

Abstract
In Anopheles dirus glutathione transferase D3-3, position 64 is occupied by a functionally conserved glutamate residue, which interacts directly with the γ-glutamate moiety of GSH (glutathione) as part of an electron-sharing network present in all soluble GSTs (glutathione transferases). Primary sequence alignment of all GST classes suggests that Glu64 is one of a few residues that is functionally conserved in the GST superfamily. Available crystal structures as well as consideration of the property of the equivalent residue at position 64, acidic or polar, suggest that the GST electron-sharing motif can be divided into two types. Electrostatic interaction between the GSH glutamyl and carboxylic Glu64, as well as with Arg66 and Asp100, was observed to extend the electron-sharing motif identified previously. Glu64 contributes to the catalytic function of this motif and the ‘base-assisted deprotonation’ that are essential for GSH ionization during catalysis. Moreover, this residue also appears to affect multiple steps in the enzyme catalytic strategy, including binding of GSH, nucleophilic attack by thiolate at the electrophilic centre and product formation, probably through active-site packing effects. Replacement with non-functionally-conserved amino acids alters initial packing or folding by favouring aggregation during heterologous expression. Thermodynamic and reactivation in vitro analysis indicated that Glu64 also contributes to the initial folding pathway and overall structural stability. Therefore Glu64 also appears to impact upon catalysis through roles in both initial folding and structural maintenance.
Keywords: active-site residue, Anopheles dirus, catalytic mechanism, electron sharing network, glutathione transferase, structural integrity
Abbreviations: adGSTD3-3, Anopheles dirus glutathione transferase Delta class homodimer of subunit 3; CDNB, 1-chloro-2,4-dinitrobenzene; DTT, dithiothreitol; FDNB, 1-fluoro-2,4-dinitrobenzene; GdmCl, guanidinium chloride; GSH, glutathione; G-site, GSH binding site; GST, glutathione transferase; PtGSTU1-1, plant Tau class GST
INTRODUCTION
GSTs (glutathione transferases; EC 2.5.1.18) are a superfamily of enzymes that contribute towards diverse cellular processes ranging from detoxification to control of gene expression [1–4]. The enzymes generally catalyse nucleophilic attack of the GSH (glutathione) sulfhydryl group to an electrophilic centre of a number of endogenous and xenobiotic compounds [1,5,6]. Conjugation of GSH to such organic molecules enhances solubility, thus facilitating their eventual elimination [5–7]. This reaction is an early step along the mercapturic acid pathway in which hydrophobic compounds are inactivated and eliminated from an organism [8]. Based on amino acid identity, substrate specificities and immunological cross-reactivity, cytosolic GSTs are currently divided into at least 12 distinct evolutionary classes, namely Alpha, Mu, Pi, Theta, Sigma, Zeta, Omega, Phi, Tau, Delta, Epsilon and Beta [6,9–15]. In addition, the number of members identified in this enzyme superfamily is increasing due to the massive growth of genomic information, which includes a number of unclassified GSTs [16].
All cytosolic GSTs adopt the same highly conserved tertiary structure [17]. GSTs are dimeric proteins (with a molecular mass of about 50 kDa) assembled from identical or structurally related subunits. Each subunit is characterized by two distinct domains and an active site. However, each active site is only fully functional when amino acid residues from both subunits are present in the final dimeric structure. The active-site of GSTs consists of two adjacent regions. The first region is the G-site (GSH binding site), formed mostly by the N-terminal domain (domain I), which adopts an α/β topology that binds GSH as the thiol substrate [7]. The second region is the non-polar H-site (hydrophobic substrate binding site), generated primarily by the C-terminal domain (domain II), which is an all-helical structure that provides structural elements for recognition of a wide range of hydrophobic co-substrates [7]. Although GSTs possess a high specificity for GSH as the nucleophile, the enzymes exhibit broad specificity with regard to structurally diverse hydrophobic substrates [17]. The catalytic strategy of GSTs for the nucleophilic substitution reaction can be divided into several steps: binding of substrates to the enzyme active site, activation of GSH by thiol deprotonation, nucleophilic attack by thiolate at the electrophilic centre, product formation and product release [18–20].
The roles of several active-site residues and functional groups of GSH have been studied intensively and a potential model has been proposed to describe deprotonation of the GSH thiol group which enhances the nucleophilicity of the reaction [20–24]. The hydroxyl group of the conserved tyrosine/serine residue at the G-site of GSTs (i.e. Tyr8 for Pi, Tyr9 for Alpha, Tyr6 for Mu and Ser9 for Delta classes) is within hydrogen-bonding distance of the thiol group of the bound GSH, and is required for correct orientation and stabilization of the deprotonated thiol anion. The deprotonated anionic GSH results from subtraction of a thiol proton by the glutamyl α-carboxylate of GSH, which acts as a catalytic base in the base-assisted deprotonation model with assistance from an electron-sharing network. The functionally conserved electron-sharing network is characterized by an ionic bridge interaction between the negatively charged glutamyl α-carboxylate of GSH, a positively charged residue and a negatively charged residue, forming a resonance motif stabilized by a network of hydrogen-bonds with surrounding residues. The network is distributed among multiple interacting amino acids that collectively provide a network function. This conserved motif's contribution to the base-assisted deprotonation is essential for the GSH ionization step of catalysis.
The functionally conserved glutamate residue, Glu64 in adGSTD3-3 (Anopheles dirus GST Delta class homodimer of subunit 3) is located in the same region as the electron-sharing network (Figure 1). Initial characterization of the electron-sharing network has provided further insight into this motif [25]. The carboxylic group of Glu64 interacts directly with the glutamyl α-amino group of GSH. Moreover, by observing the configuration of the GSH glutamyl α-carboxylate, the GSH glutamyl α-amino, Glu64 and the electron-sharing network, it is now possible to extend this conserved motif, which is maintained in all GSTs. The aim of the present paper was to ascertain the validity of Glu64 function in the electron-sharing network. We have observed previously [25] that alanine replacement of Glu64 caused the enzyme to be expressed in an insoluble form. This suggested that Glu64 is a critical residue involved in tertiary structure or initial folding of the enzyme. Therefore the contribution of this residue to structural maintenance and initial folding was also examined. The results of the present paper indicate that Glu64 is a part of the functionally conserved electron-sharing network and has roles both in catalysis as well as structural folding and maintenance.
Figure 1. Newly identified extension of an electron-sharing network in adGSTD3-3.

The electron-sharing network is an ionic interaction between negatively charged and positively charged residues stabilized by a network of hydrogen-bonds. The stereo view of corresponding three-dimensional structure of the electron-sharing network is shown in the upper panel. The lines show the putative electron movement pathway with distances between 2.5 and 3.0 Å. The lower panel shows the sequence alignment of amino acid residues in Delta, Theta, Omega, Tau, Phi, Beta, Epsilon, Pi, Alpha, Sigma, Mu and Zeta class GSTs. The newly identified functionally conserved glutamate, aspartate and glutamine in the electron-sharing network are identified by an arrow.
