

Abstract
Human β-defensin 2 (hBD-2) and hBD-3 have potent fungicidal activity in the micromolar range. Although little is known about their mechanism of action against Candida species, some similarities to the antifungal mechanism of salivary peptide histatin 5 (Hst 5) seem to exist. Since hBD-2 and hBD-3 have been reported to cause direct disruption of target cell membranes, we compared the effects of hBD-2 and hBD-3 on Candida albicans membrane integrity. Incubation of calcein-loaded C. albicans cells with a dose of hBD-2 lethal for 90% of the strains tested (LD90) resulted in a maximal dye efflux of only 10.3% ± 2.8% at 90 min, similar to that induced by Hst 5. In contrast, an LD90 of hBD-3 more than doubled calcein release from cells yet did not result in more than 24% of total release, showing that neither peptide caused gross membrane damage. As for Hst 5, killing of C. albicans cells by hBD-2 and hBD-3 was salt sensitive; however, Ca2+ and Mg2+ inhibited hBD-2 but not hBD-3 fungicidal activity. Pretreatment of C. albicans cells with sodium azide resulted in significantly decreased ATP release and susceptibility of cells to hBD-2 and hBD-3. However, hBD-3 killing was partially restored at concentrations of ≥0.8 μM, showing energy-independent mechanisms at higher doses. C. glabrata resistance to Hst 5, hBD-2, and hBD-3 is not a result of loss of expression of cell wall Ssa proteins. The candidacidal effects of hBD-2-hBD-3 and Hst 5-hBD-2 were additive, while the index of interaction between Hst 5 and hBD-3 was 0.717 (P < 0.05). Thus, the candidacidal action of hBD-2 shows many similarities to that of Hst 5 in terms of salt sensitivity, ion selectivity, and energy requirements while hBD-3 exhibits biphasic concentration-dependent mechanisms of candidacidal action complementary to those of Hst 5.
Candida albicans is the most prevalent organism causing oral candidiasis, a common sequela in persons with immunodeficiency viral infections, underlying systemic diseases, or medications that reduce salivary flow (4, 8). The toxicity of the currently used polyene antifungal drugs and the emergence of resistant candidal species to azole-based agents have initiated a search for innate peptide antibiotics as alternative drug therapies.
In the oral cavity, a large variety of antimicrobial peptides, including human defensins and histatins, contribute to the innate host defense against fungal pathogens (7). The small (3- to 5-kDa) cationic defensins represent an important peptide family among antimicrobial peptides. Human defensins are classified into α and β subfamilies on the basis of their sequence homology and the localization of six conserved cysteine residues (21). Human β-defensin 1 (hBD-1) to hBD-4 are expressed in many epithelial tissues (2, 26). In the oral cavity, hBD-1 to -3 are found in salivary glands, salivary secretions, gingival epithelia, and gingival crevicular fluid (20). In contrast to the constitutive expression of hBD-1, hBD-2 and hBD-3 are induced by interleukin-1β, tumor necrosis factor alpha, or gamma interferon or by bacterial or candidal challenge (9, 10, 11). hBD-3 is considered the most potent β-defensin characterized to date, as it kills gram-negative and gram-positive bacteria, enveloped viruses, and fungi (11). hBD-1 and hBD-2 are less potent peptides and are predominantly active against gram-negative bacteria and yeasts (14). hBD-2 and hBD-3 have potent fungicidal activity against C. albicans at micromolar concentrations, with hBD-3 being about 10 times more fungicidal than hBD-2 (14, 31).
While the broad-spectrum activity of hBDs has been well reported, their antimicrobial mechanism has been studied less extensively. Moreover, most studies have focused on the mechanism of action against bacteria while almost nothing is known about their antifungal mechanism. Recently it has been shown that hBD-3, but not hBD-2, exhibits potent antibacterial activity against Escherichia coli, Staphylococcus aureus, and Actinobacillus actinomycetemcomitans at both subphysiological and physiological salt concentrations and also in saliva and serum (11, 13, 19, 23). Membrane depolarization and permeabilization appear to be the predominant mechanisms of β-defensins against bacteria (11, 23). In contrast, hBD-2 candidacidal effects on C. albicans and C. glabrata cells have been evaluated only morphologically (9).
We have recently demonstrated that cell wall proteins Ssa1/2p are involved in the candidacidal activity of the histidine-rich salivary protein histatin 5 (Hst 5) and for β-defensins (17, 31). Hst 5 killing of C. albicans cells is initiated by binding to Ssa1/2p, followed by translocation to an intracellular compartment (17). Intracellularly, Hst 5 causes nonlytic efflux of cellular ATP and other small nucleotides and ions from the cell through mechanisms that require the Trk1p potassium transporter (1, 15, 31). Hst 5 killing of C. albicans is salt sensitive and occurs in metabolically active cells (15, 32). Therefore, we questioned whether these key features required by Hst 5 for C. albicans killing are also involved in β-defensin fungicidal activity. Here we demonstrate that, in contrast to the salt tolerance of hBD-3 against bacteria, the antifungal activities of this peptide and hBD-2 are salt sensitive, suggesting alternative mechanisms of action between bacteria and fungi. In addition, we find evidence for differences in C. albicans killing between hBD-3 and hBD-2 in which hBD-2 more resembles Hst 5.
MATERIALS AND METHODS
Materials.
C. glabrata strains 931010 and 932474 were generously provided by Janet M. Guthmiller (Dows Institute for Dental Research and Department of Periodontics, College of Dentistry, University of Iowa, Iowa City) and were used in C. glabrata resistance assays. For all other studies, C. albicans strain CAF4-2 was used. Yeast nitrogen base (YNB) and complete amino acid supplement mixture were from Q-Biogene; uridine, sodium azide, and calcein-acetoxymethyl ester (calcein-AM) were from Sigma; hBD-2 and hBD-3 were from Peptides International (Louisville, KY); and Hst 5 was synthesized by using standard solid-phase synthesis protocols and purified by reversed-phase high-performance liquid chromatography by Genemed Synthesis Inc. (San Francisco, CA). The primary structures of these peptides are shown in Table 1.
TABLE 1.
