

Abstract
Background
Malignant rhabdoid tumors (MRTs) are extremely aggressive and resist current radio- and chemotherapic treatments. To gain insight into the dysfunctions of MRT cells, the apoptotic response of a model cell line, MON, was analyzed after exposure to several genotoxic and non-genotoxic agents employed separately or in association.
Results
Fluorescence microscopy of chromatin morphology and electrophoretic analysis of internucleosomal DNA fragmentation revealed that MON cells were, comparatively to HeLa cells, resistant to apoptosis after treatment with etoposide, cisplatin (CisPt) or X-rays, but underwent some degree of apoptosis after ultraviolet (UV) C irradiation. Concomitant treatment of MON cells with X-rays or vinblastine and the phosphatidylinositol 3-kinase (PI3-K) inhibitor wortmannin resulted in synergistic induction of apoptosis. Western blot analysis showed that the p53 protein was upregulated in MON cells after exposure to all the different agents tested, singly or in combination. In treated cells, the p53 downstream effectors p21WAF1/CIP1, Mdm2 and Bax were induced with some inconsistency with regard to the accumulation of p53. Poly ADP-ribose polymerase (PARP) cleavage, indicative of ongoing apoptosis, occurred in UVC-irradiated cells and, especially, in cells treated with combinations of X-rays or vinblastine with wortmannin. However, there was moderate or no PARP cleavage in cells treated with CisPt, X-rays, vinblastine or wortmannin singly or with the combinations X-rays plus CisPt or vinblastine and CisPt plus vinblastine or wortmannin. The synergistic effect on the induction of apoptosis exerted by some agent combinations corresponded with synergy in respect of MON cell growth inhibition.
Conclusion
These results suggest abnormalities in the p53 pathway and apoptosis control in MRT cells. The Ras/PI3-K/AKT signaling pathway might also be deregulated in these cells by generating an excess of survival factors. These dysfunctions might contribute to the resistance of MRTs to current antineoplastic treatments and could warrant consideration in the search of new therapeutic approaches.
Background
MRTs occur during early childhood in soft tissues, especially kidney and the central nervous system [1]. Prognosis is poor because of the high cellular proliferation rate, propensity to metastasis and marked resistance to current radio- and chemo-therapeutic interventions [2-5].
According to cytogenetic and molecular analyses, MRTs are generally caused by biallelic alterations of the hSNF5/INI1 gene [6]. This gene encodes a member of the chromatin-remodeling SWI/SNF multiprotein complexes that activate or repress transcription of target genes [7,8]. SWI/SNF activity is also required for Rb-dependent transcriptional repression and subsequent inhibition of proliferation [9]. Moreover, the hSNF5/INI1 protein directly co-operates with several important cellular factors: c-Myc [10], Gadd34 [11] and ALL-1 [12].
Overexpression of c-Myc, a nuclear phosphoprotein that regulates DNA replication and cell division, is a consistent characteristic of rhabdoid cells [13-16]. IGF-II, IGF-IR and IGF-IIR, which promote cell proliferation and DNA synthesis through an autocrine mechanism, are also constitutively expressed in several MRT cells lines [17]. High levels and unusual distribution of p53 protein have been observed, suggesting some abnormalities in p53 status, but there is little Mdm2 mRNA expression [16]. However, p53 protein and the downstream effectors, p21WAF1/CIP1and Mdm2 were up-regulated by DNA-damaging drugs and the p53 pathway was considered to be functional [18]. There appear to be no rearrangements or amplifications in Myc, Ras, Erb B-2 and p53 genes in these cells [19].
Transfection experiments have shown that when hSNF5/INI1 protein is re-introduced into cells derived from MRTs, it inhibits the entry into S-phase [20], prevents cell proliferation, causes flat cell formation, and directly represses cyclin D1 gene [21]. Moreover, hSNF5/INI1 overexpression induced apoptosis in two of the three cell lines tested [22].
The aim of the present study was to gain further insight into the dysfunctions of MRT cells. A model cell line, MON, was evaluated in terms of its responses to the genotoxic and non-genotoxic stresses induced by physical and chemical agents with different modes of action, employed singly or in association. The treatments provoked different kinds of DNA (and protein) damage (single and double strand breaks, oxidation, alkylation, crosslinks etc.), or interfered with cellular signaling functions. Results showed (a) that MON cells may have impaired control of apoptosis and (b) that apoptosis can be strongly activated by inhibition of the PI3-K pathway under particular stress conditions.
Results
Apoptosis in response to different genotoxic and non-genotoxic stresses was assessed by monitoring the appearance of typical nuclear morphological changes and internucleosomal DNA cleavage, and by investigating some steps in the apoptotic pathway at the molecular level.
Apoptotic response
In Fig. 1 the responses of MON and HeLa cells are compared. As appraised by morphological criteria such as chromatin condensation and fragmentation, the rhabdoid cells were largely refractory to the induction of apoptosisafter exposure to etoposide (up to 40 μM, for 2 h), CisPt (up to 40 μM, for 2 h) and X-rays (up to 10 Gy). However, they showed some degree of apoptosis following UVC irradiation (20 J/m2). All these agents damaged DNA, directly or indirectly. Etoposide complexes with topoisomerase II and DNA to enhance double-strand and single-strand cleavage; CisPt forms adducts with the DNA dinucleotide d(pGpG) inducing intrastrand and interstrand crosslinks; X-rays produce single- and double-strand breaks and base modifications; UVC radiation mainly induces pyrimidine-pyrimidine dimers and 6–4 photoproducts. In contrast, HeLa cells showed significant or high levels of apoptosis in response to all treatments, even when lower drug concentrations or doses were used. It is worth noting that the doubling times of MON and HeLa cells were similar (around 24 h in the conditions used). Very few cells in either line showed a normal nuclear morphology associated with a staining by propidium iodide (1 μg/ml) after the various treatments, indicating that almost no necrosis had occurred.
