

SUMMARY
Vertebrates develop distinct asymmetries along the left-right axis, which are consistently aligned with the anteroposterior and dorsoventral axes. The mechanisms that direct this handed development of left-right asymmetries have been elusive, but recent studies of mutations that affect left-right development have shed light on the molecules involved. One molecule implicated in left-right specification is left-right dynein (LRD), a microtubule-based motor protein. In the LRD protein of the inversus viscerum (iv) mouse, there is a single amino acid difference at a conserved position, and the lrd gene is one of many genes deleted in the legless (lgl) mutation. Both iv and lgl mice display randomized left-right development. Here we extend the analysis of the lrd gene at the levels of sequence, expression and function. The complete coding sequence of the lrd gene confirms its classification as an axonemal, or ciliary, dynein. Expression of lrd in the node at embryonic day 7.5 is shown to be symmetric. At embryonic day 8.0, however, a striking asymmetric expression pattern is observed in all three germ layers of the developing headfold, suggesting roles in both the establishment and maintenance of left-right asymmetries. At later times, expression of lrd is also observed in the developing floorplate, gut and limbs. These results suggest function for LRD protein in both cilitated and non-ciliated cells, despite its sequence classification as axonemal. In addition, a targeted mutation of lrd was generated that deletes the part of the protein required for ATP binding, and hence motor function. The resulting left-right phenotype, randomization of laterality, is identical to that of iv and lgl mutants. Gross defects in ciliary structure were not observed in lrd/lrd mutants. Strikingly, however, the monocilia on mutant embryonic node cells were immotile. These results prove the identity of the iv and lrd genes. Further, they argue that LRD motor function, and resulting nodal monocilia movement, are required for normal left-right development.
Keywords: Left-right asymmetry, Situs inversus, Dynein, ATP, Mouse
INTRODUCTION
Externally, vertebrates appear bilaterally symmetrical. However, internally they display characteristic and highly conserved asymmetries along the left-right (LR) axis. These include the positioning of the heart, stomach, pancreas and liver. In wild-type animals, LR asymmetries are consistently aligned relative to the anteroposterior (AP) and dorsoventral (DV) axes, but the molecular events that link existing AP and DV positional information to establish invariant LR visceral handedness are not yet well understood.
Determination of the vertebrate LR embryonic axis occurs well before the first morphological sign of LR asymmetry, the rightward looping of the embryonic heart tube. The first signs of LR asymmetry are at the molecular level. Several genes with LR asymmetric expression patterns have been identified including sonic hedgehog (Shh), HNF 3β, activin receptor IIa (cActRIIa), cNR1 (nodal-related 1), activin βB, Pitx2 and cSnR in the chick (Levin et al., 1995, 1997; Yoshioka et al., 1998; Logan et al., 1998; Piedra et al., 1998; Isaac et al., 1997), nodal, lefty-1, lefty-2, Pitx2 and SnR in the mouse (Collignon et al., 1996; Lowe et al., 1996; Meno et al., 1996, 1998; Piedra et al., 1998; Yoshioka et al., 1998; Campione et al., 1999; Sefton et al., 1998), and Xnr-1 and Pitx2 in Xenopus (Lowe et al., 1996; Ryan et al., 1998). Although these have suggested a cascade of lateralized signaling eventually specifying LR morphological asymmetries, where and when the initial breaking of symmetry occurs is unknown. The embryonic node, known as Hensen’s node in chick and the Spemann organizer in Xenopus, has been shown to be essential to the development of LR asymmetry (Pagan-Westphal and Tabin, 1998; Hyatt and Yost, 1998) and is where the earliest asymmetric gene expression is seen. However, results from chick and Xenopus studies have suggested that positional information is initially provided to the node by adjacent tissue. Subsequently, the node signals LR instructions to more distant tissues, including the lateral plate mesoderm (Pagan-Westphal and Tabin, 1998; Hyatt and Yost, 1998). Experimental or genetic ablation of the notochord and floor plate of the neural tube indicate that these midline structures also play a key role in LR development (Danos and Yost, 1995, 1996; Lohr et al., 1997; Goldstein et al., 1998).
In the mouse, insight into laterality determination has come from analysis of the situs inversus viscerum (iv) mouse mutation (Hummel and Chapman, 1959). Adult iv/iv mutants have a roughly 50% incidence of inverted visceral organ positioning, suggesting that the gene product drives normal laterality and, in its absence, the process is randomized (Layton, 1976). The iv mutation disrupts the expression patterns of the asymmetric genes nodal (Lowe et al., 1996), lefty (Meno et al., 1996) and Pitx2 (Piedra et al., 1998; Campione et al., 1999) indicating that the iv gene plays a critical early role in the genetic hierarchy of LR development. The legless (lgl) transgene insertional mutation is allelic to the iv mutation, but has a much more severe phenotype, including hindlimb truncations and craniofacial defects (McNeish et al., 1990). Molecular analysis indicated that the lgl mutation resulted in the deletion of over 600 kb of mouse genomic DNA (Supp et al., 1996, 1997). The different phenotypes seen in iv and lgl may result from different types of mutations or the deletion of multiple genes in lgl.
A good candidate for the iv gene was cloned from the lgl deletion region, a dynein heavy chain gene which we named left-right dynein (lrd). Dyneins are microtubule-based motors that use the energy of ATP hydrolysis to generate minus-end directed motive force in the sliding of adjacent microtubules in ciliary axonemes and the transport of multiple cargoes within the cytoplasm (reviewed by Holzbaur and Vallee, 1994). A human syndrome affecting LR development further implicated dyneins in specification of the LR asymmetries. Kartagener syndrome is characterized by a roughly 50% incidence of situs inversus (complete reversal of the LR axis), as well as dynein defects in the cilia that result in chronic respiratory infections and immotile sperm (Afzelius, 1976; Splitt et al., 1996). Further suggestion that molecular motors, and perhaps cilia, are involved in LR specification comes from the targeting of the mouse genes kif3A and kif3B. Mutations of kif3A or kif3B, members of the kinesin superfamily of motor proteins, result in absence of monocilia on cells of the embryonic node and defects in LR asymmetry (Marszalek et al., 1999; Takeda et al., 1999; Nonaka et al., 1998). Interestingly, expression of lrd was noted in the embryonic node (Supp et al., 1997).
In this report, we extend our analysis of the role of lrd in LR development. The complete lrd cDNA has been cloned and sequenced, confirming the classification of lrd as an axonemal dynein heavy chain gene. This is the first report of the full coding sequence of an axonemal dynein heavy chain gene in vertebrates. A more extensive analysis of expression reveals that, in addition to symmetric expression in the embryonic node, apparently transient asymmetric expression of lrd occurs in the headfold region of 0- to 5-somite wild-type mouse embryos. At later stages, lrd is expressed symmetrically in the floorplate of the neural tube, a midline signaling center and a region of the embryo shown to be involved in LR development at earlier stages. To confirm the role of lrd in laterality, to test for possible involvement in other developmental processes and to begin to define the molecular mechanism of lrd function, a targeted mutation in the lrd gene was generated and the resulting mice characterized.