Primary structures of antimicrobial peptides used in this study
| Peptide | Primary sequence |
|---|---|
| hBD-2 | GIGDPVTCLKSGAICHPVFCPRRYKQIGTCGLPGTKCCKKP |
| hBD-3 | GIINTLQKYYCRVRGGRCAVLSCLPKEEQIGKCSTRGRKCCRRKK |
| Hst 5 | DSHAKRHHGYKRKFHEKHHSHRGY |
Candidacidal assay.
Antifungal properties of hBD-2 and hBD-3 were examined by microdilution plate assay as previously described (31), with the following modifications. Briefly, C. albicans cells were grown in YNB medium, washed twice with 10 mM sodium phosphate buffer (NaPB; Na2HPO4-NaH2PO4, pH 7.4), and resuspended in 10 mM NaPB at a concentration of 106/ml. In order to determine the salt tolerance of β-defensins, CAF4-2 cells were placed in 10 mM, 20 mM, 50 mM, or 100 mM NaPB buffer, pH 7.4. To determine ion specificity, cells were incubated in 10 mM NaPB with the addition of 0.5 mM, 1 mM, 5 mM, or 25 mM CaCl2, MgCl2, KCl, or NaCl. For the experiments evaluating the energy requirements for the antifungal activities of hBD-2 and hBD-3, cells were preincubated with 1 mM NaN3 for 2 h at 30°C with shaking before treatment with hBD-2 (2.9 to 11.6 μM) or hBD-3 (0.2 to 1.0 μM) for 1 h at 30°C. Control cells were incubated with 10 mM NaPB alone. In experiments testing C. glabrata resistance, fungicidal activity against C. glabrata 931010 and 932474 was assessed by using a peptide concentration that caused 95% killing of C. albicans strain CAF4-2 (LD90; Hst 5, 31 μM; hBD-2, 11.6 μM; hBD-3, 0.8 μM). Control cells were incubated with 10 mM NaPB alone. Cell suspensions were diluted in 10 mM NaPB, and aliquots of 500 cells were spread onto YNB agar plates and incubated for 48 h at room temperature. Candidacidal activity assays were performed in triplicate. Cell survival was expressed as a percentage of the control, and loss of viability was calculated as [1 − (colonies from peptide-treated cells/colonies from control cells)] × 100.
Synergism assay.
The Loewe additivity model was used to assess in vitro interactions among hBD-2, hBD-3, and Hst 5 by calculation of the fractional inhibitory concentration index (FICI) with PharmTools Pro V1.1 software (MCS, Elkins Park, PA). A fixed-ratio design method and combination drug analyses were used (28). Briefly, a standard candidacidal assay with C. albicans strain CAF4-2 and hBD-2 (3 to 25 μM), hBD-3 (0.3 to 2.0 μM), or Hst 5 (3.5 to 32 μM) was performed in order to establish a dose-response curve for each peptide, as well as a composite additive curve between two peptides. Killing data were analyzed by the linear-regression model (effect versus log-based doses) with PharmTools Pro V1.1 software (PharmTools, Elkins Park, PA) to calculate each 50% effective dose (ED50) and to determine the optimal proportion of each peptide for the combination assay. A fixed proportion of the less potent peptide in the combination pairs (0.737 for Hst 5-hBD-2, 0.940 for Hst 5-hBD-3, and 0.849 for hBD-2-hBD-3) was used in candidacidal assays to obtain combination dose-response data. Resulting combination dose-response data and the individual dose-response data for each peptide were used in a combination analysis (PharmTools Pro V1.1) to determine whether the combination curve differed significantly from the composite additive curve at the specified effect level. The FICI between peptide pairs was determined by the Zmix/Zadd ratio, where Zmix is the total dose calculated from the combination dose-effect curve to give the 50% effect level and Zadd is the total dose calculated from the composite additive curve to give the same response effect (28). Student's t test was used to determine the statistical significance of the synergism between peptide pairs. FICI was defined as follows: FICI ≤ 0.5 = synergy, FICI > 4.0 = antagonism, and FICI = 0.5 to 4.0 = additive interactions (no synergistic interaction) (22).
Cell permeability assay.
Cell permeability was monitored by release of the intracellular dye calcein as previously reported (6), with the following modifications. Calcein-AM loading of C. albicans cells was performed on early-stationary-phase yeast cell cultures. Cells were washed twice with 10 mM NaPB, resuspended at 107/ml, and loaded with calcein-AM at a final concentration of 5 μM for 2 h at room temperature. Cells were washed four times to remove unincorporated dye, and 100 μl of cells (106 cells) was transferred to a quartz microcuvette for analysis. Hst 5 (32 μM), hBD-2 (11.6 μM), or hBD-3 (0.8 μM) was then added to calcein-loaded cells, and the fluorescence intensity of the induced calcein release (Ir) was recorded every 5 min at excitation and emission wavelengths of 485 nm and 530 nm, respectively, in a Hitachi F-2000 fluorescence spectrophotometer. Fluorescence intensity corresponding to 100% of the potentially available internal calcein (Iint) was determined by boiling the cells for 10 min. The total fluorescence intensity of the cell population following boiling was assumed to be equivalent to the total potentially available intracellular calcein (Iint). The background fluorescence intensity (Ibkg) was determined by measuring the amount of calcein released from loaded cells without peptide treatment over 90 min. Calcein efflux was calculated as percent fluorescence release = [(Ir − Ibkg)/(Iint − Ibkg)] × 100.
Extracellular ATP bioluminescence assay.
Measurement of extracellular ATP levels was done as previously described (1), with the following modifications. C. albicans (104 cells) was mixed with 31 μM Hst 5, 11.6 μM hBD-2, or 1 μM hBD-3. Some of the cells were pretreated with 1 mM NaN3 for 2 h. Following addition of each peptide, cells were incubated for 0, 5, 15, 30, 45, and 60 min and released extracellular ATP was measured at each indicated time point with a Veritas microplate luminometer (Turner BioSystems, Inc.). Extracellular ATP concentrations were determined from ATP standard curves for each experiment and expressed as picomoles of ATP per 104 cells.
Cell fractionation and Western blotting.