Figure 1.
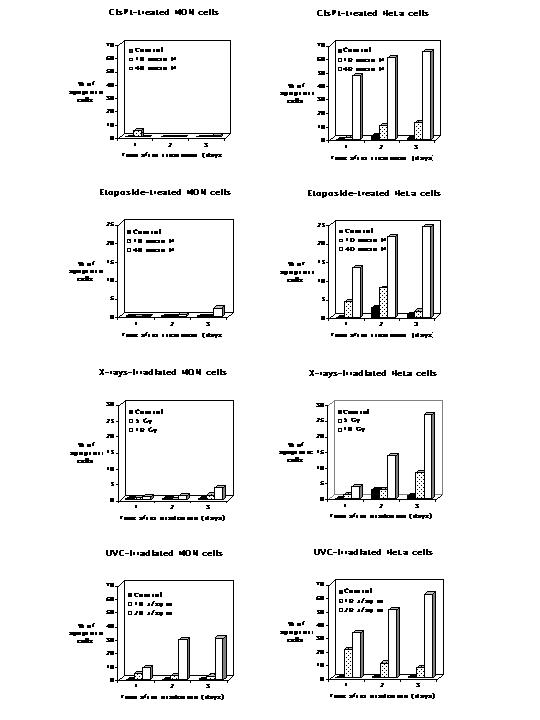
Induction of apoptosis assessed by fluorescence microscopy in HeLa and MON cells exposed for 2 h to CisPt and etoposide or irradiated with X-rays and UVC. Cells, cultured and treated in Petriperm dishes, were stained with Hoechst 33342 and visualized with an inverted epifluorescence microscope at different time intervals after the treatments. Cells with fragmented, marginated chromatin were defined as apoptotic. Quantitative analyses were performed by counting at least 1000 cells for each data point.
Electropherograms confirmed that MON cells did not enter into apoptosis after exposure to CisPt (10 μM, for 2 h), etoposide (10 μM, for 2 h) or X-rays (5 Gy) over a three-day period, but they did after UVC radiation (10–20 J/m2). In contrast, HeLa cells underwent DNA fragmentation typical of apoptosis after all the different treatments. Representative gels are shown in Fig. 2A and 2B. These various results indicate that although MON cells can become apoptotic, they are much more resistant than HeLa cells to the induction of apoptosis by a variety of agents.
Figure 2.

Analysis of internucleosomal DNA fragmentation. A: Agarose gel electrophoresis of low molecular weight DNA extracted from HeLa and MON cells exposed for 2 h to 10 μM CisPt (P) or 10 μM etoposide (E) or irradiated with 5 Gy of X-rays (X) and harvested on consecutive days thereafter. C refers to untreated controls. B: Agarose gel electrophoresis of low molecular weight DNA extracted from HeLa and MON cells exposed to 5, 10 or 20 J/m2 UVC or not irradiated (0) and harvested on consecutive days thereafter. In A and B, low molecular weight DNA was extracted from 2.106 cells at each time and analyzed by electrophoresis through a 2% agarose gel. Sizes of DNA molecular weight standards are indicated. Gels shown are representative of three separate experiments.
Cancers often show perturbation of signaling pathways and over-expression of survival signals. Experiments were therefore performed to determine whether wortmannin, a PI3-K inhibitor, affected the apoptotic response of MON cells to drug and radiation treatments. Cells were irradiated with different doses of X-rays or treated with several concentrations of vinblastine (an anticancer drug that inhibits microtubule assembly by binding tubulin) or wortmannin. These agents were employed separately or together. Fig. 3A confirms that X-irradiation up to 8 Gy did not induce any significant apoptosis in MON cells and shows that wortmannin induced moderate apoptosis at the concentrations used (up to 4 μM). However, when the two agents were applied together, the resulting degree of apoptosis was much greater than the addition of effects produced by X-rays or wortmannin singly. A similar synergistic effect was produced when vinblastine and wortmannin were combined (Fig. 3B). Vinblastine alone at 8 nM induced a moderate apoptotic response in MON cells. These findings suggest that inhibition of PI3-K may increase the susceptibility of MON cells to potential apoptotic stresses.
Figure 3.
