

Abstract
Melanoma is the cancer with the highest increase in incidence, and transformation of radial growth to vertical growth (i.e., noninvasive to invasive) melanoma is required for invasive disease and metastasis. We have previously shown that p42/p44 MAP kinase is activated in radial growth melanoma, suggesting that further signaling events are required for vertical growth melanoma. The molecular events that accompany this transformation are not well understood. Akt, a signaling molecule downstream of PI3K, was introduced into the radial growth WM35 melanoma in order to test whether Akt overexpression is sufficient to accomplish this transformation. Overexpression of Akt led to upregulation of VEGF, increased production of superoxide ROS, and the switch to a more pronounced glycolytic metabolism. Subcutaneous implantation of WM35 cells overexpressing Akt led to rapidly growing tumors in vivo, while vector control cells did not form tumors. We demonstrated that Akt was associated with malignant transformation of melanoma through at least 2 mechanisms. First, Akt may stabilize cells with extensive mitochondrial DNA mutation, which can generate ROS. Second, Akt can induce expression of the ROS-generating enzyme NOX4. Akt thus serves as a molecular switch that increases angiogenesis and the generation of superoxide, fostering more aggressive tumor behavior. Targeting Akt and ROS may be of therapeutic importance in treatment of advanced melanoma.
Introduction
Melanoma is a common skin cancer resulting in high morbidity and mortality. Melanomas are neural crest–derived malignancies, many of which arise in precursor lesions termed atypical nevi. A fraction of atypical nevi undergo further malignant change into radial growth melanoma, named for its superficial spreading growth characteristics. Radial growth melanomas undergo further genetic change, acquiring invasive, angiogenic, and metastatic capabilities, leading to recurrence, distant metastases, and death. Once melanoma has spread beyond its original location, it is usually highly resistant to chemotherapy and radiation. Thus, an improved understanding of the signaling mechanisms that contribute to invasive growth is required.
Several genes have been implicated in the development of melanoma. The most common gene involved in familial melanoma is p16ink4a, which has been implicated in at least one-third of familial melanomas. In addition, loss of p16ink4a through deletion, mutation, or promoter methylation is a common event in sporadic melanoma. More recently, B-raf has been implicated in both melanomas and atypical nevi, with a very high frequency of activating V599 lesions in both melanomas and nevi. We have previously demonstrated that activation of MAP kinase occurs in early melanoma, but not benign nevi, and that constitutive activation of MAP kinase signaling leads to malignant transformation of melanocytes but not to a very aggressive phenotype (1, 2). These findings suggest that other signaling pathways must be required for the transformation of radial growth (i.e., noninvasive) melanoma to vertical growth (i.e., invasive) melanoma. Akt is a prime candidate for a second signaling pathway that can cause aggressive growth in melanoma: (a) it confers resistance to a number of apoptotic stimuli, including Fas ligand and tumor necrosis factor apoptosis-inducing ligands (3–5); (b) it inactivates several potential tumor suppressor genes, including tuberin and the forkhead family of transcription factors (6, 7); (c) it is expressed in advanced human melanomas (8); and (d) the genetics of melanoma suggests a functional role of Akt in melanoma pathogenesis. The most commonly mutated oncogenes in melanoma are B-raf and N-ras (9–12). B-raf alone is incapable of activating Akt, while N-ras can. Thus, melanomas that have mutant B-raf often exhibit loss of PTEN, a negative regulator of Akt, allowing Akt activation in advanced lesions (13–16). In this study, we demonstrated that Akt expression allowed aggressive tumor growth of human cells in vivo.
ROS have been found in a large number of tumor types (17, 18). While exposure of primary cells to ROS results in senescence, ROS may serve as a signaling mechanism in tumor cells to promote angiogenesis, mitogenesis, and resistance to apoptosis (19–23). Introduction of oncogenes into immortalized cells has been demonstrated to increase ROS within cells, and ROS-generating genes such as NOX family members have been shown to contribute to tumorigenesis and angiogenesis in vivo (24–28). PI3K activation has been associated with transformation of melanoma (29); however, despite a plethora of downstream effectors that may be involved, the mechanisms whereby PI3K mediates its oncogenic effects have not been fully elucidated. Here we demonstrated that the introduction of a single gene, Akt, into radial growth melanoma cells was sufficient to cause vertical growth in vivo. We further demonstrated that Akt induced genes associated with resistance to ROS, including Sirt1 and rictor, and activation of the AP-1 subunit JunD. Akt thus serves as a prominent pharmacologic target in the treatment of melanoma and other malignancies.
Results
Stable WM35 cells overexpressing Akt (WM35 PKBDD) exhibited a highly significant increase in intracellular superoxide (Figure 1A). As WM35 cells overexpressing Akt had increased levels of ROS, and because we have previously shown that ROS upregulates VEGF and tumorigenesis, we compared production of VEGF in Akt-overexpressing and parental WM35 cells. In vitro growth of WM35 cells in the presence or absence of Akt was not significantly different (data not shown). Akt overexpression led to a nearly 8-fold increase in VEGF mRNA expression (Figure 1B). Increased expression of VEGF protein was observed in Akt-overexpressing WM35 cells (Figure 1C). A high level of phosphorylated Akt expression was observed in WM35 cells transduced with Akt and in tumors derived from mice injected with WM35 PKB cells (Figure 2, A and B). Implantation of Akt-overexpressing WM35 into nude mice led to the development of tumors, whereas no tumor induction was observed in vector-transfected WM35 cells (Figure 2, C and D). Akt-overexpressing WM35 tumors showed high levels of VEGF expression, particularly around the necrotic areas (Figure 2E), and smooth muscle actin immunohistochemistry demonstrated that these tumors were highly angiogenic (Figure 2F). In order to assess the mechanism of induction of ROS generation, we examined several of its potential sources. Several ROS-generating enzymes have been cloned and constitute the NOX family of enzymes. Increased expression of NOX genes has been previously shown to cause generation of ROS (28). In Akt-transformed WM35 PKBDD cells, which were cultured for long periods of time (approximately 6 months), RT-PCR revealed no elevation of the mRNAs encoding the NOX family genes (NOX1–NOX5), suggesting that upregulation of NOX genes is unlikely to account for the increased ROS observed in Akt-transformed cells. In contrast, WM35 PKB cells, which were in culture after Akt transfection for shorter periods of time (1–2 months) showed marked induction of NOX4, demonstrating at least 2 potential mechanisms that could contribute to the transforming effects of Akt (Figure 3).
Figure 1. Introduction of Akt into WM35 cells results in increased intracellular ROS.

(A) Quantitation of hydrogen peroxide by dichlorofluorescein (DCF) fluorescence and of superoxide by dihydroethidium (DHE) fluorescence. (B) Akt overexpression led to increased expression of VEGF mRNA. (C) Increased expression of VEGF protein in WM35 cells overexpressing Akt compared with parental controls. Lane 1, WM35 cells; lane 2, WM35 PKBDD cells; lane 3, pooled Akt-transduced WM35 cells. Tubulin is included as a loading control. Experiments were performed in triplicate.
Figure 2. Akt causes vertical growth melanoma in vivo.

(A) Parental WM35 cells, pooled Akt-transduced WM35 cells prior to implantation in vivo (WM35 PKB and WM35 PKBDD), and cells derived from tumors of mice injected with WM35 PKB cells (tum1–tum3) were examined for expression of phosphorylated Akt (p-Akt). The experiment was performed in triplicate. (B) Representative Western blot of phosphorylated Akt and α-tubulin; overexpression converted radial growth melanoma to vertical growth melanoma. (C and D) Mice (n = 4 per group) were injected with either vector control or Akt-expressing melanoma cells and were observed at 1 month. (E) Immunohistochemistry for VEGF. A high level of VEGF expression was noted, especially surrounding necrotic areas. (F) Smooth muscle actin immunohistochemistry, demonstrating that tumors were highly angiogenic. Note the invasion of tumor cells into vessel at the image’s center. Original magnification, ×100.