Localization of Ssa proteins in C. glabrata 931010 and C. albicans CAF4-2 was examined by two sequential cellular fractionation steps consisting of β-mercaptoethanol (β-ME) cell wall extraction, followed by cytosolic fractionation, as previously described (16). Briefly, early-log-phase cells were washed twice with 10 mM NaPB and β-ME-extractable cell wall components were released by incubation of the cell suspension in ammonium carbonate buffer (pH 8.0) containing 1% (vol/vol) β-ME for 30 min at 37°C. The supernatant containing the β-ME cell wall extract was collected following centrifugation at 500 × g. The β-ME-treated cells were then washed twice with 1 M sorbitol, and the cell pellet was disrupted in 1 volume of 0.5-mm glass beads and 1 volume of cold lysis buffer supplemented with protease inhibitors (10 mM NaPB, 1 mM phenylmethylsulfonyl fluoride, 1 mM EDTA, 1 μg/ml aprotinin, 1 μg/ml pepstatin A, 1 μg/ml leupeptin, and 1 μg/ml benzamidine). Cell lysates were prepared by vigorous vortexing at 4°C. The cytosolic fraction was collected following centrifugation at 10,000 × g for 10 min. The β-ME cell wall extracts and cytosolic proteins were normalized by protein concentration and immunoblotted with a mouse anti-HSP70 monoclonal antibody (Stressgen Biotech Corp., Victoria, British Columbia, Canada) and horseradish peroxidase-conjugated goat anti-mouse immunoglobulin G (Pierce Biotechnology Inc., Rockford, IL) as the secondary antibody.
RESULTS
hBD-3 has greater effects on C. albicans membrane permeability than hBD-2 and Hst 5.
Incubation of C. albicans with hBD-2 for 48 h has been shown to result in thinning and dissolution of the cell wall, implying that damage of the cell membrane is a cause of β-defensin-induced killing (9). Therefore, we examined the ability of hBD-2 and hBD-3 to induce yeast cell membrane destabilization by measuring the release of the fluorescent dye calcein from yeast cells in comparison with Hst 5. C. albicans cells were loaded with calcein-AM, and fluorescent emission of released free dye in response to β-defensins and Hst 5 was recorded over a period of 90 min (Fig. 1).
FIG. 1.

Time course of calcein release from C. albicans cells induced by hBD-2, hBD-3, or Hst 5. C. albicans cells were loaded for 2 h with 5 μM calcein-AM. Cells were incubated with 11.6 μM hBD-2 (closed triangles), 0.8 μM hBD-3 (closed squares), or 32 μM Hst 5 (open circles). The Ir was recorded every 5 min for 90 min at excitation and emission wavelengths of 485 and 530 nm, respectively. Calcein efflux was calculated as percent fluorescence recovery = [(Ir − Ibkg)/(Iint − Ibkg)] × 100, where Ibkg is fluorescence intensity measured before the addition of the peptide and Iint is fluorescence intensity corresponding to 100% of the potentially available calcein determined by boiling of the cells. Each experimental point represents mean ± the standard deviation of three independent experiments.
Preliminary experiments showed less than 1% of spontaneous dye release within a 90-min period from untreated cells (Ibkg). Approximately 11% calcein release was detected over a period of 90 min following treatment of cells with an LD90 of Hst 5 (32 μM) (Fig. 1, open circles), which corresponded well to our previously reported data (6). Both hBD-3 and hBD-2 induced concentration- and time-dependent calcein release from dye-loaded C. albicans cells. Incubation of calcein-loaded C. albicans cells with hBD-2 (LD90 = 11.6 μM) caused dye efflux similar to that caused by Hst 5, reaching 10.3% ± 2.8% at 90 min (Fig. 1, closed triangles). In contrast, an LD90 of hBD-3 (Fig. 1, closed squares) more than doubled (24.2% ± 1.0%) calcein release from cells compared with the equivalent dose of Hst 5 or hBD-2, showing that this peptide causes more substantial permeabilization of the cell membrane. However, none of the peptides tested caused gross membrane disturbance or lysis of C. albicans but hBD-3 induced more significant effects on membrane permeability than either Hst 5 or hBD-2.
C. albicans killing by hBD-2 and hBD-3 is salt sensitive.
A low extracellular salt concentration is required for antimicrobial activity of many peptides, including Hst 5 killing of C. albicans (5, 32). Recently it has been shown that hBD-2, but not hBD-3, bactericidal activity is salt sensitive (11, 23); however, the effect of ionic strength on hBD-2 and hBD-3 fungicidal activity is not known. Treatment of C. albicans cells with hBD-2 (11.6 μM) under our standard conditions (10 mM NaPB) resulted in 95% ± 1.5% cell killing (Fig. 2A, closed circles). However, doubling the salt concentration to 20 mM NaPB significantly decreased C. albicans susceptibility to 54% ± 7% (Fig. 2A, open circles). Further increasing the salt concentration to 50 mM reduced hBD-2 killing to 29% ± 13% (Fig. 2A, closed squares), but an increase to 100 mM NaPB only slightly reduced killing below this level to 24% ± 14% (Fig. 2A, open squares).
FIG. 2.

hBD-2 and hBD-3 killing of C. albicans is salt sensitive. C. albicans (106 cells/ml) was treated with 2.9 to 11.6 μM hBD-2 (A) or 0.2 to 1.0 μM hBD-3 (B) in the presence of 10 mM (closed circles), 20 mM (open circles), 50 mM (closed squares), or 100 mM (open squares) sodium phosphate buffer (Na2HPO4-NaH2PO4, pH 7.4) for 1 h at 30°C. Loss of cell viability is expressed as [1 − (colonies after peptide treatment/colonies after incubation with buffer only)] × 100. Each data point represents the mean ± the standard deviation of at least three independent experiments.
Reduction of hBD-3 killing with increasing salt concentrations was similar to that of hBD-2; however, the attenuation of killing was more substantial than for hBD-2 at higher (>20 mM) salt concentrations (Fig. 2B). C. albicans susceptibility to hBD-3 (1.0 μM) was decreased to 63% ± 16% in 20 mM NaPB (Fig. 2B, open circles), compared to 98.2% ± 1.5% killing with 10 mM NaPB (Fig. 2B, closed circles). Susceptibility of C. albicans cells to hBD-3 was reduced to only 18% ± 13% when cells were incubated in 50 mM NaPB (Fig. 2B, closed squares) and was completely abolished in 100 mM NaPB (Fig. 2B, open squares). Thus, hBD-2 retains ∼20% of its fungicidal activity at physiological salt concentrations (100 mM NaPB) while hBD-3 killing is completely inactivated. These results also show that candidacidal activities of both hBD-2 and hBD-3 are salt sensitive, in contrast to the salt-insensitive activity of hBD-3 against some bacterial species.
hBD-2, but not hBD-3, killing of C. albicans is ion specific.
