

Abstract
Dosing of antibacterial agents is generally based on point estimates of the effect, even though bacteria exposed to antibiotics show complex kinetic behaviors. The use of the whole time course of the observed effects would be more advantageous. The aim of the present study was to develop a semimechanistic pharmacokinetic (PK)/pharmacodynamic (PD) model characterizing the events seen in a bacterial system when it is exposed to antibacterial agents with different mechanisms of action. Time-kill curve experiments were performed with a strain of Streptococcus pyogenes exposed to a wide range of concentrations of the following antibiotics: benzylpenicillin, cefuroxime, erythromycin, moxifloxacin, and vancomycin. Bacterial counts were monitored with frequent sampling during the experiment. A simultaneous fit of all data was accomplished. The degradation of the drugs was monitored and corrected for in the model, and a link model was used to account for an effect delay. In the final PK/PD model, the total bacterial population was divided into two subpopulations: one growing drug-susceptible population and one resting insusceptible population. The drug effect was included as an increase of the killing rate of bacteria in the susceptible state, according to a maximum-effect (Emax) model. An internal model validation showed that the model was robust and had good predictability. In conclusion, for all drugs, the final PK/PD model successfully described bacterial growth and killing kinetics when the bacteria were exposed to different antibiotic concentrations. The semimechanistic model that was developed might, after further refinement, serve as a tool for the development of optimal dosing strategies for antibacterial agents.
The MIC is the most commonly used parameter to describe the efficacy of an antibacterial agent against a bacterial strain. This is an in vitro measure reflecting the efficacy of a constant antibiotic exposure to a specified bacterial inoculum after an incubation period of 16 to 20 h (19). The MIC is an estimate of the susceptibility of a bacterial strain to an antibiotic that can guide the choice of appropriate antibiotic treatment in the clinical setting. However, it is not an optimal pharmacodynamic (PD) marker since it reflects only a point estimate of the effect and does not take the time course of the effect into account. Nevertheless, the pharmacokinetic (PK)/PD relationship for antibiotics has generally been characterized by using point estimates of the pharmacodynamics (e.g., the bacterial load after 24 h of exposure) and the pharmacokinetics. This approach has led to the classification of the antibacterial effect being dependent either on the antibiotic exposure (the maximum concentration in serum/MIC or the area under the concentration-time curve/MIC) or on the time that the antibiotic concentration is kept above the MIC (4, 18). The design of dosing schedules may, however, be further optimized if it is based on models that take the whole time course of the PK/PD relation, i.e., the time course of both the drug concentration and the bacterial concentration, into consideration.
The PK/PD relationship for antibacterial drugs has been extensively studied over the years (6, 7, 21). Due to the complexity of the in vivo situation, in which the pharmacokinetics of the drug, the kinetic behavior of the bacteria, and the response of the infected host are integrated, in vivo data have so far supported only rather simple PK/PD models. In vitro studies that use time-kill curve experiments and in vitro kinetic models with the ability to simulate different concentration-time profiles offer an attractive complement to in vivo studies (8, 14). Not only are in vitro studies easier to perform, but they also allow more flexibility in the study design and produce results that are unaffected by factors that contribute to the pharmacodynamic variability in vivo, such as drug disposition, disease burden, immune defense, and bacterial heterogeneity. Data from in vitro studies have been used to support more or less complex semimechanistic PK/PD models that describe the time course of the effects of antibiotics in vitro (12, 16, 17, 20, 23-25).
PK/PD models are used to describe and compare the efficacies of different drugs and to aid in the development of optimal or improved dosing strategies. This requires the model to show good predictability even when it is applied to conditions other than those studied during model development. Incorporating prior knowledge of and experience with the system studied into the model-building process, and thereby creating mechanistically based PK/PD models, may increase the predictability of the model (15). It is well known that bacteria show different growth phases and that antibiotic-induced killing often shows an initial phase with rapid killing, followed by a decline in the killing rate with time. Until recently, little has been known about the mechanisms underlying this phenomenon. Increased knowledge regarding the production and the nature of persister cells, i.e., cells with reduced growth rates and reduced antibiotic susceptibilities, could aid in the development of more mechanism-based PK/PD models (1, 11). Mechanistically based PK/PD models aim to describe the biological system studied and the effects that drugs impose on the system separately from each other. In order to obtain the necessary resolution between bacterium-specific parameters and drug-specific parameters, it might be advantageous to simultaneously fit a PK/PD model to data for several antibiotics of different classes on the same bacterial system rather than to fit the data separately, as has been done previously.
