

Abstract
FAH (fumarylacetoacetate hydrolase) catalyses the final step of tyrosine catabolism to produce fumarate and acetoacetate. HT1 (hereditary tyrosinaemia type 1) results from deficiency of this enzyme. Previously, we prepared a partial mimic of the putative tetrahedral intermediate in the reaction catalysed by FAH co-crystallized with the enzyme to reveal details of the mechanism [Bateman, Bhanumoorthy, Witte, McClard, Grompe and Timm (2001) J. Biol. Chem. 276, 15284–15291]. We have now successfully synthesized complete mimics CEHPOBA {4-[(2-carboxyethyl)-hydroxyphosphinyl]-3-oxobutyrate} and COPHPAA {3-[(3-carboxy-2-oxopropyl)hydroxyphosphinyl]acrylate}, which inhibit FAH in slow-onset tight-binding mode with Ki values of 41 and 12 nM respectively. A high-resolution (1.35 Å; 1 Å=0.1 nm) crystal structure of the FAH·CEHPOBA complex was solved to reveal the affinity determinants for these compounds and to provide further insight into the mechanism of FAH catalysis. These compounds are active in vivo, and CEHPOBA demonstrated a notable dose-dependent increase in SA (succinylacetone; a metabolite seen in patients with HT1) in mouse serum after repeated injections, and, following a single injection (1 μmol/g; intraperitoneal), only a modest regain of FAH enzyme activity was detected in liver protein isolates after 24 h. These potent inhibitors provide a means to chemically phenocopy the metabolic defects of either HT1 or FAH knockout mice and promise future pharmacological utility for hepatocyte transplantation.
Keywords: crystal structure, fumarylacetoacetate, hepatocyte transplantation, pharmacokineties, transition-state analogue, tyrosinaemia
Abbreviations: CEHPOBA, 4-[(2-carboxyethyl)hydroxyphosphinyl]-3-oxobutyric acid; COPHPAA, 3-[(3-carboxy-2-oxopropyl)hydroxyphosphinyl]-acrylate; DMF, dimethylformamide; FAA, fumarylacetoacetate; FAH, FAA hydrolase (EC 3.7.1.2); HMPOBA, 4-(hydroxymethylphosphinyl)-3-oxo-butanoic acid; HT1, hereditary tyrosinaemia type I; LDA, lithium di-isopropylamide; MAA, maleylacetoacetate; mFAH, murine FAH; rmsd, root mean square deviation; SA, succinylacetone; SAA, succinylacetoacetate; THF, tetrahydrofuran; TMSI, trimethylsilyl iodide; TS, transition state
INTRODUCTION
FAH [FAA (fumarylacetoacetate) hydrolase] catalyses the carbon–carbon bond scission of FAA in the final step of tyrosine and phenylalanine catabolism to give fumarate and acetoacetate. Loss-of-function mutations in FAH result in HT1 (hereditary tyrosinaemia type I), the most severe of the tyrosine catabolic disorders. This disorder results in hepatocellular carcinoma, liver cirrhosis, renal tubular damage and neurological crises [1,2]. Most likely these pathologies are caused by accumulation of FAA [3], which is known to react with thiols, including glutathione, and to induce oxidative damage and apoptosis. HT1 is hallmarked by elevated SA (succinylacetone) levels presumably formed by reduction and decarboxylation of FAA through an FAH-independent pathway. A murine model of HT1 has been developed [3,4] and utilized to show that the deleterious phenotype can be reversed by the administration of transposable DNA [5], adenoviral or retroviral vectors [6] encoding normal FAH. It has been demonstrated that FAH-expressing hepatocytes injected into the liver or spleen of FAH−/− knockout mice engraft and replicate [7]; therefore we have suggested that appropriate inhibitors of FAH may be used to improve the efficiency and permanence of liver gene therapy strategies [8]. A selection regimen whereby endogenous cells are treated with an FAH inhibitor and donor cells either overexpressing an inhibitor-resistant variant or with a genetic block in upstream tyrosine catabolic enzymes may be an effective means to provide in vivo selection of donor cells.
In a previous publication [8], we described the synthesis of HMPOBA [4-(hydroxymethylphosphinyl)-3-oxo-butanoic acid], a partial analogue of the putative tetrahedral intermediate (Figure 1) lacking the ‘fumaryl’ moiety (Ki=85 μM). The crystal structure of the FAH·HMPOBA complex defined a catalytic triad, reminiscent of serine proteases, formed by Glu365, His133 and a water molecule, and revealed that the ‘fumaryl’ pocket had been occupied by an adventitious acetate ion.
Figure 1. Reaction catalysed by FAH and TS mimic inhibitors.

The tetrahedral intermediate formed during hydrolysis of FAA is mimicked by the phosphinate TS analogues 1 and 2, and the partial TS analogue HMPOBA.
Here, we present the synthesis and kinetic characterization of two complete near TS (transition state) mimics, CEHPOBA {4-[(2-carboxyethyl)hydroxyphosphinyl]-3-oxobutyric acid}, 1, and COPHPAA {3-[(3-carboxy-2-oxopropyl)-hydroxyphosphinyl]acrylate}, 2, (Figure 1) that exhibit slow-onset, tight-binding inhibition of FAH with more than three orders of magnitude improvement in Ki over the partial mimic, HMPOBA. While 2 mimics the approach to the putative TS formed during hydrolysis of FAA, 1 is analogous to the high-energy intermediate formed during FAH-catalysed hydrolysis of another physiologically relevant substrate metabolite, SAA (succinylacetoacetate) [9–11]. Further, we have solved the high-resolution crystal structure (1.35 Å; 1 Å=0.1 nm) of 1 in complex with mFAH (murine FAH) and have determined that the compound binds to FAH as an analogue of the tetrahedral intermediate at or near the TS. Additionally, we have performed in vivo pharmacokinetics using wild-type mice treated with 1 by measuring its serum half-life and identifying phenotypic hallmarks of FAH−/− knockout mice. Thus we have demonstrated that 1 leads to HT1 symptoms, such as suppression of liver FAH, increased levels of SA in serum and death.
EXPERIMENTAL
General
Synthetic products were characterized using a Hewlett–Packard 5995 chromatograph/mass spectrometer. 1H, 13C and 31P NMR spectra were recorded on a Bruker AC-300 spectrophotometer at 300.13, 100.04 and 121.49 MHz respectively. Solvents were dried by passage over activated alumina, and all reactions were performed under a positive pressure of dry nitrogen. Human FAH and mFAH were expressed in Escherichia coli and purified as described previously [8,12]. mFAH and human FAH share 88% sequence identity, with strict conservation of the active site residues. FAA was prepared from homogentisic acid as previously described [8,10,11,13].
Syntheses
3-(diethoxyphosphinyl)propionic acid, 1,1-dimethylethyl ester (product 3)
Freshly distilled 1,1-dimethylethyl 3-bromopropionate (26.8 g, 0.13 mol) was placed in a dry Schlenk flask along with o-xylene (160 ml). Solvent (10 ml) was distilled off. The solution was allowed to cool to room temperature (25 °C). Triethyl phosphite (33.4 ml, 0.2 mol) was added dropwise and the reaction mixture was heated under reflux for 40 h. Fractional distillation at reduced pressure provided 20.9 g (61% yield) of pure 3 as a colourless oil (boiling point 90–105 °C, 10 μM). 1H NMR ([2H]chloroform): δ 1.25 (6 H, t, JHCCH=6.94 Hz), δ 1.38 (9 H, s), δ 1.96 (2 H, m), δ 2.45 (2 H, m), δ 4.03 (4 H, m). 13C NMR ([2H]chloroform): δ 16.36 (d, JCCOP=6.32 Hz), δ 21.00 (d, JCP=145.22 Hz), δ 27.97 (s), δ 28.56 (d, JCCP=4.2 Hz), δ 61.66 (d, JCOP=6.32 Hz), δ 80.89 (s), δ 171.22 (d, JCCCP=18.94 Hz). 31P NMR ([2H]chloroform): δ 31.18.