Figure 3. Transfection of Akt induces NOX4 expression in melanoma cells.

Levels of NOX4 in each sample were normalized to 18S RNA. Data represent a sample set of 5. In 2 separate experiments, NOX4 was observed in the WM35 PKB cells and not in the WM35 cells. A 1.5% agarose gel was run after PCR, and single bands for NOX4 and for 18S were observed (not shown).
Implantation of Akt-overexpressing WM35 into nude mice led to the development of tumors, whereas no tumor induction was observed in vector-transfected WM35 cells (Figure 2, C and D). Thus, Akt overexpression is sufficient to transform WM35 radial growth melanoma cells to highly invasive tumors in vivo. The AP-1 subunit composition of aggressive melanoma has previously been noted to differ from nonagressive melanoma in that JunD is highly expressed in the former (30, 31). We examined activation of JunD prior to and after Akt-mediated transformation. A dramatic and consistent elevation of phosphorylated JunD was noted in Akt-derived tumors compared with cells prior to transformation and implantation (Figure 4A). A high level of JunD expression may sensitize melanoma cells to alternative forms of apoptosis, such as activating transcription factor 2 (ATF2) antagonists (31).
Figure 4. Akt inhibition downregulates VEGF, rictor, and Sirt1.

(A) Triplicate analysis of pooled WM35 cells transfected with Akt demonstrated activation (phosphorylation) of JunD in melanoma tumors. Lane 1, WM35 cells; lane 2, WM35 PKBDD cells (which have numerous mitochondrial mutations); lane 3, pooled WM35 cells freshly transfected with active PKB prior to tumor implantation; lanes 4–6, protein from individual tumors derived after injection of pooled WM35 PKB cells into mice. (B–D) Akt inhibition in A375 melanoma cells resulted in downregulation of VEGF (B), rictor (C), and Sirt1 (D) RNA. A375 cells were treated with Akt inhibitor and harvested at 2 and 24 hours. RNA levels are corrected for 18S RNA, and experiments were performed in triplicate. *P < 0.05 versus vehicle.
Melanoma cells have demonstrated resistance to multiple insults, including oxidative stress. We examined a spontaneous human melanoma cell line, A375, in the presence of a specific Akt inhibitor. Treatment of this spontaneous melanoma with the Akt inhibitor resulted in significant decreases in 3 genes that may account for this resistance. Sirt1 is a deacetylase that attenuates p53 in the presence of oxidative stress; rictor is a rapamycin-insensitive member of the mammalian target of rapamycin complex; and VEGF is likely the major angiogenic factor in melanoma. Inhibition of Akt led to significant and reproducible decreases in the mRNA levels of all 3 genes (Figure 4, B–D).
Highly malignant tumor cells have been demonstrated to possess both high levels of ROS and altered bioenergetics (17, 18). Because we saw no source for extramitochondrial ROS generation in those cells cultured for long periods of time, cells were examined for mitochondrial bioenergetic function and for the presence of mitochondrial DNA mutation. Mitochondrial dysfunction, notably in complex I, has previously been associated with generation of ROS (32–35). Complex I activity was markedly decreased in Akt-transformed cells compared with parental radial growth melanoma cells (4 units versus 160 units). Consistent with diminished activity of the mitochondrial respiratory complex, which could hamper the bioenergetic function of mitochondria in active Akt–expressing cells, we observed significantly augmented glycolysis rates in WM35 PKBDD cells compared with parental WM35 cells (Figure 5A). In agreement with these results, the expression of active Akt in the cells promoted a doubling in the expression of the glycolytic marker GAPDH (Figure 5B) without significantly affecting the expression of other bioenergetic markers of the mitochondria. As a result of these changes, the bioenergetic cellular index of melanoma cells expressing active Akt showed a 50% decrease compared with parental WM35 cells (Figure 5C). Overall, these results indicate that expression of active Akt impairs the bioenergetic function of mitochondria, at least at the level of complex I activity, with concurrent generation of ROS and activation of glycolytic metabolism in order to cope with the energetic demand required for cellular proliferation.
Figure 5. Introduction of Akt converted WM35 cells to glycolytic metabolism (Warburg effect).

(A) Glycolysis rates were determined in the presence of oligomycin. Results are mean ± SEM of 6 experiments. (B) Western blot analysis of mitochondrial β-F1-ATPase and Hsp60 and glycolytic enzyme GAPDH compared with tubulin, used as a loading control. (C) Relative expression level of the indicated mitochondrial markers as assessed by the β-F1-ATPase/Hsp60 ratio and by GAPDH relative to tubulin. The bioenergetic cellular (BEC) index showed a significant decrease in WM35 PKB cells, consistent with their conversion to a glycolytic phenotype. Results are mean ± SEM of 4 experiments. **P < 0.01, #P < 0.0005 versus WM35.
Temperature gradient capillary electrophoresis (TGCE) analysis of WM35 and WM35 PKBDD mixed heteroduplexes showed a high degree of mutation compared with the homoduplexes formed from unmixed samples of either cell line (Figure 6). The broad spectrum of changes apparent from the electropherogram tracings, as well as those identified by the mutation detection software, necessitated complete sequence analysis in order to accurately characterize the nucleotide changes. For example, in amplicon 1 as shown in Figure 6 (consisting of 440 bp of noncoding D-loop sequence), sequence analysis identified 4 single nucleotide changes, all of which were transitions, between WM35 and WM35 PKBDD cell lines. Complete sequence analysis of the entire mitochondrial DNA (mtDNA) of WM35 and WM35 PKBDD identified 85 total single nucleotide changes that affected a total of 66 codons (Table 1). These nucleotide changes resulted in a total of 50 amino acid changes between WM35 and WM35 PKBDD cells. These mutations were not randomly distributed throughout the mitochondrial genome, but rather largely clustered, especially within complex I coding genes. Complex I genes were the targets of 57 of the 85 total nucleotide changes and 38 of the 50 total amino acid substitutions. Complex IV also experienced a high mutation rate, with 13 nucleotide changes and 9 amino acid substitutions occurring within cytochrome oxidase II (COII). Complete characterizations of the nucleotide differences between WM35 and WM35 PKBDD cells are provided in Table 2 and Supplemental Table 1 (available online with this article; doi:10.1172/JCI30102DS1).
Figure 6. Amplicon 1 TGCE electropherogram tracings for WM35 cells, WM35 PKB cells, and the mixed sample.

Amplicon 1 (spanning mitochondrial positions 16278–148) is a 440-bp fragment that was one of 3 from the 17 total amplicons not subjected to restriction endonuclease treatment, yielding a single peak upon TGCE analysis, as shown for both the WM35 and the WM35 PKB homoduplexes. The mixed sample, however, consisted of a characteristic double peak, suggestive of nucleotide differences between the 2 samples that led to the formation of both homoduplex and heteroduplex species that were resolved from one another in this assay.
Table 1 .
Summary of nucleotide differences between WM35 and WM35 PKB cells

Table 2 .
Nucleotide differences between WM35 and WM35 PKB cells

We had not expected to identify such a high number of nucleotide changes, particularly nonsynonymous changes, between the 2 lineages differing only in the active expression of Akt. Therefore, it was desirable to further characterize these amino acid substitutions and determine their position within the overall 3-dimensional structure and their incorporation into specific protein domains or motifs (Table 2). Of the 50 nonsynonymous nucleotide changes, 19 involved amino acids that exhibit a low level of evolutionary conservation, and 11 of the 19 also reside in transmembrane domains and/or low-complexity protein sequence regions. These residues are not likely to be critical to the catalytic functioning of their respective mtDNA genes. An additional 13 amino acid substitutions were at positions that are generally conserved in mammals (and minimally in both mice and humans); 9 of these were in transmembrane domains and/or low-complexity sequence regions, again suggesting that the amino acid is subject to alteration with minimal functional defect. Finally, 18 amino acid changes occurred in extremely highly conserved positions, although a large proportion of them reside in transmembrane domains and/or low-complexity sequence regions (35). It is interesting to note that 11 amino acids were completely conserved among all species examined (indicating that they may be important to the protein’s structure and/or function), but they were altered between WM35 and WM35 PKBDD. All of these occurred within complex I genes, with 8 in NADH dehydrogenase subunit 2 (ND2) and 3 in ND5. These results indicate that the active expression of Akt within melanoma cells may lead to critical mutations within mitochondrially encoded complex I genes that affect the overall bioenergetic function of the organelle.