MATERIALS AND METHODS
Cloning lrd cDNA
Direct cDNA selection (Supp et al., 1997), reverse transcription polymerase chain reaction (RT-PCR) and rapid amplification of cDNA ends (RACE) (Marathon cDNA Amplification Kit, Clontech) were used to clone lrd cDNA sequences from wild-type adult mouse brain RNA. The cloned cDNA is 14,072 base pairs (bp) long, including 13,464 bp of coding sequence, 211 bp of 5′ untranslated region (UTR), and 397 bp of 3′ UTR. The putative translational start site is preceded by stop codons in all three reading frames. The coding sequence has been submitted to GenBank (accession number AF183144). For analysis of lrd cDNA in iv/iv mice, cDNAs were isolated from iv/iv mouse brain RNA using RT-PCR.
In situ hybridization
Serial section in situ hybridizations were performed as described (Supp et al., 1997) with an lrd antisense riboprobe spanning nucleotides 9,631–10,325 of the lrd cDNA sequence. Whole-mount in situ hybridizations were performed as described (Lowe et al., 1996) using an lrd antisense riboprobe spanning nucleotides 306–1,216 of the lrd cDNA sequence. Control hybridizations with sense-strand riboprobes showed no signal.
Gene targeting
The backbone targeting vector, pMJK-KO, was provided by Dr Michael Kern (S. C. Medical College). This vector contains the pMC1-TK gene for negative selection in gancyclovir, and the pGK-neo gene for positive selection in G418, subcloned into the KpnI and HindIII sites (respectively) of pBSIISK + (Stratagene). A genomic clone was isolated by screening a 129SVJ mouse lambda genomic library (Stratagene) with an lrd cDNA probe containing the first P-loop of the motor domain (Supp et al., 1997). A 6.3 kb EcoRV fragment was subcloned into the BamHI site of pMJK-KO, and a 0.8 kb SpeI-XbaI fragment was subcloned into the XhoI site (Fig. 4A). The construct was linearized by NotI digestion. Homologous recombination in ES cells and generation of gene targeted mice were performed using standard methods. Targeted ES cells and mice were genotyped using Southern blot analysis (data not shown) and PCR was used for confirmation (Fig. 4B). Primers used for PCR genotyping were: 3.7C, 5′-GGAAACATCTATAAAGGACTGGTG-3′; FAS-1, 5′-TGTTAGGACCCAAAGGTGGAAACAT-3′; and neo-1, 5′-CTTCC-TCGTGCTTTACGGTATCGCC-3′.
Fig. 4.

lrd gene targeting. (A) Gene targeting construct (top), wild-type lrd locus (middle) and structure of targeted allele (bottom). Two exons of the wild-type lrd sequence (black boxes), including the exon encoding the first P-loop, were replaced by the selectable neo gene. Shown are the locations of PCR primers (not drawn to scale) used for genotyping. (B) PCR genotyping of mice. Targeted allele yields a PCR product with neo1 and FAS-1 primers. The wild-type allele yields a PCR product with the 3.7C and FAS-1 primers. Note, the FAS-1 primer flanks the region of homology between the targeting construct and the endogenous locus. Abbreviations: RV, EcoRV; N, NcoI; X, XhoI; S, SpeI; B, BglI.
RT-PCR analysis of targeted lrd allele
RNA was prepared from entire heads and lungs of male mice by quick freezing in liquid nitrogen, grinding to a powder with a mortar and pestle and using RNAzol (Tel-Test Inc.) according to recommended protocols, except that only 35 ml of RNAzol was used for the pooled heads and lungs of two 7-week-old mice. The RNA was then further purified by phenol and chloroform extraction and ethanol precipitation. An Oligotex mRNA Midi Kit (Qiagen) batch system was used to purify mRNA, which was reverse transcribed using the Superscript Choice System for cDNA Synthesis (BRL). Reverse transcripts were PCR amplified using Taq DNA polymerase (Qiagen) and the primers LRDj1 (5′-TCCGACACGAGTGGGAGGATT-CAAGGAAAC-3′) and LRDj2 (5′-AGGAACACTGGGACAT-CATCAGTTACAATT-3′). Recommended Qiagen PCR buffer conditions, including 1× PCR buffer and 20% Q solution, were used. Samples were cycled at 66°C 1 minute, 72°C 3 minute, and 94°C for 1 minute, for 35 cycles and the products were resolved on 2% agarose gels.
Analysis of nodal cilia
Embryos were prepared for SEM by fixation for 90 minutes in 2.5% glutaraldehyde (EM) buffered with 0.1 M sodium cacodylate. SEM was performed according to standard protocols. Nodal cilia were visualized by light microscopy as previously described (Nonaka et al., 1998).
RESULTS
Complete lrd coding sequence confirms its classification as an axonemal dynein heavy chain gene
The full open reading frame of the lrd transcript has been cloned and sequenced. The lrd gene encodes a protein of 4,488 amino acids (Fig. 1A) with 60% overall amino acid sequence identity to the outer-arm β-axonemal dynein heavy chain of sea urchin, (Fig. 1B), compared with only 26% sequence identity with rat cytoplasmic dynein (Fig. 1C). The N-terminal one-third of the molecule is less conserved than the C-terminal two-thirds. This is consistent with the observation that cargo carrying and enzymatic functions that distinguish dynein family members from each other are located within the N-terminal region (Holzbaur and Vallee, 1994). The organization of the LRD motor domain is analogous to all other previously characterized dyneins. There are four centrally located P-loops spaced approximately 300 amino acids apart (Fig. 1A,E). The sequence TETTKDL, immediately following the first P-loop, further supports classification of LRD as a member of the axonemal-type dynein subclass. However, in contrast to several other axonemal-type dyneins (Kandl et al., 1995), no 5th P-loop is found in the N-terminal region of LRD. Two coiled-coil domains of 95 amino acids and 82 amino acids, separated by a 135 amino acid non-coiled region, are located 200 amino acids downstream of the 4th P-loop (Fig. 1D); this represents the likely LRD-microtubule binding site (Gee et al., 1997). RT-PCR identified an alternately spliced lrd mRNA that eliminates the third and least conserved P-loop, including amino acids 2521–2576. This smaller lrd message had an identical tissue distribution as full-length lrd (data not shown).
Fig. 1.


Sequence of LRD and homology with sea urchin axonemal dynein β heavy chain. (A) LRD amino acid sequence. The four P-loops are highlighted. Thin underline, exons deleted in targeted lrd mutation. Thick underline, exon missing in alternately spliced form of lrd.(B–E) Analysis of LRD amino acid sequence; amino acid residues in B–D are aligned with the map shown in E. (B,C) Comparisons of LRD amino acid sequence with sea urchin b-axonemal dynein and rat cytoplasmic dynein. The comparisons were done using the Compare program (GCG, University of Wisconsin), using a window of 50 and a stringency of 40. Note the higher degree of overall homology of LRD with sea urchin b-axonemal dynein (B) compared with rat cytoplasmic dynein (C). (D) Coiled-coil analysis of LRD amino acid sequence using the COILS program. Note the coiled-coil structures predicted immediately C-terminal to the 4th P-loop. (E) Representation of the lrd mRNA and amino acid sequence aligned with B–D. Thick horizontal line, sequenced mRNA. Open rectangles, translated regions; shaded boxes, P-loops. Accession number AF183144.
