

Abstract
We used the ability of Salmonella enterica serovar Typhimurium to colonize the gut of Caenorhabditis elegans to measure the fitness costs imposed by antibiotic resistance mutations. The fitness costs determined in the nematode were similar to those measured in mice, validating its use as a simple host model to evaluate bacterial fitness.
The persistence of antibiotic-resistant bacteria largely depends on the effect of the resistance mechanism on fitness (reviewed in references 3 and 4). The cost caused by resistance mutations can be decreased by second-site mutations that restore fitness without a loss of resistance (3, 17, 18). This process has been observed in many cases both in vitro (13, 17, 19, 22, 25) and in clinical settings (6, 8, 20, 26). These findings suggest that antibiotic resistance may persist in the population for a long time and that the determination of fitness parameters is of great importance for predicting the risk of resistance development.
Fitness costs are usually determined by comparing the growth rates of resistant and susceptible bacteria (3). Although mouse models have been used for these purposes, simpler invertebrates such as Caenorhabditis elegans have recently become more attractive for assessing the in vivo biological costs of antibiotic resistance (23, 24). Many bacterial genes known to be required for mammalian pathogenesis are needed also in the nematode (1, 9, 11, 15, 16, 27, 28). Some bacterial pathogens, such as Salmonella enterica serovar Typhimurium are able to establish a persistent infection in the intestine of C. elegans, reducing the life span of the host. Several genes needed for virulence in mammals are also required for pathogenesis in C. elegans (2, 10, 16, 28), implying that the invasion and proliferation of serovar Typhimurium in the host intestine depend on mechanisms common to the nematode and mammals. This makes C. elegans a relevant model for determining the infectivity and fitness of antibiotic-resistant bacteria during a host infection.
Virulence assays were performed as previously described (2) using C. elegans SS104 [glp-4 (ts)] (5), a temperature-sensitive mutant that produces progeny at 15°C but not at 25°C. At least 50 synchronized worms in larval stage L4 were transferred on solid nematode growth medium (NGM) (12) seeded with 10 μl of serovar Typhimurium mixed 1:100 with the nonpathogenic Escherichia coli strain OP50 and maintained at 25°C. Both bacterial species grow on NGM agar with no mutual inhibition. The number of viable worms was monitored every day, and the percentages of nematode survival calculated by the Kaplan-Meier method (14) were used for plotting the percent survival as a function of time. Survival kinetics were compared by using the nonparametric log-rank test and were considered statistically different when P was <0.05. We found that the time to death for 50% of the nematodes was 9 days when fed with E. coli OP50 and 7 days after ingestion of the virulent wild-type strain LT2 of serovar Typhimurium (Fig. 1). As previously shown, the glp-4 (ts) strain responded to serovar Typhimurium infections similarly to the wild-type nematode strain N2 (28). The virulence of antibiotic-resistant strains was compared to the wild-type Salmonella sp. strain LT2 (see the strain list in the supplemental material). Strains resistant to streptomycin and mupirocin were as virulent as the parental strain, whereas the fusidic acid- and actinonin-resistant mutants had a killing rate similar to that of the nonpathogenic E. coli OP50, indicating a reduction in virulence compared to the antibiotic-susceptible wild-type strain (Fig. 1).
FIG. 1.

Survival of C. elegans fed with S. enterica serovar Typhimurium JB124 (wild type; number of worms counted: n = 132), JB127 (Sm: streptomycin resistant; n = 49) (P = 0.176), JB393 (Fus: fusidic acid resistant; n = 85) (P < 0.001), JB1855 (Mup: mupirocin resistant; n = 53) (P = 0.282), DA8325 (Act: actinonin resistant; n = 62) (P < 0.0001), and E. coli OP50 (control; n = 89) (P < 0.0001) as a function of time. Survival curves compared pairwise with the one of the virulent strain JB124 are considered significantly different when P values are <0.05.