In order to determine whether mtDNA mutation is a frequent event in melanoma, we analyzed mtDNA from samples of 13 normal melanocytes, 7 melanomas, and 16 melanoma cell lines. In addition, we transfected the PKBDD construct into WM35 cells, selected them, and injected them into mice after minimal time in tissue culture. We did not observe a significant amount of mtDNA mutation in any of these samples, suggesting that Akt can transform radial growth melanoma cells in the presence or absence of extensive mtDNA mutation. Consistent with these findings from human specimens, when radial growth WM35 cells were transfected with the same Akt construct as in our initial study, extensive mtDNA mutations were not observed (data not shown). NOX4 was induced by Akt transfection, but induction of ROS in vitro was not observed (data not shown). A model accounting for both events is shown in Figure 7.
Figure 7. Schematic of the 2 mechanisms by which Akt may transform cells.

Akt may prevent cells that already have mtDNA mutations from undergoing apoptosis or senescence due to ROS. Akt may also induce NOX4, which may increase the cells’ ability to generate ROS in vivo. Both mechanisms of ROS, in the presence of Akt, may lead to NF-κB activation and resistance to apoptosis.
Discussion
Melanoma is a common cancer that in its advanced stages is resistant to most forms of chemotherapy and radiation. Melanoma progresses from an early, surgically curable stage, known as radial growth melanoma, to the invasive vertical growth melanoma. The precise biochemical changes that accompany this transition are not fully understood, but several genes have been shown to be upregulated or downregulated during this transition. Markers of angiogenesis, such as VEGF and matrix metalloproteinases, are upregulated in vertical growth melanoma, and the small G protein rho C is associated with metastatic growth of melanoma (29, 36). Integrins such as αvβ3 and αvβ5 are seen more commonly in advanced melanoma and may function in part by activating PI3K signaling in the tumor cells in response to extracellular matrix and tumor stromal cells (36, 37). NF-κB signaling is elevated in aggressive melanoma cells (38), while AP-1 subunits undergo changes in composition from c-jun/ATF2, predominating in early melanoma, to JunD in vertical growth melanoma (31, 39). Interestingly, high levels of activated (i.e., phosphorylated) JunD were observed in triplicate tumor-derived specimens in the present study, suggesting that JunD may be a direct target of Akt activation (31, 39). Normal cells respond to ROS stress by inducing p53 activity. In tumor cells that use ROS signaling, p53 activation must be diminished to allow further cell replication. One potential mechanism is through Sirt1 activation, which causes deacetylation and inactivation of p53 (40, 41). We demonstrated that Sirt1 mRNA was present in A375 human melanoma cells and was downregulated through specific Akt inhibition, implicating Sirt1 as a downstream target of Akt. However, the precise mechanism converting the radial growth phenotype to the vertical growth phenotype is unknown.
Otto Warburg first observed that tumor cells exhibit a predominantly glycolytic phenotype (42), and Akt has recently been shown to be capable of causing the switch to the glycolytic phenotype (43). We recently developed a bioenergetic index to quantify the switch to glycolytic metabolism in tumor biopsies and in archival, paraffinized sections of human tumors that illustrates the glycolytic phenotype of lung, colon, kidney, breast, esophageal, and gastric cancers (44–46). The switch to the glycolytic phenotype has been correlated with poor clinical outcome in human solid tumors of the colon and lung (45, 46). In Akt-transduced melanoma cells, we observed a mitochondrial dysfunction with concurrent upregulation of ROS production and flux of glycolysis, a situation known to affect the cellular sensitivity to apoptosis (47). Recent findings showed that cells that undergo glycolytic metabolism have increased production of ROS, including superoxide and hydrogen peroxide (48). ROS can be generated through activation of the cytochrome b558 complex, which is activated by membrane localization or amplification of NOX family genes (49, 50). Introduction of Akt into radial growth melanoma cells resulted in a marked increase in intracellular ROS. We found that Akt can increase ROS through 2 mechanisms: induction of the ROS-generating enzyme NOX4 and mtDNA mutation (Figure 7). One possibility is that sustained generation of low levels of ROS may cause mtDNA mutations in addition to genomic instability (50) and that the presence of Akt stabilizes cells that develop mtDNA mutations. Cells with induction of NOX4 did not show the same level of mtDNA mutation as did cells lacking NOX4 induction. The generation of ROS in respiration-deficient cells likely occurs at complex I (51, 52). As we have demonstrated herein, complex I activity and the mitochondrial encoded genes of this protein complex were the likely targets that impaired the normal bioenergetic function of mitochondria in Akt-transduced cells, therefore promoting their expected metabolic switch to glycolysis as the preferred energy pathway.
Previous studies have shown that neoplastic behavior in cells is associated with changes in mtDNA (50). In order to determine whether mtDNA mutations represent a necessary process in the alteration of the neoplastic phenotype, we generated new cell lines by transfecting in the pKBDD vector expressing Akt and selecting for new clones resistant to puromycin. The new clones were tested for their ability to form invasive tumors in nude mice and, indeed, they displayed the same behavioral alterations of vertical growth and the formation of large tumors as did the parental cell line WM35. We performed phenotypic studies and genotype analysis on these new clones at a low passage number. We also sought to minimize the time the clones were cultured after transformation with pKB. The ND1 and ND5 genes within the mtDNA, which were shown in our initial study to represent mutational hot spots, were amplified by PCR and sequenced.
In strong contrast to the initial studies that were carried out in cells extensively cultured after Akt transfection, the low-passage cell lines and primary clinical samples contained no acquired mtDNA changes in ND1 or ND5. Our interpretation of this finding is that the mtDNA mutations are not sufficient to support the alteration in growth seen in WM35 cells overexpressing Akt. The accrual of mtDNA mutations in the high-passage cell lines was the likely result of prolonged Akt expression, which led to increased production of ROS. This ROS imparted damage to the mtDNA in a nonrandom distribution, which likely further subjected the high-passage cells to increased oxidative stress. We conclude that although the mtDNA mutations did occur nonrandomly in the mtDNA of high-passage WM35 PKB cells, the presence of these mutations (in at least 2 of the complex I genes) was not essential for the aggressive growth phenotype, but rather was a reflection of prolonged exposure to increased oxidative stress that accumulates with prolonged Akt overexpression. Of interest, high levels of ROS were not observed in melanoma cells in which Akt had induced NOX4 in vitro. A possible reason for this observation is that in vivo growth may be necessary for maximal induction of ROS by NOX4; notably, NOX4 alone failed to transform immortalized cells in previous studies (53), implying the existence of additional factors.