3-(Chloroethoxyphosphinyl)propionic acid, 1,1-dimethylethyl ester (product 4)
Oxalyl chloride (1.4 ml, 10.4 mmol) was added dropwise to a stirred solution of 3 (1.89 g, 7.4 mmol) in dichloromethane (18 ml) at 0 °C. One drop of DMF (dimethylformamide) was added, and the reaction was allowed to warm to room temperature overnight. The volatiles were removed in vacuo to yield a yellow oil (MS m/z 183) that was suspended in THF (tetrahydrofuran; 45 ml) and used without further purification.
3-Pyrrolidin-1-yl-but-2-enoic acid, 1,1-dimethylethyl ester (product 5)
3-Oxo-butyric acid 1,1-dimethylethyl ester (9.9 ml, 60 mmol) was dissolved in 13 ml of ethyl acetate and pyrrolidine (5 ml, 60 mmol) was added dropwise with stirring. Additional ethyl acetate (5 ml) was added to aid stirring. During 4 h, a white precipitate formed and was recovered by filtration. Recrystallization from ethyl acetate yielded 7.17 g of white needles (73% yield). Melting point=112–113 °C 1H NMR ([2H]chloroform): δ 1.45 (9 H, s), δ 1.92 (4 H, m), δ 2.45 (3 H, s), δ 3.3 (4 H, bm), δ 4.4 (1 H, s). 13C NMR ([2H]chloroform): δ 16.5 (s), δ 25.2 (s), δ 28.7 (s), δ 47.8 (s), δ 77.2 (s), 85.2 (s), δ 158.8 (s), δ 169.2 (s).
4-[(2-(1,1-Dimethylethoxy)carboxyethyl)ethoxyphosphinyl]-3-oxobutyric acid, 1,1-dimethylethyl ester (product 6)
Product 5 (1.72 g, 8.1 mmol) was dried under high vacuum for 1 h and dissolved in dry THF (55 ml). The solution was cooled in a solid CO2/acetone bath, and LDA (lithium di-isopropylamide)-mono(tetrahydrofuran) (1.5 M in cyclohexane, 5.43 ml, 8.1 mmol) was added dropwise. The reaction mixture was stirred for 1 h. The phosphonochloridate, 4, from above was cooled in a CO2/acetone bath, and the anion solution transferred via cannula (dropwise over 15 min) into the stirred phosphonochloridate solution. After 4.2 h, the reaction was allowed to warm to 0 °C and transferred via cannula to a stirred solution of sodium acetate buffer (10%, 100 ml, pH 6.5). THF was removed at aspirator pressure, and the mixture was extracted with dichloromethane (3×60 ml). The combined extracts were dried (Na2SO4) and evaporated in vacuo to afford a rust-coloured oil. The oil was dissolved in methanol/water (10:1, 110 ml). Acetic acid (60 μl) was added, and the reaction mixture was allowed to stir overnight at room temperature. Additional acetic acid (1.5 ml) and water (10 ml) were added after 20 h, to complete the hydrolysis of the enamine during the course of 24 h. The mixture was extracted with dichloromethane (3×60 ml), dried (Na2SO4) and evaporated in vacuo to yield a rust-coloured oil (2.74 g, 98% crude yield). Silica gel chromatography (ethyl acetate/hexane, 1:1) afforded pure product 6 (1.3 g, 47% yield) as a mixture of keto/enol tautomers. Only data for the keto form are reported. 1H NMR ([2H]chloroform): δ 1.33 (3 H, t, JHCCH=2.12 Hz), δ 1.45 (9 H, s), δ 1.47 (9 H, s), δ 2.12 (2 H, m), δ 2.54 (2 H, m), δ 3.27 (2 H, d, JHCP=17.28 Hz), δ 4.11 (2 H, m). 13C NMR ([2H]chloroform): δ 16.46 (d, JCCOP=6.19 Hz), δ 24.16 (d, JCP=97.96 Hz), δ 27.60 (d, JCCP=3.25 Hz), δ 27.92 (s), δ 27.97 (s), δ 44.79 (d, JCP=75.54), δ 51.68 (s), δ 61.36 (d, JCOP=6.71 Hz), δ 81.17 (s), δ 82.27 (s), δ 165.92 (s), δ 196.11 (s), δ 196.18 (s). 31P NMR ([2H]chloroform): δ 46.9. MS m/z 222.1.
CEHPOBA (product 1)
Phosphinic ester, 6, (1.25 g, 3.3 mmol) was dissolved in dichloromethane (25 ml) and cyclopentene (2 ml). TMSI (trimethylsilyl iodide) (3 ml, 20 mmol) was added dropwise to the stirred solution at 25 °C. The reaction mixture was stirred for 6.3 h. Volatiles were removed in vacuo to yield a reddish oil (2.26 g, 126%). The residue was dissolved in THF and transferred via cannula to a stirred solution of KHCO3 (0.99 g, 9.9 mmol) in 1 ml of water. The resulting solution was evaporated in vacuo and volatiles removed by evaporation of the neutral solution. Traces of residual volatiles were removed by successive evaporation with acetonitrile (3×5 ml) and CCl4 (2×5 ml) to yield (1.23 g, 102% yield) of pure 1 as a caramel-like solid. Neutron activation analysis of similar deprotection products has indicated the excess mass attributable to residual iodide [8]. The solid was dissolved in water (15 ml), and the phosphorus concentration (0.20 M) determined by the method of Ames [14]. 1H NMR (H2O/2H2O, 9:1): δ 1.85 (2 H, m), δ 2.35 (2 H, m), δ 3.13 (2 H, d, JHCP=15.85 Hz), δ 3.59 (2 H, s). 13C NMR (H2O/2H2O, 9:1): δ 30.53 (d, JCP=96.81), δ 33.01 (d, JCCP=3.15 Hz), δ 50.02 (d, JCP=70.51), δ 56.99 (s), δ 177.18 (s), δ 184.54 (d, J=17.68 Hz), δ 207.92 (d, J=5.22 Hz). 31P NMR (H2O/2H2O, 9:1): δ 35.17.
