

Abstract
The effects of normal aging on the primate brain are incompletely understood. Although both human and nonhuman primates demonstrate clear functional declines in selective attention, “executive” functions, and some components of declarative memory with aging, most studies have failed to demonstrate extensive neuronal atrophy or loss as a substrate for these degenerative changes in primates. In particular, extensive age-related neuronal loss in memory-related brain regions such as the hippocampus and entorhinal cortex has not been found. However, it is possible that neuronal loss or atrophy might occur in subcortical nuclei that modulate the activity of neocortical regions, thereby accounting for altered cognitive function with aging. In the present study, we describe, to our knowledge for the first time, a significant and extensive decline in the number and size of immunolabeled neurons in subcortical cholinergic basal forebrain regions of aged rhesus monkeys, the best animal model of human aging, by using stereological methods. Notably, the loss of subcortical cholinergic neuronal markers in aged monkeys was nearly completely reversed by human nerve growth factor gene delivery. These findings (i) identify reversible cellular atrophy as a potential mechanism contributing to age-related cognitive decline in primates, (ii) suggest, when considered with other studies, that subcortical brain regions exhibit greater vulnerability to the effects of aging than cortical regions, and (iii) indicate that neurotrophin gene transfer may be an effective means of preventing neuronal atrophy or degeneration in age-related neurodegenerative disorders.
Keywords: aging, Alzheimer’s disease, nerve growth factor, stereology, cholinergic systems
Declines in cognitive function accompany normal aging in humans and nonhuman primates (1, 2). Impaired function in selective attention, “executive” functions, and some components of declarative memory constitute a regular feature of aging in both humans and in the best animal model of human aging, the rhesus monkey (1, 3). However, the structural basis, if any, for age-related declines in cognition is unclear. To date, studies of neuronal changes with aging in human and nonhuman primates have revealed modest if any reduction in neuronal number or size in several cortical brain regions, including the hippocampus (4), entorhinal cortex (5), and other neocortical regions (6–9). The failure to identify an age-related change in neuronal number has led to the suggestion that synaptic, dendritic, metabolic, or neurophysiological rather than somal structural changes underlie age-related declines in cognitive function (1, 2, 10–15). However, an alternative hypothesis is that structural degenerative changes may occur in subcortical neuronal populations that project to and regulate the function of hippocampal and cortical circuitry. To date, this possibility has not been carefully examined in primates.
Neurons located in the subcortical basal forebrain region provide virtually all cholinergic innervation to the hippocampus and neocortex (16). These basal forebrain subcortical cholinergic neurons, classified as Ch1-Ch4 neurons (16), play an important role in regulating neuronal activity in cortical circuits, and perturbation of this system has been related to deficits in attention and specific aspects of memory (17, 18). The intermediate division of the Ch4 region (Ch4i) projects to several cortical regions and is the primary source of cholinergic innervation to classic memory-mediating structures of the cerebral cortex, including the entorhinal cortex (16). To investigate whether subcortical brain regions that influence cognition exhibit vulnerability to the aged state, neuronal number and size were quantified in Ch4i in the basal forebrain by using unbiased stereological methods. Comparisons were made between aged and nonaged rhesus monkeys. In addition, a subset of aged monkeys received grafts of nerve growth factor (NGF)-secreting autologous fibroblasts to the Ch4 region to determine whether spontaneous age-related neurodegenerative events in the cholinergic basal forebrain, if present, were sensitive to and reversible by growth factor therapy. Previously it has been reported that degenerating basal forebrain cholinergic neurons are sensitive to NGF (19–25). Findings of the present study demonstrate, to our knowledge for the first time, that aging in the primate brain is accompanied by significant and quantitatively extensive neuronal atrophy in subcortical cholinergic neuronal populations. Further, the spontaneous age-related atrophy of these neurons is sensitive to and reversible by neurotrophin gene therapy.
METHODS
Four groups of rhesus monkeys were studied: four adult nonaged nonoperated subjects (mean age 9.4 ± 1.1 yr; three males and one female), three aged nonoperated subjects (mean age 25.1 ± 2.5 yr; all males), four aged monkeys that received intraparenchymal grafts of autologous fibroblasts genetically modified to produce and secrete human NGF (mean age 22.6 ± 0.8 yr; one male and three females), and four aged monkeys that received intraparenchymal control grafts of autologous fibroblasts genetically modified to produce the reporter gene β-galactosidase (β-gal) (mean age 23.3 ± 0.8 yr; three males and one female). All subjects were born at the California Regional Primate Center and spent the majority of their lives in 5-acre field enclosures containing social groups of 50–60 animals. Before the present study, animals were maintained under similar social conditions and were not involved in behavioral studies, surgical procedures, or pharmacological experiments. All animal care procedures adhered to American Association for the Accreditation of Laboratory Animal Care and institutional guidelines.
