprobes hybridizing to different regions of viral DNA. The results confirmed that the quadruple A substitution mutant synthesized only barely detectable levels of minus-strand DNA (and thus no plus-strand DNA) (data not shown). More importantly, these analyses verified that the quadruple D substitution mutant contained full-length minus-strand DNA (Fig. 5C and D, lanes 2). The apparently more severe reduction in the level of full-length minus-strand DNA in the SSDDDD mutant, as analyzed in Fig. 5 (using denatured viral DNA), than the nearly WT level of SS (full-length minus-strand) DNA in the same mutant shown in Fig. 2 (using native viral DNA) was due to the fact that the full-length minus-strand DNA detected in Fig. 5 was a combination of SS DNA plus the minus-strand DNA released from the DS DNA, mostly RC DNA, which was largely degraded in the SSDDDD mutant (during DNA extraction by exogenously added nuclease). Accordingly, when the core DNA was extracted without exogenous nuclease digestion, the decrease in the level of full-length minus-strand DNA in the SSDDDD mutant compared to that in the WT was not as great as that with nuclease digestion (Fig. 5C and D, lanes 3 and 4 versus lanes 1 and 2). The decrease of full-length minus-strand DNA in the SSDDDD mutant extracted in the absence of exogenous nuclease digestion (Fig. 5) reflected the lower level of mature RC DNA in the SSDDDD mutant, possibly due in part to the susceptibility of the mature RC DNA (and thus its minus strand) in the SSDDDD mutant to endogenous nuclease degradation (see Discussion).
FIG. 5.

Analysis of denatured core DNA. Viral core DNA was isolated from transfected LMH cells, with or without micrococcal nuclease (MNase) digestion, and heat denatured prior to Southern blot analysis. Radiolabeled specific riboprobes hybridizing to the plus (A [5′ end, nt 2651 to 415] and B [3′ end, nt 2156 to 2375])- or minus (C [5′ end, nt 2156 to 2531] and D [3′ end, nt 2651 to 3021])-strand DNA were used to detect the different regions and strands of viral DNA. The DNA marker sizes (kb) (heat denatured) are indicated. FL, full-length plus (A and B)- or minus (C and D)-strand DNA. The arrowheads in panels C and D indicate the internally deleted minus-strand DNA described in the legend to Fig. 2.
A minus-strand DNA species (ca. 2.0 kb) migrating below the full-length minus strand (marked by arrowheads in Fig. 2, 4, and 5) was present in the SSDDDD mutant at close to (ca. 80%) the WT level, regardless of whether the viral DNA was extracted with or without nuclease digestion or denatured or not prior to analysis. This minus-strand DNA species was likely derived from reverse transcription of an internally deleted pgRNA which was expressed from the complementing core protein expression construct (pCMVΔXM) and contained both of the RNA packaging signals (see Materials and Methods for details) (25) and thus could be packaged into nucleocapsids and reverse transcribed (13). In support of this explanation, this DNA species was not detected when a different complementing construct (pCDNA-Dcore), which lacked the sequences encoding the RNA packaging signals, was used to express the WT or mutant core proteins (Fig. 4B). Also, this DNA species could be detected by probes that hybridized to either the 5′ or 3′ end of the minus-strand DNA but not by a probe that hybridized to the middle of the DNA, which was deleted in the complementing construct (pCMVΔXM) (Fig. 5C and D and data not shown). The modest effect of the SSDDDD core mutation on the level of this minus-strand DNA species further indicated that this mutation did not drastically affect first (minus)-strand DNA synthesis. The packaging of the deleted pgRNA transcribed from the complementing construct and synthesis of its corresponding minus-strand DNA did not make significant contributions to the plus-strand DNA, since the deleted region contained part of an important cis-acting element, 5E (nt 2343 to 2466), which is critical for plus-strand DNA (specifically RC DNA) synthesis (12, 13). Importantly, the mutant core proteins showed the same effects on viral DNA synthesis whether they were expressed from the pCMVΔXM or pcDNA3 vector (Fig. 2 and 4B and data not shown).