The aim of the present study was to develop a general PK/PD model that incorporates mechanistic information to describe the effects of several antibiotics with different mechanisms of action on a bacterial system. Data from time-kill curve studies of Streptococcus pyogenes exposed to a wide range of concentrations of five antibiotics (benzylpenicillin, cefuroxime, erythromycin, moxifloxacin, and vancomycin) with frequent sampling for bacterial count measurements were used for model development. The model performance was validated with internal cross-validation and case deletion diagnostics.
MATERIALS AND METHODS
Bacteria and media.
S. pyogenes group A M12 strain NCTC (National Collection of Type Cultures) P1800 was used as the test organism in all experiments. The bacteria were stored at −80°C and were kept on blood agar plates (Colombia agar base with 5% horse blood) between experiments. For the time-kill curve experiments the bacteria were grown in Todd-Hewitt broth at 35°C and were seeded on blood agar plates for viable count measurements.
Antibiotics and MIC determinations.
The following five drugs were evaluated: benzylpenicillin (Bensylpenicillin; AstraZeneca), cefuroxime (Zinacef; GlaxoSmithKline), erythromycin (Abboticin; Abbott), moxifloxacin (Bayer), and vancomycin (Vancomycin Abbott; Electra-Box Pharma). Stock solutions were prepared prior to each experiment by dissolving the antibiotic in sterile distilled water (benzylpenicillin, cefuroxime, erythromycin, and moxifloxacin) or sterile phosphate-buffered saline (vancomycin) to a concentration of 10 mg/ml. The desired concentrations were obtained after appropriate dilution in Todd-Hewitt broth. The MICs for benzylpenicillin, erythromycin, moxifloxacin and vancomycin were determined by Etest (Biodisk AB, Solna, Sweden) on Iso-Sensitest agar, according to the manufacturer's instructions. For cefuroxime the MIC was determined by the macrodilution technique, according to the instructions of the Clinical and Laboratory Standards Institute (formerly NCCLS) (19). The MIC determinations were made in triplicate on separate occasions.
Time-kill curve experiments.
A total of 135 time-kill curve experiments were conducted for the study. The experiments were performed in 10-ml glass tubes with 4 ml Todd-Hewitt broth. Bacteria from a 6-h logarithmic-growth-phase culture were added to obtain a start inoculum of 106 CFU/ml. Antibiotics were added to obtain concentrations corresponding to 0.25, 0.5, 1, 2, 4, 16, and 64× MIC. To fully cover the effective concentration range, additional experiments with concentrations corresponding to 0.0625 and 0.125× MIC for benzylpenicillin, cefuroxime, and erythromycin and 1.5× MIC for vancomycin were also performed. The tubes were placed in sand to minimize temperature fluctuations during the experiment and were incubated at 35°C for 24 h. Samples for viable count determinations were taken frequently during the experiment (0, 1, 2, 4, 6, 9, 12, 15, 18, and 24 h after the start of the experiment). Each sample was diluted in series and spread on two to four blood agar plates. The numbers of CFU were counted after incubation at 35°C in 5% CO2 for 18 to 24 h. The limit of detection (LOD) was 10 CFU/ml. Drug carryover was assessed by visual inspection of the distribution of colonies on the plates. Each time-kill experiment was carried out in duplicate or triplicate on separate occasions. At least one growth control, i.e., an experiment performed without addition of antibiotics, was included each day. To fully characterize the growth dynamics, starting inocula lower than 106 CFU/ml were also used for the growth control experiments.
Determination of antibiotic concentration.
The antibiotic concentration was measured during the experiment to check for drug degradation. Determinations of effective antibiotic concentrations were made by the conventional microbiological agar diffusion method. Plates with Iso-Sensitest agar were seeded with a suspension of Bacillus stearothermophilus ATCC 3032 spores for determination of the benzylpenicillin and cefuroxime concentrations, Sarcina lutea ATCC 9341 for determination of the erythromycin concentration, Escherichia coli MB 3804 for determination of the moxifloxacin concentration, and Bacillus pumilis ATCC 14579 for determination of the vancomycin concentration. Antibiotic standards and the samples from the experiments were applied to agar wells; and the plates were incubated overnight at 35°C for plates seeded with Sarcina lutea, E. coli, and Bacillus pumilis or at 56°C for plates seeded with Bacillus stearothermophilus. All assays were performed in triplicate, and the correlation coefficient for the standard curves was always ≥0.99.
Antibiotic stability.
The stability of the antibiotics during incubation was tested in separate time-kill curve experiments. The stability study was conducted in two steps. The aim of the first step was to identify if degradation of the respective drugs occurred. If degradation occurred, a second step was conducted with the aim of characterizing the rate of degradation. In the first step, the zone diameters from sampling at the start of the experiment (0 h) and at the end of the experiment (24 h) were compared by Student's t test of log-transformed data. In this step, erythromycin, moxifloxacin, and vancomycin showed no signs of degradation during 24 h of incubation (P > 0.05). However, benzylpenicillin and cefuroxime showed statistically significant degradation (P < 0.001), and, hence, a larger stability study with more frequent sampling (0, 8, 16, and 24 h) was conducted. The concentration in the samples was analyzed by the microbiological agar diffusion method, and the degradation rate constants were determined by assuming that degradation followed first-order kinetics. For all antibiotics the stability was checked at two different concentrations.