For NGF delivery to aged subjects, autologous fibroblasts genetically modified to secrete human NGF were prepared as previously described (26, 27). Briefly, autologous fibroblasts obtained from skin biopsies were genetically modified in vitro to produce and secrete the active β-portion of human NGF. Transduction procedures used replication-incompetent retroviral vectors derived from Moloney murine leukemia virus. Transduced cells were selected by growth in G418. Production of biologically active NGF was verified by neurite outgrowth from PC12 cells (28). Production of full length NGF mRNA was determined by Northern blot. NGF protein produced from cells was assayed by NGF ELISA sensitive to 5 pg/ml (29). In vitro before grafting, autologous fibroblasts from NGF-grafted monkeys secreted a mean of 12 ± 2 ng human NGF/106cells/day. Optimal NGF-producing bulk clones were amplified to numbers sufficient for in vivo grafting. Aged control-grafted monkeys received grafts of identical autologous fibroblasts, with the exception that the NGF gene was replaced by the reporter gene Escherichia coli β-gal, which lacks neurotrophic activity. Thus, cells in NGF-grafted and control-grafted subjects differed by a single gene. Cells were harvested in vitro, and a total of 5.0 × 106 cells were grafted into each subject adjacent to the region of the intermediate division of the cholinergic basal forebrain (see surgery description below).
Surgery. Monkeys underwent preoperative MRI scans to visualize basal forebrain targets. After stereotaxic coordinates were generated from MRI scans, each monkey received intraparenchymal grafts of autologous NGF-secreting or control fibroblasts. Monkeys were preanesthetized with 25 mg/kg ketamine i.m. and were anesthetized with isofluorane. Cells were grafted into each of five sites equally spaced over the rostral-caudal extent of the Ch4 region bilaterally (10 grafts total per animal). Grafts were targeted to a position slightly dorsal to but within 500 μm of the Ch4 nucleus. Ten-microliter cell suspensions, 1.0 × 105 cells/μl, were grafted into each site with a 25-gauge Hamilton syringe at a rate of 1 μl/min, for a total of 10 million grafted cells per animal. Postoperatively, all subjects were observed closely for signs of discomfort or toxicity and received the analgesic buprenorphine. Grafted monkeys survived for 3 mo and were then sacrificed by transcardial perfusion as described below.
The duration of in vivo gene expression was examined in two additional rhesus monkeys. One monkey (age 10 yr) received a suspension graft of 25 μl of autologous fibroblasts transduced to express human NGF into the parietal cortex. The stereotaxic coordinates of the injection site were noted, and this region of the brain was freshly dissected 8 mo later and immediately frozen to −80°C until subsequent processing for NGF content by ELISA (29). A second monkey received a control graft of fibroblasts to the parietal cortex that were not genetically modified. This graft too was freshly dissected 8 mo later and immediately frozen for two-site ELISA.
Histology and Stereology. Animals were sedated with 25 mg/kg i.m. ketamine and were then deeply anesthetized with Nembutal (30 mg/kg i.p.). All reflex responses to cutaneous stimulation were verifiably absent before perfusion procedures were begun. Subjects were perfused transcardially for 1 hr with a 4% solution of paraformaldehyde in 0.1 M phosphate buffer at 4°C followed by 5% sucrose solution. Brains were stereotaxically blocked in the coronal plane.
Serial coronal sections through the brain were cut on a freezing microtome set at 40 μm. Every sixth section was processed for p75 immunoreactivity (monoclonal hybridoma cell line generated by M. Bothwell, University of Washington, dilution 1:100) as previously described (21). p75 has been reported to exhibit 95% coassociation with cholinergic neurons of the basal forebrain and labels no other neuronal types in this brain region (30). An additional 1-in-6 sections was Nissl stained for quantification of total neuronal number and to evaluate graft position and survival. Stereological procedures were used to quantify the number of cholinergic neurons in p75-immunolabeled sections and the total number of neurons in Nissl-stained sections. In both p75-immunolabeled and Nissl-stained sections, neurons were quantified in Ch4i. The following anatomical boundaries were used to demarcate Ch4i: (i) the rostral boundary of Ch4i is marked by the appearance of the ansa peduncularis as it begins to penetrate Ch4, beginning dorsomedially and traversing diagonally through Ch4i (dorsomedial to ventrolateral); (ii) the caudal boundary of Ch4i is marked by the completion of the passage of the ansa peduncularis through Ch4 at its ventrolateral extent and the subsequent grouping of cholinergic neurons into a single cluster, the posterior division (Ch4p); (iii) the ventral boundary of Ch4i at its rostral pole is formed by the Ch3 cell group. The cells of this group are smaller than those of Ch4, fusiform in shape with the long axis oriented along the ventral surface of the brain, and appear hypochromic on p75 labeling. The ventral boundary at the caudal aspect of Ch4i is formed by the ventral surface of the brain; and (iv) the dorsal boundary of Ch4i is formed by the globus pallidus. There were no significant differences among the four animal groups in the mean number of tissue sections containing Ch4i (8.6 ± 0.5) used for stereological quantification (ANOVA P = 0.86).
Stereological counts were performed on every sixth section through the entire extent of Ch4i. The stereology equation of West (31) was used to calculate total neuronal numbers:
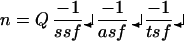 |
where n, calculated total number of neurons; Q = sum of counted points (i.e., neurons); ssf, section-sampling fraction, in this case 1/6; asf, area-sampling fraction or the fraction of the total area that is contained within counting frames, in this case 5%; and tsf, tissue sampling fraction, or the fraction of the thickness of each section within which neurons are counted, in this case 75%.
The optical fractionator method (31) was used to conduct stereology. Identical stereology parameters were used in all quantification procedures by using sampling frames pseudorandomly generated from the neurozoom stereology program (32) or the stereoinvestigator program (MicroBrightField, Colchester, VT) for p75-immunolabeled and Nissl-stained neurons, respectively: fraction (percent of area), 5%; counting frame size x, 66.46 μm; y, 53.73 μm; section thickness, 40 μm.