The analysis of the denatured viral DNA also verified that very little (1.9% of the WT level) full-length plus-strand DNA was present in the SSDDDD mutant (Fig. 5A and B, lanes 2), although omission of the nuclease digestion step during core DNA extraction again showed that some (11.2% of the WT level) (Fig. 1) full-length plus strands were synthesized by this mutant (Fig. 5A and B, lanes 4), in agreement with the suggestion that its reduced RC DNA level, as shown in Fig. 2 and 4, was due to a combination of mature NC instability and reduced synthesis of mature RC DNA. On the other hand, plus-strand DNA species of up to ca. 2 kb were detectable in the SSDDDD mutant in both the presence and absence of exogenous nuclease digestion. These short plus strands were heterogeneous and seemed to be detectable by 5′- as well as 3′-end-specific probes, suggesting that they represented a mixture of plus-strand synthesis intermediates, degradation products from more mature DS DNA, or even, potentially, plus strands initiated aberrantly downstream from the authentic initiation site. In particular, even the shortest plus-strand species (ca. 0.6 kb) was detectable by a probe specific for the 3′ end of the plus strand. However, it seemed that the same population of heterogeneous plus-strand species was also detectable in the WT (Fig. 5A and B, lanes 1 and 3). The exact nature of these short plus strands was difficult to ascertain due to their heterogeneity, but their apparent accumulation and resistance to exogenous nuclease digestion in both the WT and SSDDDD mutant NCs indicated that the quadruple D substitution mutation specifically affected the mature RC DNA (synthesis and stability) but had comparatively little effect on the short plus-strand DNAs. Taken together, these results were therefore in agreement with a requirement of core phosphorylation at the S/T-P motifs for minus-strand DNA synthesis and of the subsequent dephosphorylation at these sites during plus-strand DNA synthesis in order to stabilize mature NCs as well as to facilitate the synthesis of mature plus strands.
Substitutions at the S-K/R motifs combined with substitutions at the S/T-P motifs impaired both minus- and plus-strand DNA synthesis.
To determine if the phosphorylation state of the upstream S-K/R sites and downstream S/T-P sites could affect the role of each group in NC maturation, we combined the quadruple substitutions at the S/T-P sites with the upstream substitutions at S230 and S232 to create quintuple substitutions (SDDDDD and SAAAAA) and sextuple substitutions (AAAAAA and DDDDDD) (Fig. 1). Again, these combined substitutions did not significantly affect core protein expression (Fig. 1 and 2B, lanes 13 to 16) and only modestly affected pgRNA packaging (two- to fourfold reduction compared to the WT) (Fig. 1 and 3, lanes 11 to 14). However, Southern blot analysis of intracellular viral DNA showed that the additional A substitutions at S230 and S232 in combination with the quadruple A substitutions further decreased the amount of viral DNA synthesis, such that the barely detectable viral DNA in the SSAAAA mutant was reduced to undetectable levels in the AAAAAA mutant (Fig. 2A, lanes 14 and 16). Furthermore, the D substitutions at S230 and S232, which had little or no effect by themselves on either minus- or plus-strand DNA synthesis, led to further decreases in viral DNA levels when combined with the quadruple D substitutions (i.e., the SDDDDD and DDDDDD mutants) (Fig. 2A, lanes 13 and 15 versus lanes 3, 4, 7, and 11, and 4A, lanes 4 and 11). Interestingly, unlike the D substitutions at the S/T-P motifs, which preferentially affected plus-strand DNA accumulation, the additional D substitutions at the S-K/R motifs combined with the D substitutions at the S/T-P motifs decreased the levels of both minus- and plus-strand DNAs (Fig. 1).
Single and double S restorations at positions 245 and 259 partially rescued viral DNA synthesis.
Since previous mutagenesis studies of the individual S/T-P phosphosites indicated that S245 and S259 phosphorylation, in particular, was required for plus- and minus-strand DNA synthesis, respectively (40), we were interested in determining if restoration of these two sites, either alone or in combination, would rescue DNA synthesis in the context of the A or D substitutions at the remaining phosphosites. Interestingly, restoration of S259, but not of S245, in the D substitution background (the DDDDDS and DDDSDS mutants) led to the appearance of an additional, slower-migrating core protein species by SDS-PAGE (Fig. 2B, lanes 17, 18, 23, 24, and 27), consistent with the previously reported dominant role of S259 phosphorylation in causing core protein migrational heterogeneity (41). Interestingly, restoration of either S259, S245, or both did not cause this migrational heterogeneity in the A substitution background (Fig. 2B, lanes 19, 20, 25, 26, and 28). Since a previous study showed that phosphorylation at each of the four S/T-P sites occurs independently (41), phosphorylation at either or both of these sites in the context of the A substitutions at the other sites apparently did not lead to the putative conformational changes responsible for the core protein mobility shift by SDS-PAGE.