To determine whether an impaired ability to colonize the nematode's digestive tract could explain the lower virulence of the resistant mutants, the number of bacteria in infected C. elegans was determined at different time points by picking up five individual worms. The worms washed and resuspended in phosphate-buffered saline containing 0.1% Triton X-100 were disrupted by sonication in the presence of small glass beads. Serial dilutions of the recovered bacteria from the nematodes' gut were then made in phosphate-buffered saline and plated on Luria-Bertani (LB) agar. Colonies were counted after incubation of the plates at 37°C for 24 to 48 h. For the wild-type strain of serovar Typhimurium bacterial numbers in the nematode increased rapidly (∼2 log10) within the two first days postinfection, and maximum levels of bacteria (∼106/worm) were reached and maintained for several days until the death of the host ensued (Fig. 2). A similar increase in bacterial numbers was seen for the resistant mutants that showed a high virulence in the plate-killing assay, whereas mutants with reduced virulence according to the plate-killing assay did not increase in bacterial numbers to the same extent.
FIG. 2.
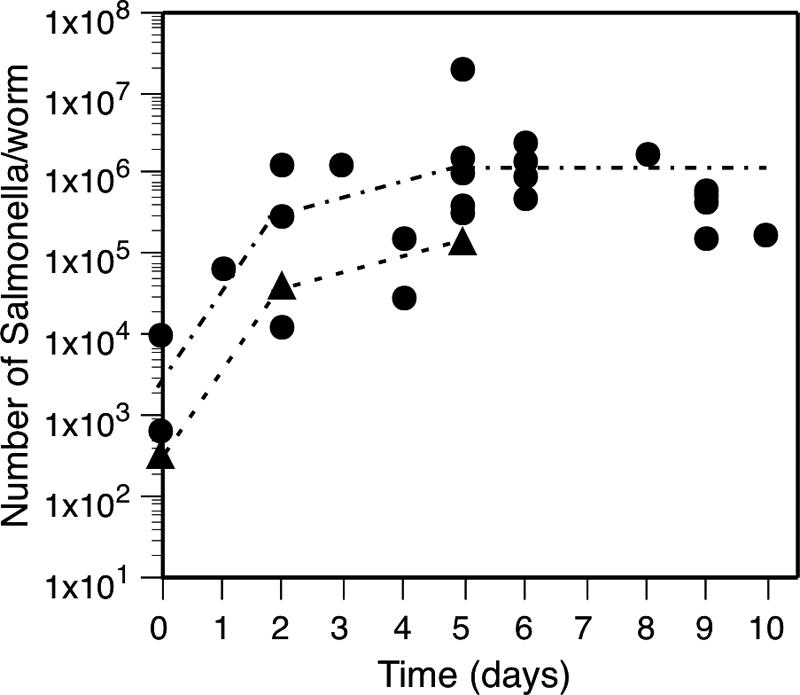
Increase in the number of wild-type serovar Typhimurium bacteria (JB124) during infection at 25°C. Each datum point represents the bacterial numbers obtained from three to five worms. Circles represent independent experiments, and triangles represent the mean number of bacteria obtained from at least 15 experiments.
Since the plate-killing assay was insensitive to small changes in bacterial fitness, we developed a competition assay. The synchronized worms in larval stage L4 were placed for several hours in contact with a 1:1 mixture of the resistant strain and the wild-type strain carrying a neutral genetic marker (8). The infected worms were then removed from the S basal medium (12) containing Salmonella, washed, and placed onto solid NGM seeded with E. coli OP50. The number of serovar Typhimurium in the worms was determined as described above. Multiplication and establishment of the bacterial populations in the gut were not affected by possible residual growth of the Salmonella strains on the agar plates (2). Competition indexes (CI) were calculated as the ratio of antibiotic-resistant bacteria divided by the reference bacteria corrected by the ratio of the two bacterial populations at the time of infection (day 0). All of the examined antibiotic-resistant strains had a lower fitness than the susceptible strain, as shown by bacterial enumerations done at 2 and 5 days postinfection (Fig. 3). The geometric mean CI at day 2 varied from 0.2 to 0.6 to ca. 0.001 (Fig. 3A). After 5 days, the mean CI decreased for most of the strains (Fig. 3B). In many cases, the antibiotic-resistant bacteria were cleared out, and no resistant cells were detected in the five worms analyzed (CI < 0.00001). From the determined CI and the generation time for serovar Typhimurium wild type in C. elegans (GA ∼6.2 h from Fig. 2), we calculated the relative fitness of each mutant (Table 1). If growth rate is the main parameter involved in competition, the mutant growth rate is kB = kA + ln(CI)/t, where kA is the growth rate of the wild-type strain (kA = ln2/GA) and t is the time of infection. The relative fitness was then calculated by dividing the generation time of the wild type (GA) by the generation time of the mutants (GB = ln2/kB). The relative fitness varied between <0.1 (corresponding to a CI of <0.008) and 0.9, a finding similar to that seen in the standard mouse model (7, 8, 21) (Table 1). In both hosts the mutation K42N in ribosomal protein S12, which confers resistance to streptomycin, decreased fitness by 50%. Likewise, the decrease in fitness for two nalidixic acid-resistant mutants carrying the mutation D82G or D87G in gyrase was similar in C. elegans and in mice: 60 and 10%, respectively. The fusidic acid-resistant strain that was unable to grow in mice also showed little growth in the nematode (Fig. 3). For strains resistant to actinonin, fitness costs appeared higher in the nematode (relative fitness of <0.1) than in the mice (relative fitness of 0.45 or 0.26). It is worth noting that for all resistant strains, the relative fitness in the two host-models was lower than the fitness measured in the laboratory medium.