These parameters were chosen to minimize the coefficient of error of the estimate while maximizing the efficiency of sampling by using the nomogram of Gunderson (33).
The stereology computer programs controlled movement from one counting frame to the next by moving a Ludl motorized stage mounted on an Olympus (New Hyde Park, NY) Vanox AHBT-3 microscope. Ch4i neurons were counted by using a ×60 high numerical aperture (1.40) oil objective (total magnification ×600). Immunolabeled cells were included in the count if (i) they were p75 positive; (ii) the cell nucleus was within the counting frame (or touching the inclusion boundary) but did not touch the exclusion boundary; and (iii) the nucleus was best in focus within the inclusion volume (i.e., the top 12.5% and bottom 12.5% were excluded and the nucleus was best in focus within the volume between the two exclusion volumes). Cells in Nissl-stained sections were counted if (i) they were hyperchromic and either polygonal or fusiform in morphology, as previously described for neurons in the Ch4i region (16); (ii) the cell nucleus was within the counting frame (or touching the inclusion boundary) but did not touch the exclusion boundary; and (iii) the nucleus was best in focus within the inclusion volume. Thus, unbiased estimates of neuronal numbers per subject were generated and compared among groups. (Immunolabeling was noted to be uniform through the z axis of sections from young and aged brains, allowing accurate comparisons between subjects.)
p75/Choline Acetyltransferase (ChAT) Double Labeling and Quantification. To determine whether changes in p75 immunolabeling were paralleled by changes in a more general cholinergic marker, ChAT, double labeling for p75/ChAT was performed. The ratio of p75/ChAT neurons in each subject was then calculated, and comparisons were made between groups. Two coronal sections per animal, chosen to correspond to points exactly 1/3 of the distance through the rostral-caudal extent of Ch4i and 2/3 of the distance through the rostral-to-caudal extent of Ch4i, were processed for p75/ChAT double immunolabeling. Double light-level immunocytochemistry was performed by using the p75 monoclonal antibody (described above) and a polyclonal ChAT antibody (Chemicon; 1:1,000 dilution). Primary antibody incubation procedures were performed sequentially on consecutive days. After incubation in appropriate secondary antibodies, p75 immunoreactive cells were visualized with Vector Red (Vector Laboratories) and ChAT immunoreactive cells were labeled with Vector SG (Vector Laboratories). Vector Red produces a red reaction product and Vector SG, a blue-gray reaction product. Thus, double-labeled cells were readily distinguished by a deep purple color reaction product (see Results). Stereological sampling of single- and double-labeled cells with neurozoom software was performed by using the criteria that cells were (i) p75 positive, ChAT positive, or both; (ii) the cell nucleus was within the counting frame (or touching the inclusion boundary) but did not touch the exclusion boundary; and (iii) the nucleus was best in focus within the inclusion volume. Cells were counted as single labeled for p75 if only the red reaction product was present, single labeled for ChAT if only the blue-gray reaction product was visible within the soma, and double labeled if the cells were purple (see Results). Numbers of p75-, ChAT-, and double-labeled cells in the Ch4i region of each subject were expressed as a ratio of total neuron numbers sampled.
All grafts were also labeled for acetylcholinesterase (AChE) histochemistry as previously described (34, 35) to detect the presence and persistence of cholinergic axonal penetration into NGF-secreting cell grafts.
Quantification of Neuronal Area.
Stereological methods were used to determine differences in p75-immunolabeled neuronal area between groups. Cells were included in the area quantification if they met the morphological criteria described above. The cycloid method was used to measure the cross-sectional area (36, 37). Briefly, a cycloid grid was overlaid on each sampled section in the neurozoom program. The computer mouse was used to “click” on intersections between sampled profiles (Ch4i neurons) and cycloids. The neurozoom program then calculated the cross-sectional area for that neuron. Cycloid spacing was set at 5 μm. Quantification was performed by using a ×60 high numerical aperture (1.40) oil objective. One hundred total neurons per animal were randomly sampled by using the neurozoom software from every p75-labeled section through the full extent of the Ch4i nucleus. The mean cross-sectional area for Ch4i neurons was calculated for each animal, and means were averaged to derive values for each group.
Statistics.
Multiple group comparisons were made by ANOVA with post hoc analysis by using Fisher’s least square difference. Biological significance was established at the 95% confidence level. Data are presented as mean ± SEM.
RESULTS
Normal Cell Numbers and Size. The number of cholinergic neurons in the normal intermediate division of the Ch4 region in four nonaged rhesus monkeys was quantified by using stereological methods and was found to contain a mean of 45,700 ± 6,200 p75-immunolabeled neurons (Figs. 1 and 2). The majority of these neurons also colocalized for ChAT: 93.9 ± 0.7% of p75-immunolabeled neurons were labeled for ChAT (Fig. 3), consistent with previous reports on colocalization of these two markers (30). Mean somal area of Ch4i neurons immunolabeled for p75 was 501 ± 9 μm2 in nonaged subjects.
Figure 1.