In agreement with a dominant role of S245, but not S259, in plus-strand DNA synthesis, restoration of S245 alone (DDDSDD), but not S259 alone (DDDDDS), in the all-D substitution background led to the accumulation of longer plus-strand DNA species (Fig. 1, 2A, lanes 17, 18, 23, and 24, and 4A, lanes 5, 6, 12, and 13; data not shown). However, the S259 restoration in combination with S245 could further enhance the effect of S245 on plus-strand accumulation, although even the double S restoration (DDDSDS) did not allow the accumulation of WT levels of mature RC DNA (Fig. 1, 2A, lane 27, and 4A, lanes 7 and 14; data not shown). Similarly, restoration of both S245 and S259 in the all-A substitution background (AAASAS) weakly restored viral DNA synthesis, although single restoration of either S245 (AAASAA) or S259 (AAAAAS) did not have any appreciable effect (Fig. 2, lanes 19, 20, 25, 26, and 28). Together, these results thus showed that S245 did indeed have a more dominant role than S259 in plus-strand maturation, as previously described (40), but a similarly dominant role for S259 in minus-strand DNA synthesis was not apparent. Furthermore, they clearly demonstrated that these two S residues alone were insufficient to facilitate WT levels of viral DNA synthesis.
Aspartate substitutions arrested minus-strand DNA elongation in the context of an RNA-DNA hybrid.
The effect of the D substitutions on DS DNA accumulation suggested that these mutations might affect plus-strand DNA (DNA-dependent DNA synthesis) preferentially and have little effect on minus-strand DNA (RNA-dependent DNA synthesis or reverse transcription). Alternatively, they could affect DNA accumulation in the context of a DS nucleic acid, regardless of strand specificity. To differentiate these two possibilities, we took advantage of the fact that certain RNase H-defective hepadnavirus mutants synthesize a DS RNA-DNA hybrid molecule (consisting of the pgRNA remaining hybridized to the newly synthesized minus-strand DNA) because the pgRNA cannot be degraded during reverse transcription. As previously reported (5), the RNase H-defective DHBV D715V mutant synthesized minus-strand DNAs of up to 2 kb in length but few full-length minus strands, even with a WT core protein (Fig. 6A and B, lanes 1). The nature of the RNA-DNA hybrids was verified by treatment with RNase H, which converted the hybrids (Fig. 7, lanes 1 and 2), migrating above the full-length SS DNA, to nascent minus-strand DNA species migrating below the full-length SS DNA, just as heat denaturation did (Fig. 7, lanes 3 and 4). As a control for RNase H activity, the WT core DNA was not changed by the treatment (Fig. 7, lanes 5 to 8), except for an apparently short RNA-DNA hybrid (*), which was converted to a faster-migrating short minus-strand DNA (**).
FIG. 6.

Effects of core mutations on minus-strand DNA synthesis in RNase H-defective NCs. Viral core DNA was isolated from transfected LMH cells as described in the legend to Fig. 2, except that the core-defective DHBV genome also harbored a mutant polymerase defective in RNase H activity (DHBV/C−/RH−). DNAs were extracted with (A, B, and C, lanes 1 to 3) or without (C, lanes 4 to 6) exogenous micrococcal nuclease (MNase) digestion and analyzed by Southern hybridization, with (B and C) or without (A) prior heat denaturation. Minus-strand DNA was detected using a radiolabeled riboprobe specific for the 5′ end of the minus strand (as in Fig. 5C), and plus-strand DNA was detected using a full-length riboprobe. The marker DNA sizes (kb), either DS (A) or denatured SS (B and C), are indicated. The numerals I through V in panel B refer to minus-strand DNA intermediates with increasing lengths. Lanes 5 in panels A and B and lanes 1 and 4 in panel C show the DNA synthesized by the WT polymerase in the context of the WT NC (WT/WT), as a control. (D) Relative amounts of minus-strand intermediates, designated I through V as in panel B, are expressed as percentages of those in the WT. Note that all comparisons in panel D were made among NCs that shared the same RNase H mutation.
FIG. 7.