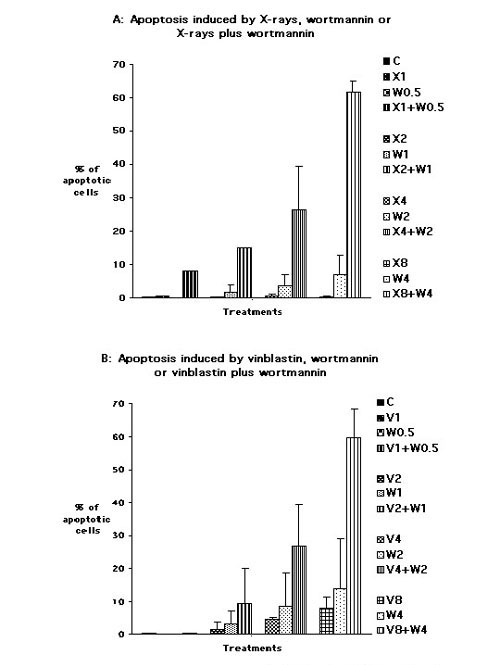
Induction of apoptosis assessed by fluorescence microscopy in MON cells following:A) irradiation with X-rays at doses of 1 (X1), 2 (X2), 4 (X4) or 8 Gy (X8), or continuous exposure to wortmannin at 0.5 (W0.5), 1 (W1), 2 (W2) or 4 μM (W4), or combined treatments with 1 Gy X-rays plus 0.5 μM wortmannin (X1+W0.5), 2 Gy X-rays plus 1 μM wortmannin (X2+W1), 4 Gy X-rays plus 2 μM wortmannin (X4+W2) and 8 Gy X-rays plus 4 μM wortmannin (X8+W4).B) continuous exposure to vinblastine at 1 (V1), 2 (V2), 4 (V4) or 8 nM (V8), or to wortmannin at 0.5 (W0.5), 1 (W1), 2 (W2) or 4 μM (W4) or to the associations 1 nM vinblastine plus 0.5 μM wortmannin (V1+W0.5), 2 nM vinblastine plus 1 μM wortmannin (V2+W1), 4 nM vinblastine plus 2 μM wortmannin (V4+W2) and 8 nM vinblastine plus 4 μM wortmannin (V8+W4). Cells, cultured and treated in Petriperm dishes, were stained with Hoechst 33342 and visualized with an inverted epifluorescence microscope 24 h after the beginning of the treatments. Fragmented, marginated chromatin defined apoptotic cells. Quantitative analyses were performed by counting at least 1000 cells for each treatment. Data in each graph represent the means values ± standard deviations of two experiments.
Western blot analysis
Western blot analysis (Fig. 4) showed that p53, a key regulator of cell cycle check points and apoptosis after DNA damage, was abundant in MON rhabdoid cells compared to HeLa cells. Moreover, it accumulated more rapidly and to a greater extent in the former than in the latter cells after UVC irradiation and CisPt treatment. The accumulation of p53 induced in MON cells by treatment with 40 μM CisPt for 2 h was of the same order than that produced by 20 J/m2 of UVC. However, the cleavage of PARP, one of the substrate of cysteines proteases activated during apoptosis, occurred in UVC-damaged MON cells but not following CisPt treatment. In HeLa cells the cleavage of PARP was observed after both UVC and CisPt damage.
Figure 4.
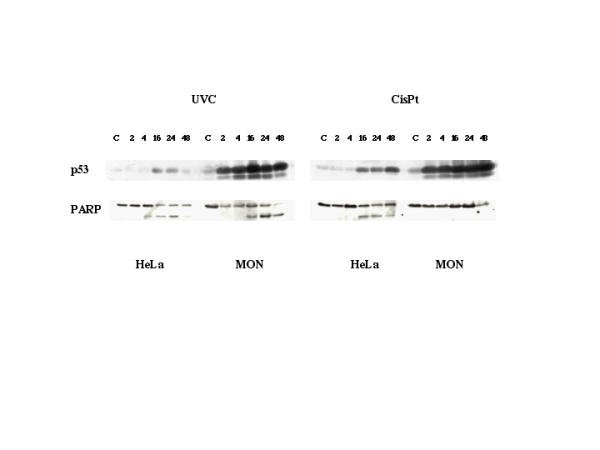
Western blot analysis of p53 accumulation and PARP cleavage in HeLa and MON cells treated with 40 μM CisPt for 2 h or irradiated with 20 J/m2 UVC and incubated for different time intervals before the sample processing. These times (hours) were noted above the lanes. C refers to untreated controls. Lysates from 5.105 cells were subjected to SDS-PAGE (8%), blotted and incubated with a monoclonal antibody to p53 or PARP. The data are representative of two (PARP) to five (p53) independent experiments.
In Fig. 5, western blot analysis was done after challenging MON cells with individual agents (X-rays, CisPt, vinblastine, wortmannin) or with different combinations of them. On the basis of the results in Fig. 3, analysis was performed after 24 h of continuous treatment. p53 was considerably upregulated by X-rays (4 Gy) and, as already shown, by CisPt (4 μM), but also by the non genotoxic agent vinblastine (8 nM). Wortmannin induced p53 protein only moderately at the concentrations used (2–4 μM). Combination of X-rays with wortmannin, CisPt or vinblastine, or of vinblastine with CisPt, resulted in higher accumulation of p53 than that provoked by any agent deployed separately. CisPt plus wortmannin, and vinblastine plus wortmannin, induced p53 to similar or somewhat lower levels than those observed after exposure to CisPt or vinblastine alone.
Figure 5.
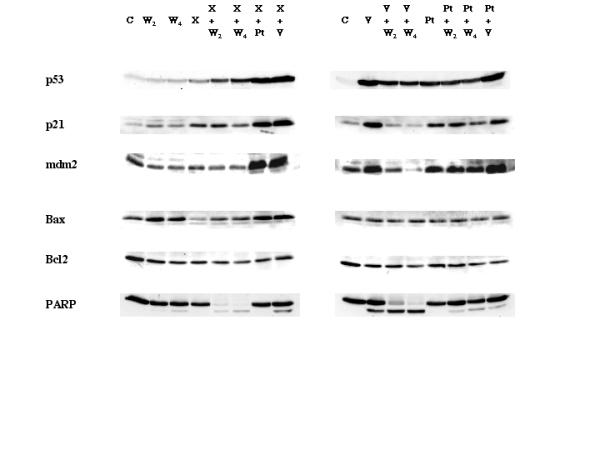
Western blot analysis of relevant proteins involved in stress response and apoptosis in MON cells irradiated with 4 Gy of X-rays (X) or exposed to 2 (W2) or 4 μM wortmannin (W4), 8 nM vinblastine (V), 4 μM CisPt (Pt) or by the combined treatments 4 Gy X-rays plus 2 (X+W2) or 4 μM wortmannin (X+W4), 4 Gy X-rays plus 4 μM CisPt (X+Pt), 4 Gy X-rays plus 8 nM vinblastine (X+V), 8 nM vinblastine plus 2 (V+W2) or 4 μM wortmannin (V+W4), 4 μM CisPt plus 2 (Pt+W2) or 4 μM wortmannin (Pt+W4), and 4 μM CisPt plus 8 nM vinblastine (Pt+V). C refers to the untreated control. Lysates from 5.105 cells were subjected to 7–14% gradient SDS-PAGE, blotted and incubated with the appropriate antibodies. The data are representative of two independent experiments.