4-[(2-{(1,1-Dimethylethoxy)carbonyl}-2-phenylselenylethyl)ethoxyphosphinyl]3-oxobutyric acid, 1,1-dimethylethyl ester (product 7)
Enamine, 5, (8.14 g, 38.5 mmol) was dried under high vacuum in a 200 ml Schlenk flask for 20 min prior to addition of dry THF (150 ml). The resulting solution was cooled in a solid CO2/acetone bath, and LDA-mono(tetrahydrofuran) (1.5 M in cyclohexane, 25.7 ml, 38.6 mmol) was added dropwise. The reaction mixture was stirred for 1 h. The phosphonochloridate, 4, (35 mmol) was prepared as above and subjected to high vacuum for 1 h where-upon THF (60 ml) was added, evaporated in vacuo, and re-dissolved in THF (260 ml). The stirred solution was cooled in a solid CO2/acetone bath, and the enamine anion was added via cannula (dropwise over 15 min) with stirring. The reaction mixture was allowed to warm to 0 °C during 1.5 h. The reaction mixture was again cooled in a solid CO2/acetone bath and LDA-mono-(tetrahydrofuran) (1.5 M in cyclohexane, 46.7 ml, 70 mmol) was added dropwise (15 min) with stirring. After 30 min, diphenyldiselenide (10.9 g, 35 mmol) was added all at once. The diselenide slowly dissolved as the reaction warmed to 0 °C during the course of 1 h. The reaction mixture was transferred via cannula to a stirred solution of sodium phosphate buffer (1.5 M; 300 ml, pH 6). THF was removed at aspirator pressure, and the residue was extracted with dichloromethane (3×120 ml). The combined extracts were dried (Na2SO4) and evaporated in vacuo to afford 26.6 g of a viscous red–brown oil. This material was dissolved in ethyl acetate/water (1:1, 50 ml) and acetic acid (3.2 ml) was added. The mixture was extracted with dichloromethane (3×120 ml), and the combined extracts were dried (Na2SO4) and evaporated in vacuo to yield 23.6 g of a red oil. Silica gel chromatography (ethyl acetate/hexane, 1:1) of this material afforded a yellow oil (7.42 g, 40% yield). 13C NMR ([2H]chloroform): δ 16.35 (1 C, d, JCCOP=6.32 Hz), δ 20.69 (1 C, s), δ 27.68 (3 C, s), δ 27.9 (3 C, s), δ 31.62 (0.5 C, d, JCP=89.45 Hz), δ 31.75 (0.5 C, d, JCP=90.49), δ 35.83 (0.5 C, d, JCCP=4.21 Hz), δ 35.95 (0.5 C, d, JCCP=4.21 Hz), δ 45.24 (0.5 C, d, JCP=76.82 Hz), δ 45.6 (0.5 C, JCP=76.82 Hz), δ 51.69 (0.5 C, s), δ 51.74 (0.5 C, s), δ 61.55 (0.5 C, d, JCOP=7.36), δ 61.72 (0.5 C, JCOP=7.37 Hz), δ 81.65 (0.5 C, s), δ 81.78 (0.5 C, s), δ 82.17 (0.5 C, s), δ 82.24 (0.5 C, s), δ 127.29 (s), δ 128.78 (s), δ 129.04 (s), δ 135.76 (s), δ 135.81 (s), δ 165.86 (s), δ 165.89 (s), δ 174.84 (s), δ 195.83 (1 C, d, JCCP=6.31 Hz). 31P NMR ([2H]chloroform): δ 44.43 (keto), δ 44.94 (keto), δ 46.67 (enol), δ 47.13 (enol).
3-[(3-{(1,1-Dimethylethyl)carbonyl}2-oxopropyl)-ethoxyphosphinyl]acrylic acid, 1,1-dimethylethyl ester (product 8)
A solution of 7 (7.42 g, 14 mmol) in THF (25 ml) was cooled to 0 °C, and acetic acid (1 ml) followed by 30% H2O2 (3.7 ml, 32 mmol) were added dropwise with stirring. The reaction was maintained at 0 °C for 3 h. The reaction mixture was poured into saturated NaHCO3 (150 ml) and extracted with dichloromethane (3×100 ml). The combined extracts were dried (Na2SO4), filtered through a bed of celite, and evaporated in vacuo to yield a pale yellow viscous oil. Silica gel chromatography (ethyl acetate/hexane, 1:1) afforded pure 8 (2.36 g, 44% yield). 1H NMR (300.13 MHz, [2H]chloroform): δ 1.27 (3 H, t); δ 1.4 (9 H, s); δ 1.44 (9 H, s); δ 3.23 (1 H, d, JHCP=18.01 Hz); δ 3.29 (1 H, d, JHCP=18.01 Hz); δ 3.5 (2 H, s); δ 4.0 (2 H, dd); δ 6.7 (2 H, dd). 13C NMR ([2H]chloroform): δ 16.33 (d, JCCOP=6.32 Hz), δ 27.86 (s), δ 27.90 (s), δ 44.54 (d, JCP=88.39), δ 51.66 (s), δ 61.92 (d, JCOP=5.26 Hz), δ 82.20 (s), δ 82.35 (s), δ 132.51 (d, JCP=124.18 Hz), δ 140.92 (d, JC=CP=5.26 Hz), δ 163.14 (d, J=23.4 Hz), δ 165.72 (s), δ 194.86 (s), δ 194.91 (d, J=6.3 Hz). 31P NMR ([2H]chloroform): δ 31.3 (keto), 33.51 (enol).
COPHPAA (product 2)
To a solution of 8 (2.36 g, 6.3 mmol) in dichloromethane (30 ml) and cyclopentene (2.5 ml) was added TMSI (5.4 ml, 37.6 mmol) dropwise with stirring at room temperature. After 12 h, the volatiles were removed in vacuo. The residue was dissolved in THF and transferred via cannula to a stirred solution of NaHCO3 (1.58 g, 18.8 mmol). Following evaporation of the neutral solution, the residual volatiles were removed by successive evaporation with acetonitrile (3×5 ml) and CCl4 (2×5 ml). The resulting solid was dissolved in water (24 ml), and the phosphorus concentration (0.26 M) was determined by the method of Ames [14]. 1H NMR (H2O/2H2O, 9:1): δ 3.22 (2 H, d, JHCP=17.6 Hz), δ 3.57 (2 H, s), δ 6.47 (2 H, JHCCH=17.5 Hz, JHCCP=18.9 Hz), δ 6.65 (2 H, JHCCH=17.5 Hz, JHCP=21.6 Hz). 13C NMR (H2O/2H2O, 9:1): δ 51.25 (d, JCP=77.87), δ 56.78 (s), δ 137.13 (d, J=125.23 Hz), δ 142.41 (d, J=3.16), δ 176.28 (d, J=21.04 Hz), δ 176.96 (s), δ 207.28 (d, J=5.26 Hz). 31P NMR (H2O/2H2O, 9:1): δ 21.15, 28.4 (minor impurity).
Kinetics of slow onset of inhibition by compounds 1 and 2
Reactions were performed in 0.2 cm path length cells and contained 500 μl total volume in 50 mM sodium phosphate buffer (pH 7.0). An uninhibited reaction contained 300 μM FAA and inhibited reactions contained 250 μM FAA along with various amounts of 1 (100, 300 and 600 μM) or 2 (100, 200 and 500 μM). Reactions were initiated by the addition of human FAH (2 μl of 5.74 μM active sites) and were monitored at 330 nm at 26 °C for up to 2 h. As a direct measure of the value of koff, FAH and inhibitor were incubated on ice for 4 h and then diluted 1000-fold into the reaction mixture. These data were globally fitted to the following mechanism for slow-onset inhibition [15] by iterative cycles of numerical integration of the corresponding differential equations followed by evaluation using the Marquardt–Levenburg algorithm. The data fit less well to a two-step model in which an initial enzyme–inhibitor complex (E·I) isomerized slowly to a tighter complex (E·I*) (see [15], Scheme 1b). The program DYNAFIT [16] was kindly made available by Dr Petr Kuzmic (BioKin Ltd, Pullman, WA, U.S.A.; http://www.biokin.com) and was employed to fit the data to the following mechanism:
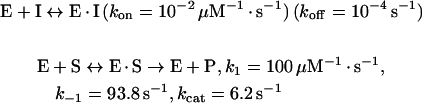 |
Scheme 1. Synthesis of FAH inhibitors 1 and 2.

Reagents and conditions: (a) ethyl phosphite, o-xylene, reflux; (b) oxalyl chloride, dichloromethane, ethyl acetate, DMF (cat.); (c) LDA (1.0 eq), THF, −78 °C; (d) THF, −78 °C; (e) acetic acid, methanol, water; (f) TMSI (6.0 eq), cyclopentene, dichloromethane; (g) KHCO3, water; (h) LDA (2.0 eq), THF, −78 °C; (i) diphenyl diselenide (1.0 eq), THF, −78° to 0 °C; (j) acetic acid, ethyl acetate, water; (k) H2O2 (2.3 eq), acetic acid, THF; (l) TMSI (6.0 eq), cyclopentene, dichloromethane; (m) NaHCO3, water.