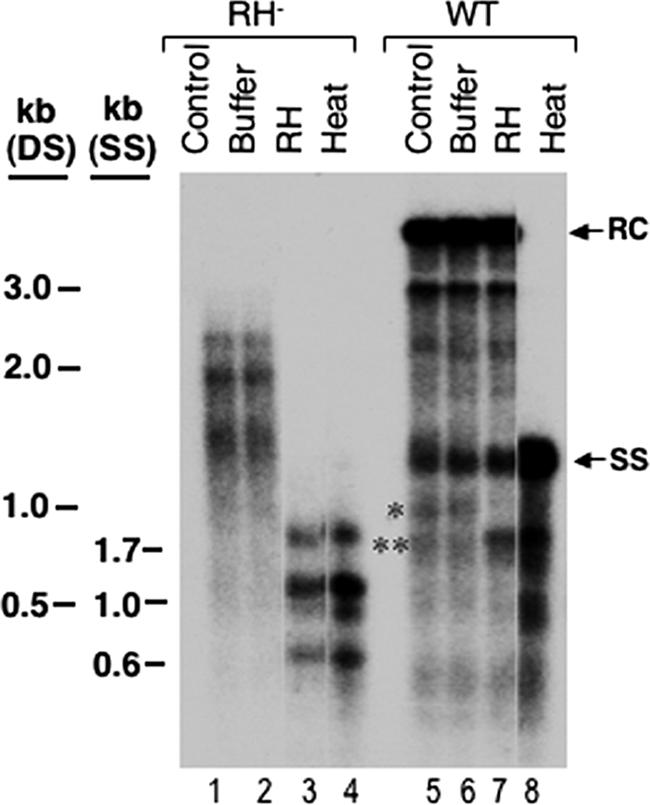
Analysis of RNA-DNA hybrids in RNase H-defective NCs. Viral core DNA isolated from the RNase H-defective mutant (RH−) or the WT was treated with RNase H (RH) (lanes 3 and 7) or buffer alone (lanes 2 and 6) or heat denatured at 95°C (lanes 4 and 8) and then analyzed by Southern blot hybridization using a riboprobe specific for the 5′ end of the minus-strand DNA (as in Fig. 5C). RC, relaxed circular DNA; SS, full-length single-stranded DNA. The marker DNA (SS, heat-denatured, single-stranded DNA; DS, double-stranded DNA) sizes are indicated. In addition to the predicted RNA-DNA hybrid present in the RH− mutant (lanes 1 and 2), an apparent RNA-DNA hybrid (*) was also present in the WT core DNA (lanes 5 and 6) and was converted to a faster-migrating short minus-strand DNA (**) after RNase H digestion (lane 7) or heating (lane 8).
Interestingly, the quadruple D substitution core mutant (SSDDDD) further blocked minus-strand elongation, leading to the accumulation of minus-strand DNA intermediates of up to approximately 1.0 kb long (to levels of more than threefold above the WT level) but a failure to synthesize longer minus strands (Fig. 6A and B, lanes 2, and D). In contrast to the instability of mature RC DNA-containing NCs induced by the SSDDDD mutation, omission of nuclease digestion during DNA extraction did not reveal longer minus-strand DNAs in the case of the RNase H mutant (Fig. 6C, lane 3 versus lane 6), suggesting that the defect here was indeed at the level of DNA synthesis rather than degradation. The apparent lack of instability of the SSDDDD mutant NC harboring the RNA-DNA hybrid might be due to the arrest of minus-strand elongation before it could reach a length sufficient to destabilize the mutant NC. The quintuple and sextuple D substitutions (SDDDDD and DDDDDD) further decreased the level but did not alter the pattern of viral DNA synthesis relative to that of the quadruple mutant (Fig. 6A and B, lanes 3 and 4, and D). The fact that the D substitution mutants at the S/T-P motifs exhibited decreased plus-strand DNA maturity in the context of WT RNase H activity but decreased minus-strand maturity in the context of the RNase H mutant suggested that these mutant capsids were sensitive to the accumulation of a DS nucleic acid, either a DS DNA or an RNA-DNA hybrid, within them. Thus, these results supported the idea that core dephosphorylation was indeed required for DNA synthesis in the context of a DS molecule, whether the template was DNA or RNA.
DISCUSSION
The mutagenesis results presented here, together with our recent biochemical analysis that revealed the dramatic, maturation-associated dephosphorylation of DHBV NCs, strongly support a model of regulation of hepadnavirus reverse transcription via sequential phosphorylation and dephosphorylation of the capsid protein (Fig. 8). First, the core protein subunits are phosphorylated either during or immediately after translation (9; J. Hu, unpublished results). Second, the phosphorylated core proteins assemble into immature nucleocapsids, packaging the pgRNA. Third, the phosphorylated NCs facilitate the synthesis of the minus-strand DNA and the initiation of the plus-strand DNA, whose elongation leads to NC dephosphorylation. Fourth, the dephosphorylated NCs, in turn, facilitate the further maturation of the plus-strand DNA and stabilization of the mature NCs.
FIG. 8.