FIG. 3.

CI values determined 2 days (A) and 5 days (B) postinfection for different strains of serovar Typhimurium (•). The detection level (dashed line) was CI < 0.00001, and competition indexes below this limit are indicated by open circles (○). Numbers in parentheses indicate the numbers of times the CI was <0.00001. Abbreviations: Sm, streptomycin resistant; Rif, rifampin resistant; Nal, nalidixic acid resistant; Fus, fusidic acid resistant; Mup, mupirocin resistant; Act, actinonin resistant.
TABLE 1.
Comparison of fitness of antibiotic-resistant strains of serovar Typhimurium in C. elegans, mice, and LB
| Strain | Antibiotic | MIC (μg/ml) | Resistance
|
Relative fitnessa
|
|||
|---|---|---|---|---|---|---|---|
| Target (gene) | Mutation | C. elegans | Mice | LB | |||
| JB1660 (wild type) | 1.0 | 1.0 | 1.0 | ||||
| JB127 | Streptomycin | >1,024 | S12 (rpsL) | K42N | 0.46 | 0.48A | 0.77 |
| SMP807 | Rifampin | >256 | RNA polymerase (rpoB) | H526Y | 0.63 | ND | 0.80 |
| SMP816 | Rifampin | >256 | RNA polymerase (rpoB) | S531L | 0.74 | ND | 0.89 |
| SMP972 | Nalidixic acid | 64 | Gyrase A (gyrA) | D82G | 0.38 | 0.45B | 0.84 |
| SMP973 | Nalidixic acid | >256 | Gyrase A (gyrA) | D87G | 0.89 | 0.91B | 1 |
| JB393 | Fusidic acid | 2,400 | EF-G (fusA) | P413L | <0.1 | No growthA | 0.41A |
| JB1850 | Mupirocin | >1,024 | IleS (ileS) | WV631L | <0.1 | <0.1 | 0.32C |
| JB1855 | Mupirocin | >1,024 | IleS (ileS) | F596L | 0.52 | ND | 0.60C |
| DA8325 | Actinonin | >1,024 | M-THFb (folD) | G129R | <0.1 | 0.45D | 0.58D |
| DA8326 | Actinonin | >1,024 | Fmt (fmt) | Nonsense | <0.1 | <0.1D | 0.24D |
| DA8340 | Actinonin | >1,024 | Fmt (fmt) | T12R | <0.1 | 0.26D | 0.38D |
Mouse experiments were performed in previous studies, and fitness was calculated from data reported earlier as indicated by the superscript letters: A, Bjorkman et al. (8); B, Bjorkman et al. (7); C, W. Paulander, S. Maisnier-Patin, and D. I. Andersson, unpublished data; D, Nilsson et al. (21). ND, not determined.
Methylene-tetrahydrofolate enzymes.