A set of parentheses indicates an initial estimate for an unconstrained parameter. The chosen values for k1 (assumed virtual diffusion-controlled rate of substrate binding, 1×108 M−1·s−1), and k−1 are consistent with the Km (1.0 μM) and kcat values determined as described below. The Ki values for inhibitors 1 and 2 were calculated for each as koff/kon.
Determination of Km and kcat for FAH
To reaction mixtures containing sodium phosphate buffer (50 mM, pH 7.0) and FAA [0.525–16.8 μM; molar absorption coefficient (ϵ)=13500 M−1·cm−1; λ=330 nm] was added 5 μl of a dilution of human FAH (∼2.87 μM active sites) to a total of 1.000 ml in a 1 cm cuvette. Initial reaction rates were measured and used to determine Km and kcat by non-linear fit to the Michaelis–Menten equation.
Crystallization, data collection and structure refinement
mFAH crystals of P21 symmetry were grown as previously described [8,12] with the exception that the protein solution was dialysed overnight against 20 mM Tris/HCl and 0.15 M NaCl (pH 7.4) containing 1 mM manganese acetate prior to final concentration. The ligand, 1, was introduced into the crystals by soaking the crystals overnight in crystallization mother-liquor to which 10 mM 1 had been added. The cryoprotectant solution for flash cooling soaked mFAH crystals was 30% PEG [poly(ethylene glycol)] 400, 0.3 M sodium acetate, 0.1 M sodium cacodylate (pH 6.5) and 10 mM 1. X-ray diffraction data were collected at the Structural Biology Center Beamline 19-ID located with the Advanced Photon Source at the Argonne National Laboratory utilizing 0.98 Å wavelength X-rays. The structure of FAH complexed with 1 was determined by direct molecular replacement using the previously published structure of mFAH (PDB no. 1QCN) [12]. The positions of the soaked ligands were clear from difference electron density maps (2Fo–Fc and Fo–Fc). The structure was subjected to iterative cycles of model building using O [17] and refinement with CNS [18] and then Refmac [19], including automated water placement. The final model contains 835 amino acids, two Na+, two Mn2+, one Ca2+, ten Ni2+, 998 water molecules and two molecules of 1.
Animal husbandry
All mice were treated according to the NIH (National Institutes of Health) Guidelines for Animal Care and with the approval of the institutional animal care and utilization committee of the Oregon Health and Sciences University. All mice were 8–12 weeks of age, housed in the Department of Comparative Medicine at Oregon Health and Sciences University, and were provided with Rodent chow (Purina 5010) and water ad libitum.
Measurement of mouse liver FAH enzyme activity
A cohort of 11 adult C57/Bl6 male mice was administered single intraperitoneal injections (1 μmol/g) of a 0.2 M aqueous solution of 1. Individual animals were anaesthetized, and killed by cervical dislocation at 0.5, 1, 2, 3, 4, 5, 6, 7, 8, 17 and 23 h following injection (an untreated control animal was also included). Livers were immediately perfused with approx. 50 ml of heparinized HBSS (Hanks balanced salt solution) and 0.5 g samples were excised and frozen immediately in 1.5 ml centrifuge tubes on liquid nitrogen. Samples were maintained at −80 °C until use. Samples were thawed and homogenized with a miniature pestle; 60% sucrose (1 ml) was added to each sample, and each sample was sonicated. Samples were centrifuged at 14000 g for 20 min, and the supernatants were transferred to clean tubes. Protein concentration was normalized by addition of 60% sucrose for all samples following determination using the Bio-Rad protein assay. FAH activity was assayed spectrophotometrically by monitoring the disappearance of FAA (330 nm) as previously described [10]. Relative FAH expression levels were determined by immunoblot with anti-FAH antibody (kindly provided by Robert M. Tanguay, Laboratory of Cellular and Developmental Genetics, Department of Medicine, Université Laval, Quebéc, Canada) as previously described [20]).
Plasma half-life determination
Plasma levels of 1 were determined by an enzyme inhibition assay. Blood collected from the above-treated mice was placed in heparinized Microtainer tubes (Becton Dickinson) and centrifuged for 3 min at 6000 g. Plasma was collected, flash frozen [N2(l)], and stored at −80 °C. Standards ranging in concentration from 10 to 1000 μM 1 were prepared using plasma from untreated animals. Reaction mixtures (500 μl) containing 5 μl of control or requisite samples and FAA (13.3 μM) in 50 mM sodium phosphate (pH 6.0) were initiated by addition of human FAH. The disappearance of substrate (FAA) was monitored at 330 nm. Data were fitted to a standard curve calculated from the initial rates of control reactions.
Determination of serum SA levels
An enzymatic assay for the combined determination of SAA and the decarboxylation product (SA) was employed [21]. C57/Bl6 mice were administered daily intraperitoneal injections of aq. 0.2 M 1 at concentrations of 0, 0.5, 1 and 2 μmol/g. Blood was collected from the retro-orbital plexus of anaesthetized animals into serum-separator Microtainer tubes (Becton Dickinson), and spun at 6000 g for 5 min. Serum was collected, flash frozen [N2(l)], and stored at −80 °C until use. The assay was prepared as described.
RESULTS AND DISCUSSION
Phosphorus-containing compounds can act as non-covalent inhibitors and TS analogues for hydrolases [8,22–25]. A previous structure–function study using the partial TS analogue HMPOBA, wherein HMPOBA (Figure 1) was soaked into the active site of FAH [8], indicated that the tetrahedral phosphorus centre of 1 and 2 might serve as a good model for the high-energy intermediate/TS implicated in the FAH-catalysed hydrolysis of SAA and FAA respectively. We have produced both of these high-energy intermediate analogues, determined their kinetic constants, solved the FAH·1 crystal structure, and have demonstrated the in vivo activity of these compounds in mice.
Synthesis of FAH inhibitors 1 and 2
The phosphinate TS mimic inhibitors, 1 and 2, were prepared from t-Bu 3-bromopropionate in five and six steps respectively (Scheme 1). Thus Michaelis–Arbuzov reaction of t-Bu 3-bromopropionate with triethylphosphite provided the phosphonate, 3, which was transformed to the corresponding phosphonochloridate, 4, upon treatment with oxalyl chloride and DMF. Multiple attempts to phosphinylate the dianion of t-Bu acetoacetate with 4 provided primarily the O-phosphonylated material. This problem was circumvented by employing the anion derived from the pyrrolidine enamine of t-Bu acetoacetate, 5. Thus when a solution in THF of the anion of 5 (from treatment of 5 with LDA at −78 °C) was added at −78 °C to a suspension of 4 the desired phosphinyl intermediate was obtained. Subsequent hydrolysis of the phosphinylated enamine with acetic acid provided 6 in 47% yield. Removal of the ethyl and t-Bu ester groups was accomplished with the use of TMSI and a mild basic hydrolytic workup to provide 1 in excellent yield (29% overall, five steps).
Somewhat surprisingly, attempted phosphinylation utilizing a vinyligous phosphonochloridate and either the dianion of t-Bu acetoacetate or the anion of 5 yielded only very minor amounts of O-phosphonylated products or no reaction respectively. Consequently, it was necessary to introduce the requisite double bond of 2 at a later stage. Thus, immediately following the reaction of 4 with the anion of 5, two additional equivalents of LDA were introduced, and the resulting dianion was allowed to react with diphenyldiselenide to give the corresponding phenylselenide α to the carboxyl-carbonyl. The crude reaction product was treated with aqueous acetic acid to convert the enamine into the corresponding ketone 7, and oxidation of 7 with H2O2 afforded 8. Only the desired E isomer was obtained, as shown by NMR [26]. Deprotection of 8 as above for the synthesis of 1 provided 2 in very good yield (8% overall, six steps).