In order to determine whether the salt-sensitive action of hBD-2 and hBD-3 on C. albicans is due to stronger buffering capacity of the medium or a specific ion effect, we tested hBD-2 and hBD-3 killing in the presence of increasing concentrations of various salts. The effect of hBD-2 and hBD-3 on C. albicans was examined in 10 mM NaPB supplemented with 0.5, 1, 5, or 25 mM CaCl2, MgCl2, NaCl, or KCl. In the presence of only 0.5 mM CaCl2 or MgCl2, cells treated with hBD-2 (11.6 μM) showed substantially increased resistance to the peptide, resulting in 69% ± 1.1% and 58.4% ± 5.2% survival, respectively (Fig. 3A, white and gray bars). Increasing the concentration of Ca2+ or Mg2+ to 5 mM resulted in nearly complete loss of activity of hBD-2, as 86% ± 5.8% of cells in 5 mM CaCl2 and 97.6% ± 1.1% of cells in 5 mM MgCl2 survived peptide treatment (Fig. 3A) while C. albicans treated with hBD-2 in 0.5 mM NaCl or KCl had only slightly reduced susceptibility to killing (24% ± 9.8% and 18% ± 4.5%, respectively). No difference in hBD-2 action among various ions was observed at higher salt concentrations, due to the generalized salt sensitivity found with hBD-2, as shown in Fig. 2. In contrast, C. albicans cells treated with an LD90 of hBD-3 (1 μM) in 10 mM NaPB supplemented with increasing concentrations of CaCl2, MgCl2, NaCl, or KCl showed no difference in susceptibility to the salts tested (Fig. 3B). To evaluate if specific anions affect hBD-3 activity, cell killing was tested in the presence of CaHPO4, MgPO4, Na2HPO4, or K2HPO4. No significant difference in susceptibility to hBD-3 was observed when Cl− was substituted for PO4− (data not shown), indicating that the susceptibility of Candida to hBD-3 is salt rather than ion dependent. These results demonstrate that hBD-2, but not hBD-3, killing is strongly affected by the presence of Ca2+ and Mg2+, an effect previously reported for another candidacidal peptide, Hst 5 (5).
FIG. 3.

hBD-2 killing of C. albicans is ion selective. C. albicans (106 cells/ml) was treated with 11.6 μM hBD-2 (A) or 1.0 μM hBD-3 (B) in the presence of 10 mM sodium phosphate buffer (Na2HPO4-NaH2PO4, pH 7.4) supplemented with CaCl2 (clear bars), MgCl2 (gray bars), NaCl (black bars), or KCl (hatched bars) for 1 h at 30°C. Loss of cell viability is expressed as [1 − (colonies after peptide treatment/colonies after incubation with buffer only)] × 100. Each data point represents the mean ± the standard deviation of at least three independent experiments.
hBD-2- and hBD-3-induced killing and ATP efflux from C. albicans cells are energy dependent.
Since the candidacidal activities of Hst 5, lactoferricin, and human neutrophil defensin (HNP-1) are energy dependent (15, 16, 18), we questioned whether hBD-2 and hBD-3 require energy for their fungicidal activities. For this purpose, we tested the candidacidal activity of hBD-2 and hBD-3 in cells pretreated for 2 h with the general metabolic inhibitor sodium azide (Fig. 4). CAF4-2 cells preincubated with sodium azide had a marked reduction in hBD-2-induced killing, reaching only 15% ± 10% at the highest concentration tested (Fig. 4A, open triangles), compared to the control cells (Fig. 4A, closed triangles). Similarly, C. albicans cells preincubated with sodium azide were highly resistant to the fungicidal activity of hBD-3 (Fig. 4B, open squares) compared with the control cells (Fig. 4B, closed squares). However, the inhibitory effect of sodium azide following hBD-3 treatment was biphasic in that 36% ± 10% of the cells were killed at higher peptide concentrations (≥0.8 μM) while lower hBD-3 doses (≤0.6 μM) resulted in less than 10% killing (Fig. 4B, open squares), suggesting that hBD-3 kills C. albicans by additional energy-independent mechanisms at higher concentrations.
FIG. 4.

β-Defensins kill C. albicans in an energy-dependent manner. C. albicans cells were incubated for 2 h with 1 mM NaN3 (open squares, open triangles) in the presence of 10 mM sodium phosphate buffer (pH 7.4) at 30°C. Control cells (closed squares, closed triangles) were treated with buffer alone. Next, the cells were incubated with hBD-2 (2.9 to 11.6 μM) or hBD-3 (0.2 to 0.8 μM) in 10 mM sodium phosphate buffer (pH 7.4) for 1 h. Survival of C. albicans cells treated with hBD-2 (A) or hBD-3 (B) is expressed as a percentage of the control, and loss of viability is calculated as [1 − (colonies from peptide-treated cells/colonies from control cells)] × 100. ATP release (C) was measured per 104 cells treated with hBD-2 (closed triangles), hBD-3 (closed squares), or Hst 5 (filled circles). ATP release per 104 cells incubated with 1 mM NaN3 prior to treatment with hBD-2 (open triangles), hBD-3 (open squares), or Hst 5 (open circles) in a representative experiment is shown.
We have previously shown that Hst 5 action results in ATP release, which could be blocked in cells pretreated with sodium azide (15). To determine whether hBD-2 and hBD-3 act in a similar manner, ATP efflux in hBD-2- and hBD-3-treated cells preincubated with sodium azide was measured. As previously reported with other C. albicans strains, treatment with lethal concentrations of Hst 5 resulted in significant release of ATP (Fig. 4C, closed circles), while preincubation of cells with 1 mM NaN3 prior to Hst 5 treatment prevented ATP release (Fig. 4C, open circles). Similarly, incubation of Candida cells with hBD-2 resulted in induction of ATP efflux in a manner similar to that of Hst 5 (Fig. 4C, closed triangles) while ATP efflux was nearly absent following 1 mM sodium azide pretreatment (Fig. 4C, open triangles). Interestingly, although hBD-3 treatment resulted in pronounced ATP efflux compared with untreated cells, maximal ATP release at 60 min of peptide treatment reached only 50% of that for Hst 5 (Fig. 4C, closed squares). Pretreatment with 1 mM sodium azide for 2 h completely blocked ATP release from hBD-3-treated cells (Fig. 4C, open squares). Thus, killing by both hBD-2 and hBD-3 is energy dependent and involves ATP release; however, the magnitude and profile of ATP efflux are most similar between Hst 5 and hBD-2.
hBD-2 and hBD-3 candidacidal activities are additive with that of Hst 5.