The survival advantage conferred to a tumor cell by a glycolytic metabolism is not fully understood, especially since ATP generation is markedly inefficient compared with respiration. We propose that glycolytic metabolism provides a survival mechanism for tumor cells through the activation of ROS–NF-κB signaling and the prevention of apoptosis. In primary cells, ROS causes cell senescence and apoptosis. This senescence is caused in part by induction of p16ink4a, and hypermethylation of p16ink4a occurs commonly in tumors induced by oxidative stress (54, 55). Further events are required to prevent apoptosis caused by ROS, such as inactivation of caspase-9/apoptotic protease–activating factor (caspase-9/Apaf) signaling, and genes in this cascade can be inactivated either functionally or epigenetically (56, 57). Caspase-9/Apaf-1–mediated apoptosis requires ATP, and decreased production of ATP as a result of glycolytic metabolism may by itself provide a survival advantage in cells that generate high levels of superoxide. Apaf-1 is hypermethylated in advanced melanoma, and caspase-9 is inactivated by Akt phosphorylation (56–58). In addition, recent findings indicate that Akt is required to inhibit apoptosis by mediating the maintenance of the glycolytic hexokinase bound to the mitochondria, thus preventing the release of cytochrome c and the execution of apoptosis (3, 59). Therefore, tumor cells (such as those found in melanoma) have defenses against senescence and apoptosis and can use ROS and a glycolytic phenotype as signaling mechanism and cell-defense devices. Recently, coexpression of hypoxia-inducible factor 1 and Akt in murine melanocytes was found to cause transformation to melanoma that was highly sensitive to rapamycin (60). Unfortunately, rapamycin analogs were not found to be active against melanoma in a human clinical trial (61). Thus, Akt likely uses other signaling pathways in human melanoma. One potential mechanism of rapamycin resistance in melanoma is through the involvement of rictor, a component of the mTOR complex that is relatively resistant to rapamcyin compared with the raptor complex. A375, a highly tumorigenic spontaneous melanoma cell line, expresses high levels of rictor mRNA. Treatment with an Akt inhibitor resulted in reduction of rictor levels, suggesting that melanoma may be rapamycin resistant, at least in part, because of high rictor expression. High expression of Sirt1 and rictor (62) may be adaptations that tumor cells make in order to survive high ROS expression, and Akt may be the immediate regulator of these proteins. We demonstrated that Akt induced ROS in human melanoma and was likely a major mechanism of Akt-induced oncogenesis in human melanoma. Manipulations that result in increased respiration, either through inhibition of Akt or by inducing synthesis of respiratory enzymes, may have therapeutic benefit in melanoma treatment.
Methods
Cell lines and protein extraction
WM35 radial growth phase melanoma cells were transfected with 40 μg of the plasmid pBABE puro or pBABE PKBDD, which encodes and expresses active Akt in the presence of 40 μl Superfect transfection reagent (Qiagen) in 2 ml complete serumless media (Cellgro; Mediatech Inc.). Both vectors encode puromycin resistance, and cells were selected in 2 μg/ml puromycin and pooled to avoid clonal variation. Expression of the gene was confirmed by Western blot analysis with an antibody specific to phosphorylated Akt. A stable clone of WM35 overexpressing Akt, WM35 PKBDD, was also studied. Cells were grown in RPMI 1640 (Invitrogen) supplemented with a mixture containing 400 μM each of the amino acids (63) alanine, asparagine, aspartic acid, glutamic acid, and proline (Merck) as well as 1 mM glutamine (EMD) plus 10% FCS (Biological Industries) and 0.6% penicillin and 1% streptomycin (Sigma-Aldrich) in 7% CO2 atmosphere (64). Cells were recovered from the plates by trypsin treatment and washed twice with PBS. The recovered cells were lysed in a buffer containing 5 mM Tris-HCl (pH 8.0), 0.5% Triton X-100, 20 mM EDTA, 1 μg/ml pepstatin, and 1 μg/ml leupeptin at 4°C for 10 minutes. After protein extraction, the samples were centrifuged (15,000 g) at 4°C for 5 minutes. The proteins from tumor tissues were obtained by homogenizing the tissue sample in the lysis buffer described above and centrifuging at 2,000 g for 10 minutes at 4°C. The supernatant-containing protein lysate was removed from the tube and used for Western blot analysis. The protein concentration in the supernatants was determined with the Bradford reagent (Bio-Rad Protein Assay) using BSA as standard. Aliquots of the supernatants were stored at –80°C until used. Complex I activity determination was performed as previously described (65). Primary cells from the Cardinal Bernardin Cancer Center at Loyola University were maintained as previously described (66). The A375 human melanoma cell line, obtained from the ATCC, was grown in DMEM with 4 mM l-glutamine adjusted to contain 90% 1.5 g/l sodium bicarbonate and 4.5 g/l glucose and 10% fetal bovine serum at 37°C.
Western blotting
Cellular proteins (10 or 15 μg protein) were fractionated on 10% SDS-PAGE and then transferred onto PVDF membranes (Millipore). Western blotting of the membranes was performed essentially as described previously (29) using the appropriate dilution of various antisera. The antibodies used in this study included anti–β-F1-ATPase (diluted 1:20,000; ref. 46), monoclonal anti-Hsp60 (diluted 1:2,000; SPA 807; Stressgene), polyclonal antibody to JunD (diluted 1:1,000; ab5799; Abcam), polyclonal antibody to phosphorylated JunD (phospho-S255, diluted 1:1,000; ab30631; Abcam), antibody to phosphorylated Akt (phospho-AktS473, diluted 1:1,000; 4058; Cell signaling), monoclonal anti–α-tubulin (diluted 1:2,000; Sigma-Aldrich), and monoclonal anti–glyceraldehyde-3-phosphate dehydrogenase (diluted 1:10,000; Abcam). Secondary horseradish peroxidase–conjugated goat anti-rabbit or anti-mouse antibody (diluted 1:3,000) was used for detection, which was accomplished using an enhanced chemiluminescence detection method (Amersham Biosciences). Quantification of immunoreactive band intensity (in AU) was accomplished using a Kodak DC120 Zoom digital camera and the Quantity One software package (version 4.3.1; Bio-Rad). In order to calculate the cellular expression level of the markers, the band intensity of β-F1-ATPase, Hsp60, and GAPDH was divided by the band intensity of α-tubulin. The β-F1-ATPase/Hsp60 ratio and bioenergetic cellular index (calculated as β-F1-ATPase/Hsp60/GAPDH; ref. 45) were also determined.
Quantitative RT-PCR
Primers for human VEGF were used for real-time RT-PCR as described by Arbiser et al. (28). Quantitative RT-PCR was performed to determine NOX4 levels using primers specific for human NOX4 or for 18S as follows: NOX4 forward, 5'-GTATGTTGCATAACAAGTTTTGG-3'; reverse, 5'-TCAGCTGAAAGACTCTTTATTGT-3'; 18S forward, 5'-GAGTATGGTTGCAAAGCTGAAAC-3'; reverse, 5'-CCAGACAAATCGCTCCACCAAC-3'. cDNA was obtained from RNA by reverse transcription using Advantage RT for PCR according to the manufacturer’s instructions (Clontech). PCR reaction was as follows: 7.3 μl cDNA, 0.1 μl 10 mM forward NOX4 or 18S primer, 0.1 μl 10 mM reverse NOX4 or 18S primer, and 7.5 μl SYBR Green PCR Master Mix (Applied Biosystems). The PCR reaction included an initial cycle of 95°C for 10 minutes, followed by 45 cycles of 95°C for 30 seconds, 56°C for 30 seconds, and 72°C for 30 seconds. Results are reported relative to the mean starting quantities of NOX4 normalized to 18S. The propagation of error was calculated using the following formula:
Equation 1 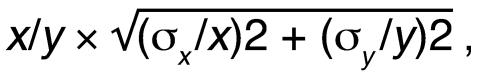
ïομ
where x represents mean NOX4, y represents mean 18S, and s represents standard deviation. Quantitative PCR for VEGF, Sirt1, and rictor was performed using Taqman Gene Expression Assays (Applied Biosystems). For the reaction, 96-well Optical Reaction Plate (ABI PRISM, no. 128; Applied Biosystems) was used. A measure of 2.5 l of template diluted 1:10 in crosslinked water was used in each well, and the experiment was performed in triplicate. VEGF(hs00173626_m1), Sirt1(HS00202021_m1), rictor(AVO3, hs01561908_m1), and corrected for 18S (Hs99999901_s1, Taqman Gene Expression Assay; Applied Biosystems) primers were used along with crosslinked molecular-grade water (Cellgro; Mediatech Inc.) and master mix (TaqMan Fast Universal PCR Master Mix; Applied Biosystems; ref. 2). The reaction was run on the 7900 Applied Biosystems Reader for Absolute Quantification for 96-well plates. Threshold cycle (Ct) values were analyzed by the Ct method, and mean and SEM were calculated.