The competition model correlated well with the mouse model, further supporting the notion that similar factors in serovar Typhimurium are required for growth in C. elegans and mice. For example, mutations in the genes fur-1, ompR, or rpoS involved in serovar Typhimurium acid tolerance important for virulence in mammals show attenuated virulence in C. elegans (16). Similarly, the PhoP/Q signal transduction system and several of the genes located in the pathogenicity island 1 are required for virulence in both mammals and C. elegans (2, 10). The identification of virulence factors that are important in both mammals and C. elegans can conceivably be explained by conserved interactions with the innate immunity systems (28) and/or similarities in the actual growth environment (e.g., nutrient levels). Conversely, the lack of a correlation between mice and C. elegans can be explained by the absence of professional phagocytic cells and adaptive immune responses in C. elegans. Thus, the use of C. elegans for host-pathogen interactions is likely to be limited to model certain stages of mammalian infection.
Supplementary Material
Acknowledgments
This study was supported by grants from the Swedish Animal Welfare Agency (Djurskyddsmyndigheten) (to S.M.-P. and D.I.A.), the Carl Tryggers Foundation (to S.M.P.), and the Swedish Research Council (to D.I.A.). We also thank the Caenorhabditis Genetic Center, which was funded by the U.S. National Institutes of Health National Center of Research Resources, for providing the C. elegans strain SS104 used in the present study.
Footnotes
Published ahead of print on 20 November 2006.
Supplemental material for this article may be found at http://aac.asm.org/.
REFERENCES
- 1.Aballay, A., and F. M. Ausubel. 2002. Caenorhabditis elegans as a host for the study of host-pathogen interactions. Curr. Opin. Microbiol. 5:97-101. [DOI] [PubMed] [Google Scholar]
- 2.Aballay, A., P. Yorgey, and F. M. Ausubel. 2000. Salmonella typhimurium proliferates and establishes a persistent infection in the intestine of Caenorhabditis elegans. Curr. Biol. 10:1539-1542. [DOI] [PubMed] [Google Scholar]
- 3.Andersson, D. I. 2003. Persistence of antibiotic resistant bacteria. Curr. Opin. Microbiol. 6:452-456. [DOI] [PubMed] [Google Scholar]
- 4.Andersson, D. I., and B. R. Levin. 1999. The biological cost of antibiotic resistance. Curr. Opin. Microbiol. 2:489-493. [DOI] [PubMed] [Google Scholar]
- 5.Beanan, M. J., and S. Strome. 1992. Characterization of a germ-line proliferation mutation in Caenorhabditis elegans. Development 116:755-766. [DOI] [PubMed] [Google Scholar]
- 6.Björkholm, B., M. Sjolund, P. G. Falk, O. G. Berg, L. Engstrand, and D. I. Andersson. 2001. Mutation frequency and biological cost of antibiotic resistance in Helicobacter pylori. Proc. Natl. Acad. Sci. USA 98:14607-14612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Björkman, J., D. Hughes, and D. I. Andersson. 1998. Virulence of antibiotic-resistant Salmonella typhimurium. Proc. Natl. Acad. Sci. USA 95:3949-3953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Björkman, J., I. Nagaev, O. G. Berg, D. Hughes, and D. I. Andersson. 2000. Effects of environment on compensatory mutations to ameliorate costs of antibiotic resistance. Science 287:1479-1482. [DOI] [PubMed] [Google Scholar]
- 9.Couillault, C., and J. J. Ewbank. 2002. Diverse bacteria are pathogens of Caenorhabditis elegans. Infect. Immun. 70:4705-4707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ernst, R. K., T. Guina, and S. I. Miller. 2001. Salmonella typhimurium outer membrane remodeling: role in resistance to host innate immunity. Microbes Infect. 3:1327-1334. [DOI] [PubMed] [Google Scholar]
- 11.Garsin, D. A., C. D. Sifri, E. Mylonakis, X. Qin, K. V. Singh, B. E. Murray, S. B. Calderwood, and F. M. Ausubel. 2001. A simple model host for identifying gram-positive virulence factors. Proc. Natl. Acad. Sci. USA 98:10892-10897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hope, I. A. 1999. Caenorhabditis elegans: a practical approach, vol. 213. Oxford University Press, New York, NY.