The downstream effectors p21WAF1/CIP and Mdm2 were expressed in treated cells to an extent roughly correlating with the amount of p53, except after treatment with X-rays plus wortmannin or with vinblastine plus wortmannin, which resulted in an induction of p21WAF1/CIP1 and of Mdm2 much lower than that of p53.
The pro-apoptotic protein Bax, also normally under the control of p53, was moderately induced by wortmannin alone and by X-rays plus CisPt or vinblastine, but was not significantly changed by vinblastine or CisPt alone or most of the other combinations. After X-rays alone, the amount of Bax was noticeably decreased. Its level was partially restored when X-rays were combined with wortmannin. The anti-apoptotic protein Bcl-2 showed a moderate decrease following all treatments.
PARP cleavage, indicative of ongoing apoptosis, was absent or very low in cells treated with X-rays, CisPt, or wortmannin alone. Vinblastine employed alone (8 nM) provoked moderate PARP cleavage. The combinations X-rays plus CisPt, X-rays plus vinblastine, CisPt plus wortmannin, and CisPt plus vinblastine had little or no effect on PARP integrity. In contrast, PARP cleavage was dramatically increased in cells exposed to vinblastine plus wortmannin and X-rays plus wortmannin.
These results confirm that MON cells (a) may be resistant to induction of apoptosis by genotoxic and stress signals, and (b) show some disconnection between the stabilization of p53 protein and the expression of some of its downstream effectors on the one hand, and the cleavage of PARP (i.e. apoptosis induction) on the other. This suggests that these cells have dysfunctions in the p53 pathway and apoptosis control. Also, western blot analysis, likewise morphological evaluation, indicates that the PI3-K inhibitor wortmannin can sensitize MON cells to some genotoxic and non-genotoxic treatments.
Cell growth inhibition assay
Finally, in order to establish whether the apoptosis-related effects of the different treatments could be relevant to MON cell proliferation, a cell growth inhibition assay was performed. The effects of the agents, separately or in combination, were evaluated after 72 h of treatment. The dose-effect relationships showed that the IC50 values were about 7.6 nM, 1.6 μM and 6.3 μM for vinblastine, CisPt and wortmannin, respectively. Also, 2.5 Gy of X-rays inhibited the proliferation of MON cells by 50%. The correlation coefficients (r values) were 0.94 or greater, indicating conformity of the data to the median-effect principle and good reproducibility. For combinations of pairs of agents, the combination index (CI) equation was employed for determining synergistic, i. e. higher than additive, and antagonistic, i. e. lower than additive, effects. Table 1 shows that the combinations X-rays plus wortmannin and vinblastine plus wortmannin inhibited MON cell growth synergistically over a fairly wide range. These same combinations had synergistic effects on the induction of apoptosis (see above). In contrast, the combinations X-rays plus CisPt or vinblastine and CisPt plus wortmannin or vinblastine, which were unable to trigger apoptosis as judged by PARP cleavage, had marked antagonistic effects on the inhibition of growth of MON cells. This was despite their damaging effects indicated by p53 accumulation (Fig. 5).
Table 1.
Variation of CIs at different effect levels
| Treatment | CI at ED50 | CI at ED75 | CI at ED90 |
| X-rays + wortmannin | + | +++ | +++ |
| X-rays + CisPt | - | -- | -- |
| X-rays + vinblastine | -- | --- | ---- |
| Vinblastine + wortmannin | ± | +++ | +++ |
| CisPt + wortmannin | -- | --- | --- |
| CisPt + vinblastine | -- | --- | ---- |
CI < 1, = 1, and > 1 indicates synergism, additivity and antagonism, respectively. More precisely, symbols +, ++ and +++ describe slight synergism (CI between 0.85 and 0.90), moderate synergism (CI between 0.7 and 0.85) and synergism (CI between 0.3 and 0.7), respectively. Symbols -, --, --- and ---- indicate slight antagonism (CI between 1.10 and 1.20), moderate antagonism (CI between 1.20 and 1.45), antagonism (CI between 1.45 and 3.3) and strong antagonism (CI > 3.3), respectively. Nearly additive effects (CI in the range 0.90 and 1.10) are represented by the symbol ±. ED50, ED75 and ED90 indicate 50%, 75% and 90% inhibition of growth, respectively. Equipotent amounts of the two agents were combined. In most experiments employing X-rays, various ratios were tested. Each CI determination resulted from at least three independent experiments evaluating at least five different doses or dilutions of the two agents.