The in vitro antimicrobial activity of β-defensins is synergistic with lysozyme, lactoferrin, and cathelicidin LL37 (2, 3, 27), suggesting that the microbicidal potency of β-defensins may be significantly increased by synergistic interactions between antimicrobial peptides. Since our experiments showed that hBD-2 and hBD-3 have differential effects with respect to salt sensitivity and ATP release from Candida cells, we tested whether these peptides also synergize Hst 5 antifungal activity. In order to determine whether hBD-2, hBD-3, and Hst 5 act in an additive or a synergistic manner, C. albicans cells were incubated with fixed proportions of hBD-2, hBD-3, and Hst 5 (Table 2) and the candidacidal activities were analyzed by a fixed-ratio method to determine any effects of peptide interactions. Although the fixed-ratio method defines synergy as combinations with an FICI of <1.0, we used a more stringent criterion in which the FICI must be ≤0.5 for synergism (22) and additive effects (or no interaction) are defined as FICI values between 0.5 and 4.0. Interactions between hBD-3 and Hst 5 showed that the combination curve differed significantly from the composite additive curve at the specified effect level (interaction index of 0.717; P < 0.05) at all of the ratios tested (Table 2), however, this combination did not reach the criterion for synergy. The candidacidal effect of hBD-2 and hBD-3 pairs was clearly additive (interaction index of 0.937; P > 0.05) (Table 2). Similarly, hBD-2 and Hst 5 also showed additive effects at all of the concentrations of peptide pairs tested (interaction index of 3.248; P < 0.05) (Table 2). Although we did not find evidence for strong synergistic action between hBD-3 and Hst 5 on C. albicans killing under the defined criterion, their interaction indexes suggest the possibility of weak synergism, in contrast to the additive effects of hBD-2 and Hst 5.
TABLE 2.
Analysis of interactions among Hst 5, hBD-2, and hBD-3 against C. albicans
| Combinationa | Proportionb | P value | FICIc |
|---|---|---|---|
| hBD-2-hBD-3 | 0.849 | >0.05 | 0.937 |
| Hst 5-hBD-2 | 0.737 | <0.05 | 3.228 |
| Hst 5-hBD-3 | 0.940 | <0.05 | 0.717 |
The respective concentrations tested (micromolar) were as follows: hBD-2-hBD-3, 0.62 and 0.11, 1.23 and 0.22, 2.46 and 0.44, and 4.92 and 0.88; Hst 5-hBD-2, 1.73 and 0.62, 3.45 and 1.23, 6.90 and 2.46, and 13.8 and 4.92; Hst 5-hBD-3, 1.73 and 0.11, 3.45 and 0.22, 6.90 and 0.44, and 13.8 and 0.88.
The proportion of the less potent peptide in the combination is shown. Proportions are based on the estimated ED50s of individual peptides.
The FICI is defined as follows: FICI ≤ 0.5 = synergy, FICI > 4.0 = antagonism, FICI = 0.5 to 4.0 = additive effects (no interaction) (22).
C. glabrata resistance to β-defensins and Hst 5 is not due to absence of cell wall Ssa1/2p.
C. glabrata has emerged as a common fungal pathogen in opportunistic fungal infections in humans with resistance to many azole drugs (24). Unlike most strains of C. albicans, several strains of C. glabrata also have substantial resistance to killing by hBD-2 and hBD-3 (14). To determine whether this resistance also extends to Hst 5, we tested a defensin-resistant strain of C. glabrata 931010 and a defensin-susceptible strain of C. glabrata 932474 in candidacidal assays with Hst 5 (Table 3). The fungicidal activity of each peptide was assessed for C. glabrata strains with an LD90 of the peptide. Defensin-susceptible strain C. glabrata 932474 was highly sensitive to hBD-2 (89.6% ± 4.8%) and moderately sensitive to hBD-3 (53.2% ± 3.2%) but displayed reduced susceptibility to Hst 5 (29.4% ± 4.2%). Thus, this C. glabrata strain had quite different susceptibilities to both hBD-2 and hBD-3, as well as to Hst 5, suggesting that there are differences in the mechanism of action among members of the β-defensin family itself, as well as between β-defensins and Hst 5. In contrast, the defensin-resistant strain of C. glabrata was equally resistant to killing by both hBD-2 and hBD-3, as well as to killing by Hst 5 (Table 3).
TABLE 3.
Fungicidal activities of Hst 5, hBD-2, and hBD-3 against Candida species
| Peptide (concn [μM])a | Mean peptide-induced cell killing (%) ± SD
|
||
|---|---|---|---|
| C. glabrata 931010 | C. glabrata 932474 | C. albicans CAF4-2 | |
| hBD-2 (11.6) | 20.2 ± 15.5 | 89.6 ± 4.8 | 95.0 ± 1.5 |
| hBD-3 (0.8) | 11.5 ± 1.4 | 53.2 ± 3.2 | 98.2 ± 1.5 |
| Hst 5 (31) | 12.2 ± 7.2 | 29.4 ± 4.2 | 97.5 ± 2.0 |
Peptide concentrations were chosen on the basis of ED90s for C. albicans.
Since a major requirement for killing of Hst 5 is initial binding to C. albicans Ssa cell wall proteins and we found that Ssa1/2 proteins also play a role in β-defensins antifungal action (31), we hypothesized that C. glabrata resistance to β-defensins and Hst 5 may be due to the absence of the heat shock protein 70 (Ssa1/2p) family on the C. glabrata cell wall. To test this possibility, we probed the cell wall of C. glabrata 931010 (which is highly resistant to hBD-2, hBD-3, and Hst 5) for the presence of Ssa proteins in comparison to sensitive C. albicans strain CAF4-2. Cell wall and cytosolic extracts were prepared from each stain and normalized by total protein concentration, and Western blots were probed with an anti-HSP70 monoclonal antibody (Fig. 5). Ssa proteins were found to be abundant in cell wall extracts of C. glabrata (Fig. 5, lane 1) and C. albicans (Fig. 5, lane 2). As expected, Ssa proteins were also present in the cytosolic fractions from C. glabrata (Fig, 5, lane 3) and C. albicans (Fig. 5, lane 4). Thus, C. glabrata also expresses Ssa protein in its cell wall and cytosol, showing that C. glabrata antifungal resistance is not a result of loss of Ssa proteins.