- 13.Johanson, U., A. Aevarsson, A. Liljas, and D. Hughes. 1996. The dynamic structure of EF-G studied by fusidic acid resistance and internal revertants. J. Mol. Biol. 258:420-432. [DOI] [PubMed] [Google Scholar]
- 14.Kaplan, E. L., and P. Meier. 1958. Nonparametric estimation from incomplete observations. J. Am. Statistical Assoc. 53:457-481. [Google Scholar]
- 15.Kurz, C. L., S. Chauvet, E. Andres, M. Aurouze, I. Vallet, G. P. Michel, M. Uh, J. Celli, A. Filloux, S. De Bentzmann, I. Steinmetz, J. A. Hoffmann, B. B. Finlay, J. P. Gorvel, D. Ferrandon, and J. J. Ewbank. 2003. Virulence factors of the human opportunistic pathogen Serratia marcescens identified by in vivo screening. EMBO J. 22:1451-1460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Labrousse, A., S. Chauvet, C. Couillault, C. L. Kurz, and J. J. Ewbank. 2000. Caenorhabditis elegans is a model host for Salmonella typhimurium. Curr. Biol. 10:1543-1545. [DOI] [PubMed] [Google Scholar]
- 17.Levin, B. R., V. Perrot, and N. Walker. 2000. Compensatory mutations, antibiotic resistance, and the population genetics of adaptive evolution in bacteria. Genetics 154:985-997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Maisnier-Patin, S., and D. I. Andersson. 2004. Adaptation to the deleterious effects of antimicrobial drug resistance mutations by compensatory evolution. Res. Microbiol. 155:360-369. [DOI] [PubMed] [Google Scholar]
- 19.Maisnier-Patin, S., O. G. Berg, L. Liljas, and D. I. Andersson. 2002. Compensatory adaptation to the deleterious effect of antibiotic resistance in Salmonella typhimurium. Mol. Microbiol. 46:355-366. [DOI] [PubMed] [Google Scholar]
- 20.Nagaev, I., J. Björkman, D. I. Andersson, and D. Hughes. 2001. Biological cost and compensatory evolution in fusidic acid-resistant Staphylococcus aureus. Mol. Microbiol. 40:433-439. [DOI] [PubMed] [Google Scholar]
- 21.Nilsson, A. I., A. Zorzet, A. Kanth, S. Dahlstrom, O. G. Berg, and D. I. Andersson. 2006. Reducing the fitness cost of antibiotic resistance by amplification of initiator tRNA genes. Proc. Natl. Acad. Sci. USA 103:6976-6981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Reynolds, M. G. 2000. Compensatory evolution in rifampin-resistant Escherichia coli. Genetics 156:1471-1481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ruiz-Diez, B., P. Sanchez, F. Baquero, J. L. Martinez, and A. Navas. 2003. Differential interactions within the Caenorhabditis elegans-Pseudomonas aeruginosa pathogenesis model. J. Theor. Biol. 225:469-476. [DOI] [PubMed] [Google Scholar]
- 24.Sanchez, P., J. F. Linares, B. Ruiz-Diez, E. Campanario, A. Navas, F. Baquero, and J. L. Martinez. 2002. Fitness of in vitro selected Pseudomonas aeruginosa nalB and nfxB multidrug resistant mutants. J. Antimicrob. Chemother. 50:657-664. [DOI] [PubMed] [Google Scholar]
- 25.Schrag, S. J., V. Perrot, and B. R. Levin. 1997. Adaptation to the fitness costs of antibiotic resistance in Escherichia coli. Proc. R. Soc. Lond. B Biol. Sci. 264:1287-1291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sherman, D. R., K. Mdluli, M. J. Hickey, T. M. Arain, S. L. Morris, C. E. Barry III, and C. K. Stover. 1996. Compensatory ahpC gene expression in isoniazid-resistant Mycobacterium tuberculosis. Science 272:1641-1643. [DOI] [PubMed] [Google Scholar]
- 27.Sifri, C. D., J. Begun, F. M. Ausubel, and S. B. Calderwood. 2003. Caenorhabditis elegans as a model host for Staphylococcus aureus pathogenesis. Infect. Immun. 71:2208-2217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tenor, J. L., B. A. McCormick, F. M. Ausubel, and A. Aballay. 2004. Caenorhabditis elegans-based screen identifies Salmonella virulence factors required for conserved host-pathogen interactions. Curr. Biol. 14:1018-1024. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.