Discussion
MRTs combine aggressiveness and resistance to therapeutic treatments and have a very discouraging prognosis. In a search for factors responsible for the MRT phenotype, some aspects of the apoptotic pathway were investigated in MON rhabdoid cells. Normally, ATM and ATR, activated in response to DNA damage or stress, phosphorylate p53 and block Mdm2, which targets p53 for destruction. This results in increased levels and conformational changes of p53 and subsequent suppression of proliferation by cell cycle arrest or apoptosis [reviewed in [23]]. The choice between these outcomes is dependent on the extent of damage, as well as on environmental and intrinsic cellular factors. In the present study, comparison of MON cells to HeLa cells revealed a particularly low susceptibility of MON cells to apoptosis following treatments with several DNA damaging agents. Nevertheless, p53 was abundant and was upregulated after treatment with all genotoxic and non-genotoxic agents examined, singly and in various combinations, whereas PARP cleavage did not occur systematically. The lack of correlation between p53 and cell death is illustrated by the fact that treatment of cells with X-rays plus wortmannin, which leads to a massive destruction of PARP, resulted in an induction of p53 lower than that produced by X-rays plus CisPt, after which no PARP cleavage was observed.
These observations indicate that rhabdoid cells may have dysfunctions in the p53 pathway and in the control of apoptosis, at least after some types of damage. In fact, the transcription stimulating activity of p53, eliciting the expression of responsive genes such as p21WAF1/CIP1and Mdm2, appeared to be perturbed in treated MON cells. That was the case after treatment of cells with X-rays plus wortmannin and, especially, with vinblastine plus wortmannin, which resulted in a lower induction of p21WAF1/CIP1 and Mdm2 than expected on the basis of p53 accumulation. These treatments were able to drive cells into apoptosis. Since p21WAF1/CIP1 blocks cell cycle progression through its negative activity on various cyclin dependent kinases, it could be hypothesized that the relatively reduced induction, or the diminution of p21WAF1/CIP1, antagonizes the establishment of a secure G1 arrest and thus facilitates apoptosis in these treated cells.
Anomalies in the control of apoptosis in MON cells are probably also illustrated by the lack of positive correlation between increase in the Bax/Bcl2 ratio and cell death. Members of the Bcl-2 family normally interact to regulate programmed cell death. Pro-apoptotic members of this family include Bax, Bad etc. Bcl2 and others proteins act as apoptotic inhibitors [reviewed in [24]]. In MON cells, the Bax/Bcl2 balance did not appear to be a determining factor for apoptotic outcome since, although this ratio was increased by PARP-cleavage-stimulating treatments (X-rays or vinblastine plus wortmannin), it was equally enhanced in cells exposed to wortmannin alone, X-rays plus CisPt, or CisPt plus vinblastine, after which no PARP cleavage occurred.
The results also showed that the apoptotic outcome in MON cells can be triggered by treatments combining the PI3-K inhibitor wortmannin with another agent. Among the Ras effector signaling pathways, the PI3-K/AKT pathway facilitates G1 to S phase progression and plays a major role in protecting cells from apoptosis by inhibiting BAD and thus cytochrome C release, inactivating caspase-9 and -3, and targeting p53 for destruction [25]. In MON cells the apoptotic function of p53 is largely abrogated possibly because the Ras signaling pathway, perhaps stimulated by autocrine growth factors, generates an antiapoptotic state that results in resistance to antineoplastic treatments. Wortmannin, by inhibiting the PI3-K, may decrease the expression of survival factors such as AKT. This is not sufficient to provoke apoptosis (wortmannin alone has little effect at the concentrations tested). However, apoptosis is triggered in the presence of other damage or stress signals such as X-rays (unable by themselves to induce significant apoptosis in MON cells) or vinblastine (moderately effective in provoking apoptosis at the concentration used). Nevertheless, inhibition the PI3-K/AKT signaling pathway does not systematically result in increased susceptibility to apoptosis after all types of damage, as shown by the treatment combining wortmannin and CisPt. The damage dependence of the wortmannin effect has no clear explanation. It suggests a complex crosstalk between survival factors, nature of damage and apoptotic signals in MRT cells.
Interestingly, the synergistic effects on apoptosis of some agent associations, such as wortmannin plus X-rays or vinblastine, are reflected in the inhibition of MON cell growth. The considerable antagonistic effects of several other combinations of CisPt, X-rays, vinblastin and wortmannin are also worth noting. The establishment of synergistic and antagonistic interactions between agents may be relevant for future clinical choices of therapeutic strategies.
Conclusions
In conclusion, these results obtained on a model cell line suggest that perturbations of the p53 pathway and a reduced apoptotic response in rhabdoid tumor cells might contribute to the resistance of MRTs to antineoplastic treatments. The Ras/PI3-K/AKT signaling pathway seems to be involved in the dysfunctions induced in these cells by the mutation in the hSNF5/INI1 gene and probably results in increased cell survival factors. Some combinations that are synergistic or antagonistic towards the inhibition of cell growth and the induction of apoptosis have been identified and it is hoped that this approach can provide some suggestions for new rational design of therapeutic protocols.
Methods
Chemicals
CisPlatinium (II) diamine dichloride, etoposide, vinblastine and wortmannin were purchased from Sigma-Aldrich, St Quentin Fallavier, France. Stock solutions were made in the appropriate solvents and stored in aliquots at -20°C. Further dilutions were made in culture medium immediately before use.
Irradiations
X-rays were delivered by a Philips MG 325 (Philips industrial X-Ray, Hamburg, Germany) employed at 260 kV, 13 mA with a 0.5 mm Cu and 1 mm Al filter. The dose rate was 1 Gy/min.
UVC: a Philips germicide tube was used with a maximal emission at 254 nm. Cells were irradiated after removal of the culture medium from the plates. The dose rate was 0.5 J/m2/s. Doses given in this paper are incident doses.