The lrd cDNA was also cloned and sequenced from iv/iv mice. Comparison of the coding sequence of the wild-type and iv alleles revealed only the previously reported difference, a glutamic acid to a lysine change in the conserved motor domain (Supp et al., 1997).
Expression of lrd
We previously showed that lrd expression at embryonic day 7.5 (E7.5) is limited to the node (Supp et al., 1997). The nature of this expression was examined further. Serial section in situ hybridizations, using both frontal and transverse embryo orientations, showed expression in the node to be symmetric about the LR axis (data not shown). This symmetric distribution of lrd transcript was confirmed by whole-mount in situ hybridizations (Fig. 2A).
Fig. 2.

Expression of lrd in E8 and E8.5 embryos localized by whole-mount in situ hybridization. All are ventral views, so the right side of the embryo is to the left of the figure. (A) Early E8 embryo, showing symmetric lrd expression in the node. (B) Slightly older E8 embryo showing lrd expression in the node and in the right anterior. (C,D) E8.25 embryos showing strong anterior asymmetric expression on the left (C), or on the right (D). The posterior of both embryos is curled away and not visible. (E) Transverse section through the anterior region of the embryo in D at the level indicated by the line. Expression is strongest in the endoderm and head mesenchyme but is also apparent in the overlying neuroectoderm. (F) E8.5 embryo showing strong asymmetric anterior expression. (G) E8.5 embryo with symmetric anterior expression.
Expression studies with whole-mount embryo preparations revealed a striking pattern of lrd transcript distribution in E8.0–E8.5 embryos (0–7 somites). Of 146 embryos examined, 44% (n=64) showed lrd expression in the headfold region of the embryo, while 56% (n=82) showed no lrd expression in this region. Of the 64 embryos showing headfold lrd expression, 40 embryos had bilateral expression and 24 had asymmetric expression, with both left- or right-sided lrd expression patterns observed (Fig. 2B-D,F). There was no apparent correlation between developmental stage, as measured by somite count, and expression pattern. Sectioning revealed that this anterior expression was present in the gut endoderm and cephalic mesenchyme, and in the basal aspect of the overlying neurectoderm (Fig. 2E). In embryos with asymmetric expression, there was a sharp boundary between expressing and non-expressing cells at the midline of the embryo (Fig. 2E). Asymmetric expression in the anterior of the embryo is apparently a brief event, since the majority of embryos examined had either symmetric or no expression of lrd in this region (Fig. 2G).
Further analysis by serial section in situ hybridization showed continued lrd expression in E8.5 endoderm near the foregut/midgut junctional area (Fig. 3A,B). At this developmental stage, no signal was seen in the neural groove or developing heart. No lrd expression was detected in the notochord at any stage. At E9.5, lrd expression was observed in the endodermal lining of the midgut and hindgut (Fig. 3C). Expression in these regions was essentially undetectable by E10.5. At this stage, lrd expression appeared in the floorplate of the developing brain and neural tube, in a LR symmetrical pattern (Fig. 3D). At E10.5, floorplate expression was restricted to the anterior of the embryo, with no expression in the posterior neural tube (Fig. 3E). By E12.5, expression in the floorplate extended to the posterior of the neural tube (Fig. 3F). In addition, lrd expression was observed in mesenchymal cells ventral to the notochord (Fig. 3F, inset). This region does not appear to correspond to any particular embryonic structure. At E12.5 there was also strong expression in the epithelium lining the roof of the fourth ventricle of the brain (Fig. 3G). Expression in the developing limb was observed at E12.5 (Fig. 3H), and may correspond to regions of cartilage condensation. In newborn mice, expression was seen in several ciliated cell types, including the epithelial lining of the nasal cavity (Fig. 3I) and the ependymal lining of the third ventricle of the brain (Fig. 3J).
Fig. 3.
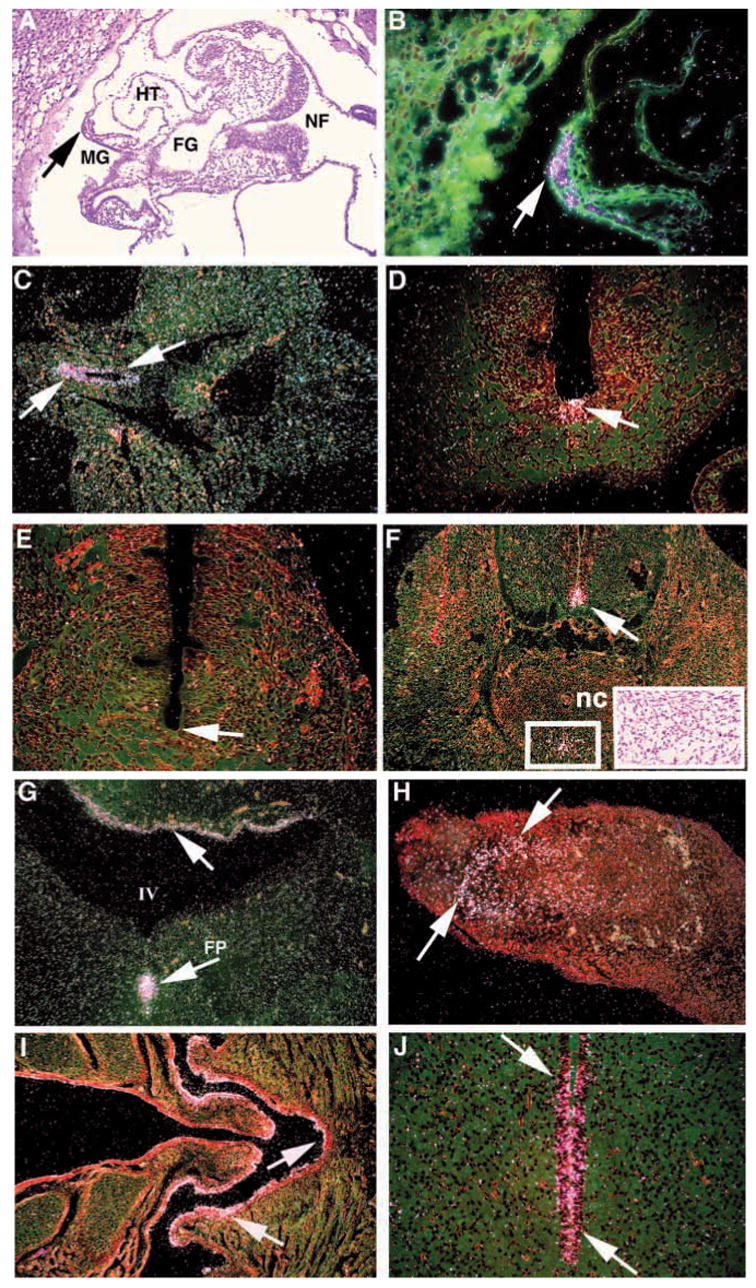
Expression of lrd localized by serial section in situ hybridizations. (A–J) Hybridization to antisense lrd riboprobe; no signal was detected with a sense control probe (not shown). (A) Bright-field transverse section of an E8.5 embryo, showing the locations of the heart tube (HT), midgut region (MG), foregut (FG) and neural fold (NF). (B) Dark-field, magnification of A, showing lrd expression the endoderm adjacent to developing midgut (arrow). (C) E9.5 embryo showing expression in the hindgut (arrows). (D) Transverse section through the abdominal region of an E10.5 embryo showing expression in the floor plate of the anterior neural tube (arrow). (E) The posterior neural tube shows no hybridization (arrow). (F) Transverse section through the abdominal region of an E 12.5 embryo showing expression in the floorplate of the neural tube (arrow) and a region of cells (boxed) ventral to the notochord (nc). These midline cells are distinguished only by their mesenchymal character (insert, bright field). (G) Expression of lrd in E12.5 brain (arrows), in the epithelium lining the fourth ventricle (IV) and in the floorplate (FP). (H) lrd expression in mesenchyme of the limb of an E12.5 embryo (arrows). (I,J) Transverse sections through the head of a newborn mouse showing lrd expression (arrows) in the ciliated epithelium lining of the nasal cavity (I) and the ependymal lining of the third ventricle of the brain (J).