Determination of glycolysis
Cells were seeded and allowed to grow until reaching 60% confluence in supplemented RPMI 1640 media. In order to determine the rates of glycolysis, the cells were preincubated for 30 minutes with 6 μM oligomycin. After replacing the culture medium supplemented with the inhibitor of the mitochondrial H+-ATP synthase, a 0.1-ml aliquot of the media was taken after 2 hours of incubation. The collected samples were precipitated in 6% perchloric acid, neutralized, and used for the enzymatic determination of lactate as described previously (66).
Dichlorofluorescein diacetate and dihydroethidium assays
Cells were allowed to grow until reaching 80% confluency. Cells were washed once with 5 ml HBSS and digested with 0.5 ml 0.25% trypsin in 2.7 mM EDTA for 2 minutes at room temperature followed by the addition of DMEM without phenol red. Cells were dislodged from the plate by gentle pipetting and transferred to eppendorf tubes. Cells were pelleted in a Hermle Z18M tabletop centrifuge at 300 g for 2 minutes. The media was gently removed from the pellet by pipetting, and cells were resuspended in 1 ml DMEM without phenol red and divided into 2 0.5-ml aliquots. In the first aliquot, 0.5 ml of 4 μM dichlorofluorescein diacetate in DMEM (without phenol red) was added for a final dichlorofluorescein diacetate concentration of 2 μM, and the cells were incubated in the dark on a rotating wheel at room temperature for 45 minutes. In the second aliquot, 0.5 ml of 20 μM dihydroethidium in DMEM (without phenol red) was added for a final dihydroethidium concentration of 10 μM, and the cells were incubated at room temperature in the dark for 30 minutes. Cells were then immediately transferred to ice and analyzed on a flow cytometer (BD Biosciences).
Superoxide dismutase assays
Cells were washed 3 times in PBS (pH 7.0) plus 2.7 mM KCl, 1.5 mM KH2PO4, 8 mM NaHPO4, and 136.9 mM NaCl, scraped from the dishes, and centrifuged at 82 g for 5 minutes. The supernatant was discarded, and the pellet was resuspended in 50 mM PBS (pH 7.8). Cells were sonicated 3 times at 28% amplitude, 30 seconds each time on ice using a Vibra Cell Sonicator with a cup horn (Sonics and Materials Inc.). Protein concentrations were measured by the Bradford method (Bio-Rad; ref. 67) and standardized with BSA. The protein concentrations for the glutathione assay were based on the method described by Lowry (68).
Western blots were performed according to the method described by Laemmli (69). Total cellular proteins were electrophoresed in a 12.5% SDS polyacrylamide gel for MnSOD and CuZnSOD and 8% SDS polyacrylamide gel for catalase (CAT). Proteins were transferred onto nitrocellulose membranes (Whatman Schleicher & Schuell), blocked in 5% dry milk in TTBS (0.01 M Tris, 0.15 M NaCl buffer, pH 8.0, and 0.1% Tween 20) at room temperature for 1 hour. After washing 3 times with TTBS, the membrane was incubated with primary antibody (diluted 1:1,000) in TTBS at room temperature for 1 hour for MnSOD, CuZnSOD or overnight at 4°C for CAT. After washing the membrane 3 times with TTBS, the blots were incubated with goat anti-rabbit IgG conjugated with horseradish peroxidase diluted 1:10,000 in TTBS for 1 hour. After incubating the membrane in TTBS for 40 minutes, the washed blot was treated with chemiluminescence (ECL Plus) Western blot detection solution (Amersham Biosciences) and exposed to X-ray film (BioMaxMR; Eastman Kodak Company).
The SOD activity gel assay is based on the inhibition of the reduction of nitroblue tetrazolium (NBT) by SOD (70). Equal amounts of protein (100 μg) from different samples were subjected to 12% native polyacrylamide gels by electrophoresis in nondenaturing running buffer (pH 8.3). For SOD band visualization, after electrophoresis the gel was incubated in 2.43 mM NBT, 28 μM riboflavin-5-phosphate, 28 mM tetramethylethylenediamine, and 0.75 mM NaCN in double distilled H2O for 20 minutes under dark conditions. The gels were illuminated under a fluorescent light until achromatic SOD bands and a satisfactory blue background appeared.
The CAT activity gel assay was carried out as previously described by Sun et al. (71). Equal amounts of protein (100 μg) from different samples were subjected to 8% native polyacrylamide gels by electrophoresis in nondenaturing running buffer (pH 8.3). For CAT band visualization, after electrophoresis the gel was incubated in 0.003% hydrogen peroxide for 10 minutes and then staining with 2% ferric chloride and 2% potassium ferricyanide until achromatic CAT bands began to form.
Mitochondrial DNA analysis
DNA isolation and PCR amplification.
Genomic DNA was isolated from melanoma cell culture specimens of WM35 and WM35 PKBDD using Clontech’s NucleoSpin Tissue Kit. In addition, samples of 13 normal melanocytes, 7 melanomas, and 16 melanoma cell lines were analyzed. The entire mtDNA from both cell lines was amplified by 17 pairs of oligonucleotide primers (Sigma-Aldrich), producing 17 overlapping amplicons. PCR reactions were performed in 50-μl volumes containing 10 ng of template DNA; 0.5 μM of the forward and reverse oligonucleotide primers (for complete list of primer pairs, see Supplemental Table 1 of ref. 72); 0.2 mM of each dNTP (New England Biolabs Inc.); 1.5 mM MgCl2; magnesium-free buffer (50 mM KCl and 10 mM Tris-HCl); and 2.5 U Taq DNA polymerase. Each sample was subjected to the following amplification conditions for 32 cycles: an initial denaturation at 95°C for 5 minutes, 45 seconds’ denaturation at 95°C, 1 minute at the annealing temperature of 57°C, and elongation at 72°C for 2 minutes. This was followed by a 5-minute final extension at 72°C and subsequent cooling to 4°C. For analysis, 5 μl of each sample was fractionated on a 2% agarose gel with 0.05 μg/ml ethidium bromide. PCR products were directly visualized with UV fluorescence. Of the remaining 45 μl of each PCR product, 22.5 μl was prepared as described below and used for sequencing, while the other 22.5 μl was digested and prepared for TGCE analysis.
TGCE analysis.
The 17 PCR amplicons for each of the WM35 and WM35 PKBDD cell lines were subjected to restriction endonuclease digestion as described previously (see Supplemental Table 1 of ref. 72). The digested products were visualized by gel electrophoresis on a 2% agarose gel with 0.05 μg/ml ethidium bromide. The intent was to use TGCE as a screening method to detect mtDNA sequence regions that contained nucleotide differences between WM35 cells and WM35 PKBDD cells. Each of the 17 digested PCR amplicons from the WM35 template was mixed with an equal quantity of the corresponding WM35 PKBDD PCR amplicon. In a 10-μl total reaction volume, the samples were initially denatured at 95°C for 3 minutes, followed by annealing via a stepwise reduction in temperature as previously described (72).
Automated TGCE analysis was carried out using the Reveal Mutation Discovery System (SpectruMedix). This system separated the restriction digests of the 17 amplicons from WM35 and WM35 PKBDD cells into homoduplexes in identical sequence regions and heteroduplexes in sequence regions mutated between the 2 cell lines. The parameters for the thermal ramp, injection, capillary length, and gel formation were as recommended by the manufacturer and have been enumerated previously (72). The resulting data were analyzed by Revelation Mutation Discovery software (version 2.4; SpectruMedix).
Sequence analysis.