Mode of inhibition of FAH by compounds 1 and 2
Both 1 and 2 competitively inhibit FAH in a time-dependent manner, and both materials function in a manner consistent with slow-onset tight-binding inhibition. The progress curves for the reactions (Figure 2) show a continued decrease in reaction rate over time, not caused by either depletion of substrate or build-up of products. This result indicates a slow formation of the enzyme–inhibitor complex, and the fit of the data to the simplest model for slow-onset inhibition [15] is very good. The relevant kinetic and thermodynamic constants that describe these processes are given in Table 1. The Ki values for both inhibitors are similar, but the kinetic parameters of inhibition are substantially different. The nearly 3-fold faster kon for 1 suggests that it forms an enzyme–inhibitor complex more rapidly than does 2. However, the FAH·2 complex dissociates about ten times more slowly, as indicated by the relative koff values. Hence, despite a slower rate of formation, the FAH·2 complex is significantly tighter, such that 2 is the more potent inhibitor of FAH, as reflected by the relative Ki values. Given that 2 is more closely related to the favoured substrate (FAA) by virtue of the fumaryl double bond, it was expected that binding of 2 would be entropically favoured, and the results are consistent with this assumption.
Figure 2. Slow-onset inhibition of FAH by compound 1 (left panel) and compound 2 (right panel), and recovery from inhibition after dilution.

For compound 1, 0.50 ml reaction mixtures contained 50 mM sodium phosphate buffer (pH 7.0), 250 μM FAA and 1 (100, 300 and 600 μM) as indicated. Reactions were initiated by addition of 2 μl of 5.74 μM sites of human FAH at 26 °C. A control reaction (labelled ‘0’) contained 300 μM FAA and no 1. The trace labelled ‘Off’ was obtained by pre-incubation of a mixture that contained 25.8 μM FAH and 300 μM 1 in 50 mM sodium phosphate buffer (pH 7.0) for 4 h at 4 °C followed by dilution of 0.5 μl of this mixture into a 0.50 ml reaction mixture that contained 260 μM FAA in 50 mM sodium phosphate buffer (pH 7.0). The experimental data are presented as circles and appear as dense traces. The thinner lines depict the simulated data (DYNAFIT) using the fitted kinetic constants and Ki values given in Table 1. For compound 2, the same experimental method was applied except that slightly different concentrations of this inhibitor (0, 100, 200 and 500 μM, as indicated) were employed and the [FAA] was uniformly 361 μM. The trace marked ‘Off’ was obtained by addition at t=0 of 0.5 μl of a mixture containing 134 μM 2 and 34.5 μM FAH in 50 mM sodium phosphate buffer (pH 7.0) (pre-incubated for 4.5 h at 4 °C) into a 0.50 ml reaction mixture that contained 361 μM FAA in 50 mM sodium phosphate buffer (pH 7.0). The data were subjected to global fitting analysis.
Table 1. Kinetic parameters for inhibition of FAH by phosphinate analogues 1 and 2.
Values given in parentheses are confidence intervals at P=0.05.
| Compound | koff (s−1) | kon (M−1·s−1) | Ki (nM) |
|---|---|---|---|
| 1 | 2.82 (0.03)×10−5 | 6.90 (0.03)×102 | 41 (0.4) |
| 2 | 2.90 (0.15)×10−6 | 2.52 (0.015)×102 | 12 (0.6) |
Crystal structure of the FAH·1 complex
We chose to pursue 1 rather than 2 for in vivo and crystallographic studies, as the compound is more efficiently synthesized, possesses similar binding affinity, and lacks a reactive double bond. The crystal structure of mFAH complexed with 1 was determined to a resolution of 1.35 Å by soaking crystals of the enzyme in a solution of the TS analogue. The structure was solved using molecular replacement and the previously determined structure of murine apo-FAH (PDB no. 1QCN) [12] and refined to a crystallographic residual of 0.168 (Rfree=0.188; Table 2).
Table 2. Data collection and refinement statistics for the FAH·1 complex.
| Parameter | Value |
|---|---|
| X-ray source | APS 19-ID; 0.98 Å wavelength |
| PDB code | 2HZY |
| Space group | P21 |
| Cell dimensions | |
| a (Å) | 64.14 |
| b (Å) | 109.47 |
| c (Å) | 67.49 |
| β (°) | 102.35 |
| Resolution (Å) | 30–1.35 |
| 〈I〉/〈σ〉 | 19.9 (2.6) |
| Rsym | 0.060 (0.444) |
| Reflections (unique) | 822020 (188128) |
| Completeness (%) | 94.6 (79.7) |
| R/Rfree | 0.168/0.188 |
| Average B-factor (Å2) | |
| Overall | 17.8 |
| Protein | 16.3 |
| Inhibitor | 13.9 |
| Number of atoms | 7503 |
| Protein | 6460 |
| Ligand+ions | 30+15 |
| Solvent | 998 |
| rmsd (root mean square deviation) | |
| From ideal bond lengths (Å) | 0.022 |
| From ideal bond angles (°) | 1.79 |
| Ramachandran (%, favoured/allowed) | 92.2/7.8 |
The crystal structure of FAH·1 shows features consistent with the proposed mechanism of FAH [8,12]. The manner in which 1 binds to the catalytic metal positions the tetrahedral phosphinyl group (the ‘fumaryl carbonyl’ group of FAA) such that the pro-S oxygen is close to the side chains of Lys253, Gln240 and Arg237, and the pro-R oxygen is near His133. The side chains of the former residues are positioned to enable the stabilizing interactions for the negatively charged oxygen of the putative tetrahedral TS/intermediate. The position of the pro-R oxygen atom is found to be occupied in all FAH structures by either a ligand oxygen atom or a water molecule. The water molecule is proposed to function as the active site nucleophile, as part of the catalytic triad including His133 and Glu364.
As has been found previously, there are two metals bound in the FAH active site. The metal cation to which the ligands bind was unambiguously identified as Mn2+, which assumes a distorted octahedral geometry (Figure 3a). Additionally, FAH retains maximal activity following multi-day dialysis only in the presence of Mn2+ (Supplementary Table 1 at http://www.BiochemJ.org/bj/402/bj4020251add.htm). The other metal cation lies within a pocket defined by Asp233, Trp234, Lys253, Gly256 and Thr257. It makes contacts with either side chain or main chain oxygen atoms of these five residues to form a distorted square-based pyramid (Figure 3a). On the basis of refined isotropic temperature factors this cation was previously identified as Mg2+ [8]. This assignment may be correct, but there is evidence to support an alternate identification as Na+. Sodium and magnesium ions possess an identical complement of electrons, such that when refined as either Mg2+ or Na+, the temperature factors of these peaks correlate strongly to those of the atoms in the surrounding residues. However, the metal binding pocket carries one net negative charge, rather than two, supporting the assignment as a sodium ion. In addition, the distances between the cation and the co-ordinated oxygen atoms vary between 2.4 and 2.6 Å, which are long for Mg–O distances, but closer to those found for Na–O [27]. Bond valence calculations using WASP [28] further support the Na+ assignment.
Figure 3. Stereo diagrams of metal-binding sites (a) and active site (b) in FAH.

(a) Stereo diagram of the metal-binding sites in FAH. The catalytic Mn2+ ion to which the FAH inhibitor 1 is bound is coloured yellow and the structural sodium ion is coloured green. (b) Stereo diagram of the active site in FAH. A stereo diagram of the active site is shown with Mn2+ coloured yellow and C, N and O coloured grey, blue and red respectively. A 2Fo–Fc electron density map for the FAH inhibitor 1 and Mn2+ is contoured in purple at a level of 2σ. Broken lines indicate important hydrogen bonds that stabilize the complex.