Pharmacokinetic model.
In time-kill curve experiments, a bacterial system is exposed to constant concentrations of antibiotics. However, some degradation of the drug might occur during the incubation period; and a first-order degradation rate constant (kdeg), as determined from the stability experiments, was included in the PK model. The presence of a time delay between drug addition and the observed effect was explored by introducing an effect compartment (Ce), with the effect delay characterized by a first-order rate constant (ke) (22). The effect compartment was introduced without affecting the mass balance of the kinetic compartment (C). A schematic illustration of the PK model is shown in Fig. 1.
FIG. 1.

Schematic illustration of the PK/PD model. The PK model is a one-compartment model (C) with first-order elimination due to degradation of the drug (kdeg) and a biophase compartment (Ce) with a first-order rate constant (ke) accounting for a possible delay in the observed effect. The PD model includes one proliferating and drug-susceptible compartment (S) and one resting and drug-insusceptible compartment (R). The bacterial system is described with first-order rate constants for multiplication of the bacteria in the susceptible compartment (kgrowth), for the degradation of bacteria in both compartments (kdeath), and for the transfer between the compartments (kSR and kRS). The total bacterial content in the system (S + R) stimulates the transfer from the normally growing stage into the resting stage (kSR). The antibiotic concentration in the biophase compartment is assumed to stimulate the killing rate of bacteria in the susceptible stage according to an Emax model (DRUG).
Semimechanistic PK/PD model building.
The concentration of bacteria (B) in an inoculum over time without drug exposure can be described according to the general equation
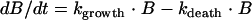 |
(1) |
This equation explains the exponential growth of bacteria seen in time-kill curve experiments as the net result of a growth rate (kgrowth) and the rate constant for natural cell death (kdeath). However, this equation does not describe the decrease in the net growth rate when the system is approaching the stationary phase. To account for this and to enhance the mechanistic input into the model, the bacterial system was described in this model by implementing the idea that phenotypic switching between normally growing cells and persister cells with reduced growth rates occurs (1). A model in which the total bacterial population was divided into two subpopulations, one growing population (S) and one resting population (R), was used (Fig. 1). In the early logarithmic growth phase, the majority of the bacteria are assumed to be in the growing stage (S) and to have a net growth rate determined by kgrowth and kdeath. Bacteria in the growing stage are transformed into a resting stage when the total bacterial content in the system reaches high values, i.e., when the system is approaching the stationary phase. In this resting stage, the bacteria show no net growth, although they are still assumed to have the same natural death rate (kdeath) as bacteria in the growing stage. The transformation from the growing stage into the resting stage should be triggered by a high total bacterial level in the system. It was found that the transformation could be well described by using a linear function in which the transfer rate (kSR) is equal to a proportionality constant times the bacterial concentration in the system (S + R). In the parameterization of the model, a more easily comprehensible parameter, Bmax, which is the bacterial concentration in the system at stationary phase, instead of the proportionality constant was estimated. During time-kill curve experiments, the bacterial system is exposed to a constant antibiotic pressure and no dilution of bacteria at high inocula is present. Thus, the transfer back to the susceptible stage (kRS) was assumed to be negligible and was therefore fixed to 0. This parameter is presented in Fig. 1 and in the equations in order to make them more general. According to the discussion above, the kinetic behavior of the bacterial system in the absence of antibiotic exposure was modeled by using the following equations:
 |
(2) |
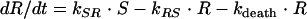 |
(3) |
The antimicrobial effect was assumed to be nonlinearly dependent on the concentration of the antibacterial agent in the effect compartment and was modeled by using an ordinary sigmoidal Emax model (equation 4), where Emax represents the maximal achievable effect with a certain drug treatment; EC50 is the antibiotic concentration that produces 50% of the maximum effect; and γ is the sigmoidicity factor, which defines the shape of the concentration-effect relationship. The antibacterial effect could hypothetically be included to either inhibit the growth rate or enhance the rate of killing of the bacteria and could be included as either an additive or a proportional effect. For all alternatives, the drug effect (DRUG) was incorporated only to affect bacteria being in the growing susceptible stage (S). The drug effect was incorporated into equation 2 according to equations 5 to 7.