Cell culture
The MRT cells MON [genetically characterized in ref. [6]], generously supplied by O. Delattre (Institut Curie, Paris), and HeLa cells were cultured as monolayers in RPMI-1640 supplemented with 10% fetal calf serum and 20 μg/ml gentallin in a humidified 5% CO2 atmosphere at 37°C. The cultures tested negative for mycoplasma species.
Cell growth inhibition assay
The physical and chemical agents were evaluated for their effects separately and in pairs. After counting with a cell counter (Beckman Coulter Inc., Palo Alto, CA, USA), 2.5 × 103 rhabdoid cells were seeded in 100 μl of medium in 96-well microtitre plates and incubated for 24 h at 37° prior to adding the drugs or irradiating. The final volume of medium, with or without drugs, was 200 μl per well and incubation was continued for three days, which ensured logarithmic growth of control cells throughout the experiment. The effects of the treatments were determined in terms of growth inhibition by measuring the cellular protein content according to the method of Skehan et al. [26]. Cells were fixed with trichloroacetic acid and then stained for 30 min. with 0.4% sulforhodamine B in 1% acetic acid. Unbound dye was removed by acetic acid washes, and the protein-bound dye was extracted with Tris base (pH 10) before determining its absorbance at 540 nm in a 96-well microplate reader Victor2 (PerkingElmer Life Siences, Boston, MA, USA).
Median-effect principle for dose-effect analysis
The multiple drug effect analysis of Chou and Talalay [27] was used to calculate combined drug effects. Dose-effect curves for each agent and for combinations, in multiply diluted concentrations or scaled irradiation doses, were plotted using the median-effect equation fa/fu = (D/Dm)m, in which D is the dose, Dm is the dose required for 50% effect, fa is the fraction affected by dose D, fu is the unaffected fraction and m is a coefficient of the sigmoidicity of the dose-effect curve. The conformity of the data to the median-effect principle was determined by the linear correlation coefficient r. The combination index (CI) equation for mutually non-exclusive drugs CI = (D)1/(Dx)1 + (D)2/(Dx)2 + (D)1(D)2/(Dx)1(Dx)2, in which drug 1, (D)1, and drug 2, (D)2, in combination inhibit x%, and (Dx)1 and (Dx)2 are the doses of drug 1 and drug 2 alone, respectively, inhibiting x%, was employed for measuring synergism and antagonism. CI < 1, = 1, and > 1 indicates synergism, additive effect and antagonism, respectively. Quantitations by computerized analysis [28] were done using the Calcusyn software (Biosoft, Cambridge, UK).
Determination of apoptosis
Apoptosis was evaluated by both DNA fluorescence microscopy of nuclear changes, as described elsewhere [29], and DNA fragmentation analysis. For the latter, low molecular weight DNA was extracted according to Hermann et al. [30] with minor modifications. At specific times after treatment, non-adherent and adherent cells were combined, rinsed with phosphate buffered saline (PBS), counted and pelleted. Aliquots of 2.106cells were lysed in 1% Nonidet P-40 (NP40) in 20 mM EDTA, 50 mM Tris-HCl (pH 7.5). After centrifugation for 5 min at 1,600 × g, the supernatant was collected and the extraction repeated on the pellet. The supernatants were combined, adjusted to 1% SDS and then treated for 2 h at 56°C with RNAse A (final concentration, 1 μg/μl). Proteinase K was added (final concentration, 1 μg/μl), and the mixture was incubated for 2 h at 37°C. Ammonium acetate was then added (final concentration, 1 M), and the DNA was precipitated by the addition of 2.5 volumes of ethanol and an overnight incubation at -20°C. After centrifugation (14,000 × g for 20 min at 4°C), the pellet was washed with 70% ethanol, dissolved in 30 μl of gel-loading buffer and separated by electrophoresis through a 2% agarose gel using 40 mM Tris-acetate, 2 mM EDTA as running buffer. DNA was visualized by ethidium bromide staining (1 μg/ml) and photographed under UV illumination.
Western blot analysis
Whole-cell protein extracts were made from HeLa and MON cells irradiated with UVC or X-rays or treated with different compounds. At specific times after treatment, non-adherent and adherent cells were collected, combined, rinsed with PBS, counted and pelleted. Aliquots of 5.105 cells were lysed in RIPA buffer (50 mM Tris-HCl pH 7.5, 150 mM NaCl, 1% NP40, 0.5% sodium deoxycholate, 0.1% SDS) containing 1 mM phenyl methyl sulfonyl fluoride, 1 μg/ml aprotinin, 1 μg/ml pepstatin, 1 μg/ml leupeptin and 50 mM Na fluoride for 30 min at 4 °C and then boiled for 5 min. Alternatively, for PARP analysis, cell pellets were resuspended in a defined volume of reducing loading buffer (62.5 mM Tris-HCl pH 6.8, 6 M urea, 10% glycerol, 2% SDS, 0.003% bromophenol blue, 5% 2-mercaptoethanol (freshly added)), sonicated on ice with a cup horn tip to break DNA, and incubated for 15 min at 65°C. Proteins were separated by SDS-polyacrylamide gel electrophoresis (PAGE) and transferred on to polyvinylidene fluoride membranes. Ponceau red staining was performed to check that closely comparable amounts of proteins had been loaded and transferred in each lane. Immunoblots were incubated with the indicated primary antibodies for 2 h at room temperature. Antibodies against Mdm2 (SMP14, mouse monoclonal), Bcl-2 (100, mouse monoclonal), Bax (P-19, goat polyclonal and B-9, mouse monoclonal), p21WAF1/CIP1 (C-19, goat polyclonal and F-5, mouse monoclonal) and p53 (DO-1, mouse monoclonal), were from Santa Cruz Biotechnology, Inc. Santa Cruz, CA, USA. C2-10 mouse anti-PARP monoclonal antibody was from PharMingen International (Becton Dickinson, Grenoble, France). After washing, the blots were incubated with the appropriate secondary antibodies conjugated to horseradish peroxidase for 1 h at room temperature, washed again and developed with an enhanced chemiluminescence kit (Amersham Pharmacia Biotech, Litle Chalfont, UK) according to the manufacturer's instructions.