Binding of 1 within the FAH active site is mediated predominantly by hydrogen bond and/or charge interactions between the ligand oxygen atoms and amino acid side chains of FAH, some of which are mediated by the catalytic metal Mn2+ (Figure 3b). Supplementary Table 2 (http://www.BiochemJ.org/bj/402/bj4020251add.htm) details the hydrogen bond and metal co-ordination distances between 1 and FAH in each subunit of the FAH homodimer. Little difference in bond length is observed between the subunits; therefore, average lengths are noted. The carboxyl oxygen atoms of the ‘succinyl’ moiety make hydrogen bonds to the terminal nitrogen atoms of Arg142 (2.8 Å) and the OH of Tyr128 (2.6 Å). The pro-S phosphinyl oxygen is positioned equidistant from the side chain atoms Gln240 Nϵ2, Arg237 NH1 and Lys253 Nζ with distances of 2.9, 2.8 and 2.9 Å respectively. The pro-R phosphinoyl oxygen atom accepts a hydrogen bond from His133 Nϵ2 (2.6 Å). The ligand binds to the catalytic metal cation through the carbonyl oxygen (O5) and the carboxyl oxygen (O1) of the inhibitor β-diketo moiety with distances of 2.2 and 2.1 Å respectively, while the adjacent carboxyl oxygen atom (O3) hydrogen-bonds to a water molecule (3.0 Å) that is also co-ordinated to Thr350 O and the main chain nitrogen Ser130 N, both with distances of 3.0 Å (results not shown). Potential van der Waals attractions exist between the ligand and enzyme. The closest of these interactions include those between the methylene carbon (C1) and Leu247 Cδ2 (3.6 Å) and Val137 Cγ1 (3.9 Å). In addition, Tyr128 Cϵ1 is within 3.6 Å of the adjacent inhibitor methylene carbon.
In FAH·1 the mobile 140/147 loop lies close to the active site, with the side chains of Arg142 and Phe141 penetrating more deeply into the cavity than previously seen for FAH structures. Arg142 makes strong contacts with the carboxylate terminus of the bound ligand and forms part of a predicted hydrogen bond network including Tyr128 and Tyr144 [8] that may be largely responsible for anchoring the loop in place. In addition, the more withdrawn position of this loop has the effect of restricting solvent access to the active site. Chain B of the FAH homodimer is most well defined with respect to this loop. Therefore the following measurements were made with respect to chain B. The Arg142 Cα atom is displaced by 3.4 Å from its position in the apoenzyme structure and its side chain is displaced by 3.5 Å further into the active site pocket [12]. Similarly, the Arg142 Cα atom and side chain are displaced by 3.2 and 3.7 Å respectively from their position in the FAH·HMPOBA complex (PDB no. 1HYO) [8] (Supplementary Figure 1 at http://www.BiochemJ.org/bj/402/bj4020251add.htm). Both FAH·HMPOBA and apoenzyme structures exhibit similar loop orientations with the Arg142 Cα atom and side chain displaced by less than 0.5 Å. The Cα atom of Phe141 in the FAH·1 complex is displaced some 2.9 Å from its position in the apoenzyme and 2.8 Å from its position in the FAH·HMPOBA complex. The aromatic ring is positioned perpendicular to the end of the Arg142 side chain and is within van der Waals contact distance of the carboxyl oxygen of 1 (Figure 3b). The FAH·HMPOBA complex did not reveal a significant main chain movement relative to the apoenzyme (Phe141 Cα atom displaced by less than 0.2 Å), although the aromatic ring was rotated to a position parallel to that observed in the FAH·1 complex. The hydroxyl oxygen atoms of Tyr128 are displaced by less than 0.5 Å between FAH·1 and FAH·HMPOBA complexes, but are both displaced by 1.5 Å relative to the apoenzyme. Interestingly, there is a 180° bond rotation about the phosphorus–methylene bond of the ‘succinyl’ moiety of 1, different in each subunit of the FAH homodimer. The carboxylate, however, occupies an analogous position for interaction with Arg142 in either subunit.
Hydrogen-bonding contacts between Glu199 and the pro-R oxygen atom of the substrate and the side chain of Arg237 appear to anchor the position of the side chain and exclude water from the active site. The position of Glu199, which was found displaced by 2.0 Å in the FAH·HMPOBA complex from its position in apo-FAH, shows less than a 0.8 Å shift in position between the FAH·1 complex and the apoenzyme structure. In addition, this structure lacks the water molecule positioned 3.5 Å from Glu199 in the FAH·HMPOBA complex (Supplementary Figure 1). The side chain of Glu199 is believed to aid in the positioning of the activated water molecule and was found twisted away when a water molecule was present in the active site [8]. In the FAH·1 structure, Glu199 makes a hydrogen-bonding contact to the pro-S oxygen atom and is positioned in a similar position to that found in the apoenzyme structure and is likely to represent the conformation adopted following attack of the activated water molecule of FAA to generate the TS of the reaction.
Further positioning and stabilization of 1 is possible through the methylene carbon atoms that lie in a position to make van der Waals contacts with a band of hydrophobic side chains from Tyr128, Val137 and Leu247. The active site is solvent-inaccessible when 1, and presumably the physiological substrate FAA or SAA, is bound. There is no water molecule bound where it might mediate any proton transfer to an incipient carbanion. Direct proton transfer could be effected from a protonated carboxy group of fumarate or alternatively from Lys253. Lys253 is anchored 4.5 Å from the critical ‘fumaryl carbonyl’ (phosphorus) by interaction with Ser235, although in a previous structure Ser235 was swung away, possibly releasing Lys253 to rotate to a position from which the nitrogen could become the proton donor to the carbanion [8]. Alternatively, a protonated fumarate carboxyl is already in a position to directly donate a proton to the carbanion and the now negatively charged fumarate could then accept the proton from His133, which would complete the cycle of returning the catalytic residues to their original states. On the other hand, a water-mediated proton transfer from His133 to Lys253 could accomplish the same if Lys253 were to function as a general acid and become the proton donor to the carbanion.
As previously reported, the conformation of the oxyanion hole is sensitive to the nature of the substrate occupying the position of the tetrahedral alkoxy centre [12]. The network of interactions between Arg237, Gln240 and Lys253 is most extensive in the FAH·1 compared with previous structures, and the network also allows multiple contacts to the ligand. Changes in the conformation of the oxyanion hole, which are necessary to accommodate the changes from substrate to product during the enzymatic reaction, are principally brought about by the flexibility of Gln240 as it interacts with the differing structures of the substrate, TS and products [8,12].
FAA is produced from MAA (maleylacetoacetate) by the glutathione-dependent enzyme MAA isomerase that involves Z to E isomerization of the maleyl group of MAA, the penultimate reaction of the tyrosine catabolic pathway. The fact that MAA does not act as a substrate for FAH can be ascribed to the steric constraints imposed by the active site, as shown in the FAH·1 structure. In MAA the cis-carboxylate group would lie in very close proximity to the side chain of Gln240. Although Gln240 has been observed in different conformations [8,12], the hydrogen-bonding interaction that the side chain could make with the carbonyl oxygen of MAA would tend to counter any such flexibility. Also, with the carboxylate group pointing down into the active site pocket, MAA would be unable to make the crucial interactions with Arg142 that close the active site and anchor Phe141 deep within. Thus part of the discrimination between potential substrates may be the amount of binding energy associated with substrate binding that can be utilized to drive the catalytic reaction and the conformational changes associated with the side chains that interact with the TS for the reaction. The ion-pair interaction between the ‘succinyl’ carboxylate of 1 and Arg142, as well as the resultant hydrogen-bonding network involving Tyr128 and Tyr144, are likely to be the predominant interactions between FAH and 1 that result in a more than 2000-fold decrease in Ki over HMPOBA which lacks the requisite carboxylate. Interestingly, the ‘succinyl’ moiety of 1 is planar in the FAH·1 structure, as would also be the case were the analogue 2 bound. The entropic penalty for rigidifying the ‘succinyl’ moiety is eliminated in 2, and therefore may account for its decreased Ki.