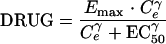 |
(4) |
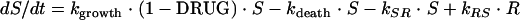 |
(5) |
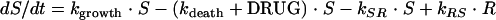 |
(6) |
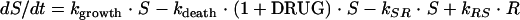 |
(7) |
To summarize the PK/PD model, the bacterial population was divided into growing drug-susceptible cells and resting drug-insusceptible cells and was characterized by the bacterium-specific parameters kgrowth, kdeath, and Bmax. The drug effect was incorporated as a stimulatory effect to increase the rate constant for the death of the growing bacterial cells by using an Emax model with the drug-specific parameters Emax, EC50, and γ.
Data analysis.
The data were analyzed by using the first-order conditional estimation method algorithm with ADVAN9 within the population analysis software NONMEM, version VIβ (3). In addition to describing the mean tendencies for the population, the use of this approach makes it possible to allow for variability between experiments, between experimental days, as well as within individual experiments (residual variability). All data from all experiments were fitted simultaneously in a single analysis.
All raw data were included in the data analysis; i.e., no averaging or exclusion of data was carried out prior to the analysis. Thus, more than one observation per sampling time point was included in the analysis. To avoid the bias that can occur due to correlations between these replicate samples, the residual error was divided into and estimated as two components: one consistent difference between all replicates (ɛ) and one replicate-specific difference (ɛrepl) (10). For values below the LOD, the first value in a consecutive series was entered as LOD/2 and all other values below the LOD were omitted (2). The data were log transformed before the start of the analysis.
As described above, the possibility of variability between single experiments and experimental days was considered. Even though the starting inocula were prepared by a standardized procedure, variations might originate from the fact that some starting inocula are in true logarithmic growth phase while others are approaching stationary phase. The mixture module within NONMEM was used (3) to describe this potential variation between the starting inocula. The inocula for each day were allowed to be allocated to those either in logarithmic growth phase, with all bacteria being in the susceptible stage (mix 1), or in late logarithmic phase approaching stationary phase, with both susceptible and resting bacteria (mix 2). The parameters estimated in the model were the fraction of the total number of experiments belonging to mix 1 (fmix1) and the fraction of bacteria in the starting inoculum being resting cells in experiments belonging to mix 2 (fpers). The fraction of bacteria in the resting stage was assumed to be the same for all experiments allocated to mix 2.
Model performance was assessed by evaluation of diagnostic plots, the objective function value, and the precision of parameter estimates. To discriminate between nested models, the difference in the objective function value (−2 log likelihood) was used. The criterion for inclusion of a parameter was a decrease in the objective function value of 10.83 (P < 0.001). Graphical evaluations were performed with the program Xpose, version 3.1 (9).
Model validation.
An internal model validation was performed by using internal cross-validation (XV) and case deletion diagnostics (CDD). During XV, data from experiments with the same concentration were excluded and the model parameters were estimated from the remaining data. The excluded experiments were thereafter predicted by the model by using the parameter values from the model when those data had been excluded. The procedure was repeated until the data from each set of concentrations had been excluded. The observed values were plotted versus the predicted values and presented graphically.
The CDD procedure was divided into two parts. During the first part of the CDD, data from 1 day's experiments at a time were excluded from the full data set. The parameter values were reestimated and compared with the estimates from the model based on the full data set. The procedure was repeated until the data from each day had been excluded once from the full data set. During the second part of the CDD, data from one of the experiments (one tube) at a time were excluded and the parameter values were reestimated and compared with the estimates from the model developed by using the full data set. The procedure was repeated until data from all experiments had been excluded once from the full data set. The relative difference between the CDD models and the model with the full data set was calculated and presented graphically.
RESULTS
MICs.
The MICs of the five antibiotics are presented in Table 1. Overall, the results obtained were consistent with the values reported previously (5, 13).
TABLE 1.
MIC values, concentrations used in the time-kill curve experiments as multiples of the MIC, and number of observations per drug
| Drug | MIC (mg/liter) | Concn (multiples of MIC) | No. of observations |
|---|---|---|---|
| Benzylpenicillin | 0.012 | 0.0625, 0.125, 0.25, 0.5, 1, 2, 4, 16, 64 | 455 |
| Cefuroxime | 0.0313 | 0.0625, 0.125, 0.25, 0.5, 1, 2, 4, 16, 64 | 427 |
| Erythromycin | 0.125 | 0.0625, 0.125, 0.25, 0.5, 1, 2, 4, 16, 64 | 455 |
| Moxifloxacin | 0.125 | 0.25, 0.5, 1, 2, 4, 16, 64 | 376 |
| Vancomycin | 0.25 | 0.25, 0.5, 1, 1.5, 2, 4, 16, 64 | 409 |
Time-kill curve experiments.