Model of sequential core phosphorylation and dephosphorylation regulating hepadnavirus reverse transcription. (1) Core phosphorylation during or after translation; (2) NC assembly and pgRNA packaging; (3) complete minus-strand DNA synthesis and initial stage of plus-strand DNA synthesis; (4) core dephosphorylation followed by plus-strand DNA maturation. K1 and K2, unknown cellular kinases; Pase, unknown cellular phosphatase. See the text for details.
Previous studies have shown that phosphorylation at the three SP motifs of the HBV core C-terminal domain is critical for HBV pgRNA packaging, whereas the phosphorylation state of the analogous four S/T-P motifs of the DHBV core protein has only a slight impact on DHBV pgRNA packaging (9, 20, 40). Our results also showed that the phosphorylation state at the S/T-P or the two additional upstream S-K/R motifs had only a modest effect (maximum of three- to fourfold reduction when all six phosphosites were replaced with either A or D) on DHBV pgRNA packaging in cultured cells. It is currently unclear what may account for this differential requirement in these two related viruses. Although it remains possible that this may simply reflect an intrinsic difference between the two viruses, it is conceivable that core phosphorylation may play a much more critical role in DHBV pgRNA packaging under in vivo conditions.
With respect to DNA synthesis, our results showed that the recently identified S230 and S232 (the S-K/R motif) core phosphorylation is required for reverse transcription. Although single A substitutions at these sites had only a modest effect on viral DNA synthesis, as also noted previously (40), a double A substitution at these two sites showed a drastic effect, suggesting that core phosphorylation at either of these two sites can partially substitute for that at the other and thus plays a redundant role in reverse transcription. Similarly, the quadruple A substitution at the four downstream S/T-P sites almost completely abolished any DNA synthesis, whereas the single S259A and S245A mutants were previously shown to have partial defects in DNA synthesis and the single T239A or S257A mutant did not show any defect (40). This again indicates a partially redundant role of core phosphorylation at these S/T-P sites, like the case for the S-K/R sites, in viral DNA synthesis. The hierarchy of each individual phosphosite in regulating DNA synthesis remains to be clarified. The inhibitory effect on minus-strand DNA synthesis by the single S259A mutation, but not the other single substitutions, suggested that S259 phosphorylation may play a dominant role in minus-strand DNA synthesis (40), although our results clearly showed that it alone is insufficient to facilitate reverse transcription. It is also possible that phosphorylation at the different sites may be coordinated. Since the two different motifs of phosphosites most likely employ distinct kinases, it follows that at least two different host kinases (Fig. 8), and probably two different signaling pathways involving these kinases, are required to facilitate viral reverse transcription.
The precise stage in minus-strand DNA synthesis that was blocked by the A substitutions is not yet certain. Since the initiation of minus-strand DNA synthesis via protein priming can occur in the absence of any core protein (34), it is unlikely that these core mutations inhibited protein priming. Instead, they may block minus-strand template switching or early elongation (33). How core phosphorylation may facilitate minus-strand DNA synthesis also remains to be elucidated. The fact that the D substitutions were compatible with full-length minus-strand DNA synthesis indicates that the negative charges afforded by the phosphate groups may somehow be involved. However, negative charge alone is unlikely to account for all of the effects of core phosphorylation, as the amounts of SS DNA synthesized by the D mutants, especially the SDDDDD and DDDDDD mutants, were less than that of the WT, suggesting that an additional effect of core phosphorylation, e.g., a subtle conformational change of the NC, which may not be mimicked by the D substitutions, may be important for optimal minus-strand DNA synthesis. Circumstantial evidence in support of the conformational hypothesis came from the observation that the D mutations did not cause retarded mobility by SDS-PAGE, which has been shown to result from phosphorylation-induced core protein conformational changes (41).
The dramatic and selective effect of the quadruple D substitutions at the S/T-P motifs on the accumulation of the plus-strand DNA strongly supports a role of core dephosphorylation at these sites specific for plus-strand accumulation. One effect of the D substitutions was clearly the destabilization of mature NCs such that the mature RC DNA within them became susceptible to exogenous nuclease digestion during extraction. Some mature RC DNA might also be degraded by endogenous nucleases while being synthesized (before harvesting), which could be partially responsible for the decreased RC DNA of the SSDDDD mutant that was isolated even without exogenous nuclease treatment. However, we suggest that the second effect of the D substitutions could be a block of plus-strand DNA elongation. Thus, the same core mutation blocked minus-strand DNA elongation in the context of an RNA-DNA hybrid by the RNase H mutant in the absence of any apparent NC destabilization, such that the levels of short DNA strands (shorter than 1 kb) accumulated to levels above those of the WT, while those of long DNA strands (longer than 1.5 kb) were diminished. These results thus strongly indicate that dephosphorylation of the S/T-P motifs is required for minus-strand DNA elongation in the context of an RNA-DNA hybrid, and probably also for plus-strand elongation in the context of a DS DNA. The marked accumulation of short minus-strand DNA intermediates as RNA-DNA hybrids in the case of the RNase H mutant and the accumulation of some short plus strands by the SSDDDD core mutant in the case of the WT polymerase also suggest that the initial stage of second-strand DNA synthesis does not require core dephosphorylation at the S/T-P motifs (Fig. 8).