Deletion of the catalytic first P-loop of LRD by gene targeting
Previous studies suggested, but did not prove, the involvement of lrd in LR development. For example, while it was cloned from the lgl deletion, this region is large and encompasses many genes (Supp et al., 1996). In addition, the lrd gene in the iv mouse was found to carry a base difference that results in a coding change at a highly conserved position in the motor domain of the protein (Supp et al., 1997). Nevertheless, the original mouse stock on which the spontaneous iv mutation arose is no longer available for comparison, so it remained possible that this base change represented a polymorphism. Finally, expression of lrd was found in the node of the embryo (Supp et al., 1997), which again suggested a role in LR axis formation, but did not prove it. To confirm the role of lrd in LR specification, to determine if lrd is required for limb or gut development and to test models for the mechanism of lrd function, a targeted mutation was generated.
Several studies have indicated that, of the multiple P-loop motifs in the motor domain of dyneins, only the first P-loop is actually involved in ATP hydrolysis (Gibbons et al., 1991; Gee et al., 1997). To generate a protein unable to catalyze ATP hydrolysis and therefore without motor activity, we deleted two exons of the lrd gene that encompass the first P-loop (Fig. 4A) by homologous recombination in ES cells, and used these cells to generate heterozygous mice. Mice were genotyped by PCR (Fig. 4B). RT-PCR analysis of heterozygous mice showed the expected products for wild-type and targeted alleles (Fig. 5). The targeting vector was designed such that mRNA splicing would generate a transcript with the correct reading frame that lacked the first P-loop. To confirm correct processing, mutant lrd RT-PCR products were subcloned and sequenced. Six of seven clones analyzed showed precisely correct splicing around the deleted exons, using the predicted splice donor and acceptor sequences of the flanking exons. The seventh clone skipped the exon immediately 3′ of the deleted exons but also failed to generate a frameshift mutation. Taken together, the RT-PCR studies strongly suggest that a significant fraction of lrd transcripts from the knockout allele were properly processed around the two deleted exons to retain the reading frame.
Fig. 5.
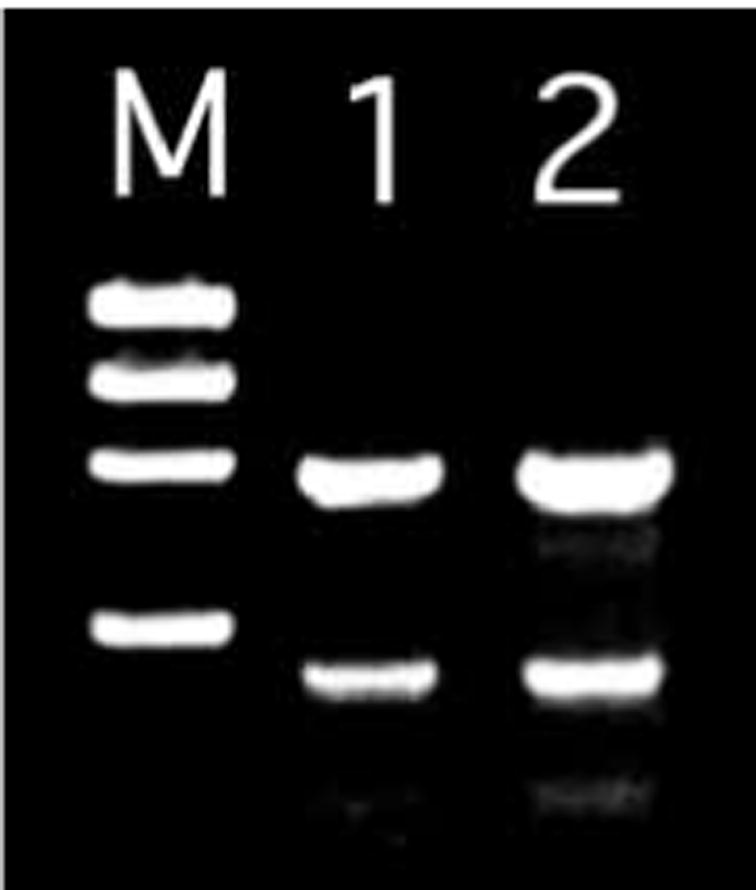
Molecular analysis of RNA splicing of lrd targeted allele. RT-PCR analysis of +/lrd RNA was performed using primers flanking the targeted deletion, and resulted in an 844 bp product from the wild-type allele and a 547 bp product from the mutant allele (lanes 1,2). Doubling of the input RT (lane 2) product resulted in an approximate doubling of the PCR product bands, suggesting that the reaction conditions were within the linear range. M, φX174 RF DNA/HaeIII molecular weight standard.
Randomized LR development in targeted lrd/lrd mutants
Mice heterozygous for the lrd targeted mutation were overtly normal. Of an initial set of 85 progeny of matings between lrd heterozygotes which were genotyped at weaning, 16 mice (19%) were found to be lrd−/− homozygotes, 50 (59%) were heterozygotes and 19 (22%) were wild type. This is not statistically different from the expected 1:2:1 ratio, indicating an absence of embryonic lethality. Gross examination of neonates revealed that approximately half of homozygous mutants had inverted right-sided stomachs. No phenotype was detected in heterozygotes. To better define the LR phenotype, 31 lrd homozygous mutant mice were dissected and examined for laterality of the lungs, heart, stomach, liver and spleen. Fifteen had normal laterality for these organs, with a single-lobed lung on the left and a multiple-lobed lung on the right, heart apex on the left, normal liver lobe pattern, and stomach and spleen on the left. Twelve animals showed a complete reversal of positioning of these organs. In addition, two animals showed heterotaxia, a discordant positioning of individual organs within the same animal, with the heart and liver normal but the stomach and spleen reversed. One of these had normal lungs and the other had an isomerism, with both left and right lungs showing a single-lobed (left-sided) pattern. The remaining two animals had left lung isomerism with one having otherwise normal situs, and other having reversed situs. These findings are similar to those obtained from the analysis of liveborn iv mutant mice.