Time-kill curves for the five antibiotics used in the study are shown in Fig. 2. As expected, bacteria exposed to no antibiotics or low concentrations of antibiotics were found to grow exponentially until approaching a maximum bacterial concentration. When the bacteria were exposed to higher concentrations, all antibiotics except vancomycin caused a biphasic killing curve with a decline in the killing rate over time, indicating the presence of persister cells less susceptible to the antibiotics. The growth rate was found to be similar for the growth controls starting with different inoculum sizes (data not shown).
FIG. 2.

Time-kill curves for S. pyogenes exposed to five antibiotics at concentrations ranging from 0 to 64 times the MIC. Data from single experiments with each concentration studied are shown. Lines represent predictions based on the PK/PD model that was developed.
Pharmacokinetic model.
During the experiments benzylpenicillin and cefuroxime showed significant degradation. The degradation was assumed to proceed according to first-order kinetics, and degradation rates of 0.020 h−1 and 0.026 h−1 for benzylpenicillin and cefuroxime, respectively, were incorporated into the model. The concentrations of erythromycin, moxifloxacin, and vancomycin were assumed to be constant during the experiments. The inclusion of a model component describing a possible time delay between the addition of a drug and cell killing significantly improved the fits for benzylpenicillin, cefuroxime, and moxifloxacin. However, the data did not support the estimation of a time delay for erythromycin or vancomycin.
Semimechanistic PK/PD model.
The final PK/PD model well described the growth and killing of the bacterial system studied both without drug exposure and with exposure to a wide range of concentrations of the five antibacterial agents used in the study (Fig. 3 to 5 ). No difference in the goodness of fit between the alternative approaches for the inclusion of the drug effect was seen (equations 5 to 7), and the antibiotic effect was included in this model as an additive part to the natural death rate of the bacteria (i.e., in accordance with equation 6). Estimates and relative standard errors are presented in Table 2 for the bacterium-specific parameters and in Table 3 for the drug-specific parameters. A sigmoidal Emax model gave a significantly better fit than the ordinary Emax model (in which γ is equal to 1) for all five antibiotics. For erythromycin, γ was estimated to be less than 1, indicating a more shallow concentration-effect relationship for erythromycin than for benzylpenicillin, cefuroxime, and moxifloxacin. As shown in Fig. 2, vancomycin had a very steep concentration-effect relation, indicating an all-or-nothing effect, and the sigmoidicity factor was estimated to be very high (>50). Such high values might result in mathematical problems in the minimization, and the sigmoidicity factor for vancomycin was fixed to the lowest value that did not have a detrimental effect on the fit. In this case, the value was found to be 20.
FIG. 3.

Goodness-of-fit plots with observed and model-predicted bacterial concentrations. Lines of identity are included.
FIG. 5.

Results from cross validation. Goodness-of-fit plots with observed and model-predicted bacterial concentrations are shown. Lines of identity are included.
TABLE 2.
Estimates of bacterium-specific parameters with typical values and relative standard error
| Parameter | Estimate | Relative standard error (%) |
|---|---|---|
| kgrowth (h−1) | 1.35 | 5.4 |
| kdeath (h−1) | 0.179 | 6.5 |
| Bmax (CFU/ml) | 4.15 × 108 | 9.2 |
| fmix1 | 0.747 | 16 |
| fpers | 0.0529 | 48 |
| ɛ (%) | 98 | 20 |
| ɛrepl (%) | 47 | 9.3 |
TABLE 3.
Estimates of drug-specific parameters with typical values and relative standard errors
| Drug | Emax (h−1) | EC50 (mg/liter) | γ | ke (h−1) |
|---|---|---|---|---|
| Benzylpenicillin | 2.44 (8.6)a | 0.00438 (7.7) | 1.29 (10) | 1.00 (9.6) |
| Cefuroxime | 3.30 (6.1) | 0.00829 (6.6) | 1.69 (8.5) | 0.861 (17) |
| Erythromycin | 2.03 (6.4) | 0.0276 (15) | 0.769 (19) | 100b |
| Moxifloxacin | 3.20 (4.6) | 0.0747 (3.0) | 1.59 (7.2) | 0.644 (20) |
| Vancomycin | 1.36 (5.5) | 0.384 (0.9) | 20b | 100b |
Values in parentheses are relative standard errors (in percent).
Fixed parameter.
Allowing variability between the different experiments or different experimental days did not result in a change of the parameter estimates or improvement of the overall fit of the data. However, the implementation of a mixture model to allow for variations in the fractions of bacteria being in the resting stage at the start of the experiment significantly improved the fit. Three of 23 starting inocula were allocated to belong to mix 2, i.e., the mixture population with both susceptible and resting bacteria at the start. Approximately 5% of the cells in the starting inoculum allocated to mix 2 were estimated to be resting cells.