p75 immunolabeling in aged and nonaged monkeys. (a) Ch4i region in nonaged monkey exhibits normal distribution of cholinergic neurons. Ch4id: Ch4 nucleus, intermediate-dorsal component; Ch4iv: Ch4 nucleus, intermediate-ventral component. M, medial. Bar = 450 μm in a–d. (Inset) Normal p75-immunolabeled neurons and neurite density at 20-fold higher magnification. (b) Ch4i region in aged nongrafted monkey, showing fewer neurons that appear more pale than those of nonaged subjects. Inset indicates lower neuronal and neurite density than observed in nonaged monkey. (c) In contrast, p75-labeled neurons in monkeys with control grafts exhibit no evident morphological response to the adjacent graft. Neurons resemble those of nongrafted aged subjects in extent of loss of neuronal labeling and neurite density. (d) Ch4i region in aged NGF-grafted monkey shows restoration of neuronal and neuritic density. NGF-secreting fibroblast graft (G) is dark because of dense penetration by p75-labeled cholinergic axons. Many neuronal soma orient in the direction of the NGF-secreting cell source and extend axons into the graft. Inset demonstrates these changes at 20-fold higher magnification.
Figure 2.

Neuronal quantification. Quantification of neuronal number (a) and size (b) in nonaged and aged monkeys. Aged monkeys exhibit a significant decline in the number (ANOVA P < 0.05) and size (P < 0.05) of p75-immunolabeled neurons compared with nonaged monkeys. Aged monkeys that have received grafts of control fibroblasts that express the reporter gene β-gal show losses in cellular markers identical to those observed in nongrafted subjects. In contrast, aged recipients of NGF-secreting cell grafts show significant amelioration of losses in immunolabeled cell numbers and size. Post hoc testing reveals significant losses only in the aged nongrafted and β-gal-grafted groups (∗).
Figure 3.

Technique of double labeling for p75/ChAT. (a) A neuron in the cholinergic basal forebrain is single labeled for p75. (b) A different neuron in the same histological slice is single labeled for ChAT. (c) A third neuron in the same histological slice is double labeled for p75 and ChAT. Bar = 20 μm in a–c.
Nissl counts of all neurons in the Ch4i region were also performed by using stereological methods. Cells (60,300 ± 3,400) with neuronal morphology were present within the same anatomical boundaries in which p75-labeled neurons were quantified (Table 1). Thus, at least 74.7 ± 6.6% of neurons in Ch4i are cholinergic.
Table 1.
Stereological quantification of Nissl-stained neurons in Ch4i region
| Group | No. Nissl-stained neurons (SEM) |
|---|---|
| Nonaged | 60,300 ± 3,400 |
| Aged intact | 57,200 ± 3,900 |
| Aged β-gal grafted | 55,000 ± 5,600 |
| Aged NGF grafted | 53,100 ± 2,400 |
ANOVA P = 0.62
Normal Aging Results in a Decline in Cell Number and Size.
Aging resulted in a significant decline in both the number and size of neurons immunolabeled for p75 (Figs. 1, 2). The number of p75-labeled neurons was significantly reduced in three nonoperated aged monkeys by 43% compared with nonaged monkeys, to 26,000 ± 2,100 neurons (ANOVA P < 0.05). The size of remaining p75-labeled neurons also significantly declined by 10.4% in nonoperated aged monkeys, to 449 ± 9 μm2 (ANOVA P < 0.05). Thus, aging resulted in a significant reduction in the number and size of basal forebrain cholinergic neurons labeled for a specific marker of basal forebrain cholinergic neurons, the p75 low-affinity neurotrophin receptor. The shrinkage in size of remaining neurons suggested that cholinergic neurons were undergoing a general state of atrophy rather than isolated down-regulation of p75 expression. This general state of cholinergic atrophy was confirmed by ChAT/p75 double immunolabeling, which revealed that declines in p75 expression were paralleled by declines in ChAT expression: in aged primates, 92.6 ± 1.2% of p75-labeled neurons also labeled for ChAT. However, the number of Nissl-stained neurons did not significantly decline in aged unoperated subjects compared with nonaged monkeys (57,200 ± 3,900 neurons, P = 0.62; Table 1), indicating that reductions in immunolabeled cell numbers reflected atrophy rather than death.
Reversibility of Age-Related Cholinergic Neuronal Atrophy by NGF Gene Transfer. All NGF-grafted and β-gal-grafted animals showed surviving cellular transplants (Fig. 1c and d). Age-related declines in p75-labeled neuronal number and size were reversed in the four recipients of NGF-secreting cell grafts (Figs. 1, 2). Mean p75-immunolabeled neuronal number in aged recipients of NGF-secreting cells was restored to within 7.7% of values in nonaged subjects (42,200 ± 2,600 neurons), an amount that was not significantly different from nonaged subjects on post hoc analysis. Similarly, neuronal size in recipients of NGF-secreting cells recovered to within 3.5% of measurements in nonaged subjects (484 ± 9 μm2), an amount that also did not differ from nonaged subjects on post hoc analysis. The four aged recipients of control β-gal-secreting cell grafts exhibited significant declines of 43.9% in neuronal number (25,700 ± 4,400) and 13% in neuronal size (435 ± 19 μm2) compared with nonaged monkeys, amounts that were indistinguishable from findings in the unoperated aged subjects. These findings in the aged grafted-control group indicate that the grafting procedure itself neither worsened nor improved age-associated changes in neuronal parameters.
p75/ChAT double labeling revealed that 89.5 ± 0.5% of neurons immunolabeled for p75 were also labeled for ChAT in the NGF graft recipient group, compared with 92.6 ± 0.3% of neurons in the control graft group. This finding supports the observation that NGF restores overall cholinergic function rather than merely up-regulating p75 expression: if p75 expression alone had been augmented by NGF grafts, an increase in the ratio of p75 to ChAT would occur, rather than the slight decrease that was actually observed. Once again, Nissl counts in aged subjects suggested that neurons in cholinergic brain regions were atrophic but not dead: the number of Nissl-stained neurons in aged NGF-treated animals (53,100 ± 2,400) and aged β-gal-grafted animals (55,000 ± 5,600) did not differ significantly from numbers in nonaged subjects (ANOVA = 0.62). Collectively, these data indicate that the stage of aging examined in this study is associated with neuronal atrophy, and that cellularly delivered neurotrophic factors ameliorate this atrophy.