Gross malformations of the limbs and face, such as those seen in lgl/lgl mutant mice (McNeish et al., 1990), were not observed in the targeted lrd/lrd homozygous mutants. Skeletal staining of lrd/lrd mutants was performed to look for more subtle malformations, and none were found.
Nodal cilia of the lrd mutants are present but immotile
Adult ciliary function is normal in lrd mutants. Male lrd/lrd mice are fertile, indicating the presence of motile, functioning sperm. In addition, tracheal cilia are present and beat (data not shown). The absence of defects in sperm tails and tracheal cilia was previously reported for the iv/iv mutant mice (Handel and Kennedy, 1984).
The nodes of E7.5 lrd/lrd and iv/iv mutants were examined by scanning electron microscopy, and normal monocilia were observed (Fig. 6). The cilia in these mutant embryos were the same size and distribution as seen in wild-type embryos. The nodal cilia of freshly dissected embryos were also examined by light microscopy. It was observed that the wild-type cilia of the node exhibited a ‘vortical’ motion as reported by Nonaka et al. (1998). Of interest, the lrd mutant cilia were rigid and immotile (http://genome.chmcc.org/cilia/ and http://www.biologists.com/Development/movies/dev3019.html).
Fig. 6.

Monocilia on the nodes of E7.5 wild-type and lrd mutant embryos. Scanning electron micrographs of embryonic nodes showing the presence of cilia on the ventral cell surfaces. Wild type is shown on the left and mutant (homozygous for targeted lrd allele) on the right. Embryos homozygous for the iv spontaneous mutant allele of lrd also had node cilia that appeared indistinguishable from wild type (data not shown). The cilia are approximately 1.5 μm in length.
DISCUSSION
lrd is required only for LR development
Previous results, including the expression of lrd in the node and the presence of a mis-sense base change in the lrd gene of the iv mouse strain, strongly suggested but did not prove lrd function in LR determination. The randomized laterality observed in lrd targeted mice demonstrates conclusively that lrd is required for normal LR development and confirms that the iv and lrd genes are the same. The three mutant alleles of lrd can therefore now be referred to as lrdiv, lrdlgl and lrdΔP1.
Loss of LRD motor function appears to only affect LR development. In this regard, the lrd mutation is unique among those affecting LR asymmetries. Mutations of lefty-1 (Meno et al., 1998), kif3A (Marszalet et al., 1999; Takeda et al., 1999), kif3B (Nonaka et al., 1998), Ft (Heymer et al., 1997), inv (Yokoyama et al., 1993), Mgat (Metzler et al., 1994; L. A. L. and M. R. K., unpublished), SIL (Izraeli et al., 1999), and nt (Melloy et al., 1998) all result in prenatal or postnatal lethality that is often accompanied by severe midline and/or anteroposterior defects. The expression of lrd in the developing limbs and the truncated hindlimbs in mice homozygous for the lgl deletion had suggested a possible function for lrd in limb development. It remains possible that the lrd transcripts from the targeted lrdΔP1 allele retain function in limb development but not LR axis formation, resulting in a less severe phenotype than the lrdlgl allele, which has a deletion of the entire lrd gene. A more likely explanation is that additional gene(s) deleted by the lgl transgene insertional mutation are responsible for the lgl/lgl mutant limb defects.
Targeted lrdΔP1 homozygous mutant mice did not exhibit the poor reproductive performance or the relatively high resorption rate and resultant small litter size observed with mice of the lrdiv strain (Brown et al., 1989). This may represent a modification of the phenotype due to genetic background differences, or it may be due to subtle differences between the targeted lrdΔP1 allele and the spontaneous lrdiv mutation.
Motile tracheal cilia were present in lrdΔP1 mutant mice, and males were fertile, indicating normal sperm motility. In contrast, human males with Kartagener syndrome, which display randomized visceral asymmetry, are generally sterile with immotile sperm and suffer respiratory tract infections due to immotile tracheal cilia (Afzelius, 1976). The results of the lrd gene targeting in this report indicate that a dynein functional disturbance could be directly responsible for the laterality defects of Kartagener patients. The differences in the lrd mutant and Kartagener phenotypes, however, suggests the involvement of distinct dynein genes, or dramatically different mutant alleles.
lrd may be involved in both establishment and maintenance of LR asymmetry
Expression of lrd in the node at the late primitive streak/early head fold stage is consistent with a role in LR determination. Other genes implicated in LR development are also expressed in and around the node at these stages, including nodal (Lowe et al., 1996; Collignon et al., 1996), lefty (Meno et al., 1996), HNF3β (Collignon et al., 1996; Dufort et al., 1998), Shh (Echelard et al., 1993; Izraeli et al., 1999) and kif3B (Nonaka et al., 1998). The striking pattern of lrd expression observed in slightly older embryos is altogether novel. Roughly 50% of embryos at this stage (0–5 somites) showed expression of lrd in all three germ layers, at the anterior of the embryo in the region of the erupting head folds. Some embryos displayed left-sided expression, some showed right-sided expression and some had bilateral expression. This period of expression, especially the asymmetric expression, is apparently fleeting since it is observed only in a fraction of embryos analyzed. Direct correlation between sidedness of lrd expression and somite number was not observed. Overlapping this period, but beginning slightly later, there is lateral asymmetric gene expression of nodal, lefty-2 and Pitx2 in the left lateral plate mesoderm and SnR in the right lateral plate mesoderm. lrd expression, however, is not in the lateral plate, but is more anterior, dorsal to the precardiac region. It is tempting to speculate that this transient asymmetric domain of lrd expression functions in the transfer of LR patterning information from the midline node to influence the rightward looping of the heart tube. This lrd expression also suggests a possible role in the establishment of anterior central nervous system asymmetries.
The only defect detected in the lrdΔP1/lrdΔP1mutant gut was randomized laterality. This suggests that the observed lrd expression in the developing gut may be involved in LR specification. Although the gut tube begins to close at E8.5, the gut does not initiate handed asymmetric coiling until after mid-gestation. Expression of lrd in the gut endoderm is only found up to E9.5, suggesting that lrd may not directly function in the lateralization of the gut, but rather acts through an indirect mechanism as proposed above for heart looping.
Expression of lrd in E10.5–12.5 embryos was found in the floorplate of the neural tube. This midline structure has been implicated in LR specification, although at earlier stages of embryonic development. For example, extirpation of the notochord, hypochord and floorplate from Xenopus embryos, at stages when the neural tube is still open, randomizes LR asymmetries and disrupts the normal left-sided expression of Xnr-1 (Lohr et al., 1997). Furthermore, several mutations in zebrafish (Danos and Yost, 1996; Goldstein et al., 1998) causing midline defects and mutations in mice causing defects in either the node, notochord or neural tube (Metzler et al., 1994; Heymer et al., 1997; Melloy et al., 1998; King et al., 1998; Izraeli et al., 1999; Takeda et al., 1999) lead to randomization of LR asymmetries. In addition, mutation of lefty-1, which is expressed asymmetrically in the floorplate, results in isomerisms (Meno et al., 1998). The likely role of these midline structures including the floorplate, early in the development of the LR axis, is either in signaling (Lohr et al., 1997; Izraeli et al., 1999) or as a midline barrier (Meno et al., 1998; Vogan and Tabin, 1999). Expression of lrd in the floorplate at later stages, after morphological asymmetries have been established, may indicate a heretofore unrealized role for the floorplate in maintaining laterality or in patterning later occurring asymmetries.