An additive residual error model was used to describe the random variability. However, since the data were log transformed in the data analysis, this resembles a constant coefficient of variation error structure of the original data. ɛrepl was estimated to be 47%, and ɛ was estimated to be 98%, reflecting the wide range of bacterial concentrations rather than a poor fit. The exclusion of plates in which the number of colonies counted was less than or equal to 5 did, as expected, improve the replicate variability (36 versus 47%). However, it did not improve the overall residual error (95 versus 98%), nor did it change the parameter estimates significantly.
Model validation.
The cross-validation showed that the model has good predictability (Fig. 5). When experiments from one day were excluded in the first part of the CDD, no parameter estimate except for the fraction of bacteria being in the resting stage for the experiments belonging to mix 2 changed substantially (see data for fpers in Fig. 6). However, the mixture model estimated the same starting inocula allocated to belong to mix 2, regardless of the day for which the data were excluded. The second part of the CDD, in which data for one experiment at a time were excluded from the data set, revealed that one of the parameters, i.e., ke for benzylpenicillin, was strongly influenced by one of the experiments (Fig. 7). When the data from that single experiment were excluded from the analysis, ke increased drastically, indicating that no time delay was evident from the other experiments. The model was therefore refitted with the ke for benzylpenicillin fixed to a high value (100 h−1). This procedure resulted in an increase in the objective function value of 18 units and no change or only a limited change in the values for the remaining parameters (EC50 underwent the largest change, i.e., 11%). For that reason, the estimated ke was kept in the final model.
FIG. 6.

Results of CDD, part 1. Data from one day's experiments at a time were excluded from the full data set. The parameter values were reestimated and compared with the estimates from the full model. The definitions of the suffixes are as follows: MXF, moxifloxacin; PEN, benzylpenicillin; VAN, vancomycin; CXM, cefuroxime; ERY, erythromycin.
FIG. 7.

Results of CDD, part 2. Data from one experiment at a time were excluded, and the parameter values were reestimated and compared with the estimates from the full model. The definitions of the suffixes are as follows: MXF, moxifloxacin; PEN, benzylpenicillin; VAN, vancomycin; CXM, cefuroxime; ERY, erythromycin.
DISCUSSION
From the results of the time-kill curve experiments performed in this study, it can be seen that the bacterial system shows different growth and killing phases. All antibiotics except vancomycin, which produced only a modest bactericidal effect, resulted in biphasic bacterial concentration-time curves with a rapid initial killing rate followed by a decline in the killing rate with time. This phenomenon may be due to the phenotypic switching occurring between normally growing cells and persister cells with reduced growth rates and reduced antibiotic susceptibilities. In this study we therefore developed a PK/PD model consisting of two bacterial stages representing normally growing antibiotic-susceptible cells and resting insusceptible cells, respectively. The degradation observed for benzylpenicillin and cefuroxime was corrected for in the PK part of the model and did not explain the decrease in the bacterial killing rate. The final model accurately described the time course of the different events seen in a bacterial inoculum when the bacteria are exposed to wide ranges of concentrations of antibacterial agents belonging to different classes.
Previously, the PK/PD models most commonly used to characterize the in vitro kinetics of a bacterial system exposed to antibiotics have been models that do not take the different growth and/or killing phases into account. These models have been shown to describe the data accurately only when the first hours of the time-kill curve experiments were studied (12, 20) or when in vitro kinetic systems were used to simulate concentration-time profiles mimicking the pharmacokinetics in humans, in which the rapid initial killing rate is followed by regrowth of bacteria due to dilution of the antibiotics (12, 25). The experimental design in these studies limits the ability to detect and characterize the presence of persister cells. Even though the models described these data well, the usefulness of such models may be limited in terms of making predictions or simulations beyond the conditions studied.
A few PK/PD models that have been further extended with regard to their semimechanistic complexities have been developed in order to describe the different growth and killing phases of a bacterial system. One approach has been to include a concentration- and/or a time-dependent adaptation factor that influences either the growth constant (17) or the susceptibility of the bacteria, i.e., EC50 (23). This is an empirical approach that, on the basis of our experimental data, did not prove to be robust when the approach was to fit all data simultaneously. The change in the killing rate over time has also been described as the result of a true genetic heterogeneity in the total bacterial population, with a number of subpopulations with different susceptibilities to drug treatment being present in the starting inoculum (16, 17). A high starting inoculum (∼108 CFU/ml) was used in those studies in order to observe a heterogeneous bacterial population. In our experiments, the standard methodology with a lower bacterial concentration in the starting inoculum (∼106 CFU/ml) was used, and the presence or development of true genetic resistance was not thought to be the explanation for the decrease in the killing rate. Our model has structural similarities to the model proposed by Yano et al. (24). Their model also described drug-susceptible and -insusceptible cells. However, it did not include the transition of growing cells turning into persister cells when they reached stationary phase, as our model does.