Cholinergic Sprouting Toward the Intraparenchymal NGF Source.
Cholinergic neurons in aged monkeys exhibited somal orientation toward NGF-secreting grafts and extended p75-labeled axons into all of these grafts (Fig. 1c). This neurotropic response was not evident in subjects that received control grafts (Fig. 1d). Thus, a parenchymal NGF source elicits cholinergic process outgrowth in aged subjects in addition to ameliorating somal atrophy. Grafts of genetically modified cells were identifiable in living monkeys on postoperative MRI scans (Fig. 4).
Figure 4.

Cell grafts can be visualized and monitored in the living primate. (a) Grafts of NGF-expressing cells in bilateral basal forebrain cholinergic nuclei are visible on MRI as circular regions of attenuated signal (arrows). (b) The same slice after histological processing reveals healthy graft (G) tissue that is robustly penetrated by p75-labeled cholinergic axons. Bar = 450 μm.
Duration of in Vivo Transgene Expression.
Eight months after in vivo grafting, a freshly dissected NGF graft and surrounding host tissue contained 25 ng human NGF/gm tissue, exceeding by 25-fold the highest physiological levels of NGF production in the cortex. Fresh dissection of a control graft revealed no detectable human NGF. In addition, human NGF-secreting grafts in all four of the aged NGF-grafted subjects were densely penetrated by cholinergic axons on AChE labeling, suggesting that NGF production persisted through the 3-mo experimental period in these subjects (data not shown). Control grafts were rarely penetrated by AChE-labeled axons.
DISCUSSION
Findings of the present study indicate that aging in primates is associated with extensive atrophy of a population of subcortical neurons that modulate cortical activity. The normal number of immunolabeled cholinergic cells in the intermediate division of the primate basal forebrain cholinergic complex is 45,700 ± 6,200 neurons, and the normal size of these neurons is 501 ± 9 μm2. Loss of immunolabeling for p75 occurs in 43% of cholinergic neurons with aging, with parallel reductions in the transmitter ChAT. Remaining cholinergic neurons exhibit a 10% shrinkage in area. These changes in subcortical neurons contrast with reports of either limited or no change in cell number and size with aging in cortical regions in primates, including the hippocampus, entorhinal cortex, presubiculum, and frontal cortex (e.g., refs. 1, 4, 6–9). However, these age-related changes in subcortical cholinergic neurons represent atrophy rather than death, because they are reversible by delivery of the NGF gene at the moderate stage of aging represented by this study.
Interestingly, a recently published stereological study also found significant age-related declines in a different subcortical neuronal system in aged rhesus monkeys: 40% of nigral neurons exhibit spontaneous degeneration with aging (38). Taken together with findings of the present study, the possibility emerges that subcortical systems may be selectively vulnerable to aging. Indeed, preliminary data from the same set of animals examined in the present study indicate a lack of cell loss with aging in entorhinal cortical layers II–III. The possibility that subcortical neural systems are selectively vulnerable to aging compared with cortical systems can be further explored by examining age-related changes in other neuronal populations such as the locus coeruleus and nucleus raphae. These studies are in progress.
Previous studies have reported age-related reductions in markers of basal forebrain cholinergic systems in rodents, although the extent of these reductions has varied considerably between studies. Some of these differences are likely attributable to prescreening of the study population for cognitive performance, the types of markers utilized to study cell number (immunolabeling vs. Nissl stains), methods of neuronal quantification (stereological vs. nonstereological), and the species of rodent. For example, Bäckman et al. (39) reported cell shrinkage with aging in rodents, but no reduction in number of p75-immunolabeled cholinergic neurons. Other studies in aged rodents have reported losses of immunolabeled basal forebrain cholinergic neurons, but only if rats are divided into “aged-impaired” and “aged-unimpaired” groups on the basis of spatial memory performance. Under such circumstances, aged-impaired subjects exhibit reductions in numbers of AChE-stained, ChAT-immunolabeled, or p75-immunolabeled neurons (20, 40, 41), and these reductions vary in extent from 16% (40) to 40% (20). On the other hand, two recent stereological studies of the rodent basal forebrain (42, 43) have reported cell atrophy with aging that more closely approximates findings of the present primate study. Smith and Booze (42) reported a 30% reduction in the number of ChAT-immunolabeled and Nissl-stained neurons in the basal forebrain of rodents that were not prescreened for spatial memory performance, whereas Martinez-Serrano et al. reported a 40% reduction in the number of p75-labeled cholinergic neurons in aged rodents that were preselected for impaired spatial memory function (44). The extent of cholinergic neuronal atrophy in aged primates observed in the present experiment is most consistent with the more severe reports of atrophy in rodents, because (i) primate reductions in cellular immunolabeling occurred in a cohort of subjects unselected for behavioral function, (ii) the neuronal atrophy was consistent among all primate subjects, and (iii) a large proportion (43%) of the primate cholinergic neurons exhibited atrophy.