MATERIALS AND METHODS
Site-directed mutagenesis
The plasmid pET3a-adgstD3, described previously [26], was used as the template to generate the single point mutations via PCR-based site-directed mutagenesis. The functionally conserved active-site residue Glu64 was replaced by an alanine, leucine, valine, lysine, glutamine, asparagine or aspartate residue by using PCR primers based on the wild-type adgstD3 gene sequence (Genbank accession number AF273039). Mutants were screened randomly by restriction enzyme digestion analysis. Mutant plasmids could be distinguished from the wild-type template by digestion with restriction enzymes corresponding to the restriction recognition site introduced by the mutagenic primers. The full-length GST coding sequence carrying E64A, E64L, E64V, E64K, E64Q, E64D and E64N mutations were verified by the dideoxy chain termination method.
Heterologous expression and purification
The proteins were expressed from the pET3a-adgstD3 vector in Escherichia coli BL21 (DE3) pLysS cells. The cells were grown to D600=0.5 and expression was induced by addition of 0.1 mM isopropyl β-D-thiogalactoside. Following a 3 h induction, cells were collected by centrifugation at 10000 g for 20 min at 4 °C and lysed by sonication using an Ultrasonic processor XL (Misonix) at power level 3 for 10 s, paused for 1 min and repeated three times at 4 °C. The soluble recombinant GST proteins were purified by GSTrap™ affinity chromatography (Amersham Biosciences) or S-hexylglutathione agarose (Sigma–Aldrich) affinity chromatography. The protein concentration was determined by the Bradford method [27] using BSA as a standard.
Steady-state kinetics
Steady-state kinetics were studied for wild-type and mutant enzymes at varying concentrations of CDNB (1-chloro-2,4-dinitrobenzene) and GSH in 0.1 M phosphate buffer pH 6.5. The reaction was monitored at 340 nm, ϵ 9600 M−1cm−1. Apparent kinetic constants, kcat, Km and kcat/Km were determined by fitting the collected data to a Michaelis–Menten equation by non-linear regression analysis using GraphPad Prism (GraphPad software).
pH dependence of kinetic constants
The pH dependence of kcat/KmCDNB was obtained as stated above by recording the enzymatic reaction in the following buffers: 0.1 M sodium acetate buffers (from pH 5.0 to 5.5) and 0.1 M potassium phosphate buffer (from pH 6.0 to 8.5). The pH was altered in increments of 0.5, and control experiments showed no discontinuities from buffer types. pKa values were obtained by fitting the data to eqn (1) [20]:
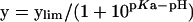 |
(1) |
Fluoride/chloride leaving group replacement
The second order kinetic constants at pH 6.5 for the spontaneous reaction of GSH with CDNB and FDNB (1-fluoro-2,4-dinitrobenzene) and the catalytic-centre activities at pH 6.5 for adGSTD3-3 with CDNB and FDNB as co-substrates were obtained as described previously [28].
Viscosity effect on the kinetic parameters
The effect of viscosity on kinetic parameters was obtained by using 0.1 M potassium phosphate buffer pH 6.5 with various glycerol concentrations. Viscosity values (η) at 25 °C were calculated as described previously [29].
Structural studies
A Jasco J-714 spectropolarimeter was used for CD measurements in the far-UV region from 190–260 nm. Spectra were recorded using 0.3 mg/ml of protein in 2-mm path length cuvettes. Intrinsic fluorescence emission spectra were measured with a Jasco FP-6300 spectrofluorimeter. The excitation wavelength was 295 nm and the λmax and the fluorescence intensity of emission spectra were analysed at a protein concentration of 0.2 mg/ml.
Kinetics of thermal denaturation
Heat inactivation of the wild-type and Glu64 mutant enzymes was monitored at different temperatures. Enzymes (40 μM) were incubated in 0.1 mM potassium phosphate buffer pH 6.5, 1 mM EDTA and 5 mM DTT (dithiothreitol). The inactivation time-courses were determined by withdrawing suitable aliquots at different time points to assay the remaining activity using the first time point as 100% native protein. An equation describing a single exponential decay with a rate constant of thermal unfolding ku was fitted to the data according to eqn (2) [30]:
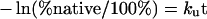 |
(2) |
The free-energy of activation of thermal unfolding (ΔGu) was calculated according to Eyring theory as eqn (3) [30]:
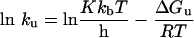 |
(3) |
where kb is the Boltzmann constant; T, absolute temperature Kelvin; h, Plank's constant; R, the gas constant; and K is the transmission factor, which was set to unity. The difference of free-energy of activation of the thermal denaturation between wild-type (wt) and each mutant (mut) protein (ΔΔGu) was calculated according to eqn (4) [30]:
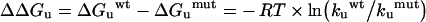 |
(4) |
Substitution of Equation 5:
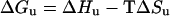 |
(5) |
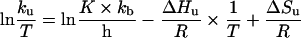 |
(6) |
Both activation enthalpy ΔHu and entropy ΔSu were determined from the temperature dependence of ku.
Temperature dependence of refolding in vitro
The in vitro refolding of the wild-type and Glu64 mutant enzymes were monitored at different temperatures. Enzyme (20 μM) was first denatured in 4 M GdmCl (guanidinium chloride) in renaturation buffer (0.2 M potassium phosphate buffer, 1 mM EDTA and 5 mM DTT, pH 7.0) at room temperature (25 °C) for 30 min and then rapidly diluted (defining time 0) 1:40 (v/v) into renaturation buffer at 18, 25 and 33 °C. The final GdmCl concentration was 0.1 M during refolding. All refolding experiments were carried out by rapid addition of the denatured enzyme to renaturation buffer. The recovery of activity of the proteins was monitored as a function of time by withdrawal of appropriate aliquots of the renaturation mixture and immediately assaying for activity at 25 °C. Refolding rate constants were determined by nonlinear regression analysis of the experimental data by using SigmaPlot 2001 for Windows version 7.0 (SPSS). The refolding rates of all variants were independent, in the range 10 to 30 μM, of the protein concentration. At greater enzyme concentrations all variants were characterized by reduced refolding yield. The values reported represent the means for three different experimental data sets. Under these conditions, an equation describing a single exponential reaction can be fitted to the data:
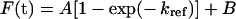 |
(7) |
where F(t) is the activity at time t; A, the amplitude; kref, the rate constant; and B, the reactivation value at time 0 [31]. The effect of mutation on the energy of the transition state of folding can be calculated using transition-state theory in a similar manner to that reported by Jackson et al. [32]. The stability of the transition state of a mutant protein relative to that of the wild-type is calculated from:
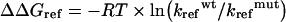 |
(8) |
where ΔΔGref is the difference in energy of the transition state of folding relative to the unfolded state between wild-type (wt) and mutant (mut) proteins and kref, the rate constants of refolding [32].