Suspected nucleotide changes between WM35 and WM35 PKBDD amplicons identified by TGCE analysis were quantified and clarified by direct sequence analysis. Equal amounts (22.5 μl) of each of the PCR products representing the original 17 overlapping amplicons (undigested) were prepared for sequencing as follows: 0.25 μl exonuclease I, 0.5 μl shrimp alkaline phosphatase (SAP), and 0.25 μl of SAP buffer were added to each of the PCR products, followed by incubation for 30 minutes at 37°C, 30 minutes at 80°C, and cooling to 4°C. The PCR products were then sequenced using an Applied Biosystems 3100 automated DNA sequencer by the chain-termination method.
Computational analyses.
The 17 overlapping sequences were used in the manual creation of contigs covering over 98% of the entire mtDNA sequences for WM35 and WM35 PKBDD cells. These 2 complete sequences were aligned with the Revised Cambridge Reference mitochondrial sequence (Mitomap; http://www.mitomap.org) using the MegAlign module of Lasergene software (version 6 for Windows; DNASTAR), with the Clustal V algorithm and the default settings, followed by manual refinement. Mitoanalyzer was used to determine the amino acid changes that accompanied corresponding nucleotide changes between WM35 and WM35 PKBDD. Homologene (http://www.ncbi.nlm.nih.gov/HomoloGene) was used to attain access to homologous eukaryotic sequences for each mitochondrial gene that was found to be affected by nonsynonymous amino acid changes. These sequences were aligned and analyzed for evolutionary conservation via T-Coffee software (35). Finally, Pfam (version 16.0; ref. 73) domain structures were used to determine the specific domains of the protein’s 3-dimensional conformation that are involved in specific amino acid changes.
Immunofluorescence
Cells on glass coverslips were rinsed quickly in ice-cold PBS, fixed in freshly prepared 4% paraformaldehyde in PBS for 10 minutes at room temperature, permeabilized in 0.05% Triton X-100 in PBS for 5 minutes, and rinsed sequentially in PBS, 50 mmol/l NH4Cl, and PBS for 10 minutes each. After incubation for 1 hour in blocking buffer (PBS plus 3% BSA), cells were incubated with primary antibody — goat polyclonal anti-p47phox (sc-7660; Santa Cruz Biotechnology Inc.), rabbit polyclonal anti-p47phox (sc14015; Santa Cruz Biotechnology Inc.), mouse monoclonal anti-p47phox (610354; BD Biosciences), rabbit polyclonal anti–p47phox R360, or mouse monoclonal anti–p47phox 43.12 (both kind gifts of M. Quinn, Montana State University, Bozeman, Montana, USA) — for 1 hour at room temperature, rinsed in PBS/BSA, and then incubated in either FITC-conjugated goat anti-rabbit or goat anti-mouse IgG (Jackson ImmunoResearch Laboratories Inc.) for 1 hour at room temperature. Cells on coverslips were mounted onto glass slides in Vectashield (Vector Laboratories) and examined using the 488-nm line of the argon ion laser with a 515/30 nm band pass filter in the confocal laser scanning imaging system (MRC-1024; Bio-Rad) or using a Zeiss Axioskop microscope equipped with fluorescence optics and an AxioCam CCD camera.
In vivo growth
Control and activated Akt–expressing WM35 cells (1 × 106 cells per group) were injected subcutaneously into 5- to 6-week-old male nude mice. Tumor volume was calculated as 0.52(w2 × l), where w represents the shortest dimension. In addition, pBabe PKB or control pBabe puro was transiently transfected using lipofectamine into WM35 to derive cells that had not been extensively cultured after PKB overexpression. Both vectors encode puromycin resistance, and cells were selected in 2 μg/ml puromycin and pooled 72 hours after transfection. Subsequently, 6 × 105 transfected cells were pooled and then injected subcutaneously in the presence of 0.3 ml Matrigel/mouse, as well as an equivalent number of vector control cells.
Statistics
All experiments were performed in triplicate. Statistical analyses were performed by 2-tailed Student’s t test for nonpaired samples. A P value less than 0.05 was considered statistically significant.
Supplementary Material
Acknowledgments
J.L. Arbiser was supported by a Veterans Administration Merit Award, NIH grant R01 AR02030, and grants from the Jamie Rabinowitch-Davis Foundation and the Minsk Foundation. B. Nickoloff was supported by NIH grants CA59327 and CA27502. J.M. Cuezva was supported by grant BMC2001-0710 from the Ministerio de Ciencia y Tecnología of Spain. B. Govindarajan was supported by a Dermatology Foundation Career Development Award.
Footnotes
Nonstandard abbreviations used: CAT, catalase; COII, cytochrome oxidase II; mtDNA, mitochondrial DNA; ND, NADH dehydrogenase; TGCE, temperature gradient capillary electrophoresis.
Conflict of interest: The authors have declared that no conflict of interest exists.
Citation for this article: J. Clin. Invest. 117:719–729 (2007). doi:10.1172/JCI30102
References
- 1.Cohen C., et al. Mitogen-actived protein kinase activation is an early event in melanoma progression. Clin. Cancer Res. 2002;8:3728–3733. [PubMed] [Google Scholar]
- 2.Govindarajan B., et al. Malignant transformation of melanocytes to melanoma by constitutive activation of mitogen-activated protein kinase kinase (MAPKK) signaling. J. Biol. Chem. 2003;278:9790–9795. doi: 10.1074/jbc.M212929200. [DOI] [PubMed] [Google Scholar]
- 3.Majewski N., et al. Hexokinase-mitochondria interaction mediated by Akt is required to inhibit apoptosis in the presence or absence of Bax and Bak. Mol. Cell. 2004;16:819–830. doi: 10.1016/j.molcel.2004.11.014. [DOI] [PubMed] [Google Scholar]
- 4.Rathmell J.C., et al. Akt-directed glucose metabolism can prevent Bax conformation change and promote growth factor-independent survival. Mol. Cell. Biol. 2003;23:7315–7328. doi: 10.1128/MCB.23.20.7315-7328.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Peli J., et al. Oncogenic Ras inhibits Fas ligand-mediated apoptosis by downregulating the expression of Fas. EMBO J. 1999;18:1824–1831. doi: 10.1093/emboj/18.7.1824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Plas D.R., Thompson C.B. Akt activation promotes degradation of tuberin and FOXO3a via the proteasome. J. Biol. Chem. 2003;278:12361–12366. doi: 10.1074/jbc.M213069200. [DOI] [PubMed] [Google Scholar]
- 7.Arbiser J., et al. Solenopsin, the alkaloidal component of the fire ant (Solenopsis invicta), is a naturally occurring inhibitor of phosphatidylinositol-3-kinase signaling and angiogenesis. Blood. 2006;109:560–565. doi: 10.1182/blood-2006-06-029934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dai D.L., Martinka M., Li G. Prognostic significance of activated Akt expression in melanoma: a clinicopathologic study of 292 cases. J. Clin. Oncol. 2005;23:1473–1482. doi: 10.1200/JCO.2005.07.168. [DOI] [PubMed] [Google Scholar]