To our knowledge, this is the first stereological report of age-related changes in cholinergic systems with aging in primates, but a previous nonstereological study also examined age-related changes in a different component of the primate basal forebrain cholinergic system, the medial septal nucleus (Ch1 region) (45). Stroessner-Johnson et al. reported a nonsignificant reduction of 15% in cholinergic neuron number in the medial septum of a small sample of aged monkeys, with a more marked reduction in numbers of caudally located cholinergic neurons compared with rostrally located neurons (45). A 7% increase in cholinergic somal area was also reported in two aged cognitively impaired subjects (45). Although Armstrong et al. also found hypertrophy of cholinergic neurons in aged rodents, cellular hypertrophy occurred in cognitively unimpaired subjects (41) rather than in the aged cognitively impaired subjects reported in the primate study of Stroessner-Johnson et al. (45). In the present primate study, neuronal atrophy was consistently present in all aged subjects in the Ch4i region. Differences in neuronal area measurements between studies probably reflect variations in cholinergic subdivisions, species, methodologies, and sample sizes.
Spontaneous atrophy of aging neurons in primates was reversed by neurotrophin therapy in this study. Delivery of NGF to the region of the cholinergic soma restored the number of immunolabeled neurons to within 8% of values observed in young animals and restored somal size to within 3% of normal. The fact that reductions in neuronal number and size are reversed by neurotrophin therapy supports the contention that these neurons are atrophic rather than dead, at least at this stage of aging, as mentioned above. Two alternative mechanisms of NGF-induced increases in cholinergic cell numbers in the aged primate brain can also be postulated: that neurotrophin delivery induces neurogenesis in the cholinergic basal forebrain, or that NGF delivery induces a switch to the cholinergic phenotype in previously noncholinergic neurons. These two possibilities are unlikely, however, because (i) previous studies of neurogenesis in the mammalian brain have not demonstrated neuronal precursor cells in the cholinergic basal forebrain, (ii) previous studies with NGF protein intracerebroventricular delivery have failed to provide evidence of a switch of neurons to a cholinergic phenotype (19–24), and (iii) quantification of Nissl-stained neurons revealed no significant loss in the present study. Thus, the most likely explanation for the reversibility of cell atrophy is that neurons are beginning a process of degeneration but have not died and can still be returned to a normal transmitter phenotype. Similar reversal of age-associated neuronal atrophy by NGF has been reported in aged rodents (20, 40, 46).
The present findings have potential implications for the treatment of chronic age-related neurodegenerative conditions such as Alzheimer’s disease (AD). Extensive neuronal degeneration occurs in the cholinergic basal forebrain in AD (47, 48). Findings of the present study indicate that intraparenchymal delivery of NGF by ex vivo gene therapy can reverse neuronal atrophy over a broad group of primate cholinergic neurons, without inducing some of the adverse effects that have been observed after intracerebroventricular infusions of NGF. For example, intracerebroventricular NGF infusions in rodents and primates cause weight loss (49) and sprouting of other NGF-sensitive systems such as sympathetic axons and sensory axons (50, 51); these adverse effects were not observed in the present study (data not shown). Gene expression from the vector system used in this study persisted for at least 8 mo in vivo in the primate brain and for at least 14 mo in the rodent central nervous system (52), indicating that intraparenchymal NGF gene delivery may be a practical means of providing NGF to the brain for extended time periods. The cause of cholinergic neuronal degeneration in AD is unknown, although some evidence exists to suggest that defects in NGF trafficking may contribute to cholinergic neuronal decline (53, 54). If true, this defect could be bypassed by NGF delivery directly to the cholinergic neuronal soma. Yet even if abnormalities in NGF trafficking or availability in AD do not occur, the possibility remains that NGF delivery would enhance cholinergic activity and neurotransmission (55).
Acknowledgments
This work was supported by grants from the National Institutes of Health (AG10435, NS37083), Department of Veterans Affairs, and the California Regional Primate Research Center Base.