RESULTS AND DISCUSSION
A functionally conserved electron-sharing network can be observed in the same region for all GSTs, but with slightly different residue positions [22]. However, a primary sequence alignment of the known GST classes suggests that Glu64 is one of a few residues that is functionally conserved in the GST superfamily. Observation of available crystal structures; adGSTD3-3, hGSTP1-1 (human Pi GST1), hGSTA1-1 (human Alpha GST1), rGSTM1-1 (rat Mu GST1), hGSTO1-1 (human Omega GST1), hGSTT2-2 (human Theta GST2), squid Sigma GST and wheat Tau GST, as well as consideration of the property of the equivalent residue at position 64, acidic or polar, suggests that the GST electron-sharing motif can be divided into two types (Figure 2). Electron-sharing network type I, consisting of GSTs from Delta, Theta, Omega, Pi and Tau classes, contain an acidic amino acid (glutamate or aspartate) at this position. The equivalent residue is the only amino acid in an acceptable distance range to form an ionic interaction between its own negatively charged side chain and the positively charged GSH glutamyl α-amino (Figure 2A). On the other hand, electron-sharing network type II GSTs from the Alpha, Mu, Pi and Sigma classes have a polar amino acid (glutamine) side-chain that interacts directly with the GSH glutamyl α-amino. In addition, type II networks have a strictly conserved acidic amino acid (Asp98 in the Pi class, Asp101 in the Alpha class, Asp105 in the Mu class and Asp97 in the Sigma class) that participates in an ionic interaction (Figure 2B). In the Pi class GSTs, replacement of this acidic residue (Asp98 with asparagine) was shown to increase the pKa for GSH by approx. 0.5 pH unit [33]. These results suggest the importance of a negative charge involvement with the positively charged GSH glutamyl α-amino to fulfil the function of the electron-sharing network in the ionization process [33]. Dividing the electron-sharing network into two types is supported by studies of binding, activation and ionization of GSH, including the fate of the thiol proton in Pi, Alpha, Mu and Delta class GSTs [23]. It has been reported that GST classes Pi, Alpha and Mu, which are classified as electron-sharing network type II GSTs, display similarities in the multi-step mechanism for binding of the substrate and also yield a similar fate for the thiol proton. The Delta class GST, which we propose belongs to electron-sharing network type I, shows a difference in proton extrusion that implies a different activation mechanism for GSH. Moreover, the modality of proton output is also preserved in Pi, Alpha and Mu class enzymes. This mechanistic difference suggests that Delta GST is distantly related to Pi, Alpha and Mu GSTs in evolutionary terms.
Figure 2. Stereo views of examples of electron-sharing networks for type I (A) and type II (B) of GSTs.

Categorization is based on properties of the equivalent amino acid at position 64 and configuration of the electron-sharing network. (A) Electron-sharing network type I is composed of only one conserved acidic amino acid (circled), which forms an ionic interaction. Wheat Tau GSTI (PDB accession number 1GWC). (B) Electron-sharing network type II contains two strictly conserved polar and acidic residues (circled by solid and dotted lines respectively), which participate in an ionic bridge interaction. Human Pi hGSTP1-1 (PDB accession number 1PKW).
To investigate the role of the functionally conserved Glu64 residue in adGSTD3-3, this residue was replaced with seven amino acids; alanine, leucine, valine, lysine, glutamine, aspartate or asparagine. Evidence suggested that the replacements were temperature-sensitive therefore protein expression was performed at 18, 25 and 37 °C. The E64A, E64L, E64V and E64K mutants were expressed as insoluble proteins at all temperatures. Attempts at refolding these four proteins were unsuccessful. The yields of the E64Q, E64D and E64N mutants were less than wild-type and clearly decreased with increasing temperature (results not shown). This probably reflects the fact that the E64Q, E64D and E64N molecules fail to achieve the native folding at near physiological temperatures. Therefore, proteins expressed at the more permissive temperature of 18 °C were utilized in this study.
In the present study, a structural role for Glu64 for stability and folding was examined. The similarity of the far-UV CD spectra of all the Glu64 mutants indicated that the secondary structure content of the proteins are essentially unaffected by the mutations (Figure 3). The intrinsic fluorescence spectra show differences between the wild-type and all of the Glu64 mutants (Figure 4). The spectra of the Glu64 mutants were slightly red-shifted compared with the wild-type protein (λmax at 331 nm) to give a λmax at 332 nm for E64Q, 335 nm for E64D and 334 nm for E64N. The normalized intensities of the fluorescence of the mutants were slightly less than that of the wild-type. Changes in amino acid side-chains around Trp63 resulted in an alteration in fluorescence intensity, and a red-shifted spectrum is observed as the protein unfolds to random coil [34]. Consequently, the differences in both λmax and fluorescence intensity suggest that the mutation of Glu64 causes the tryptophan residue to be more exposed to the solvent. Heat inactivation of wild-type and Glu64 mutants was performed at different temperatures (Figure 5). Furthermore, the E64Q and E64N mutants were much more unstable than the wild-type or E64D mutant. It is important to note that the single Glu64 replacement with polar residues (glutamine and asparagine) became unstable at a lower temperature than the negatively charged residues (wild-type and E64D) and their inactivation generated the formation of protein aggregates under the experimental conditions used. Thermal denaturation of all variants was irreversible, showing that inactivation kinetics could not be used to determine thermodynamic parameters at equilibrium. However, making use of the temperature dependence of the unfolding rate, the application of Eyring formalism provides the thermodynamic parameters of the activation barrier of the thermal denaturation [30]. The heat inactivation of all variants is described by straight lines in an Eyring plot. This indicates that the temperature dependence of both the unfolding activation enthalpy (ΔH) and activation entropy (ΔS) is negligible. The temperature dependence of ΔG is reflected in the slope of the linear fit, dependent on ΔH. Table 1 summarizes the kinetic and thermodynamic constants for the activation barrier of thermal denaturation for the wild-type and Glu64 mutant enzymes. The large energy changes of Glu64 mutants compared with the wild-type (ΔΔH) are almost completely compensated for by an accompanying reduction in ΔS (that is, ΔΔS). At 42 °C, this corresponds to lower values of the unfolding free-energy for all the Glu64 mutant enzymes (ΔΔG). The Glu64 mutants were remarkably more destabilized than the wild-type enzyme, especially for the polar amino acids glutamine and asparagine (Table 1). This corresponds to an unfolding free-energy difference (ΔΔG) for the polar amino acid replacements compared with the wild-type, being greater than with the acidic residue aspartate. The Gibbs free-energy for the unfolding process of wild-type and Glu64 mutants were comparable, with general estimations of Gibbs free-energy for small globular proteins based on the summation of increments of the different stabilizing forces