Models for lrd function in LR development
Several models for LR axis determination have been proposed (Brown and Wolpert, 1990; Klar, 1994; Srivistava, 1997; Levin and Mercola, 1998; Nonaka et al., 1998; Vogan and Tabin, 1999). These models can be grouped into two categories: those that require LRD motive force and those that do not. One model, based on the binding of a Gli-family transcription factor by the kinesin-like molecule costal2 in Drosophila, invokes a cytoplasmic anchoring function for LRD (Srivastava, 1997). According to this model, LRD is held in the cytoplasm by its binding to microtubules, and LRD in turn binds to a transcription factor, perhaps Zic3, a transcription factor mutated in X-linked heterotaxias in humans (Gebbia et al., 1997). Binding of LRD to Zic3 would hold it in the cytoplasm, until a signal (Shh for the costal2-Gli pathway in Drosophila) triggers its release. Upon release the transcription factor would translocate to the nucleus and modulate gene expression patterns. According to this model, LRD would respond to an asymmetric signal and represent a component of a signal transduction pathway. This model requires LRD binding to microtubules and the putative transcription factor, but not motive force. Srivastava (1997) suggested that the mis-sense mutation in the motor domain encoding region of the lrdiv allele perturbed transcription factor binding and hence disrupted LR determination function. The lrdΔP1 allele carries a mutation that alters a distinct domain of the LRD protein that is also required for motor activity. It is quite unlikely that both of these mutations would eliminate the proposed transcription factor binding domain, and neither alters the known microtubule binding domain. The results suggest, therefore, that LRD must have motor activity to perform normal LR axis specification.
Other models have been proposed that require LRD motive force for LR specification. For example, an attractive model originally proposed by Brown and Wolpert (1990) and recently updated by Levin and Mercola (1998) proposes a handed asymmetric molecule, represented by the letter ‘F’, aligning in a specific fashion with respect to the AP and DV axes and thereby orienting the LR axis. In this model, LR asymmetries would result from an asymmetric movement of molecules or cellular components directed by the ‘F’ molecule. The results presented here support this model and suggest that the function of the hypothetical ‘F’ molecule is supplied by node monocilia.
The ‘nodal flow’ model also requires LRD motive force. It was observed that, in wild-type embryos, the monocilia of the node undergo ‘vortical’ motion that results in a leftward ‘nodal flow’ of extraembryonic fluid (Nonaka et al., 1998). This could hypothetically create a LR asymmetry of a soluble morphogen in the extracellular fluid of the node (Nonaka et al., 1998). In this report, we have shown, as was predicted by others (Nonaka et al., 1998; Vogan and Tabin, 1999), that LRD motor function is required for normal motility of the node monocilia.
The phenotypes of mice with targeted mutations in the kif3A and kif3B genes support but do not prove the ‘nodal flow’ model. These mice have absent node monocilia and defective LR development (Marszalek et al., 1999; Takeda et al., 1999; Nonaka et al., 1998). The KIF3A and KIF3B proteins, together with the non-motor KAP3 protein, form the heterotrimeric kinesin complex. This complex functions as a plus-end directed microtubule motor, and the heterotrimeric kinesin complex has been shown to be required for ciliary assembly in sea urchin and Chlamydomonas (Morris and Scholey, 1999). Gene targeting demonstrated the requirement for KIF3A and KIF3B in assembly of node monocilia in mice and implicated these cilia in the development of LR asymmetry. Nevertheless, monocilia are unusual in structure and have been proposed to have multiple functions in addition to motility. Monocilia have a 9+0 microtubule organization instead of the more common 9+2. Such cilia have been generally considered non-motile and have been suggested to have a sensory function, or to indicate cell polarity (Wheatley et al., 1996), or to play a midline barrier role (Vogan and Tabin, 1999). In the kif3A/B mutants, the node monocilia are entirely absent, removing all ciliary function, not just motility. In lrd mutants, the node monocilia are present but immotile, presumably more precisely perturbing nodal flow.
The lrd and kif3A/B mutations perturb nodal cilia and LR development, but the LR phenotypes are not identical. In kif3B mutants, embryonic turning is not randomized as in lrdΔP1 or lrdiv mutants. Instead most kif3B−/− mice fail to turn altogether and die by E10. More interesting, in kif3B mutants, expression of lefty is absent or bilateral (symmetric), in contrast to the randomly asymmetric pattern observed in lrdiv mutants. Mutants lacking kif3A also die by E10 and display neural tube degeneration and mesodermal and caudal dysgenesis in addition to absence of node cilia and LR randomization (Marszalek et al., 1999; Takeda et al., 1999). The different developmental defects can be at least partly explained by much broader developmental roles for kif3A and kif3B. In striking contrast to the kinesin mutations, there are only two discernable abnormalities in lrdΔP1 mutant mice: the node cilia in E7.5 embryos are rigid rods that do not exhibit normal vortical motion, and there is random development of LR asymmetry, without other developmental defects. The observed ‘rigor’ state of the lrdΔP1 mutant cilia is consistent with the report that cytoplasmic dynein with a defective P-loop irreversibly binds microtubules (Gee et al., 1997). The distinct LR phenotypes argue that the lrd and kif3A/kif3B mutations do not both work exclusively through identical blocks of nodal flow.
The data presented here strongly support the hypothesis that node cilia are motile and that it is motility of these cilia that is required to generate left-right asymmetry. Thus, ‘F’ is the node monocilium: a chiral macromolecular structure that is oriented relative to the existing AP and DV axes. This structure can convert its inherent chirality to organismal handedness by directional movement of a soluble substance at the node powered by the left-right dynein motor.
Footnotes
The authors wish to thank John Dunlop and Kyle Vogan for scanning electron micrographs, Diane Bodenmiller, Jen Lachey, Pam Groen, Kathy Saalfeld and Lisa Artmayer for technical support, and Alicia Emley for photographic assistance. Also, special thanks to the Webmaster, Bruce Aronow, for creating the web movies of wild-type and mutant cilia. This work was supported by NIH grants HD24517 to S. S. P., HD36439-01to M. B., HD07200 to D. M. S., AHA (OH-WV Affiliate) grant SW-96-43-S to D. P. W. and a grant from the Hood foundation to M. B.