Interestingly, our results showed that the two upstream S-K/R sites, in contrast to the four downstream S/T-P phosphosites, did not have to be dephosphorylated to allow mature plus-strand accumulation. However, the dephosphorylation at the S-K/R sites, which also accompanies NC maturation (27), may still play a role in optimizing viral DNA synthesis, as the quintuple and sextuple D substitution mutants showed further reduced levels of DNA synthesis compared with that of the quadruple D substitution mutant. Interestingly, a previous study with single D substitutions individually at each of the four S/T-P sites did not reveal any deleterious effect on viral DNA synthesis (40), suggesting that the role of core dephosphorylation at these sites during plus-strand DNA elongation is also redundant, as is core phosphorylation during minus-strand DNA synthesis.
How core dephosphorylation may facilitate second-strand DNA synthesis and mature NC stability remains to be elucidated. One possibility is that the accumulation of the additional nucleic acid negative charge inside the NCs as second-strand elongation progresses needs to be balanced by the removal of the negatively charged phosphate groups from the core protein before any further plus-strand elongation, and consequently further accumulation of negative charges, can take place. In the absence of NC dephosphorylation, continued plus-strand elongation either cannot proceed or, if it does occur, apparently leads to the destabilization of the mature NC. During normal minus-strand DNA synthesis, the accumulation of minus-strand DNA is balanced by the degradation of the pgRNA such that no core dephosphorylation at this stage would be needed, so that full-length SS (minus-strand) DNA can be synthesized by the D substitution mutants. The inhibitory effect of the D mutants on minus-strand DNA elongation in the context of RNase H mutation could be explained by the fact that the pgRNA is not degraded while the minus-strand DNA is being made, resulting in an abnormal accumulation of negative charges within the NCs and a block to further minus-strand DNA elongation. This so-called “charge balance” hypothesis was also partially supported by mutational studies that artificially manipulated the number of charged residues in the HBV core C-terminal domain (21). It is also possible that NC dephosphorylation leads to a subtle conformational change that is, in turn, required for plus-strand maturation. A recent cryo-electron microscopy study has indeed revealed some structural differences between the mature, virion-derived HBV NCs and the recombinant NCs expressed in bacteria (29). In addition, the phosphorylation state of the core protein may modulate its nucleic acid binding activity (23), which may also be involved in regulating reverse transcription.
The present study illustrates how the dynamics of a posttranslational modification of the capsid protein is employed to regulate the ordered progression of hepadnavirus reverse transcription within its capsid shell. Although it is possible, in theory, that the effects of the core substitution mutations at the phosphosites may be unrelated to the phosphorylation state of the core proteins, a caveat common to all mutational analyses of this type, the differential effect of the A and D substitutions at the S/T-P motifs on the first and second steps of viral DNA synthesis, respectively, combined with our recent biochemical evidence of maturation-associated NC dephosphorylation, strongly support the notion that sequential phosphorylation and dephosphorylation of hepadnavirus NCs facilitate the ordered progression of viral reverse transcription. These results have broad implications for other complex macromolecular processes, including retroviral reverse transcription (3, 11), that similarly must occur within an enclosed shell or scaffold. The results presented here clearly demonstrate that rather than acting simply as a passive vessel, the capsid shell plays an active role, which is further subjected to host regulation, in the entire reverse transcription process.
Acknowledgments
We thank William Mason for the antibody against the DHBV core and Jesse Summers for plasmid constructs and a critical reading of the manuscript.
This work was supported by Public Health Service grant R01 AI43453 (J.H.) from the National Institutes of Health.
Footnotes
Published ahead of print on 29 November 2006.