ABBREVIATIONS
- Ch4i
the intermediate division of the Ch4 region
- NGF
nerve growth factor
- β-gal
β-galactosidase
- ChAT
choline acetyltransferase
- AChE
acetylcholinesterase
References
- 1.Morrison J H, Hof P R. Science. 1997;278:412–419. doi: 10.1126/science.278.5337.412. [DOI] [PubMed] [Google Scholar]
- 2.Peters A, Morrison J H, Rosene D L, Hyman B T. Cereb Cortex. 1998;8:295–300. doi: 10.1093/cercor/8.4.295. [DOI] [PubMed] [Google Scholar]
- 3.Burke D M, Mackay D G. Philos Trans R Soc London. 1997;352:1845–1856. doi: 10.1098/rstb.1997.0170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.West M J, Coleman P D, Flood D G, Troncoso J C. Lancet. 1994;344:769–772. doi: 10.1016/s0140-6736(94)92338-8. [DOI] [PubMed] [Google Scholar]
- 5.Gomez-Isla T, Price J L, McKeel D W, Morris J C, Growdon J H, Hyman B T. J Neurosci. 1996;16:4491–4500. doi: 10.1523/JNEUROSCI.16-14-04491.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pakkenberg B, Gunderson H J. J Comp Neurol. 1997;384:312–320. [PubMed] [Google Scholar]
- 7.Simic G, Kostovic I, Winblad B, Bogdanovic N. J Comp Neurol. 1997;379:482–494. doi: 10.1002/(sici)1096-9861(19970324)379:4<482::aid-cne2>3.0.co;2-z. [DOI] [PubMed] [Google Scholar]
- 8.West M J, Gunderson H J. J Comp Neurol. 1990;296:1–22. doi: 10.1002/cne.902960102. [DOI] [PubMed] [Google Scholar]
- 9.West M J. Neurobiol Aging. 1993;14:287–293. doi: 10.1016/0197-4580(93)90113-p. [DOI] [PubMed] [Google Scholar]
- 10.de Jong G I, Naber P A, Van der Zee E A, Thompson L T, Disterhoft J F, Luiten P G. Neurobiol Aging. 1996;17:459–465. doi: 10.1016/0197-4580(96)00030-9. [DOI] [PubMed] [Google Scholar]
- 11.Peters A, Rosene D L, Moss M B, Kemper T L, Abraham C R, Tigges J, Albert M S. J Neuropathol Exp Neurol. 1996;55:861–874. doi: 10.1097/00005072-199608000-00001. [DOI] [PubMed] [Google Scholar]
- 12.Chang P L, Isaacs K R, Greenough W T. Neurobiol Aging. 1991;12:517–522. doi: 10.1016/0197-4580(91)90082-u. [DOI] [PubMed] [Google Scholar]
- 13.Coleman P D, Flood D G. Neurobiol Aging. 1987;8:521–545. doi: 10.1016/0197-4580(87)90127-8. [DOI] [PubMed] [Google Scholar]
- 14.Barnes C A, Suster M S, Shen J, McNaughton B L. Nature (London) 1997;388:272–275. doi: 10.1038/40859. [DOI] [PubMed] [Google Scholar]
- 15.Gazzaley A H, Siegel S J, Kordower J H, Mufson E J, Morrison J H. Proc Natl Acad Sci USA. 1996;93:3121–3125. doi: 10.1073/pnas.93.7.3121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mesulam M M, Mufson E J, Levey A I, Wainer B H. J Comp Neurol. 1983;214:170–197. doi: 10.1002/cne.902140206. [DOI] [PubMed] [Google Scholar]
- 17.Voytko M L, Olton D S, Richardson R T, Gorman L K, Tobin J R, Price D L. J Neurosci. 1994;14:167–186. doi: 10.1523/JNEUROSCI.14-01-00167.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wainer B H, Steininger T L, Roback J D, Burke-Watson M A, Mufson E J, Kordower J. Prog Brain Res. 1993;98:9–30. doi: 10.1016/s0079-6123(08)62378-x. [DOI] [PubMed] [Google Scholar]
- 19.Hefti F. J Neurosci. 1986;6:2155–2162. doi: 10.1523/JNEUROSCI.06-08-02155.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fischer W, Wictorin K, Björklund A, Williams L R, Varon S, Gage F H. Nature (London) 1987;329:65–68. doi: 10.1038/329065a0. [DOI] [PubMed] [Google Scholar]
- 21.Tuszynski M H, U, H S, Amaral D G, Gage F H. J Neurosci. 1990;10:3604–3614. doi: 10.1523/JNEUROSCI.10-11-03604.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tuszynski M H, Gage F H. Proc Natl Acad Sci USA. 1995;92:4621–4625. doi: 10.1073/pnas.92.10.4621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Koliatsos V E, Nauta H J, Clatterbuck R E, Holtzman D M, Mobley W C, Price D L. J Neurosci. 1990;10:3801–3813. doi: 10.1523/JNEUROSCI.10-12-03801.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Holtzman D M, Li Y, Chen K, Gage F H, Epstein C J, Mobley W C. Neurology. 1993;43:2668–2673. doi: 10.1212/wnl.43.12.2668. [DOI] [PubMed] [Google Scholar]
- 25.Kordower J H, Winn S R, Liu Y-T, Mufson E J, Sladek J R, Hammang J P, Baetge E E, Emerich D F. Proc Natl Acad Sci USA. 1994;91:10898–10902. doi: 10.1073/pnas.91.23.10898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tuszynski M H, Senut M C, Ray J, U, H-S, Gage F H. Neurobiol Dis. 1994;1:67–78. doi: 10.1006/nbdi.1994.0009. [DOI] [PubMed] [Google Scholar]
- 27.Tuszynski M H, Roberts J, Senut M C, U, H-S, Gage F H. Gene Ther. 1996;3:305–314. [PubMed] [Google Scholar]