- 9.Edlundh-Rose E., et al. NRAS and BRAF mutations in melanoma tumours in relation to clinical characteristics: a study based on mutation screening by pyrosequencing. Melanoma Res. 2006;16:471–478. doi: 10.1097/01.cmr.0000232300.22032.86. [DOI] [PubMed] [Google Scholar]
- 10.Landi M.T., et al. MC1R germline variants confer risk for BRAF-mutant melanoma. Science. 2006;313:521–522. doi: 10.1126/science.1127515. [DOI] [PubMed] [Google Scholar]
- 11.Curtin J.A. Distinct sets of genetic alterations in melanoma. N. Engl. J. Med. 2005;353:2135–2147. doi: 10.1056/NEJMoa050092. [DOI] [PubMed] [Google Scholar]
- 12.Pollock P.M., Meltzer P.S. A genome-based strategy uncovers frequent BRAF mutations in melanoma. Cancer Cell. 2002;2:5–7. doi: 10.1016/s1535-6108(02)00089-2. [DOI] [PubMed] [Google Scholar]
- 13.Tsao H., Goel V., Wu H., Yang G., Haluska F.G. Genetic interaction between NRAS and BRAF mutations and PTEN/MMAC1 inactivation in melanoma. J. Invest. Dermatol. 2004;122:337–341. doi: 10.1046/j.0022-202X.2004.22243.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Haluska F.G., et al. Genetic alterations in signaling pathways in melanoma. Clin. Cancer Res. 2006;12(Suppl.):2301s–2307s. doi: 10.1158/1078-0432.CCR-05-2518. [DOI] [PubMed] [Google Scholar]
- 15.Liu Z.J., et al. Notch1 signaling promotes primary melanoma progression by activating mitogen-activated protein kinase/phosphatidylinositol 3-kinase-Akt pathways and up-regulating N-cadherin expression. Cancer Res. 2006;66:4182–4190. doi: 10.1158/0008-5472.CAN-05-3589. [DOI] [PubMed] [Google Scholar]
- 16.Stahl J.M., et al. Deregulated Akt3 activity promotes development of malignant melanoma. Cancer Res. 2004;64:7002–7010. doi: 10.1158/0008-5472.CAN-04-1399. [DOI] [PubMed] [Google Scholar]
- 17.Meyskens F.L., Jr., Buckmeier J.A., McNulty S.E., Tohidian N.B. Activation of nuclear factor-kappa B in human metastatic melanoma cells and the effect of oxidative stress. Clin. Cancer Res. 1999;5:1197–1202. [PubMed] [Google Scholar]
- 18.Szatrowski T.P., Nathan C.F. Production of large amounts of hydrogen peroxide by human tumor cells. Cancer Res. 1991;51:794–798. [PubMed] [Google Scholar]
- 19.Irani K., et al. Mitogenic signaling mediated by oxidants in Ras-transformed fibroblasts. Science. 1997;275:1649–1652. doi: 10.1126/science.275.5306.1649. [DOI] [PubMed] [Google Scholar]
- 20.Finkel T. Redox-dependent signal transduction. FEBS Lett. 2000;476:52–54. doi: 10.1016/s0014-5793(00)01669-0. [DOI] [PubMed] [Google Scholar]
- 21.Archer H., Bar-Sagi D. Ras and Rac as activators of reactive oxygen species (ROS). Methods Mol. Biol. 2002;189:67–73. doi: 10.1385/1-59259-281-3:067. [DOI] [PubMed] [Google Scholar]
- 22.Nagata Y., et al. PTEN activation contributes to tumor inhibition by trastuzumab, and loss of PTEN predicts trastuzumab resistance in patients. Cancer Cell. 2004;6:117–127. doi: 10.1016/j.ccr.2004.06.022. [DOI] [PubMed] [Google Scholar]
- 23.Hu M.C.T., et al. I kappa B kinase promotes tumorigenesis through inhibition of forkhead FOXO3a. Cell. 2004;117:225–237. doi: 10.1016/s0092-8674(04)00302-2. [DOI] [PubMed] [Google Scholar]
- 24.Yang J.Q., et al. V-Ha-Ras overexpression induces superoxide production and alters levels of primary antioxidant enzymes. Antioxid. Redox. Signal. 2001;3:697–709. doi: 10.1089/15230860152543032. [DOI] [PubMed] [Google Scholar]
- 25.Yang J.Q., et al. v-Ha-ras mitogenic signaling through superoxide and derived reactive oxygen species. Mol. Carcinog. 2002;33:206–218. doi: 10.1002/mc.10037. [DOI] [PubMed] [Google Scholar]
- 26.Suh Y.A., et al. Cell transformation by the superoxide-generating oxidase Mox1. Nature. 1999;401:79–82. doi: 10.1038/43459. [DOI] [PubMed] [Google Scholar]
- 27.Arnold R.S., et al. Hydrogen peroxide mediates the cell growth and transformation caused by the mitogenic oxidase Nox1. Proc. Natl. Acad. Sci. U. S. A. 2001;98:5550–5555. doi: 10.1073/pnas.101505898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Arbiser J.L., et al. Reactive oxygen generated by Nox1 triggers the angiogenic switch. Proc. Natl. Acad. Sci. U. S. A. 2002;99:715–720. doi: 10.1073/pnas.022630199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chudnovsky Y., Adams A.E., Robbins P.B., Lin Q., Khavari P.A. Use of human tissue to assess the oncogenic activity of melanoma-associated mutations. Nat. Genet. 2005;37:745–749. doi: 10.1038/ng1586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yang S., McNulty S., Meyskens F.L., Jr. During human melanoma progression AP-1 binding pairs are altered with loss of c-Jun in vitro. Pigment Cell Res. 2004;17:74–83. doi: 10.1046/j.1600-0749.2003.00114.x. [DOI] [PubMed] [Google Scholar]
- 31.Ronai Z., et al. ATF2 confers radiation resistance to human melanoma cells. Oncogene. 1998;16:523–531. doi: 10.1038/sj.onc.1201566. [DOI] [PubMed] [Google Scholar]
- 32.Esposito L.A., Melov S., Panov A., Cottrell B.A., Wallace D.C. Mitochondrial disease in mouse results in increased oxidative stress. Proc. Natl. Acad. Sci. U. S. A. 1999;96:4820–4825. doi: 10.1073/pnas.96.9.4820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Brown M.D., et al. Novel mtDNA mutations and oxidative phosphorylation dysfunction in Russian LHON families. Hum. Genet. 2001;109:33–39. doi: 10.1007/s004390100538. [DOI] [PubMed] [Google Scholar]
- 34.Sherer T.B., et al. An in vitro model of Parkinson’s disease: Linking mitochondrial impairment to altered alpha-synuclein metabolism and oxidative damage. J. Neurosci. 2002;22:7006–7015. doi: 10.1523/JNEUROSCI.22-16-07006.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Notredame C., Higgins D.G., Heringa J.2000. . T-Coffee: a novel method for fast and accurate multiple sequence alignment. J. Mol. Biol. 302205–217. [DOI] [PubMed] [Google Scholar]
- 36.Hsu M.Y., et al. Adenoviral gene transfer of beta3 integrin subunit induces conversion from radial to vertical growth phase in primary human melanoma. Am. J. Pathol. 1998;153:1435–1442. doi: 10.1016/s0002-9440(10)65730-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dhawan P., Singh A.B., Ellis D.L., Richmond A. Constitutive activation of Akt/protein kinase B in melanoma leads to up-regulation of nuclear factor-kappa B and tumor progression. Cancer Res. 2002;62:7335–7342. [PubMed] [Google Scholar]
- 38.Bhoumik A., et al. An ATF2-derived peptide sensitizes melanomas to apoptosis and inhibits their growth and metastasis. J. Clin. Invest. 2002;110:643–650. doi: 10.1172/JCI200216081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Datta R., et al. Involvement of reactive oxygen intermediates in the induction of c-jun gene transcription by ionizing radiation. Biochemistry. 1992;31:8300–8306. doi: 10.1021/bi00150a025. [DOI] [PubMed] [Google Scholar]
- 40.Luo J., et al. Negative control of p53 by Sir2alpha promotes cell survival under stress. Cell. 2001;107:137–148. doi: 10.1016/s0092-8674(01)00524-4. [DOI] [PubMed] [Google Scholar]