In vivo activities of compounds 1 and 2
In addition to being a potent near-TS mimic inhibitor of FAH, 1 inactivated liver FAH for up to 24 h following administration and produced a pronounced increase in combined SA and SAA levels with repeated daily administration to C57/Bl6 mice. Rapid plasma clearance of 1 was evident following a single intraperitoneal injection (1 μmol/g). The concentration of 1 fell to undetectable levels <20 μM within 2 h of injection (Figure 4a). Hepatic FAH enzyme activity, however, was effectively inhibited as indicated by only a modest regain of activity (18% of control activity) after 24 h, suggesting that 1 is bioavailable and enters hepatocytes readily. Anti-FAH Western blots indicated the activity decrease was not due to degradation of FAH, as the concentration of FAH was virtually identical at all time points (results not shown). Daily intraperitoneal injections of 1 (0.5–2 μmol/g) resulted in a dose-dependent increase in the combined levels of SA and SAA (Figure 4b), both metabolites of FAA found in afflicted organisms. Doses of 2.0 and 1.0 μmol/g daily resulted in animal deaths on days 3 and 16 respectively. Similar studies using equimolar 2 (1 μmol/g daily for 6 days, intraperitoneal) indicate a comparable decrease in FAH liver enzyme activity at 24 h; however, the combined SA and SAA increase was 63% of that demonstrated with 1 (results not shown). We are currently using 1 for hepatocyte selection studies, as it is more active in vivo, and does not contain a Michael acceptor that could react with intracellular thiols.
Figure 4. CEHPOBA pharmacokinetics.

(a) Plasma concentration of the FAH inhibitor 1 (-◆-) and FAH activity in homogenates of perfused mouse liver (-○-) following a single intraperitoneal injection of 1 (1 μmol/g). (b) Total serum combined SA and SAA in mice receiving daily intraperitoneal injections of the FAH inhibitor 1 (-●-, 2.0 μmol/g; -▲-, 1.0 μmol/g; -○-, 0.5 μmol/g; -■-, 0 μmol/g).
The increase in SA was comparable with that observed in genetically FAH-deficient mice. Importantly, hepatocytes that express FAH have a very strong in vivo selective growth advantage both in mice [7] and humans [29]. For this reason it is possible to achieve near complete liver repopulation with either transplanted [30] or genetically corrected hepatocytes [5]. In addition to hepatocytes that express FAH, cells that do not produce FAA due to mutations in enzymes upstream in tyrosine catabolism are also strongly selected. Liver cells deficient in homogentisic acid dioxygenase [31] or 4-OH-phenylpyruvate dioxygenase [32] are completely protected from FAH deficiency. These genetic findings suggest several approaches for the clinical use of FAH inhibitors in liver repopulation. Thus these approaches could be used in the context of either cell transplantation or stable gene therapy. First, it may be possible to overexpress wild-type FAH or an inhibitor-resistant but enzymatically active mutant of FAH in hepatocytes one wishes to select. A similar strategy has already been used successfully for haemopoietic gene therapy [33]. Alternatively, genetic knockdown of enzymes upstream of FAH in the tyrosine catabolic pathway would prevent the accumulation of FAA and render hepatocytes resistant to FAH inhibitors. The advent of gene transfer vectors expressing shRNA (small-hairpin RNAs) is making such approaches feasible in the liver in vivo [34].
Online data
Acknowledgments
This study was supported by grants from the Medical Research Foundation of Oregon and the NIGMS (National Institute of General Medical Sciences) (R15GM60032) to R.W.M. and by the NIH grants to M.G. [NIDDK (National Institute of Diabetes and Digestive and Kidney Diseases) DK-51592] and to D.E.T. and T.D.H. (NIDDK DK-54738). We thank Ms Kristen Parks (Arthur F. Scott Laboratory of Chemistry, Reed College, Portland, OR, U.S.A.) and Aimin Lin (Department of Biochemistry and Molecular Biology, Indiana University, Indianapolis, IN, U.S.A.) for their contributions to this study, and Kevan Shokat (Department of Cellular and Molecular Pharmacology, University of California San Francisco, San Francisco, CA, U.S.A.) for helpful discussion and support. Use of the Advanced Photon Source at Argonne National Laboratory (Argonne, IL, U.S.A.) was supported by the U.S. Department of Energy, Basic Energy Sciences, Office of Science, under contract W-31-109-Eng-38.
References
- 1.Kvittingen E. A., Brodtkorb E. The pre- and post-natal diagnosis of tyrosinemia type I and the detection of the carrier state by assay of fumarylacetoacetase. Scand. J. Clin. Lab. Invest. Suppl. 1986;184:35–40. [PubMed] [Google Scholar]
- 2.Mitchell G., Larochelle J., Lambert M., Michaud J., Grenier A., Ogier H., Gauthier M., Lacroix J., Vanasse M., Larbrisseau A., et al. Neurologic crises in hereditary tyrosinemia. N. Engl. J. Med. 1990;322:432–437. doi: 10.1056/NEJM199002153220704. [DOI] [PubMed] [Google Scholar]
- 3.Grompe M., al-Dhalimy M., Finegold M., Ou C. N., Burlingame T., Kennaway N. G., Soriano P. Loss of fumarylacetoacetate hydrolase is responsible for the neonatal hepatic dysfunction phenotype of lethal albino mice. Genes Dev. 1993;7:2298–2307. doi: 10.1101/gad.7.12a.2298. [DOI] [PubMed] [Google Scholar]
- 4.Grompe M., Lindstedt S., al-Dhalimy M., Kennaway N. G., Papaconstantinou J., Torres-Ramos C. A., Ou C. N., Finegold M. Pharmacological correction of neonatal lethal hepatic dysfunction in a murine model of hereditary tyrosinaemia type I. Nat. Genet. 1995;10:453–460. doi: 10.1038/ng0895-453. [DOI] [PubMed] [Google Scholar]
- 5.Montini E., Held P. K., Noll M., Morcinek N., Al-Dhalimy M., Finegold M., Yant S. R., Kay M. A., Grompe M. In vivo correction of murine tyrosinemia type I by DNA-mediated transposition. Mol. Ther. 2002;6:759–769. doi: 10.1006/mthe.2002.0812. [DOI] [PubMed] [Google Scholar]
- 6.Overturf K., al-Dhalimy M., Ou C. N., Finegold M., Tanguay R., Lieber A., Kay M., Grompe M. Adenovirus-mediated gene therapy in a mouse model of hereditary tyrosinemia type I. Hum. Gene Ther. 1997;8:513–521. doi: 10.1089/hum.1997.8.5-513. [DOI] [PubMed] [Google Scholar]
- 7.Overturf K., Al-Dhalimy M., Tanguay R., Brantly M., Ou C. N., Finegold M., Grompe M. Hepatocytes corrected by gene therapy are selected in vivo in a murine model of hereditary tyrosinaemia type I. Nat. Genet. 1996;12:266–273. doi: 10.1038/ng0396-266. [DOI] [PubMed] [Google Scholar]