Acknowledgments
Acknowledgements
This work is dependent upon the support of the ARC and CNRS. The assistance of Michèle Guggiari is acknowledged.
References
- Beckwith JB, Palmer NF. Histopathology and prognosis of Wilms tumors: results from the First National Wilms' Tumor Study. Cancer. 1978;41:1937–1948. doi: 10.1002/1097-0142(197805)41:5<1937::aid-cncr2820410538>3.0.co;2-u. [DOI] [PubMed] [Google Scholar]
- Weeks DA, Beckwith JB, Mierau GW, Luckey DW. Rhabdoid tumor of kidney. A report of 111 cases from the National Wilms' Tumor Study Pathology Center. Am J Surg Pathol. 1989;13:439–458. [PubMed] [Google Scholar]
- Kodet R, Newton WA, Jr, Sachs N, Hamoudi AB, Raney RB, Asmar L, Gehan EA. Rhabdoid tumors of soft tissues: a clinicopathologic study of 26 cases enrolled on the Intergroup Rhabdomyosarcoma Study. Hum Pathol. 1991;22:674–684. doi: 10.1016/0046-8177(91)90289-2. [DOI] [PubMed] [Google Scholar]
- Burger PC, Yu IT, Tihan T, Friedman HS, Strother DR, Kepner JL, Duffner PK, Kun LE, Perlman EJ. Atypical teratoid/rhabdoid tumor of the central nervous system: a highly malignant tumor of infancy and childhood frequently mistaken for medulloblastoma: a Pediatric Oncology Group study. Am J Surg Pathol. 1998;22:1083–1092. doi: 10.1097/00000478-199809000-00007. [DOI] [PubMed] [Google Scholar]
- Biegel JA, Kalpana G, Knudsen ES, Packer RJ, Roberts CW, Thiele CJ, Weissman B, Smith M. The role of INI1 and the SWI/SNF complex in the development of rhabdoid tumors: meeting summary from the workshop on childhood atypical teratoid/rhabdoid tumors. Cancer Res. 2002;62:323–328. [PubMed] [Google Scholar]
- Versteege I, Sevenet N, Lange J, Rousseau-Merck MF, Ambros P, Handgretinger R, Aurias A, Delattre O. Truncating mutations of hSNF5/INI1 in aggressive paediatric cancer. Nature. 1998;394:203–206. doi: 10.1038/28212. [DOI] [PubMed] [Google Scholar]
- Peterson CL. SWI/SNF complex: dissection of a chromatin remodeling cycle. Cold Spring Harb Symp Quant Biol. 1998;63:545–552. doi: 10.1101/sqb.1998.63.545. [DOI] [PubMed] [Google Scholar]
- Tyler JK, Kadonaga JT. The "dark side" of chromatin remodeling: repressive effects on transcription. Cell. 1999;99:443–446. doi: 10.1016/s0092-8674(00)81530-5. [DOI] [PubMed] [Google Scholar]
- Zhang HS, Gavin M, Dahiya A, Postigo AA, Ma D, Luo RX, Harbour JW, Dean DC. Exit from G1 and S phase of the cell cycle is regulated by repressor complexes containing HDAC-Rb-hSWI/SNF and Rb-hSWI/SNF. Cell. 2000;101:79–89. doi: 10.1016/S0092-8674(00)80625-X. [DOI] [PubMed] [Google Scholar]
- Cheng SW, Davies KP, Yung E, Beltran RJ, Yu J, Kalpana GV. c-MYC interacts with INI1/hSNF5 and requires the SWI/SNF complex for transactivation function. Nat Genet. 1999;22:102–105. doi: 10.1038/8811. [DOI] [PubMed] [Google Scholar]
- Adler HT, Chinery R, Wu DY, Kussick SJ, Payne JM, Fornace AJ, Jr, Tkachuk DC. Leukemic HRX fusion proteins inhibit GADD34-induced apoptosis and associate with the GADD34 and hSNF5/INI1 proteins. Mol Cell Biol. 1999;19:7050–7060. doi: 10.1128/mcb.19.10.7050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rozenblatt-Rosen O, Rozovskaia T, Burakov D, Sedkov Y, Tillib S, Blechman J, Nakamura T, Croce CM, Mazo A, Canaani E. The C-terminal SET domains of ALL-1 and TRITHORAX interact with the INI1 and SNR1 proteins, components of the SWI/SNF complex. Proc Natl Acad Sci U S A. 1998;95:4152–4157. doi: 10.1073/pnas.95.8.4152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gansler T, Gerald W, Anderson G, Gramling TS, Williams CH, Sens D, Garvin AJ. Characterization of a cell line derived from rhabdoid tumor of kidney. Hum Pathol. 1991;22:259–266. doi: 10.1016/0046-8177(91)90160-q. [DOI] [PubMed] [Google Scholar]
- Ota S, Crabbe DC, Tran TN, Triche TJ, Shimada H. Malignant rhabdoid tumor. A study with two established cell lines. Cancer. 1993;71:2862–2872. doi: 10.1002/1097-0142(19930501)71:9<2862::aid-cncr2820710930>3.0.co;2-d. [DOI] [PubMed] [Google Scholar]