FIG. 5.
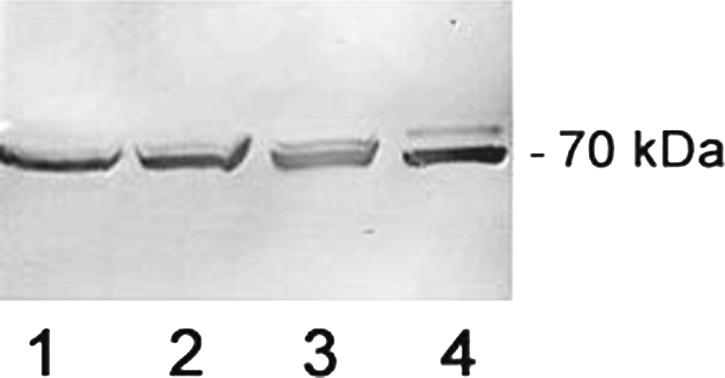
Both C. glabrata and C. albicans have Ssa proteins in their cell wall and cytoplasmic fractions. Cell wall proteins were extracted from C. glabrata 931010 (lane 1) and C. albicans CAF4-2 (lane 2) by β-ME digestion. Cytosolic fractions of C. glabrata (lane 3) and C. albicans (lane 4) were obtained by glass bead disruption of β-ME-digested cells, and soluble proteins were recovered. All cellular fractions were normalized by protein content, and Western blots were probed with an anti-HSP70 monoclonal antibody.
DISCUSSION
Human defensins are of significant interest because of their antimicrobial and immunomodulatory activities, coupled with their inherent immunological compatibility. While the antimicrobial spectrum of the β-defensins has been well evaluated, very little is known about the mechanism of action of defensins against oral pathogens, especially C. albicans. Gross morphological changes have been observed in Candida cells treated with β-defensins that are suggestive of membrane disruption (9). However, we found no evidence of membrane permeabilization in C. albicans cells treated with hBD-2 and only small leakage of calcein from cells treated with hBD-3 at concentrations close to normal salivary levels in vivo (300 to 600 ng/ml) (29, 30). About 24% calcein release from C. albicans cells was detected following treatment with hBD-3, while less than 12% release was found with hBD-2 and Hst 5 when an LD90 of each peptide was used. However, the effect of hBD-3 on membrane permeability was indistinguishable from that of hBD-2 or Hst 5 when an LD50 of hBD-3 was applied (12.2%), showing that membrane permeabilization is highly sensitive to the hBD-3 dosage. It should also be noted that after 30 min of incubation with the peptide (the time required for killing of 90% of the cells), only about 3% calcein was released by Hst 5 while C. albicans cells treated with hBD-3 released only 7% of their total intracellular calcein. Since Hst 5 exerts its primary effects on C. albicans viability within 30 min, subsequent time points generally represent secondary effects associated with cell death (15). As both β-defensins and Hst 5 exhibit similar time- and concentration-dependent profiles of calcein release, our results show that β-defensins do not cause gross membrane disruption and membrane destabilization. Thus, previously reported observations of membrane permeabilization are likely to be secondary effects associated with β-defensin-induced cell death.
Our findings on C. albicans membrane permeability differ from a previous study that showed thinning of the cell walls and loss of cytoplasmic material from cells following hBD-2 treatment (9). This difference may be due to the use of different β-defensin sources (recombinant versus commercial peptide) or to different treatment conditions. It is noteworthy that the candidacidal potency of recombinant hBD-2 is about 10-fold higher than that of recombinant hBD-3 (1 μM versus 10 μM) but commercially available hBD-2 is 10 times less fungicidal than hBD-3 (9, 14, 31). Thus, recombinant hBD-2 differs from the commercially available peptide used in the present study, which might be the reason for differences in results. In our experiments, calcein release from C. albicans cells was measured within 90 min after incubation with an LD90 of hBD-2, hBD-3, or Hst 5. The previously observed cell wall effects of hBD-2 may be due to the prolonged (48-h) peptide treatment and concentrations of the peptide exceeding the LD90 by 10-fold (9). Interestingly, previously reported cell wall perforation and explosion-like effects on plasma membranes of S. aureus (11) were also monitored following incubation with hBD-3 concentrations exceeding the LD90 by more than 10-fold, thereby producing robust cellular effects that are not likely to occur at physiological peptide concentrations. As hBD-3 effects on bacterial cells were evaluated only morphologically with supralethal peptide concentrations, additional studies are needed to determine whether the primary bacterial target of β-defensins is indeed the membrane.
The greater potential effects of an LD90 of hBD-3 on membrane permeability could be due in part to the higher net cationic charge of hBD-3 (11) or its ability to form dimers (25). Electrostatic-charge-based mechanisms of membrane permeabilization rather than formation of bilayer-spanning pores have been widely proposed for β-defensins (12, 25). However, the equivalent salt sensitivity between hBD-2 and hBD-3 in the killing of C. albicans cells argues against electrostatic interactions being crucial for hBD-3 effects on membrane permeability. In addition, a 17-amino-acid peptide derived from the N terminus of hBD-3 (net charge, +4) retains the activity of the full peptide (net charge, +11) (13). Thus, peptide conformation, charge, and secondary structure maintained by cysteine residues do not appear to be critical for C. albicans killing (13). In contrast, hBD-3 killing of E. coli, Pseudomonas aeruginosa, and Enterococcus faecium is affected by peptide net charge or secondary structure (13). In this regard, our results showing energy dependence of hBD-2 and hBD-3 killing also suggest that charge-based membrane interactions that do not require cellular energy are unlikely to contribute substantially to cell killing. Similar to the previously reported salt-dependent antibacterial effect of hBD-2 (2), our results show that C. albicans killing following treatment with hBD-3 is salt sensitive. This observation was rather surprising since it is known that the peptide retains antibacterial activity at physiological salt concentrations (11). These results clearly indicate a difference in the bactericidal and candidacidal mechanism of hBD-3 action. Whether β-defensins' ionic-strength sensitivity is due to altered attachment to the Candida cell surface or to other downstream events in the mechanism of killing remains to be elucidated. In this regard, we found that C. glabrata resistance to hBD-2, hBD-3, and Hst 5 is not due to lack of expression of Ssa1/2p on the cell wall; however, these experiments did not rule out the possibility that C. glabrata Ssa proteins have altered binding affinities that result in defects in the cellular uptake of peptides. Additional experiments are ongoing to answer this question.