References
- Afzelius BA. A human syndrome caused by immotile cilia. Science. 1976;193:317–319. doi: 10.1126/science.1084576. [DOI] [PubMed] [Google Scholar]
- Brown NA, Hoyle CI, McCarthy A, Wolpert L. The development of asymmetry: the sidedness of drug-induced limb abnormalities is reversed in situs inversus mice. Development. 1989;107:637–642. doi: 10.1242/dev.107.3.637. [DOI] [PubMed] [Google Scholar]
- Brown NA, Wolpert L. The development of handedness in left/right asymmetry. Development. 1990;109:1–9. doi: 10.1242/dev.109.1.1. [DOI] [PubMed] [Google Scholar]
- Campione M, Steinbeisser H, Schweickert A, Deissler K, van Bebber F, Lowe LA, Nowotshcin S, Viebahn C, Haffter P, Kuehn MR, Blum M. The homeobox gene Pitx2: mediator of asymmetric left-right signaling in vertebrate heart and gut looping. Development. 1999;126:1225–1234. doi: 10.1242/dev.126.6.1225. [DOI] [PubMed] [Google Scholar]
- Collignon J, Varlet I, Robertson EJ. Relationship between asymmetric nodal expression and the direction of embryonic turning. Nature. 1996;381:155–158. doi: 10.1038/381155a0. [DOI] [PubMed] [Google Scholar]
- Danos MC, Yost HJ. Linkage of cardiac left-right asymmetry and dorsal-anterior development in Xenopus. Development. 1995;121:1467–1474. doi: 10.1242/dev.121.5.1467. [DOI] [PubMed] [Google Scholar]
- Danos MC, Yost HJ. Role of notochord in specification of cardiac left-right orientation in zebrafish and Xenopus. Dev Biol. 1996;177:96–103. doi: 10.1006/dbio.1996.0148. [DOI] [PubMed] [Google Scholar]
- Dufort D, Schwartz L, Harpal K, Rossant J. The transcription factor HNF3beta is required in visceral endoderm for normal primitive streak morphogenesis. Development. 1998;125:3015–3025. doi: 10.1242/dev.125.16.3015. [DOI] [PubMed] [Google Scholar]
- Echelard Y, Epstein DJ, St-Jacques B, Shen L, Mohler J, McMahon JA, McMahon AP. Sonic hedgehog, a member of a family of putative signaling molecules, is implicated in the regulation of CNS polarity. Cell. 1993;75:1417–1430. doi: 10.1016/0092-8674(93)90627-3. [DOI] [PubMed] [Google Scholar]
- Gebbia M, Ferrero GB, Pilia G, Bassi MT, Aylsworth AS, Penman-Splitt M, Bird LM, Bamforth JS, Burn J, Schlessinger D, Nelson DL, Casey B. X-linked situs abnormalities result from mutations in ZIC3. Nature Gen. 1997;17:305–308. doi: 10.1038/ng1197-305. [DOI] [PubMed] [Google Scholar]
- Gee MA, Heuser JE, Vallee RB. An extended microtubule-binding structure within the dynein motor domain. Nature. 1997;390:636–639. doi: 10.1038/37663. [DOI] [PubMed] [Google Scholar]
- Gibbons IR, Gibbons BH, Mocz G, Asai DJ. Multiple nucleotide-binding sites in the sequence of dynein b heavy chain. Nature. 1991;352:640–643. doi: 10.1038/352640a0. [DOI] [PubMed] [Google Scholar]
- Goldstein AM, Ticho BS, Fishman MC. Patterning the heart’s left-right axis: from zebrafish to man. Dev Genet. 1998;22:278–287. doi: 10.1002/(SICI)1520-6408(1998)22:3<278::AID-DVG9>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- Handel MA, Kennedy JR. Situs inversus in homozygous mice without immotile cilia. J Hered. 1984;75:498. doi: 10.1093/oxfordjournals.jhered.a109995. [DOI] [PubMed] [Google Scholar]
- Heymer J, Kuehn M, Ruther U. The expression pattern of nodal and lefty in the mouse mutant Ft suggests a function in the establishment of handedness. Mech Dev. 1997;66:5–11. doi: 10.1016/s0925-4773(97)00084-1. [DOI] [PubMed] [Google Scholar]
- Holzbaur ELF, Vallee RB. Dyneins: molecular structure and cellular function. Annu Rev Cell Biol. 1994;10:339–372. doi: 10.1146/annurev.cb.10.110194.002011. [DOI] [PubMed] [Google Scholar]
- Hummel KP, Chapman DB. Visceral inversion and associated anomalies in the mouse. J Hered. 1959;50:9–13. [Google Scholar]
- Hyatt BA, Yost HJ. The left-right coordinator: the role of Vg1 in organizing left-right axis formation. Cell. 1998;93:37–46. doi: 10.1016/s0092-8674(00)81144-7. [DOI] [PubMed] [Google Scholar]
- Isaac A, Sargent MG, Cooke J. Control of vertebrate left-right asymmetry by a Snail-related zinc finger gene. Science. 1997;275:1301–1304. doi: 10.1126/science.275.5304.1301. [DOI] [PubMed] [Google Scholar]
- Izraeli S, Lowe LA, Bertness V, Good DJ, Dorward DW, Kirsch IR, Kuehn MR. The SIL gene is required for mouse embryonic axial development and left-right specification. Nature. 1999;399:691–694. doi: 10.1038/21429. [DOI] [PubMed] [Google Scholar]
- Kandl KA, Forney JD, Asai DJ. The dynein genes of Paramecium tetraurelia: the structure and expression of ciliary β and cytoplasmic heavy chains. Mol Biol Cell. 1995;6:1549–1562. doi: 10.1091/mbc.6.11.1549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King T, Beddington RSP, Brown NA. The role of the brachyury gene in heart development and left-right specification in the mouse. Mech Dev. 1998;79:29–37. doi: 10.1016/s0925-4773(98)00166-x. [DOI] [PubMed] [Google Scholar]
- Klar AJS. A model for specification of the left-right axis in vertebrates. Trends Genet. 1994;10:392–396. doi: 10.1016/0168-9525(94)90055-8. [DOI] [PubMed] [Google Scholar]
- Layton WM. Random determination of a developmental process. J Hered. 1976;67:336–338. doi: 10.1093/oxfordjournals.jhered.a108749. [DOI] [PubMed] [Google Scholar]
- Levin M, Johnson RL, Stern CD, Kuehn M, Tabin C. A molecular pathway determining left-right asymmetry in chick embryogenesis. Cell. 1995;82:803–814. doi: 10.1016/0092-8674(95)90477-8. [DOI] [PubMed] [Google Scholar]
- Levin M, Pagan S, Roberts DJ, Cooke J, Kuehn MR, Tabin CJ. Left/right patterning signals and the independent regulation of different aspects of situs in the chick embryo. Dev Biol. 1997;189:57–67. doi: 10.1006/dbio.1997.8662. [DOI] [PubMed] [Google Scholar]
- Levin M, Mercola M. The compulsion of chirality: toward an understanding of left-right asymmetry. Genes Dev. 1998;12:763–769. doi: 10.1101/gad.12.6.763. [DOI] [PubMed] [Google Scholar]