- 8.Bateman R. L., Bhanumoorthy P., Witte J. F., McClard R. W., Grompe M., Timm D. E. Mechanistic inferences from the crystal structure of fumarylacetoacetate hydrolase with a bound phosphorus-based inhibitor. J. Biol. Chem. 2001;276:15284–15291. doi: 10.1074/jbc.M007621200. [DOI] [PubMed] [Google Scholar]
- 9.Fernandez-Canon J. M., Baetscher M. W., Finegold M., Burlingame T., Gibson K. M., Grompe M. Maleylacetoacetate isomerase (MAAI/GSTZ)-deficient mice reveal a glutathione-dependent nonenzymatic bypass in tyrosine catabolism. Mol. Cell. Biol. 2002;22:4943–4951. doi: 10.1128/MCB.22.13.4943-4951.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Knox W. E., Edwards S. W. Homogentisate oxidase of liver. J. Biol. Chem. 1955;216:479–487. [PubMed] [Google Scholar]
- 11.Mahuran D. J., Angus R. H., Braun C. V., Sim S. S., Schmidt D. E., Jr Characterization and substrate specificity of fumarylacetoacetate fumarylhydrolase. Can. J. Biochem. 1977;55:1–8. doi: 10.1139/o77-001. [DOI] [PubMed] [Google Scholar]
- 12.Timm D. E., Mueller H. A., Bhanumoorthy P., Harp J. M., Bunick G. J. Crystal structure and mechanism of a carbon–carbon bond hydrolase. Structure. 1999;7:1023–1033. doi: 10.1016/s0969-2126(99)80170-1. [DOI] [PubMed] [Google Scholar]
- 13.Hsiang H. H., Sim S. S., Mahuran D. J., Schmidt D. E., Jr Purification and properties of a diketo acid hydrolase from beef liver. Biochemistry. 1972;11:2098–2102. doi: 10.1021/bi00761a016. [DOI] [PubMed] [Google Scholar]
- 14.Ames B. N. Assay of inorganic phosphate, total phosphate and phosphatases. Methods Enzymol. 1966;8:115–118. [Google Scholar]
- 15.Morrison J. F., Walsh C. T. The behavior and significance of slow-binding enzyme inhibitors. Adv. Enzymol. Relat. Areas Mol. Biol. 1988;61:201–301. doi: 10.1002/9780470123072.ch5. [DOI] [PubMed] [Google Scholar]
- 16.Kuzmic P. Program DYNAFIT for the analysis of enzyme kinetic data: application to HIV proteinase. Anal. Biochem. 1996;237:260–273. doi: 10.1006/abio.1996.0238. [DOI] [PubMed] [Google Scholar]
- 17.Jones T. A., Zou J. Y., Cowan S. W., Kjeldgaard M. Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Crystallogr. Sect. A Found. Crystallogr. 1991;47:110–119. doi: 10.1107/s0108767390010224. [DOI] [PubMed] [Google Scholar]
- 18.Brunger A. T., Adams P. D., Clore G. M., DeLano W. L., Gros P., Grosse-Kunstleve R. W., Jiang J. S., Kuszewski J., Nilges M., Pannu N. S., et al. Crystallography & NMR system: a new software suite for macromolecular structure determination. Acta Crystallogr. Sect. D Biol. Crystallogr. 1998;54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 19.Murshudov G. N., Vagin A. A., Dodson E. J. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. Sect. D Biol. Crystallogr. 1997;53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 20.Labelle Y., Puymirat J., Tanguay R. M. Localization of cells in the rat brain expressing fumarylacetoacetate hydrolase, the deficient enzyme in hereditary tyrosinemia type 1. Biochim. Biophys. Acta. 1993;1180:250–256. doi: 10.1016/0925-4439(93)90046-4. [DOI] [PubMed] [Google Scholar]
- 21.Grenier A., Lescault A. Succinylacetone. In: Bergmeyer H. V., editor. Methods of Enzymatic. Analysis. 3rd edn. volume 8. Weinheim: Verlag Chemie; 1985. pp. 73–79. [Google Scholar]
- 22.Bartlett P. A., Marlowe C. K. Phosphonamidates as transition-state analogue inhibitors of thermolysin. Biochemistry. 1983;22:4618–4624. doi: 10.1021/bi00289a002. [DOI] [PubMed] [Google Scholar]
- 23.Fraser M. E., Strynadka N. C., Bartlett P. A., Hanson J. E., James M. N. Crystallographic analysis of transition-state mimics bound to penicillopepsin: phosphorus-containing peptide analogues. Biochemistry. 1992;31:5201–5214. doi: 10.1021/bi00137a016. [DOI] [PubMed] [Google Scholar]
- 24.Hanson J. E., Kaplan A. P., Bartlett P. A. Phosphonate analogues of carboxypeptidase A substrates are potent transition-state analogue inhibitors. Biochemistry. 1989;28:6294–6305. doi: 10.1021/bi00441a022. [DOI] [PubMed] [Google Scholar]
- 25.Phillips M. A., Kaplan A. P., Rutter W. J., Bartlett P. A. Transition-state characterization: a new approach combining inhibitor analogues and variation in enzyme structure. Biochemistry. 1992;31:959–963. doi: 10.1021/bi00119a003. [DOI] [PubMed] [Google Scholar]
- 26.Melppelder F. H., Beck H. C. The nuclear magnetic resonance spectra of diethyl Z- and E-2-(methoxycarbonyl)vinylphosphonate. Rec. Trav. Chim. 1975;94:149–150. [Google Scholar]
- 27.Castagnetto J. M., Hennessy S. W., Roberts V. A., Getzoff E. D., Tainer J. A., Pique M. E. MDB: the Metalloprotein Database and Browser at The Scripps Research Institute. Nucleic Acids Res. 2002;30:379–382. doi: 10.1093/nar/30.1.379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nayal M., Di Cera E. Valence screening of water in protein crystals reveals potential Na+ binding sites. J. Mol. Biol. 1996;256:228–234. doi: 10.1006/jmbi.1996.0081. [DOI] [PubMed] [Google Scholar]
- 29.Kvittingen E. A., Rootwelt H., Berger R., Brandtzaeg P. Self-induced correction of the genetic defect in tyrosinemia type I. J. Clin. Invest. 1994;94:1657–1661. doi: 10.1172/JCI117509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lagasse E., Connors H., Al-Dhalimy M., Reitsma M., Dohse M., Osborne L., Wang X., Finegold M., Weissman I. L., Grompe M. Purified hematopoietic stem cells can differentiate into hepatocytes in vivo. Nat. Med. 2000;6:1229–1234. doi: 10.1038/81326. [DOI] [PubMed] [Google Scholar]
- 31.Manning K., Al-Dhalimy M., Finegold M., Grompe M. In vivo suppressor mutations correct a murine model of hereditary tyrosinemia type I. Proc. Natl. Acad. Sci. U.S.A. 1999;96:11928–11933. doi: 10.1073/pnas.96.21.11928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Endo F., Kubo S., Awata H., Kiwaki K., Katoh H., Kanegae Y., Saito I., Miyazaki J., Yamamoto T., Jakobs C., et al. Complete rescue of lethal albino c14CoS mice by null mutation of 4-hydroxyphenylpyruvate dioxygenase and induction of apoptosis of hepatocytes in these mice by in vivo retrieval of the tyrosine catabolic pathway. J. Biol. Chem. 1997;272:24426–24432. doi: 10.1074/jbc.272.39.24426. [DOI] [PubMed] [Google Scholar]
- 33.Davis B. M., Humeau L., Dropulic B. In vivo selection for human and murine hematopoietic cells transduced with a therapeutic MGMT lentiviral vector that inhibits HIV replication. Mol. Ther. 2004;9:160–172. doi: 10.1016/j.ymthe.2003.11.003. [DOI] [PubMed] [Google Scholar]
- 34.Arbuthnot P., Carmona S., Ely A. Exploiting the RNA interference pathway to counter hepatitis B virus replication. Liver Int. 2005;25:9–15. doi: 10.1111/j.1478-3231.2004.0966.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.