that give values in the range 42–84 kJ/mol [35]. Due to the large decrease in activation enthalpy (ΔΔH, 131–220 kJ/mol), the destabilizing effects of the Glu64 mutants were clearly significant to the structural properties of the enzymes. In addition the magnitude of TΔΔS at 42 °C, which is the energy difference from decreases in the entropy of the thermal unfolding pathway (198.4, 126.0 and 211.0 kJ/mol for E64Q, E64D and E64N respectively), indicates that major changes in the conformational freedom characterizes the denaturation of the wild-type protein compared with those occurring during the thermal inactivation of the Glu64 mutants [36]. This means that the wild-type adGSTD3-3, because it is more rigid than the Glu64 mutants, tolerates larger perturbations of its structure before the unfolding transition state is reached. The replacement of polar amino acids, both glutamine and asparagine, give greater values of all thermodynamic constants than for the acidic amino acids glutamate and aspartate. This data suggests that the negatively charged residue forming an ionic bridge interaction in this position plays an essential role for the overall stability of the protein. Recent studies proposed that Glu64 is a critical residue involved in the determination of the direction of the polypeptide chain during folding [25]. One consequence of replacing Glu64 with other amino acid residues results in a completely impaired folding property (for E64A, E64L, E64V and E64K) or temperature-sensitive folding (for E64Q, E64D and E64N). To test this hypothesis, reactivation yields of adGSTD3-3 and its mutants at different temperatures were determined. The denaturant 4 M GdmCl was sufficient to completely unfold 20 μM enzyme, as shown by CD spectrum (results not shown). The reactivation yields of the Glu64 mutants were very different from the wild-type protein at increasing temperatures of refolding (Table 2 and Figure 6). The reactivation yield of the wild-type enzyme was unaffected by temperatures from 18–33 °C, whereas the yield of the mutants, although to different extents, decreased markedly with increasing temperature. All data sets were fit to a single exponential equation for the refolding kinetics (Figure 6). At each temperature, as shown in Table 2, the temperature-dependent refolding rates show a similar tendency in the wild-type and Glu64 mutants that was expected, that is, the enzymes refold at higher temperature more rapidly than at lower temperature. The refolding rates of both E64Q and E64D mutants were slightly different from the wild-type at all temperatures. These differences in refolding rates reflect changes in free-energy of activation of folding for a mutation. Estimated free-energy values (ΔΔGref) at 18 °C that were significantly different from zero indicate that interactions of the residue at position 64 toward neighbouring residues are present in the transition state of the reactivation process. The in vitro refolding experiments suggest that the thermal stability of the final structure of the mutants reflects differences in the conformational properties of a productive folding intermediate. Reactivation in vitro of all Glu64 mutants was thermosensitive, and so the refolding yields of the Glu64 mutants, although to different extents, decreased markedly with increasing temperature. The analysis of the reactivation at 18 °C indicates that amino acid replacement of Glu64 destabilizes the transition of folding. It should be noted that a single exponential equation could be fitted to refolding data for all adGSTD3-3 variants indicating that no significant amount of intermediate is accumulated during the reactivation of wild-type and variants, and that none of the mutations had a major change on the refolding pathway. Upon mutation of adGSTD3-3, rate constants decreased for all mutants (Table 2). This is because the removal of the negative charge or size decrease of the side-chain at this position destabilized the transition state and thereby increased the activation energy for folding. In particular, the ΔΔGref value for E64D is greater than E64Q, suggesting that the functional group size is important for the Glu64 position, which contributes to stabilizing the transition state. Previous investigations of the equivalent residue, Glu66, in PtGSTU1-1 (plant Tau class GST) demonstrated that alanine replacement made the enzyme unstable at 50 °C, retaining only about 15% of its activity compared with 95% for the wild-type enzyme [37]. In contrast, the reactivation yield of E66A was 2-fold greater than the wild-type. The effect of the mutation at this equivalent residue in PtGSTU1-1 was less than found in adGSTD3-3, an insect Delta class GST. This result might be explained by data for Glu64 in adGSTD4-4, an alternatively spliced product derived from the same gene as adGSTD3-3. Glu64 is not only located at the dimeric interface of the enzyme, but has also been identified as a ‘lock’ residue in the Delta class specific ‘lock-and-key clasp’ motif, which is not found in plant Tau class GST [38]. The lock-and-key motif, including the Delta class specific lock-and-key clasp motif, is located at the intersubunit interface and plays a crucial role in the structural stability of dimeric GSTs [39,40]. These data therefore confirm a critical structural role for the functionally conserved Glu64 residue for both overall protein stability and initial folding process.
Figure 3. Far-UV CD spectra of adGSTD3-3 and mutants.

CD spectra were measured in far-UV region from 190–260 nm. Spectra were recorded at a protein concentration of 0.3 mg/ml with cuvettes with a 2-μm path-length. D3, wild-type adGSTD3-3.
Figure 4. Intrinsic fluorescence spectra of wild-type and mutant adGSTD3-3.

Intrinsic fluorescence emission spectra were measured with the excitation wavelength 295 nm. The λmax and the fluorescence intensity of emission spectra were analysed at a protein concentration of 0.2 mg/ml. The λmax are 331 nm for wild-type (D3), 332 nm for E64Q, 335 nm for E64D and 334 nm for E64N. The intensity is shown in arbitrary units. The results are means±S.D. for experiments performed in triplicate.
Figure 5. Thermal stability of wild-type adGSTD3-3 (A), E64Q (B), E64D (C) and E64N (D) mutants at different temperatures.

Each enzyme (40 μM) was incubated at various temperatures in 0.1 M potassium phosphate buffer, pH 6.5, 1 mM EDTA and 5 mM DTT. Appropriate aliquots from an incubation mixture were assayed at 25 °C to monitor residual activity. The lines represent fits according to eqn (2), as described in the Materials and methods section.
Table 1. Kinetic and thermodynamic parameters for the activation barrier of thermal denaturation for wild-type and Glu64 mutants of adGSTD3-3.
| Enzyme | ku (min−1) | ku/ku,wt* | ΔGu* (kJ/mol) | ΔΔGu (kJ/mol) | ΔHu† (kJ/mol) | ΔΔHu† (kJ/mol) | ΔSu† (kJ/mol·K) | ΔΔSu† (kJ/mol·K) |
|---|---|---|---|---|---|---|---|---|
| Wild-type | 0.040±0.001 | 1 | 85.69±0.09 | 0 | 439.00±4.90 | 0 | 1.120±0.015 | 0 |
| E64Q | 1.583±0.028 | 39.58 | 76.08±0.05 | 9.63 | 231.38±13.02 | 207.62 | 0.490±0.041 | 0.630 |
| E64D | 0.256±0.008 | 6.40 | 80.85±0.08 | 4.86 | 307.51±1.28 | 131.49 | 0.720±0.004 | 0.400 |
| E64N | 1.590±0.045 | 39.75 | 76.07±0.08 | 9.64 | 218.52±10.92 | 220.48 | 0.450±0.035 | 0.670 |
Table 2. Kinetics of the reactivation and percentage recovery of wild-type adGSTD3-3 and Glu64 mutants during refolding at different temperatures and changes in free energy of the transition state of folding at 18 °C.