Despite the salt-sensitive killing of C. albicans following hBD-2 or hBD-3 treatment, we observed that hBD-2, but not hBD-3, killing of Candida is cation selective, indicating pronounced differences in their killing mechanisms. C. albicans sensitivity to hBD-2 was significantly decreased in the presence of as little as 0.5 mM CaCl2 and MgCl2, showing that these ions have a pronounced inhibitory effect on hBD-2 action in Candida. Interestingly, we have previously reported similar results with Hst 5 (5), suggesting some similarities in the mechanisms of action of hBD-2 and Hst 5. In addition, the candidacidal activity of hBD-2 had features very similar to those of the cytotoxic action of Hst 5 with respect to active concentrations, magnitude of induction of nonlytic ATP efflux, and calcein release. Thus, even though hBD-2 and Hst 5 possess unique pathways in terms of their action on the Trk1p potassium transporter, they clearly share other steps in the mechanism of candidacidal action. These similarities are also highlighted by FICI values showing additive effects of hBD-2 and Hst 5 but an FICI value approaching that for synergistic action for the combination of hBD-3 with Hst 5.
hBDs are a family containing at least six related members; however, it is evident that the family members hBD-2 and hBD-3 have distinct mechanisms of action with respect to fungicidal activity. We have found that hBD-3 elicits a greater magnitude of calcein release but lower levels of ATP release compared to hBD-2 and Hst 5. The candidacidal action of hBD-2 shares many similarities with that of Hst 5 in terms of salt sensitivity, ion selectivity, and energy requirements but also has unique pathways in its mechanism. These results suggest the relevance of the use of combinations of candidacidal peptides such as Hst 5 and hBD-3 for therapeutic applications on the basis of complementary mechanisms of action.
Acknowledgments
This work was supported by NIH grant DE10641 from the National Institute of Dental and Craniofacial Research (to M.E.).
Footnotes
Published ahead of print on 30 October 2006.
REFERENCES
- 1.Baev, D., A. Rivetta, S. Vylkova, J. N. Sun, G. F. Zeng, C. L. Slayman, and M. Edgerton. 2004. The TRK1 potassium transporter is the critical effector for killing of Candida albicans by the cationic protein, histatin 5. J. Biol. Chem. 279:55060-55072. [DOI] [PubMed] [Google Scholar]
- 2.Bals, R., X. Wang, Z. Wu, T. Freeman, V. Bafna, M. Zasloff, and J. M. Wilson. 1998. Human beta-defensin 2 is a salt-sensitive peptide antibiotic expressed in human lung. J. Clin. Investig. 102:874-880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chen, X., F. Niyonsaba, H. Ushio, D. Okuda, I. Nagaoka, S. Ikeda, K. Okumura, and H. Ogawa. 2005. Synergistic effect of antibacterial agents human beta-defensins, cathelicidin LL-37 and lysozyme against Staphylococcus aureus and Escherichia coli. J. Dermatol. Sci. 40:124-132. [DOI] [PubMed] [Google Scholar]
- 4.Dawes, C. 2004. Factors influencing salivary flow rate and composition, p. 32-49. In W. M. Edgar, C. Dawes, and D. M. O-Mullane (ed.), Saliva and oral health, 3rd ed. British Dental Association, London, United Kingdom.
- 5.Dong, J., S. Vylkova, X. S. Li, and M. Edgerton. 2003. Calcium blocks fungicidal activity of human salivary histatin 5 through disruption of binding with Candida albicans. J. Dent. Res. 82:748-752. [DOI] [PubMed] [Google Scholar]
- 6.Edgerton, M., S. E. Koshlukova, T. E. Lo, B. G. Chrzan, R. M. Straubinger, and P. A. Raj. 1998. Candidacidal activity of salivary histatins. Identification of a histatin 5-binding protein on Candida albicans. J. Biol. Chem. 273:20438-20447. [DOI] [PubMed] [Google Scholar]
- 7.Edgerton, M., and S. E. Koshlukova. 2000. Salivary histatin 5 and its similarities to the other antimicrobial proteins in human saliva. Adv. Dent. Res. 14:16-21. [DOI] [PubMed] [Google Scholar]
- 8.Epstein, J. B., and B. Polsky. 1998. Oropharyngeal candidiasis: a review of its clinical spectrum and current therapies. Clin. Ther. 20:40-57. [DOI] [PubMed] [Google Scholar]
- 9.Feng, Z., B. Jiang, J. Chandra, M. Ghannoum, S. Nelson, and A. Weinberg. 2005. Human beta-defensins: differential activity against candidal species and regulation by Candida albicans. J. Dent. Res. 84:445-450. [DOI] [PubMed] [Google Scholar]
- 10.Garcia, J. R., F. Jaumann, S. Schulz, A. Krause, J. Rodrigues-Jimenez, U. Forssmann, K. Adermann, E. Kluver, C. Vogelmeier, D. Becker, R. Hedrich, W. G. Forsmann, and R. Bals. 2001. Identification of a novel, multifunctional beta-defensin (human beta-defensin 3) with specific antimicrobial activity. Its interaction with plasma membranes of Xenopus oocytes and the induction of macrophage chemoattraction. Cell Tissue Res. 306:257-264. [DOI] [PubMed] [Google Scholar]
- 11.Harder, J., J. Bartels, E. Christophers, and J. M. Schroder. 2001. Isolation and characterization of human beta-defensin-3, a novel human inducible peptide antibiotic. J. Biol. Chem. 276:5707-5713. [DOI] [PubMed] [Google Scholar]
- 12.Hoover, D. M., K. R. Rajashankar, R. Blumenthal, A. Puri, J. J. Oppenheim, O. Chertov, and J. Lubkowski. 2000. The structure of human beta-defensin-2 shows evidence of higher order oligomerization. J. Biol. Chem. 275:32911-32918. [DOI] [PubMed] [Google Scholar]