- 41.Vaziri H., et al. hSIR2(SIRT1) functions as an NAD-dependent p53 deacetylase. Cell. 2001;107:149–159. doi: 10.1016/s0092-8674(01)00527-x. [DOI] [PubMed] [Google Scholar]
- 42.Warburg O., Geissler A.W., Lorenz S. Genesis of cancer metabolism through vitamin B1 deficiency (thiamine deficiency) [In German]. Z. Naturforsch. B. 1970;25:332–333. [PubMed] [Google Scholar]
- 43.Elstrom R.L., et al. Akt stimulates aerobic glycolysis in cancer cells. Cancer Res. 2004;64:3892–3899. doi: 10.1158/0008-5472.CAN-03-2904. [DOI] [PubMed] [Google Scholar]
- 44.Cuezva J.M., et al. Mitochondrial biogenesis in the liver during development and oncogenesis. J. Bioenerg. Biomembr. 1997;29:365–377. doi: 10.1023/a:1022450831360. [DOI] [PubMed] [Google Scholar]
- 45.Cuezva J.M., et al. The bioenergetic signature of cancer: A marker of tumor progression. Cancer Res. 2002;62:6674–6681. [PubMed] [Google Scholar]
- 46.Cuezva J.M., et al. The bioenergetic signature of lung adenocarcinomas is a molecular marker of cancer diagnosis and prognosis. Carcinogenesis. 2004;25:1157–1163. doi: 10.1093/carcin/bgh113. [DOI] [PubMed] [Google Scholar]
- 47.Jeong D.W., Kim T.S., Cho I.T., Kim I.Y. Modification of glycolysis affects cell sensitivity to apoptosis induced by oxidative stress and mediated by mitochondria. Biochem. Biophys. Res. Commun. 2004;313:984–991. doi: 10.1016/j.bbrc.2003.12.033. [DOI] [PubMed] [Google Scholar]
- 48.Lambeth J.D., Cheng G., Arnold R.S., Edens W.A. Novel homologs of gp91phox. Trends Biochem. Sci. 2000;25:459–461. doi: 10.1016/s0968-0004(00)01658-3. [DOI] [PubMed] [Google Scholar]
- 49.Brar S.S., et al. An NAD(P)H oxidase regulates growth and transcription in melanoma cells. Am. J. Physiol. Cell Physiol. 2002;282:C1212–C1224. doi: 10.1152/ajpcell.00496.2001. [DOI] [PubMed] [Google Scholar]
- 50.Petros J.A., et al. mtDNA mutations increase tumorigenicity in prostate cancer. Proc. Natl. Acad. Sci. U. S. A. 2005;102:719–724. doi: 10.1073/pnas.0408894102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Betarbet R., et al. Chronic systemic pesticide exposure reproduces features of Parkinson’s disease. Nat. Neurosci. 2000;3:1301–1306. doi: 10.1038/81834. [DOI] [PubMed] [Google Scholar]
- 52.Bijur G.N., Jope R.S. Rapid accumulation of Akt in mitochondria following phosphatidylinositol 3-kinase activation. J. Neurochem. 2003;87:1427–1435. doi: 10.1046/j.1471-4159.2003.02113.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Geiszt M., Kopp J.B., Varnai P., Leto T.L. Identification of Renox, an NAD(P)H oxidase in kidney. Proc. Natl. Acad. Sci. U. S. A. 2000;97:8010–8014. doi: 10.1073/pnas.130135897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Govindarajan B., et al. Reactive oxygen-induced carcinogenesis causes hypermethylation of p16(Ink4a) and activation of MAP kinase. Mol. Med. 2002;8:1–8. [PMC free article] [PubMed] [Google Scholar]
- 55.Ito K., et al. Regulation of oxidative stress by ATM is required for self-renewal of haematopoietic stem cells. Nature. 2004;431:997–1002. doi: 10.1038/nature02989. [DOI] [PubMed] [Google Scholar]
- 56.Soengas M.S., et al. Inactivation of the apoptosis effector Apaf-1 in malignant melanoma. Nature. 2001;409:207–211. doi: 10.1038/35051606. [DOI] [PubMed] [Google Scholar]
- 57.Cardone M.H., et al. Regulation of cell death protease caspase-9 by phosphorylation. Science. 1998;282:1318–1321. doi: 10.1126/science.282.5392.1318. [DOI] [PubMed] [Google Scholar]
- 58.Hersey P., Zhang X.D. How melanoma cells evade trail-induced apoptosis. Nat. Rev. Cancer. 2001;1:142–150. doi: 10.1038/35101078. [DOI] [PubMed] [Google Scholar]
- 59.Majewski N., Nogueira V., Robey R.B., Hay N. Akt inhibits apoptosis downstream of BID cleavage via a glucose-dependent mechanism involving mitochondrial hexokinases. Mol. Cell. Biol. 2004;24:730–740. doi: 10.1128/MCB.24.2.730-740.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Bedogni B., et al. The hypoxic microenvironment of the skin contributes to Akt-mediated melanocyte transformation. Cancer Cell. 2005;8:443–454. doi: 10.1016/j.ccr.2005.11.005. [DOI] [PubMed] [Google Scholar]
- 61.Margolin K., et al. CCI-779 in metastatic melanoma - A phase II trial of the California Cancer Consortium. Cancer. 2005;104:1045–1048. doi: 10.1002/cncr.21265. [DOI] [PubMed] [Google Scholar]
- 62.Sarbassov D.D., et al. Rictor, a novel binding partner of mTOR, defines a rapamycin-insensitive and raptor-independent pathway that regulates the cytoskeleton. Curr. Biol. 2004;14:1296–1302. doi: 10.1016/j.cub.2004.06.054. [DOI] [PubMed] [Google Scholar]
- 63.Liang J., et al. PKB/Akt phosphorylates p27, impairs nuclear import of p27 and opposes p27-mediated G1 arrest. Nat. Med. 2002;8:1153–1160. doi: 10.1038/nm761. [DOI] [PubMed] [Google Scholar]
- 64.Niers L., van den Heuvel L., Trijbels F., Sengers R., Smeitink J. Prerequisites and strategies for prenatal diagnosis of respiratory chain deficiency in chorionic villi. J. Inherit. Metab. Dis. 2003;26:647–658. doi: 10.1023/b:boli.0000005605.57420.b4. [DOI] [PubMed] [Google Scholar]
- 65.Qin J.Z., et al. Proteasome inhibitors trigger NOXA-mediated apoptosis in melanoma and myeloma cells. Cancer Res. 2005;65:6282–6293. doi: 10.1158/0008-5472.CAN-05-0676. [DOI] [PubMed] [Google Scholar]
- 66.Cuezva J.M., Burkett E.S., Kerr D.S., Rodman H.M., Patel M.S. The newborn of diabetic rat. 1. Hormonal and metabolic changes in the postnatal period. Pediatr. Res. 1982;16:632–637. doi: 10.1203/00006450-198208000-00009. [DOI] [PubMed] [Google Scholar]
- 67.Bradford M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976;72:248–254. doi: 10.1006/abio.1976.9999. [DOI] [PubMed] [Google Scholar]
- 68.Lowry O.H., Rosebrough N.J., Farr A.L., Randall R.J. Protein measurement with the Folin phenol reagent. J. Biol. Chem. 1951;193:265–275. [PubMed] [Google Scholar]
- 69.Laemmli U.K. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 70.Beauchamp C., Fridovich I. Superoxide dismutase: improved assays and an assay applicable to acrylamide gels. Anal. Biochem. 1971;44:276–287. doi: 10.1016/0003-2697(71)90370-8. [DOI] [PubMed] [Google Scholar]
- 71.Sun Y., Elwell J.H., Oberley L.W.1988. . A simultaneous visualization of the antioxidant enzymes glutathione-peroxidase and catalase on polyacrylamide gels. Free Radic. Res. Commun. 567–75. [DOI] [PubMed] [Google Scholar]
- 72.Girald-Rosa W., Vieugels R.A., Musiek A.C., Sligh J.E. High-throughput mitochondrial genome screening method for nonmelanoma skin cancer using multiplexed temperature gradient capillary electrophoresis. Clin. Chem. 2005;51:305–311. doi: 10.1373/clinchem.2004.040311. [DOI] [PubMed] [Google Scholar]
- 73.Finn R.D., et al. Pfam: clans, web tools and services. Nucl. Acids Res. 2006;34(Database issue):D247–D251. doi: 10.1093/nar/gkj149. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.