Statistics performed using one-way ANOVA and Tukey–Kramer multiple comparisons test. Values significantly different from wild-type are shown by * P<0.001. Nd, not determined, low enzymatic activity precluded performing this experiment.
| 18 °C | 25 °C | 37 °C | |||||
|---|---|---|---|---|---|---|---|
| Enzyme | kref (min−1) | % recovery | kref (min−1) | % recovery | kref (min−1) | % recovery | ΔΔGref at 18 °C (kJ/mol) |
| Wild-type | 0.169±0.020 | 60.9±0.4 | 0.518±0.053 | 35.1±3.4 | 0.957±0.027 | 35.1±0.6 | 0 |
| E64Q | 0.074±0.003 | 63.8±0.7 | 0.289±0.004 | 40.0±0.3 | 1.073±0.124 | 10.6±0.3 | −1.987±0.371* |
| E64D | 0.059±0.005 | 93.0±6.5 | 0.242±0.024 | 57.7±1.1 | 0.981±0.030 | 34.5±1.3 | −2.521±0.129* |
| E64N | Nd | Nd | Nd | Nd | Nd | Nd | Nd |
Figure 6. Kinetics of reactivation of wild-type adGSTD3-3 (●), E64Q (■) and E64D (▲) during refolding at different temperatures: 18 °C (A), 25 °C (B) and 33 °C (C).

Purified enzyme (20 μM), heterologously expressed at 18 °C, was first denatured in 4 M GdmCl for 30 min. This denaturant concentration was sufficient to completely unfold the proteins, as indicated by the loss of secondary structure shown by CD. Successive aliquots of unfolded enzyme were diluted (defining time 0) 1:40 into renaturation buffer at the different temperatures. The final GdmCl concentration was 0.1 M during the refolding. Appropriate aliquots from this incubation mixture were immediately assayed for catalytic activity at 25 °C.
Steady-state kinetic constants were obtained with various concentrations of GSH and CDNB substrate. Michaelis–Menten kinetic analysis was performed using non-linear regression (Table 3). All of the mutations showed significantly increased Km values for GSH. Individually, the mutants; E64Q, E64D and E64N had values 26-, 34- and 29-fold greater than wild-type. Conversely, no significant changes were found in the Km values for CDNB substrate when compared with the wild-type enzyme. The differences in kcat values with CDNB observed for E64D and E64N decreased approximately 1.5- and 25-fold respectively. The kinetic studies of soluble Glu64 mutant enzymes demonstrated that replacement with a glutamine residue, a similar size and property functional group, preserved the catalytic activity, whereas replacement with the slightly smaller amino acids aspartate and asparagine reduced kcat, especially the arparagine replacement, which nearly abolished enzyme activity. This suggests that the volume of the amino acid at this position affects packing of the active-site, which directly impacts upon enzyme catalysis. However, all the mutants showed a substantially lower affinity (greater Km) towards GSH indicating that the Glu64 position impacts upon the binding of GSH possibly through active-site rearrangement. Catalytic efficiency can be related to the difference in free-energy change for formation of transition states in the mutant and wild-type enzymes (ΔΔG), as calculated from eqn (9) [41]:
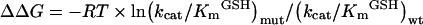 |
(9) |
These calculated values are 8.03 kJ/mol for E64Q, 9.94 kJ/mol for E64D and 16.33 kJ/mol for E64N at 25 °C, indicative of a decreased stabilization of the transition state for the Glu64 mutations. Stabilization of the transition state may occur through a pre-organized active-site contributing to catalysis through multiple mechanisms: binding interaction with GSH, activation of GSH by thiol deprotonation or nucleophilic attack at the electrophilic centre (σ-bond formation) by the thiolate. Deficiency in the pre-organized environment (the changes that occur along the reaction pathway from reactants to the transition state) would decrease the rate of catalysis by incompletely providing a stabilization of the transition state [42]. Therefore, the effect on the rate-limiting step in the catalytic mechanism was examined. The pH dependence of kcat/KmCDNB reflects a kinetically relevant ionization of the GST–GSH complex. Therefore, an apparent pKa value of 6.36 was determined for the wild-type adGSTD3-3. To differentiate the influence of the functional group of Glu64 on the GSH thiol ionization, the pKa values for Glu64 mutants were measured by this kinetic approach (Table 3). An increased pKa for bound GSH of approx. 0.6 and 1 pH unit greater than that found for wild-type were observed for the E64Q and E64N mutants. It has been shown previously that a crucial function of the electron-sharing network is to lower the pKa of the thiol group of the bound GSH [22]. Our results show that Glu64 replacement with the polar amino acids glutamine and asparagine increased the pKa values by about 0.5–1.0 pH unit. This consequence appears to be due to the deletion of the negative charge, resulting in loss of ionic interaction within the electron-sharing network. This is supported by the replacement of the critical glutamate residue with the negatively charged aspartate, which has no affect on the ionization process. Therefore, an acidic amino acid in this position is essential to form an ionic interaction to fulfil the function of the electron-sharing network in the ionization process. However, the formation of a σ-complex intermediate in the enzyme-catalysed reaction was affected by the replacement with glutamine and aspartate. It is well established that the bimolecular nucleophilic substitution reactions proceed through a σ-complex intermediate [43]. Thus, the rate-limiting formation of a spontaneous σ-complex intermediate can be increased by replacement of chlorine in the CDNB molecule with the more electronegative fluorine. The ratio of the catalytic rate of GSH with FDNB and CDNB was comparable to the ratio of the second-order rate constants for a spontaneous uncatalysed reaction. That is, kcatFDNB/kcatCDNB=40 is similar to kcFDNB/kcCDNB=47, which indicates that the σ-complex formation is the rate-limiting step. Although the kcatFDNB/kcatCDNB of all Glu64 mutants reflected varying insensitivity to the nature of the leaving group, there were two mutants E64Q and E64D that exhibited significant differences in the catalytic efficiency (kcat/Km) (Table 4). For the E64N mutant, it appears that an alteration of the relative turn-over number is a consequence of changes in binding affinity towards different substrate leaving groups rather than a reflection of the rate of σ-complex formation. Differences in relative catalytic efficiency (kcat/Km) for the fluoride/chloride leaving group replacement may not strongly support the idea that the rate-determining step to σ-complex formation for E64Q and E64D was changed. Although it appears that transition state stability of the enzyme–intermediate complex, influenced by electron density and distribution in the σ-complex, was partially altered by these two amino acid replacements [43,44].