REFERENCES
- 1.Birnbaum, F., and M. Nassal. 1990. Hepatitis B virus nucleocapsid assembly: primary structure requirements in the core protein. J. Virol. 64:3319-3330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bottcher, B., S. A. Wynne, and R. A. Crowther. 1997. Determination of the fold of the core protein of hepatitis B virus by electron cryomicroscopy. Nature 386:88-91. [DOI] [PubMed] [Google Scholar]
- 3.Bowzard, J. B., J. W. Wills, and R. C. Craven. 2001. Second-site suppressors of Rous sarcoma virus Ca mutations: evidence for interdomain interactions. J. Virol. 75:6850-6856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chang, L. J., R. C. Hirsch, D. Ganem, and H. E. Varmus. 1990. Effects of insertional and point mutations on the functions of the duck hepatitis B virus polymerase. J. Virol. 64:5553-5558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chen, Y., and P. L. Marion. 1996. Amino acids essential for RNase H activity of hepadnaviruses are also required for efficient elongation of minus-strand viral DNA. J. Virol. 70:6151-6156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Conway, J. F., N. Cheng, A. Zlotnick, P. T. Wingfield, S. J. Stahl, and A. C. Steven. 1997. Visualization of a 4-helix bundle in the hepatitis B virus capsid by cryo-electron microscopy. Nature 386:91-94. [DOI] [PubMed] [Google Scholar]
- 7.Ganem, D., and A. M. Prince. 2004. Hepatitis B virus infection—natural history and clinical consequences. N. Engl. J. Med. 350:1118-1129. [DOI] [PubMed] [Google Scholar]
- 8.Ganem, D., and R. J. Schneider. 2001. Hepadnaviridae, p. 2923-2969. In D. M. Knipe and P. M. Howley (ed.), Fields virology. Lippincott Williams & Wilkins, Philadelphia, PA.
- 9.Gazina, E. V., J. E. Fielding, B. Lin, and D. A. Anderson. 2000. Core protein phosphorylation modulates pregenomic RNA encapsidation to different extents in human and duck hepatitis B viruses. J. Virol. 74:4721-4728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gerelsaikhan, T., J. Tavis, and V. Bruss. 1996. Hepatitis B virus nucleocapsid envelopment does not occur without genomic DNA synthesis. J. Virol. 70:4269-4274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gonsky, J., E. Bacharach, and S. P. Goff. 2001. Identification of residues of the Moloney murine leukemia virus nucleocapsid critical for viral DNA synthesis in vivo. J. Virol. 75:2616-2626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Havert, M. B., L. Ji, and D. D. Loeb. 2002. Analysis of duck hepatitis B virus reverse transcription indicates a common mechanism for the two template switches during plus-strand DNA synthesis. J. Virol. 76:2763-2769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Havert, M. B., and D. D. Loeb. 1997. cis-Acting sequences in addition to donor and acceptor sites are required for template switching during synthesis of plus-strand DNA for duck hepatitis B virus. J. Virol. 71:5336-5344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hu, J., and C. Seeger. 1996. Hsp90 is required for the activity of a hepatitis B virus reverse transcriptase. Proc. Natl. Acad. Sci. USA 93:1060-1064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jilbert, A. R., T. T. Wu, J. M. England, P. M. Hall, N. Z. Carp, A. P. O'Connell, and W. S. Mason. 1992. Rapid resolution of duck hepatitis B virus infections occurs after massive hepatocellular involvement. J. Virol. 66:1377-1388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kawaguchi, T., K. Nomura, Y. Hirayama, and T. Kitagawa. 1987. Establishment and characterization of a chicken hepatocellular carcinoma cell line, LMH. Cancer Res. 47:4460-4464. [PubMed] [Google Scholar]
- 17.Kock, J., M. Kann, G. Putz, H. E. Blum, and F. Von Weizsacker. 2003. Central role of a serine phosphorylation site within duck hepatitis B virus core protein for capsid trafficking and genome release. J. Biol. Chem. 278:28123-28129. [DOI] [PubMed] [Google Scholar]
- 18.Kock, J., M. Nassal, K. Deres, H. E. Blum, and F. von Weizsacker. 2004. Hepatitis B virus nucleocapsids formed by carboxy-terminally mutated core proteins contain spliced viral genomes but lack full-size DNA. J. Virol. 78:13812-13818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kock, J., S. Wieland, H. E. Blum, and F. von Weizsacker. 1998. Duck hepatitis B virus nucleocapsids formed by N-terminally extended or C-terminally truncated core proteins disintegrate during viral DNA maturation. J. Virol. 72:9116-9120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lan, Y. T., J. Li, W. Liao, and J. Ou. 1999. Roles of the three major phosphorylation sites of hepatitis B virus core protein in viral replication. Virology 259:342-348. [DOI] [PubMed] [Google Scholar]