In order to fully characterize the bacterial system, we chose to expose the same bacterial strain to wide ranges of concentrations of five antibiotics of different classes. We monitored the bacterial concentration with frequent sampling for viable count determinations and simultaneously fitted a model to all data in order to separate as well as possible between bacterium-specific parameters that describe the kinetic behavior of the underlying bacterial system and the drug-specific parameters that describe the effect imposed on the system. Furthermore, model validation showed that the model has good predictability and robustness. This is, to our knowledge, the first time that a model describing the relationship between the pharmacokinetics (exposure) of several drugs of different classes and the pharmacodynamic effect on a certain bacterial strain has been simultaneously fitted to all data, thereby providing a general description of the bacterial system studied. This model might improve the possibility to compare the pharmacodynamic effects of different drugs on the bacterium in question. New agents may be evaluated and compared by carrying out additional experiments. Due to the amount of information already included in the model, such experiments may be less comprehensive.
In the present study model development was based on data from time-kill curve experiments performed with constant antibiotic concentrations. This type of study is commonly used in drug development to characterize the efficacy of an antibacterial agent. However, the design of the present study might have had an impact on the final model structure due to the limited information on different aspects of the system studied that were available. Further experiments with different mechanistic information and different concentration-time profiles are therefore needed to fully elucidate the validity of the proposed model. However, the general structure of the model and the separation of bacterium-specific and drug-specific parameters make the model easy to apply to data obtained from experimental settings other than those used in this study.
Parameter estimates from in vitro PK/PD models (i.e., EC50 and γ) have previously been linked to the empirical classification of antibacterial effects as either concentration or time dependent (17, 23, 24). The aim of the PK/PD model described in this study was to characterize as well as possible the whole time course of events seen in a bacterial system when exposed to antibiotics. By combining this knowledge about the PK/PD relationship with in silico methods, the dosing of antibiotics could be improved beyond application of the prevailing empirical classification. By using simulations of dosing strategies based on mechanistically based models, it is possible not only to evaluate and compare experimentally tested dosing strategies but also to evaluate other, not necessarily previously tested, dosing strategies. Furthermore, the concentration-effect relationship characterized in the PK/PD model could be combined with knowledge of the PKs for different populations, drug toxicity, and antibiotic resistance in simulation studies to search for the most optimal usage of the antibacterial agent in the clinical setting.
In summary, in the present study a general semimechanistic PK/PD model has been developed for the in vitro antibiotic effects of five antibiotics (benzylpenicillin, cefuroxime, erythromycin, moxifloxacin, and vancomycin) against an S. pyogenes strain. The model structure may be applied to other strains and antibiotics and might provide a tool for the development of improved dosing regimens.
FIG. 4.

Weighted residuals versus time. Included are horizontal lines for WRES=0 (solid lines) and loess smooths (broken lines).
Acknowledgments
We gratefully thank Anita Perols for excellent technical assistance.
Footnotes
Published ahead of print on 23 October 2006.
REFERENCES
- 1.Balaban, N. Q., J. Merrin, R. Chait, L. Kowalik, and S. Leibler. 2004. Bacterial persistence as a phenotypic switch. Science 305:1622-1625. [DOI] [PubMed] [Google Scholar]
- 2.Beal, S. L. 2001. Ways to fit a PK model with some data below the quantification limit. J. Pharmacokinet. Pharmacodyn. 28:481-504. [DOI] [PubMed] [Google Scholar]
- 3.Beal, S. L., and L. B. Sheiner. 1989;-1998. NONMEM users guides. GloboMax, Inc., Hanover, MD.