- Logan M, Pagan-Westphal SM, Smith DM, Paganessi L, Tabin CJ. The transcription factor Pitx2 mediates situs-specific morphogenesis in response to left-right asymmetric signals. Cell. 1998;94:307–317. doi: 10.1016/s0092-8674(00)81474-9. [DOI] [PubMed] [Google Scholar]
- Lohr JL, Danos MC, Yost HJ. Left-right asymmetry of a nodal-related gene is regulated by dorsoanterior midline structures during Xenopus development. Development. 1997;124:1465–1472. doi: 10.1242/dev.124.8.1465. [DOI] [PubMed] [Google Scholar]
- Lowe LA, Supp DM, Sampath K, Yokoyama T, Wright CVE, Potter SS, Overbeek P, Kuehn MR. Conserved left-right asymmetry of nodal expression and alterations in murine situs inversus. Nature. 1996;381:158–161. doi: 10.1038/381158a0. [DOI] [PubMed] [Google Scholar]
- Marszalek JR, Ruiz-Lozano P, Roberts E, Chien KR, Goldstein LS. Situs inversus and embryonic ciliary morphogenesis defects in mouse mutants lacking the KIF3A subunit of kinesin-II. Proc Natl Acad Sci USA. 1999;96:5043–5048. doi: 10.1073/pnas.96.9.5043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNeish JD, Thayer J, Walling K, Sulik KK, Potter SS, Scott WJ. Phenotypic characterization of the transgenic mouse insertional mutation, legless. J Exp Zool. 1990;253:151–162. doi: 10.1002/jez.1402530205. [DOI] [PubMed] [Google Scholar]
- Melloy PG, Ewart JL, Cohen MF, Desmond ME, Kuehn MR, Lo CW. No turning, a mouse mutation causing left-right and axial patterning defects. Dev Biol. 1998;193:77–89. doi: 10.1006/dbio.1997.8787. [DOI] [PubMed] [Google Scholar]
- Meno C, Saijoh Y, Fujii H, Ikeda M, Yokoyama T, Yokoyama M, Toyoda Y, Hamada H. Left-right asymmetric expression of the TGFβ-family member lefty in mouse embryos. Nature. 1996;381:151–155. doi: 10.1038/381151a0. [DOI] [PubMed] [Google Scholar]
- Meno C, Shimono A, Saijoh Y, Yashiro K, Mochida K, Ohishi S, Noji S, Kondoh H, Hamada H. Lefty-1 is required for left-right determination as a regulator of lefty-2 and nodal. Cell. 1998;94:287–297. doi: 10.1016/s0092-8674(00)81472-5. [DOI] [PubMed] [Google Scholar]
- Metzler M, Gertz A, Sarkar M, Schachter H, Schrader JW, Marth JD. Complex asparagine-linked oligosaccharides are required for morphogenic events during post-implantation development. EMBO J. 1994;13:2056–2065. [Google Scholar]
- Morris RL, Scholey JM. Heterotrimeric kinesin-II is required for the assembly of motile 9+2 ciliary axonemes on sea urchin embryos. J Cell Biol. 1999;138:1009–1022. doi: 10.1083/jcb.138.5.1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nonaka S, Tanaka Y, Okada Y, Takeda S, Harada A, Kanai Y, Kido M, Hirokawa N. Randomization of left-right asymmetry due to loss of nodal cilia generating leftward flow of extraembryonic fluid in mice lacking KIF3B motor protein. Cell. 1998;95:829–837. doi: 10.1016/s0092-8674(00)81705-5. [DOI] [PubMed] [Google Scholar]
- Pagan-Westphal SM, Tabin CJ. The transfer of left-right positional information during chick embryogenesis. Cell. 1998;93:25–35. doi: 10.1016/s0092-8674(00)81143-5. [DOI] [PubMed] [Google Scholar]
- Piedra ME, Icardo JM, Albajar M, Rodriguez-Rey JC, Ros MA. Pitx2 participates in the late phase of the pathway controlling left-right asymmetry. Cell. 1998;94:319–324. doi: 10.1016/s0092-8674(00)81475-0. [DOI] [PubMed] [Google Scholar]
- Ryan AK, Blumberg B, Rodriquez-Esteban C, Yonei-Tamura S, Tamura K, Tsukui T, de la Pena J, Sabbagh W, Greenwald J, Choe S, Norris DP, Roberston EJ, Evans EM, Rosenfeld MG, Belmonte JCI. Pitx2 determines left-right asymmetry of internal organs in vertebrates. Nature. 1998;394:545–551. [Google Scholar]
- Sefton M, Sanchez S, Nieto MA. Conserved and divergent roles for members of the Snail family of transcription factors in the chick and mouse embryo. Development. 1998;125:3111–3121. doi: 10.1242/dev.125.16.3111. [DOI] [PubMed] [Google Scholar]
- Splitt MP, Burn J, Goodship J. Defects in the determination of left-right asymmetry. J Med Genet. 1996;33:498–503. doi: 10.1136/jmg.33.6.498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srivastava D. Left, right... which way to turn? Nat Genet. 1997;17:305–308. doi: 10.1038/ng1197-252. [DOI] [PubMed] [Google Scholar]
- Supp DM, Witte DP, Branford WW, Smith EP, Potter SS. Sp4, a member of the Sp1-family of zinc finger transcription factors, is required for normal murine growth, viability, and male fertility. Dev Biol. 1996;176:284–299. doi: 10.1006/dbio.1996.0134. [DOI] [PubMed] [Google Scholar]
- Supp DM, Witte DP, Potter SS, Brueckner M. Mutation of an axonemal dynein affects left-right asymmetry in inversus viscerum mice. Nature. 1997;389:963–966. doi: 10.1038/40140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeda S, Yonekawa Y, Tanaka Y, Okada Y, Nonaka S, Hirokawa N. Left-right asymmetry and kinesin superfamily protein KIF3A: new insights in determination of laterality and mesoderm induction by kif3A−/− mice analysis. J Cell Biol. 1999;145:825–836. doi: 10.1083/jcb.145.4.825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogan KJ, Tabin CJ. A new spin on handed asymmetry. Nature. 1999;397:295–298. doi: 10.1038/16796. [DOI] [PubMed] [Google Scholar]
- Wheatley DN, Wang AM, Strugnell GE. Expression of primary cilia in mammalian cells. Cell Biol Inter. 1996;20:73–81. doi: 10.1006/cbir.1996.0011. [DOI] [PubMed] [Google Scholar]
- Yokoyama T, Copeland NG, Jenkins NA, Montgomery CA, Elder FFB, Overbeek PA. Reversal of left-right asymmetry; a situs inversus mutation. Science. 1993;260:679–682. doi: 10.1126/science.8480178. [DOI] [PubMed] [Google Scholar]
- Yoshioka H, Meno C, Koshiba K, Sugihara M, Itoh H, Ishimaru Y, Inoue T, Ohuchi H, Semina EV, Murray JC, Hamada H, Noji S. Pitx2, a bicoid-type homeobox gene, is involved in a lefty-signaling pathway in determination of left-right asymmetry. Cell. 1998;94:299–330. doi: 10.1016/s0092-8674(00)81473-7. [DOI] [PubMed] [Google Scholar]