- 28.Greene L A, Aletta J M, Rukenstein A, Green S H. Methods Enzymol. 1987;147:207–216. doi: 10.1016/0076-6879(87)47111-5. [DOI] [PubMed] [Google Scholar]
- 29.Weskamp G, Otten U. J Neurochem. 1987;48:1779–1786. doi: 10.1111/j.1471-4159.1987.tb05736.x. [DOI] [PubMed] [Google Scholar]
- 30.Kordower J H, Bartus R T, Bothwell M, Schatteman G, Gash D M. J Comp Neurol. 1988;277:465–486. doi: 10.1002/cne.902770402. [DOI] [PubMed] [Google Scholar]
- 31.West M J, Gunderson H J. J Comp Neurol. 1996;296:1–22. doi: 10.1002/cne.902960102. [DOI] [PubMed] [Google Scholar]
- 32.Nimchinsky E A, Hof P R, Young W G, Morrison J H. J Comp Neurol. 1996;374:136–160. doi: 10.1002/(SICI)1096-9861(19961007)374:1<136::AID-CNE10>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]
- 33.Gunderson H J G. J Microsc (Oxford) 1987;147:229–263. doi: 10.1111/j.1365-2818.1987.tb02837.x. [DOI] [PubMed] [Google Scholar]
- 34.Amaral D G, Kurz J. J Comp Neurol. 1985;240:37–59. doi: 10.1002/cne.902400104. [DOI] [PubMed] [Google Scholar]
- 35.Hedreen J C, Bacon J C, Price D L. J Histochem Cytochem. 1985;33:134–140. doi: 10.1177/33.2.2578498. [DOI] [PubMed] [Google Scholar]
- 36.Gunderson H J G, Bendtsen T F, Korbo L, Marcussen N, Moller A, Nielsen K, Nyengaard J R, Pakkenberg B, Sorensen F B, Vesterby A, et al. Acta Pathol Microbiol Scand Suppl. 1988;96:379–394. doi: 10.1111/j.1699-0463.1988.tb05320.x. [DOI] [PubMed] [Google Scholar]
- 37.Peterson D A, Jones D G. J Neurosci Methods. 1993;46:107–120. doi: 10.1016/0165-0270(93)90146-i. [DOI] [PubMed] [Google Scholar]
- 38.Emborg M E, Ma S Y, Mufson E J, Levey A I, Taylor M D, Brown W D, Holden J E, Kordower J H. J Comp Neurol. 1998;401:253–265. [PubMed] [Google Scholar]
- 39.Bäckman C, Rose G M, Hoffer B J, Henry M A, Bartus R T, Friden P, Granholm A C. J Neurosci. 1996;16:5437–5442. doi: 10.1523/JNEUROSCI.16-17-05437.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chen K S, Gage F H. J Neurosci. 1995;15:2819–2825. doi: 10.1523/JNEUROSCI.15-04-02819.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Armstrong D M, Sheffield R, Buzsaki G, Chen K S, Hersh L B, Nearing B, Gage F H. Neurobiol Aging. 1993;14:457–470. doi: 10.1016/0197-4580(93)90104-j. [DOI] [PubMed] [Google Scholar]
- 42.Smith M L, Booze R M. Neuroscience. 1995;67:679–688. doi: 10.1016/0306-4522(95)00076-u. [DOI] [PubMed] [Google Scholar]
- 43.Martinez-Serrano A, Fischer W, Björklund A. Neuron. 1995;15:473–484. doi: 10.1016/0896-6273(95)90051-9. [DOI] [PubMed] [Google Scholar]
- 44.Fischer W, Chen K S, Gage F H, Björklund A. Neurobiol Aging. 1992;13:9–23. doi: 10.1016/0197-4580(92)90003-g. [DOI] [PubMed] [Google Scholar]
- 45.Stroessner-Johnson H M, Rapp P R, Amaral D G. J Neurosci. 1992;12:1936–1944. doi: 10.1523/JNEUROSCI.12-05-01936.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Martínez-Serrano A, Fischer W, Söderström S, Ebendal T, Björklund A. Proc Natl Acad Sci USA. 1996;93:6355–6360. doi: 10.1073/pnas.93.13.6355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Coyle J T, Price P H, Delong M R. Science. 1983;219:1184–1189. doi: 10.1126/science.6338589. [DOI] [PubMed] [Google Scholar]
- 48.Bartus R, Dean R L, Beer C, Lippa A S. Science. 1982;217:408–417. doi: 10.1126/science.7046051. [DOI] [PubMed] [Google Scholar]
- 49.Williams L R. Exp Neurol. 1991;113:31–37. doi: 10.1016/0014-4886(91)90143-z. [DOI] [PubMed] [Google Scholar]
- 50.Isaacson L G, Saffran B N, Crutcher K A. Neurobiol Aging. 1990;11:51–55. doi: 10.1016/0197-4580(90)90062-5. [DOI] [PubMed] [Google Scholar]
- 51.Winkler J, Ramirez G A, Kuhn H G, Peterson D A, Day-Lollini P A, Stewart G R, Tuszynski M H, Gage F H, Thal L J. Ann Neurol. 1996;40:128–139. doi: 10.1002/ana.410410114. [DOI] [PubMed] [Google Scholar]
- 52.Tuszynski M H, Gabriel K, Gage F H, Suhr S, Meyer S, Rosetti A. Exp Neurol. 1996;137:157–173. doi: 10.1006/exnr.1996.0016. [DOI] [PubMed] [Google Scholar]
- 53.Mufson E J, Conner J M, Kordower J H. NeuroReport. 1995;6:1063–1066. doi: 10.1097/00001756-199505090-00028. [DOI] [PubMed] [Google Scholar]
- 54.Scott S A, Mufson E J, Weingartner J A, Skau K A, Crutcher K A. J Neurosci. 1995;15:6213–6221. doi: 10.1523/JNEUROSCI.15-09-06213.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Rylett R J, Goddard S, Schmidt B M, Williams L R. J Neurosci. 1993;13:3956–3963. doi: 10.1523/JNEUROSCI.13-09-03956.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]