- 21.Le Pogam, S., P. K. Chua, M. Newman, and C. Shih. 2005. Exposure of RNA templates and encapsidation of spliced viral RNA are influenced by the arginine-rich domain of human hepatitis B virus core antigen (HBcAg 165-173). J. Virol. 79:1871-1887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Liao, W., and J. H. Ou. 1995. Phosphorylation and nuclear localization of the hepatitis B virus core protein: significance of serine in the three repeated SPRRR motifs. J. Virol. 69:1025-1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Machida, A., O. Tsuda, A. Yoshikawa, Y. Hoshi, T. Tanaka, S. Kishimoto, Y. Akahane, Y. Miyakawa, and M. Mayumi. 1991. Phosphorylation in the carboxyl-terminal domain of the capsid protein of hepatitis B virus: evaluation with a monoclonal antibody. J. Virol. 65:6024-6030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Melegari, M., S. K. Wolf, and R. J. Schneider. 2005. Hepatitis B virus DNA replication is coordinated by core protein serine phosphorylation and HBx expression. J. Virol. 79:9810-9820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ostrow, K. M., and D. D. Loeb. 2002. Characterization of the cis-acting contributions to avian hepadnavirus RNA encapsidation. J. Virol. 76:9087-9095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Perlman, D., and J. Hu. 2003. Duck hepatitis B virus virion secretion requires a double-stranded DNA genome. J. Virol. 77:2287-2294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Perlman, D. H., E. A. Berg, P. B. O'Connor, C. E. Costello, and J. Hu. 2005. Reverse transcription-associated dephosphorylation of hepadnavirus nucleocapsids. Proc. Natl. Acad. Sci. USA 102:9020-9025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pugh, J., A. Zweidler, and J. Summers. 1989. Characterization of the major duck hepatitis B virus core particle protein. J. Virol. 63:1371-1376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Roseman, A. M., J. A. Berriman, S. A. Wynne, P. J. Butler, and R. A. Crowther. 2005. A structural model for maturation of the hepatitis B virus core. Proc. Natl. Acad. Sci. USA 102:15821-15826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Schlicht, H. J., R. Bartenschlager, and H. Schaller. 1989. The duck hepatitis B virus core protein contains a highly phosphorylated C terminus that is essential for replication but not for RNA packaging. J. Virol. 63:2995-3000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Summers, J., and W. S. Mason. 1982. Replication of the genome of a hepatitis B-like virus by reverse transcription of an RNA intermediate. Cell 29:403-415. [DOI] [PubMed] [Google Scholar]
- 32.Summers, J., P. M. Smith, and A. L. Horwich. 1990. Hepadnavirus envelope proteins regulate covalently closed circular DNA amplification. J. Virol. 64:2819-2824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wang, G. H., and C. Seeger. 1993. Novel mechanism for reverse transcription in hepatitis B viruses. J. Virol. 67:6507-6512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang, G. H., and C. Seeger. 1992. The reverse transcriptase of hepatitis B virus acts as a protein primer for viral DNA synthesis. Cell 71:663-670. [DOI] [PubMed] [Google Scholar]
- 35.Wang, X., N. Grammatikakis, and J. Hu. 2002. Role of p50/CDC37 in hepadnavirus assembly and replication. J. Biol. Chem. 277:24361-24367. [DOI] [PubMed] [Google Scholar]
- 36.Wei, Y., J. E. Tavis, and D. Ganem. 1996. Relationship between viral DNA synthesis and virion envelopment in hepatitis B viruses. J. Virol. 70:6455-6458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wu, T. T., L. D. Condreay, L. Coates, C. Aldrich, and W. Mason. 1991. Evidence that less-than-full-length pol gene products are functional in hepadnavirus DNA synthesis. J. Virol. 65:2155-2163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wynne, S. A., R. A. Crowther, and A. G. Leslie. 1999. The crystal structure of the human hepatitis B virus capsid. Mol. Cell 3:771-780. [DOI] [PubMed] [Google Scholar]
- 39.Yu, M., and J. Summers. 1991. A domain of the hepadnavirus capsid protein is specifically required for DNA maturation and virus assembly. J. Virol. 65:2511-2517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yu, M., and J. Summers. 1994. Multiple functions of capsid protein phosphorylation in duck hepatitis B virus replication. J. Virol. 68:4341-4348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yu, M., and J. Summers. 1994. Phosphorylation of the duck hepatitis B virus capsid protein associated with conformational changes in the C terminus. J. Virol. 68:2965-2969. [DOI] [PMC free article] [PubMed] [Google Scholar]