Table 3. Steady-state kinetic constants and pKa values for the thiol group of GSH of wild-type and mutants of adGSTD3-3 for the CDNB conjugation reaction at pH 6.5 and 25 °C.
The enzyme activities were measured at various concentrations of CDNB and GSH in 0.1 M phosphate buffer pH 6.5. The pKa was obtained by using 0.1 M sodium acetate buffers (from pH 5.0 to 5.5) and 0.1 M potassium phosphate buffer (from pH 6.0 to 8.5).The reaction was monitored at 340 nm, ϵ 9600 M−1cm−1. Statistics were performed using one-way ANOVA and Tukey–Kramer multiple comparisons test. Values significantly different from wild-type are shown by * P<0.001. The wild-type values have been reported previously [25].
| Enzyme | kcat (s−1) | KmGSH (mM) | KmCDNB (mM) | kcat/KmGSH (s−1/mM) | kcat/KmCDNB (s−1/mM) | pKa |
|---|---|---|---|---|---|---|
| Wild-type | 35.4 | 0.27±0.05 | 0.14±0.01 | 131 | 246 | 6.36±0.11 |
| E64Q | 36.3 | 7.10±0.28* | 0.43±0.03 | 5.11 | 83.6 | 6.97±0.14* |
| E64D | 21.6 | 9.12±0.20* | 0.28±0.01 | 2.37 | 75.8 | 6.42±0.12 |
| E64N | 1.4 | 7.89±0.19* | 0.33±0.01 | 0.18 | 4.4 | 7.37±0.07* |
Table 4. Effect of fluoride/chloride leaving group substitution on the rate of catalysis.
The ratios of kinetic constants for the conjugation reaction catalyzed by adGSTD3-3 enzymes for GSH with CDNB or FDNB as co-substrates were calculated at pH 6.5. Statistics performed using one-way ANOVA and Tukey–Kramer multiple comparisons test. Values significantly different from wild-type are shown by * P<0.001. The wild-type values have been reported previously [25].
| Enzyme | kcatFDNB/kcatCDNB | (kcat/Km)FDNB/(kcat/Km)CDNB |
|---|---|---|
| Wild-type | 2.40±0.08 | 6.72±0.34 |
| E64Q | 6.97±0.24* | 14.36±1.22* |
| E64D | 8.40±0.08* | 10.31±0.38* |
| E64N | 5.92±0.05* | 6.58±0.05 |
The effect of viscosity on kinetic parameters was examined to elucidate the rate-limiting step in the catalytic reaction. A decrease in the rate constant with increasing medium viscosity should reflect the weight of diffusion events on catalysis [45]. It would indicate that the rate-limiting step is related to diffusion-controlled motions of the protein or the dissociation of the product. A plot of the reciprocal of the relative catalytic constant (kcato/kcat) against the relative viscosity (η/ηo) should be linear. The slope should be equal to unity when the product release or structural transition is limited by a strictly diffusional barrier. If the slope approaches zero either the chemistry or another non-diffusion barrier is rate-limiting. For wild-type adGSTD3-3, a plot of the inverse relative rate constant (kcato/kcat) versus the relative viscosity (η/ηo) gives a linear dependence with a slope (1.14±0.01) very close to unity (Figure 7). In contrast the E64Q mutant enzyme yields plots that are fully viscosity independent, with a slope approaching zero (0.05±0.01). The other mutants, E64D and E64N, exhibited kcat values with different degrees of viscosity dependence compared with the wild-type enzyme. The viscosity experiment showed that the rate-limiting step catalysed by adGSTD3-3 is a non-physical step (Figure 7). However, structural alterations in the Glu64 mutants decreased the viscosity effects on the enzyme to intermediate values (0<slope<1), especially for the replacement with the polar residues glutamine and asparagine. This indicates that the rate-limiting step is not strictly dependent on a diffusion barrier or other viscosity-dependent motions and that conformational changes of the engineered proteins contribute to the rate-limiting step [45]. This suggests that the structural flexibility of functionally important regions of the engineered enzymes have been altered. Previously, we have observed [22] that changing the residues in the electron-sharing network can influence the topology of the active-site, which affects both the catalytic mechanism as well as the structural maintenance of the enzyme. The results of the present study demonstrate that the functionally conserved Glu64, which is now identified as being part of the electron-sharing network, impacts upon enzyme catalysis not only through its negative charge but also through structural effects.
Figure 7. Viscosity effect on kinetic parameters of wild-type adGSTD3-3 and mutant enzymes.

The effect of viscosity on kinetic constants was assayed by using 0.1 M potassium phosphate buffer, pH 6.5, with various glycerol concentrations. Dependence of the reciprocal of the relative turn-over number (kcato/kcat) on the relative viscosity (η/ηo) for CDNB as a co-substrate. The experiment was performed in triplicate and the lines were calculated by linear regression analysis. The slopes of the linear regression lines are 1.14±0.01 for wild-type, 0.05±0.01 for E64Q, 0.85±0.01 for E64D and 0.42±0.02 for E64N.
In conclusion, the results of the present paper, as well as the high level of functional conservation of the residue at position 64 among all classes of GSTs supports the hypothesis that Glu64 is part of a functionally conserved electron-sharing network. The present paper now extends the network identified previously to include three critical residues that form ionic bridge interactions. These are between a negatively charged/polar active site residue (glutamate, aspartate or glutamine), a positively charged GSH glutamyl α-amino, a negatively charged GSH glutamyl α-carboxylate, a positively charged active-site residue (primarily arginine) and a negatively charged active-site residue (glutamate or aspartate) stabilized by hydrogen-bonding networks with surrounding residues (serine, threonine and/or water mediated contact). Glu64 in the electron-sharing network contributes to the function of this motif and the base-assisted deprotonation model which are essential for the GSH ionization process in the catalytic mechanism. However, this residue also appears to affect additional steps in the enzyme catalytic strategy including binding of GSH to the enzyme active site, nucleophilic attack by thiolate at the electrophilic centre and product formation. Therefore, the Glu64 position also appears to impact upon catalysis through roles in both initial folding and structural maintenance.
Acknowledgments
This work was supported by the Thailand Research Fund. P.W. was supported by a Royal Golden Jubilee Scholarship and a scholarship from the Senior Research Fellowship of Professor Emeritus Sakol Panyim.