- 4.Craig, W. A. 1995. Interrelationship between pharmacokinetics and pharmacodynamics in determining dosage regimens for broad-spectrum cephalosporins. Diagn. Microbiol. Infect. Dis. 22:89-96. [DOI] [PubMed] [Google Scholar]
- 5.Critchley, I. A., D. F. Sahm, C. Thornsberry, R. S. Blosser-Middleton, M. E. Jones, and J. A. Karlowsky. 2002. Antimicrobial susceptibilities of Streptococcus pyogenes isolated from respiratory and skin and soft tissue infections: United States LIBRA surveillance data from 1999. Diagn. Microbiol. Infect. Dis. 42:129-135. [DOI] [PubMed] [Google Scholar]
- 6.Drusano, G. L. 2004. Antimicrobial pharmacodynamics: critical interactions of ‘bug and drug.’ Nat. Rev. Microbiol. 2:289-300. [DOI] [PubMed] [Google Scholar]
- 7.Frimodt-Moller, N. 2002. How predictive is PK/PD for antibacterial agents? Int. J. Antimicrob. Agents 19:333-339. [DOI] [PubMed] [Google Scholar]
- 8.Gustafsson, I., E. Lowdin, I. Odenholt, and O. Cars. 2001. Pharmacokinetic and pharmacodynamic parameters for antimicrobial effects of cefotaxime and amoxicillin in an in vitro kinetic model. Antimicrob. Agents Chemother. 45:2436-2440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jonsson, E. N., and M. O. Karlsson. 1999. Xpose—an S-PLUS based population pharmacokinetic/pharmacodynamic model building aid for NONMEM. Comput. Methods Programs Biomed. 58:51-64. [DOI] [PubMed] [Google Scholar]
- 10.Karlsson, M. O., S. L. Beal, and L. B. Sheiner. 1995. Three new residual error models for population PK/PD analyses. J. Pharmacokinet. Biopharm. 23:651-672. [DOI] [PubMed] [Google Scholar]
- 11.Keren, I., N. Kaldalu, A. Spoering, Y. Wang, and K. Lewis. 2004. Persister cells and tolerance to antimicrobials. FEMS Microbiol. Lett. 230:13-18. [DOI] [PubMed] [Google Scholar]
- 12.Liu, P., K. H. Rand, B. Obermann, and H. Derendorf. 2005. Pharmacokinetic-pharmacodynamic modelling of antibacterial activity of cefpodoxime and cefixime in in vitro kinetic models. Int. J. Antimicrob. Agents 25:120-129. [DOI] [PubMed] [Google Scholar]
- 13.Lorian, V. 1989. In vitro simulation of in vivo conditions: physical state of the culture medium. J. Clin. Microbiol. 27:2403-2406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.MacGowan, A., and K. Bowker. 2002. Developments in PK/PD: optimising efficacy and prevention of resistance. A critical review of PK/PD in in vitro models. Int. J. Antimicrob. Agents 19:291-298. [DOI] [PubMed] [Google Scholar]
- 15.Marshall, S., F. Macintyre, I. James, M. Krams, and N. E. Jonsson. 2006. Role of mechanistically-based pharmacokinetic/pharmacodynamic models in drug development: a case study of a therapeutic protein. Clin. Pharmacokinet. 45:177-197. [DOI] [PubMed] [Google Scholar]
- 16.Meagher, A. K., A. Forrest, A. Dalhoff, H. Stass, and J. J. Schentag. 2004. Novel pharmacokinetic-pharmacodynamic model for prediction of outcomes with an extended-release formulation of ciprofloxacin. Antimicrob. Agents Chemother. 48:2061-2068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mouton, J. W., A. A. Vinks, and N. C. Punt. 1997. Pharmacokinetic-pharmacodynamic modeling of activity of ceftazidime during continuous and intermittent infusion. Antimicrob. Agents Chemother. 41:733-738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mueller, M., A. de la Pena, and H. Derendorf. 2004. Issues in pharmacokinetics and pharmacodynamics of anti-infective agents: kill curves versus MIC. Antimicrob. Agents Chemother. 48:369-377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.NCCLS. 2003. Methods for dilution antimicrobial susceptibility tests for bacteria that grow aerobically, 6th ed. Document M7-A6. NCCLS, Villanova, PA.
- 20.Regoes, R. R., C. Wiuff, R. M. Zappala, K. N. Garner, F. Baquero, and B. R. Levin. 2004. Pharmacodynamic functions: a multiparameter approach to the design of antibiotic treatment regimens. Antimicrob. Agents Chemother. 48:3670-3676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schuck, E. L., and H. Derendorf. 2005. Pharmacokinetic/pharmacodynamic evaluation of anti-infective agents. Expert Rev. Anti-Infect. Ther. 3:361-373. [DOI] [PubMed] [Google Scholar]
- 22.Sheiner, L. B., D. R. Stanski, S. Vozeh, R. D. Miller, and J. Ham. 1979. Simultaneous modeling of pharmacokinetics and pharmacodynamics: application to d-tubocurarine. Clin. Pharmacol. Ther. 25:358-371. [DOI] [PubMed] [Google Scholar]
- 23.Tam, V. H., A. N. Schilling, and M. Nikolaou. 2005. Modelling time-kill studies to discern the pharmacodynamics of meropenem. J. Antimicrob. Chemother. 55:699-706. [DOI] [PubMed] [Google Scholar]
- 24.Yano, Y., T. Oguma, H. Nagata, and S. Sasaki. 1998. Application of logistic growth model to pharmacodynamic analysis of in vitro bactericidal kinetics. J. Pharm. Sci. 87:1177-1183. [DOI] [PubMed] [Google Scholar]
- 25.Zhi, J., C. H. Nightingale, and R. Quintiliani. 1986. A pharmacodynamic model for the activity of antibiotics against microorganisms under nonsaturable conditions. J. Pharm. Sci. 75:1063-1067. [DOI] [PubMed] [Google Scholar]