- Garvin AJ, Re GG, Tarnowski BI, Hazen-Martin DJ, Sens DA. The G401 cell line, utilized for studies of chromosomal changes in Wilms' tumor, is derived from a rhabdoid tumor of the kidney. Am J Pathol. 1993;142:375–380. [PMC free article] [PubMed] [Google Scholar]
- Rosson GB, Hazen-Martin DJ, Biegel JA, Willingham MC, Garvin AJ, Oswald BW, Wainwright L, Brownlee NA, Wright CF. Establishment and molecular characterization of five cell lines derived from renal and extrarenal malignant rhabdoid tumors. Mod Pathol. 1998;11:1228–1237. [PubMed] [Google Scholar]
- Suzuki A, Ohta S, Shimada M. Gene expression of malignant rhabdoid tumor cell lines by reverse transcriptase-polymerase chain reaction. Diagn Mol Pathol. 1997;6:326–332. doi: 10.1097/00019606-199712000-00004. [DOI] [PubMed] [Google Scholar]
- Rosson GB, Vincent TS, Oswald BW, Wright CF. Drug resistance in malignant rhabdoid tumor cell lines. Cancer Chemother Pharmacol. 2002;49:142–148. doi: 10.1007/s00280-001-0398-y. [DOI] [PubMed] [Google Scholar]
- Hirose M, Yamada T, Abe T, Hirose T, Shimizu E, Yamamoto Y, Kagami S, Takano S, Yamaguchi T, Kuroda Y. Establishment and characterization of two cultured cell lines derived from malignant rhabdoid tumors of the kidney. Int J Cancer. 1996;67:218–223. doi: 10.1002/(SICI)1097-0215(19960717)67:2<218::AID-IJC12>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- Versteege I, Medjkane S, Rouillard D, Delattre O. A key role of the hSNF5/INI1 tumour suppressor in the control of the G1-S transition of the cell cycle. Oncogene. 2002;21:6403–6412. doi: 10.1038/sj.onc.1205841. [DOI] [PubMed] [Google Scholar]
- Zhang ZK, Davies KP, Allen J, Zhu L, Pestell RG, Zagzag D, Kalpana GV. Cell cycle arrest and repression of cyclin D1 transcription by INI1/hSNF5. Mol Cell Biol. 2002;22:5975–5988. doi: 10.1128/MCB.22.16.5975-5988.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ae K, Kobayashi N, Sakuma R, Ogata T, Kuroda H, Kawaguchi N, Shinomiya K, Kitamura Y. Chromatin remodeling factor encoded by ini1 induces G1 arrest and apoptosis in ini1-deficient cells. Oncogene. 2002;21:3112–3120. doi: 10.1038/sj.onc.1205414. [DOI] [PubMed] [Google Scholar]
- Vogelstein B, Lane D, Levine AJ. Surfing the p53 network. Nature. 2000;408:307–310. doi: 10.1038/35042675. [DOI] [PubMed] [Google Scholar]
- Hengartner MO. The biochemistry of apoptosis. Nature. 2000;407:770–776. doi: 10.1038/35037710. [DOI] [PubMed] [Google Scholar]
- Kennedy SG, Wagner AJ, Conzen SD, Jordan J, Bellacosa A, Tsichlis PN, Hay N. The PI3-Kinase/Akt signaling pathway delivers an anti-apoptotic signal. Genes Dev. 1997;11:701–713. doi: 10.1101/gad.11.6.701. [DOI] [PubMed] [Google Scholar]
- Skehan P, Storeng R, Scudiero D, Monks A, McMahon J, Vistica D, Warren JT, Bokesch H, Kenney S, Boyd MR. New colorimetric cytotoxicity assay for anticancer-drug screening. J Natl Cancer Inst. 1990;82:1107–1112. doi: 10.1093/jnci/82.13.1107. [DOI] [PubMed] [Google Scholar]
- Chou TC, Talalay P. Generalized equations for the analysis of inhibitions of Michaelis-Menton and higher order kinetic systems with two or more mutually exclusive and non-exclusive inhibitors. Eur J Biochem. 1981;115:207–216. doi: 10.1111/j.1432-1033.1981.tb06218.x. [DOI] [PubMed] [Google Scholar]
- Chou TC, Tan QH, Sirotnak FM. Quantitation of the synergistic interaction of edatrexate and cisplatin in vitro. Cancer Chemother Pharmacol. 1993;31:259–264. doi: 10.1007/BF00685668. [DOI] [PubMed] [Google Scholar]
- Nocentini S, Moreno G, Petit PX, Guggiari M, Salet C, Demerseman P, Dodin G. Induction of mitochondrial dysfunction and apoptosis in HeLa cells by bis-pyridinium oximes, a newly synthesized family of lipophilic biscations. Biochem Pharmacol. 1997;53:1543–1552. doi: 10.1016/S0006-2952(97)00092-0. [DOI] [PubMed] [Google Scholar]
- Herrmann M, Lorenz HM, Voll R, Grunke M, Woith W, Kalden JR. A rapid and simple method for the isolation of apoptotic DNA fragments. Nucleic Acids Res. 1994;22:5506–5507. doi: 10.1093/nar/22.24.5506. [DOI] [PMC free article] [PubMed] [Google Scholar]