- 13.Hoover, D. M., Z. Wu, K. Tucker, W. Lu, and J. Lubkowski. 2003. Antimicrobial characterization of human beta-defensin 3 derivatives. Antimicrob. Agents Chemother. 47:2804-2809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Joly, S., C. Maze, P. B. McCray, and J. M. Guthmiller. 2004. Human beta-defensins 2 and 3 demonstrate strain-selective activity against oral microorganisms. J. Clin. Microbiol. 42:1024-1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Koshlukova, S. E., T. L. Lloyd, M. W. Araujo, and M. Edgerton. 1999. Salivary histatin 5 induces non-lytic release of ATP from Candida albicans leading to cell death. J. Biol. Chem. 274:18872-18879. [DOI] [PubMed] [Google Scholar]
- 16.Lehrer, R. I., T. Ganz, D. Szklarek, and M. E. Selsted. 1988. Modulation of the in vitro candidacidal activity of human neutrophil defensins by target cell metabolism and divalent cations. J. Clin. Investig. 81:1829-1835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li, X. S., M. S. Reddy, D. Baev, and M. Edgerton. 2003. Candida albicans Ssa1/2p is the cell envelope binding protein for human salivary histatin 5. J. Biol. Chem. 278:28553-28561. [DOI] [PubMed] [Google Scholar]
- 18.Lupetti, A., A. Paulusma-Annema, M. M. Welling, S. Senesi, J. T. van Dissel, and P. H. Nibbering. 2000. Candidacidal activities of human lactoferrin peptides derived from the N terminus. Antimicrob. Agents Chemother. 44:3257-3263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Maisetta, G., G. Batoni, S. Esin, G. Raco, D. Bottai, F. Favilli, W. Florio, and M. Campa. 2005. Susceptibility of Streptococcus mutans and Actinobacillus actinomycetemcomitans to bactericidal activity of human β-defensin 3 in biological fluids. Antimicrob. Agents Chemother. 49:1245-1248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mathews, M., H. P. Jia, J. M. Guthmiller, G. Losh, S. Graham, G. K. Johnson, B. F. Tack, and P. B. McCray, Jr. 1999. Production of β-defensin antimicrobial peptides by the oral mucosa and salivary glands. Infect. Immun. 67:2740-2745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Morrison, G. M., C. A. Semple, F. M. Kilanowski, R. E. Hill, and J. R. Dorin. 2003. Signal sequence conservation and mature peptide divergence within subgroups of the murine beta-defensin gene family. Mol. Biol. Evol. 20:460-470. [DOI] [PubMed] [Google Scholar]
- 22.Odds, F. C. 2003. Synergy, antagonism, and what the chequerboard puts between them. J. Antimicrob. Chemother. 52:1. [DOI] [PubMed] [Google Scholar]
- 23.Ouhara, K., H. Komatsuzawa, S. Yamada, H. Shiba, T. Fujiwara, M. Ohara, K. Sayama, K. Hashimoto, H. Kurihara, and M. Sugai. 2005. Susceptibilities of periodontopathogenic and cariogenic bacteria to antibacterial peptides, β-defensins and LL37, produced by human epithelial cells. J. Antimicrob. Chemother. 55:888-896. [DOI] [PubMed] [Google Scholar]
- 24.Pfaller, M. A., L. Boyken, R. J. Hollis, S. A. Messer, S. Tendolkar, and D. J. Diekema. 2005. In vitro susceptibilities of clinical isolates of Candida species, Cryptococcus neoformans, and Aspergillus species to itraconazole: global survey of 9,359 isolates tested by Clinical and Laboratory Standards Institute broth microdilution methods. J. Clin. Microbiol. 43:3807-3810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schibli, D. J., H. N. Hunter, V. Aseyev, T. D. Starner, J. M. Wiencek, P. B. McCray Juniorperiod, B. F. Tack, and H. J. Vogel. 2002. The solution structures of the human beta-defensins lead to a better understanding of the potent bactericidal activity of HBD3 against Staphylococcus aureus. J. Biol. Chem. 277:8279-8289. [DOI] [PubMed] [Google Scholar]
- 26.Schneider, J. J., A. Unholzer, M. Schaller, M. Schafer-Korting, and H. C. Korting. 2005. Human defensins. J. Mol. Med. 83:587-595. [DOI] [PubMed] [Google Scholar]
- 27.Singh, P. K., B. F. Tack, P. B. McCray Juniorperiod, and M. J. Welsh. 2000. Synergistic and additive killing by antimicrobial factors found in human airway surface liquid. Am. J. Physiol. Lung Cell Mol. Physiol. 279:799-805. [DOI] [PubMed] [Google Scholar]
- 28.Tallarida, R. J. 2000. Calculation for combination drug analysis, p. 57-75. In R. J. Tallarida (ed.), Drug synergism and dose-effect data analysis, 1st ed. Chapman & Hall/CRC Press, Boca Radon, FL.
- 29.Tanida, T., T. Okamoto, A. Okamoto, H. Wang, T. Hamada, E. Ueta, and T. Osaki. 2003. Decreased excretion of antimicrobial proteins and peptides in saliva of patients with oral candidiasis. J. Oral Pathol. Med. 32:586-594. [DOI] [PubMed] [Google Scholar]
- 30.Tao, R., R. J. Jurevic, K. K. Coluton, M. T. Tsutsui, M. C. Roberts, J. R. Kimball, N. Welsh, J. Berndt, and B. A. Dale. 2005. Salivary antimicrobial peptide expression and dental caries experience in children. Antimicrob. Agents Chemother. 49:3883-3888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Vylkova, S., X. S. Li, J. C. Berner, and M. Edgerton. 2006. Distinct antifungal mechanisms: β-defensins require Candida albicans Ssa protein, while Trk1p mediates activity of cysteine-free cationic peptides. Antimicrob. Agents Chemother. 50:324-331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Xu, Y., I. Ambudkar, H. Yamagishi, W. Swaim, T. J. Walsh, and B. C. O'Connell. 1999. Histatin 3-mediated killing of Candida albicans: effect of extracellular salt concentration on binding and internalization. Antimicrob. Agents Chemother. 43:2456-2462. [DOI] [PMC free article] [PubMed] [Google Scholar]