References
- 1.Sheehan D., Meade G., Foley V. M., Dowd C. A. Structure, function and evolution of glutathione transferases: implications for classification of non-mammalian members of an ancient enzyme superfamily. Biochem. J. 2001;360:1–16. doi: 10.1042/0264-6021:3600001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cho S.-G., Lee Y. H., Park H.-S., Ryoo K., Kang K. W., Park J., Eom S.-J., Kim M. J., Chang T.-S., Choi S.-Y., et al. Glutathione S-transferase Mu modulates the stress-activated signals by suppressing apoptosis signal-regulating kinase 1. J. Biol. Chem. 2001;276:12749–12755. doi: 10.1074/jbc.M005561200. [DOI] [PubMed] [Google Scholar]
- 3.Gate L., Majumdar R. S., Lunk A., Tew K. D. Increased myeloproliferation in glutathione S-transferase π deficient mice is associated with a deregulation of JNK and janus kinase/STAT. J. Biol. Chem. 2004;279:8608–8616. doi: 10.1074/jbc.M308613200. [DOI] [PubMed] [Google Scholar]
- 4.Ronai Z. Coordinated regulation of stress kinases by GSTp. Chem. Biol. Interact. 2001;133:285–286. [Google Scholar]
- 5.Hayes J. D., Pulford D. J. The glutathione S-transferase supergene family: regulation of GST and the contribution of the isoenzymes to cancer chemoprotection and drug resistance. CRC Crit. Rev. Biochem. Mol. Biol. 1995;30:445–600. doi: 10.3109/10409239509083491. [DOI] [PubMed] [Google Scholar]
- 6.Mannervik B., Danielson U. H. Glutathione transferases: structure and catalytic activity. CRC Crit. Rev. Biochem. 1988;23:283–337. doi: 10.3109/10409238809088226. [DOI] [PubMed] [Google Scholar]
- 7.Armstrong R. N. Structure, catalytic mechanism, and evolution of the glutathione transferases. Chem. Res. Toxicol. 1997;10:2–18. doi: 10.1021/tx960072x. [DOI] [PubMed] [Google Scholar]
- 8.Habig W. H., Pabst M. J., Jakoby W. B. Glutathione S-transferases. The first enzymatic step in mercapturic acid formation. J. Biol. Chem. 1974;249:7130–7139. [PubMed] [Google Scholar]
- 9.Ahmad H., Wilson D. E., Fritz R. R., Singh S. V., Medh R. D., Nagle G. T., Awasthi Y. C., Kurosky A. Primary and secondary structural analyses of glutathione S-transferase π from human placenta. Arch. Biochem. Biophys. 1990;278:398–408. doi: 10.1016/0003-9861(90)90277-6. [DOI] [PubMed] [Google Scholar]
- 10.Board P., Baker R. T., Chelvanayagam G., Jermiin L. S. Zeta, a novel class of glutathione transferases in a range of species from plants to humans. Biochem. J. 1997;328:929–935. doi: 10.1042/bj3280929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mannervik B., Ålin P., Guthenberg C., Jensson H., Tahir M. K., Warholm M., Jörnvall H. Identification of three classes of cytosolic glutathione transferase common to several mammalian species: correlation between structural data and enzymatic properties. Proc. Natl. Acad. Sci. U.S.A. 1985;82:7202–7206. doi: 10.1073/pnas.82.21.7202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mannervik B., Awasthi Y. C., Board P. G., Hayes J. D., Di Ilio C., Ketterer B., Listowsky I., Morgenstern R., Muramatsu M., Pearson W. R., et al. Nomenclature for human glutathione transferases. Biochem. J. 1992;282:305–306. doi: 10.1042/bj2820305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Meyer D. J., Coles B., Pemble S. E., Gilmore K. S., Fraser G. M., Ketterer B. Theta, a new class of glutathione transferases purified from rat and man. Biochem. J. 1991;274:409–414. doi: 10.1042/bj2740409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Motoyama N., Dauterman W. C. Molecular weight, subunits, and multiple forms of glutathione S-transferase from the house fly. Insect Biochem. 1978;8:337–348. [Google Scholar]
- 15.Pemble S. E., Taylor J. B. An evolutionary perspective on glutathione transferases inferred from class-Theta glutathione transferase cDNA sequences. Biochem. J. 1992;287:957–963. doi: 10.1042/bj2870957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ding Y., Ortelli F., Rossiter L. C., Hemingway J., Ranson H. The Anopheles gambiae glutathione transferase supergene family: annotation, phylogeny and expression profiles. BMC Genomics. 2003;4:35–50. doi: 10.1186/1471-2164-4-35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wilce M. C. J., Parker M. W. Structure and function of glutathione S-transferases. Biochim. Biophys. Acta. 1994;1205:1–18. doi: 10.1016/0167-4838(94)90086-8. [DOI] [PubMed] [Google Scholar]
- 18.Armstrong R. N., Rife C., Wang Z. Structure, mechanism and evolution of thiol transferases. Chem. Biol. Interact. 2001;133:167–169. [Google Scholar]
- 19.Caccuri A. M., Ascenzi P., Antonini G., Parker M. W., Oakley A. J., Chiessi E., Nuccetelli M., Battistoni A., Bellizia A., Ricci G. Structural flexibility modulates the activity of human glutathione transferase P1-1. Influence of a poor co-substrate on dynamics and kinetics of human glutathione transferase. J. Biol. Chem. 1996;271:16193–16198. doi: 10.1074/jbc.271.27.16193. [DOI] [PubMed] [Google Scholar]
- 20.Caccuri A. M., Antonini G., Nicotra M., Battistoni A., Lo Bello M., Board P. G., Parker M. W., Ricci G. Catalytic mechanism and role of hydroxyl residues in the active site of theta class glutathione S-transferases. Investigation of Ser-9 and Tyr-113 in a glutathione S-transferase from the Australian sheep blowfly, Lucilia cuprina. J. Biol. Chem. 1997;272:29681–29686. doi: 10.1074/jbc.272.47.29681. [DOI] [PubMed] [Google Scholar]
- 21.Gustafsson A., Pettersson P. L., Grehn L., Jemth P., Mannervik B. Role of the glutamyl α-carboxylate of the substrate glutathione in the catalytic mechanism of human glutathione transferase A1-1. Biochemistry. 2001;40:15835–15845. doi: 10.1021/bi010429i. [DOI] [PubMed] [Google Scholar]
- 22.Winayanuwattikun P., Ketterman A. J. An electron-sharing network involved in the catalytic mechanism is functionally conserved in different glutathione transferase classes. J. Biol. Chem. 2005;280:31776–31782. doi: 10.1074/jbc.M502612200. [DOI] [PubMed] [Google Scholar]
- 23.Caccuri A. M., Antonini G., Board P. G., Parker M. W., Nicotra M., Lo Bello M., Federici G., Ricci G. Proton release on binding of glutathione to Alpha, Mu and Delta class glutathione transferases. Biochem. J. 1999;344:419–425. [PMC free article] [PubMed] [Google Scholar]
- 24.Tan K.-L., Chelvanayagam G., Parker M. W., Board P. G. Mutagenesis of the active site of the human Theta-class glutathione transferase GSTT2-2: catalysis with different substrates involves different residues. Biochem. J. 1996;319:315–321. doi: 10.1042/bj3190315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Winayanuwattikun P., Ketterman A. J. Catalytic and structural contributions for glutathione binding residues in a delta class glutathione S-transferase. Biochem. J. 2004;382:751–757. doi: 10.1042/BJ20040697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jirajaroenrat K., Pongjaroenkit S., Krittanai C., Prapanthadara L., Ketterman A. J. Heterologous expression and characterization of alternatively spliced glutathione S-transferases from a single Anopheles gene. Insect Biochem. Mol. Biol. 2001;31:867–875. doi: 10.1016/s0965-1748(01)00032-7. [DOI] [PubMed] [Google Scholar]
- 27.Bradford M. M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 28.Caccuri A. M., Ascenzi P., Lo Bello M., Federici G., Battistoni A., Mazzetti P., Ricci G. Are the steady state kinetics of glutathione transferase always dependent on the deprotonation of the bound glutathione? New insights in the kinetic mechanism of GST P1-1. Biochem. Biophys. Res. Commun. 1994;200:1428–1434. doi: 10.1006/bbrc.1994.1610. [DOI] [PubMed] [Google Scholar]
- 29.Wolf A. V., Brown M. G., Prentiss P. G. Boca Raton: CRC Press; 1985. Handbook of Chemistry and Physics. [Google Scholar]
- 30.Kong G. K. W., Polekhina G., McKinstry W. J., Parker M. W., Dragani B., Aceto A., Paludi D., Principe D. R., Mannervik B., Stenberg G. Contribution of glycine 146 to a conserved folding module affecting stability and refolding of human glutathione transferase P1-1. J. Biol. Chem. 2003;278:1291–1302. doi: 10.1074/jbc.M209581200. [DOI] [PubMed] [Google Scholar]
- 31.Stenberg G., Dragani B., Cocco R., Principe D. R., Mannervik B., Aceto A. A conserved ‘hydrophobic staple motif’ plays a crucial role in the refolding of human glutathione transferase P1-P1. Chem. Biol. Interact. 2001;133:49–50. doi: 10.1074/jbc.275.14.10421. [DOI] [PubMed] [Google Scholar]
- 32.Jackson S. E., el Masry N., Fersht A. R. Structure of the hydrophobic core in the transition state for folding of chymotrypsin inhibitor 2: a critical test of the protein engineering method of analysis. Biochemistry. 1993;32:11270–11278. doi: 10.1021/bi00093a002. [DOI] [PubMed] [Google Scholar]
- 33.Widersten M., Kolm R. H., Björnestedt R., Mannervik B. Contribution of five amino acid residues in the glutathione binding site to the function of human glutathione transferase P1-1. Biochem. J. 1992;285:377–381. doi: 10.1042/bj2850377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dirr H. W., Wallace L. A. Role of the C-terminal helix 9 in the stability and ligandin function of class a glutathione transferase A1-1. Biochemistry. 1999;38:15631–15640. doi: 10.1021/bi991179x. [DOI] [PubMed] [Google Scholar]
- 35.Jones M. N. Biochemical Thermodynamics (Studies in Modern Thermodynamics) In: Jones M. N., editor. Oxford: Elsevier; 1979. pp. 75–115. [Google Scholar]
- 36.Haynie D. T. Biological thermodynamics. Cambridge: Cambridge University Press; 2001. Statistical thermodynamics; pp. 185–222. [Google Scholar]
- 37.Zeng Q.-Y., Wang X.-R. Catalytic properties of glutathione-binding residues in a τ class glutathione transferase (PtGSTU1) from Pinus tabulaeformis. FEBS Lett. 2005;579:2657–2662. doi: 10.1016/j.febslet.2005.03.086. [DOI] [PubMed] [Google Scholar]
- 38.Wongsantichon J., Ketterman A. J. An intersubunit lock-and-key ‘clasp’ motif in the dimer interface of delta class glutathione transferase. Biochem. J. 2006;394:135–144. doi: 10.1042/BJ20050915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sayed Y., Wallace L. A., Dirr H. W. The hydrophobic lock-and-key intersubunit motif of glutathione transferase A1-1: implications for catalysis, ligandin function and stability. FEBS Lett. 2000;465:169–172. doi: 10.1016/s0014-5793(99)01747-0. [DOI] [PubMed] [Google Scholar]
- 40.Stenberg G., Abdalla A.-M., Mannervik B. Tyrosine 50 at the subunit interface of dimeric human glutathione transferase P1-1 is a structural key residue for modulating protein stability and catalytic function. Biochem. Biophys. Res. Commun. 2000;271:59–63. doi: 10.1006/bbrc.2000.2579. [DOI] [PubMed] [Google Scholar]
- 41.Dirr H. W., Little T., Kuhnert D. C., Sayed Y. A conserved N-capping motif contributes significantly to the stabilisation and dynamics of the C-terminal region of class alpha glutathione transferases. J. Biol. Chem. 2005;280:19480–19487. doi: 10.1074/jbc.M413608200. [DOI] [PubMed] [Google Scholar]
- 42.Garcia-Viloca M., Gao J., Karplus M., Truhlar D. G. How enzymes work: analysis by modern rate theory and computer simulations. Science. 2004;303:186–195. doi: 10.1126/science.1088172. [DOI] [PubMed] [Google Scholar]
- 43.Chen W.-J., Graminski G. F., Armstrong R. N. Dissection of the catalytic mechanism of isozyme 4–4 of glutathione S-transferase with alternative substrates. Biochemistry. 1988;27:647–654. doi: 10.1021/bi00402a023. [DOI] [PubMed] [Google Scholar]
- 44.Graminski G. F., Zhang P., Sesay M. A., Ammon H. L., Armstrong R. N. Formation of the 1-(S-glutathionyl)-2,4,6-trinitrocyclohexadienate anion at the active site of glutathione S-transferase: evidence for enzymic stabilization of σ-complex intermediates in nucleophilic aromatic substitution reactions. Biochemistry. 1989;28:6252–6258. doi: 10.1021/bi00441a017. [DOI] [PubMed] [Google Scholar]
- 45.Johnson W. W., Liu S., Ji X., Gilliland G. L., Armstrong R. N. Tyrosine 115 participates both in chemical and physical steps of the catalytic mechanism of a glutathione S-transferase. J. Biol. Chem. 1993;268:11508–11511. [PubMed] [Google Scholar]