

Abstract
Drug activity is often studied in well controlled and characterized cellular environments in vitro. However, the biology of cells in culture is only a part of the tissue behavior in vivo. Quantitative studies of the dose response to drugs in vivo have been limited by the inability to reliably determine or predict the concentrations achieved in tissues. We developed a method to study the dose response of injured arteries to exogenous heparin in vivo by providing steady and predictable arterial levels of drug. Controlled-release devices were fabricated to direct heparin uniformly and at a steady rate to the adventitial surface of balloon-injured rat carotid arteries. We predicted the distribution of heparin throughout the arterial wall by using computational simulations of intravascular drug binding and transport, and we correlated these concentrations with the biologic response of the tissues. This allowed the estimation of the arterial concentration of heparin required to maximally inhibit intimal hyperplasia after injury in vivo, 0.3 mg/ml. This estimation of the required concentration of drug seen by a specific tissue is independent of the route of administration and holds for all forms of drug release. In this way we may now be able to evaluate the potential of widely disparate forms of drug release and to finally create some rigorous criteria by which to guide the development of particular delivery strategies for local diseases.
Many studies have shown that heparin inhibits smooth muscle cell (SMC) proliferation in culture (1–6) and intimal hyperplasia in models of arterial injury (7–12). Yet, the clinical efficacy of heparin in modulating the response to vascular injury is mixed at best. No study with systemic administration has demonstrated a beneficial effect, and two recent investigations§ differed as to whether locally applied heparin prevented restenosis in stented coronary arteries. Differences in drug administration regimens in these and prior clinical attempts could have caused differential outcomes. Resolving why some pharmacologic strategies succeed and others fail involves understanding the often subtle local pharmacokinetics and pharmacodynamics. For a pharmacologic strategy to succeed, drugs must achieve sufficient levels in target tissues quickly enough, and for a sufficient duration, for biologic effect to emerge (13). The drug level in tissues after local delivery, however, is a complex function of transport from sites of administration to sites of action, as balanced by metabolism and clearance everywhere along the way. The mechanisms of intra- and extra-arterial heparin transport have been recently quantified, assembled into local arterial pharmacokinetic models, and validated in animals (14–18). The quantitative pharmacodynamics, the dose response of a drug on a tissue, however, is far more difficult to resolve, and the clinical implementation of local delivery strategies has been stunted by poorly defined goals, the concentration of drug in the target tissue needed to achieve the desired biologic effect. For example, the tissue concentration of heparin sufficient to maximally inhibit intimal hyperplasia in injured arteries is unknown. We, therefore, sought to measure the dose response of the injured arterial wall to exogenous heparin administration.
Although several reports have shown that exogenous heparin can limit intimal hyperplasia in a dose-dependent manner in animal models of arterial injury, the amount needed differed with routes of delivery, and ultimate tissue concentration could not be calculated (7–12). Every method of administration transfers a unique amount of drug to the target tissue, and at a characteristic efficiency. For example, to achieve the same net tissue concentration and biologic response in a specific arterial segment, more drug may need to be administered from a subcutaneous osmotic pump than from a polymeric delivery device implanted immediately adjacent to an injured artery (12). For an in vivo dose response to be generalizable and extrapolatable from one delivery strategy to another, it must be defined in terms of tissue concentration and not merely the amount administered.
Dose responses have been elucidated in cell culture preparations, wherein a well described and controlled pharmacologic environment exists (1–6). These data are invaluable but reflect an essentially static situation, in the absence of drug consumption or degradation, without the complex pharmacokinetics or concentration gradients across target tissues observed in vivo. We now quantitatively examine the dose response of injured arteries to heparin, in a preparation that provides the control and characterization of cell culture while preserving the arterial environment. In previous studies we measured the forces that govern heparin transport and binding in the arterial wall (14, 15), and we created kinetic models of drug distribution (18). In the present study we injured rat carotid arteries and applied heparin from polymeric matrices that constrain the released drug to the adventitial surface, in both an axially and circumferentially symmetric manner, and measured the proliferative response at 2 weeks. We then combined quantification of the biologic response with determination of tissue drug concentration to measure the heparin levels in tissue required to maximally inhibit the proliferative response to vascular injury. Our techniques finally address the fundamental question surrounding local delivery as to what concentrations of drug in tissue should site-specific strategies aim to achieve.
MATERIALS AND METHODS
Dose Response in Cell Culture.
The dose response of serum-stimulated sparsely plated bovine vascular SMCs to heparin (Hepar, Franklin, OH) was characterized in vitro. Bovine aortic SMCs were harvested from collagenase-treated, scored arterial segments under organ culture conditions (19, 20). Clonality was verified through consistent cell morphology, immunohistochemical staining for α-actin, and the absence of staining for von Willebrand factor (19, 20). Cells were maintained in DMEM (GIBCO) supplemented with penicillin (100 units/ml), streptomycin (100 μg/ml), glutamine (100 mM), and 5% calf serum (GIBCO) and cultured up to passage 5.
SMCs were seeded (4 × 103 cells per ml, 1 ml per well) in 12-well tissue culture plates (Costar) in DMEM supplemented with 5% calf serum, allowed to attach overnight, and growth arrested in 0.05% calf serum in DMEM for 3 days. At the initiation of the 5-day inhibition assay, heparin was added over a range from 0 to 100 μg/ml in 5% calf serum in DMEM. Experiments were performed in quadruplicate. At the end of the 5-day culture the cells were removed through trypsinization and counted with a ZF1 Coulter Counter.
Fabrication of Heparin-Releasing Devices.
Perivascular drug-eluting devices were formed into hollow cylindrical collars that released heparin, with axial and circumferential symmetry and near-steady kinetics, to the adventitial surface of a segment of rat carotid artery for 2 weeks. The same lot of heparin was used in these and the cell culture experiments. Ethylene–vinyl acetate copolymer (EVAc, 40% vinyl acetate, ELVAX-40p, DuPont) matrices were loaded with the vasoactive agent, heparin, and the inert doping compound, bovine serum albumin (BSA; FisherBiotech). In all collar devices, the final heparin–BSA mixture to EVAc mass ratio was 1:2. This constant ratio ensured consistent release kinetics of solute, while different rates of heparin release were achieved by doping with variable amounts of BSA (21). Heparin and BSA were dissolved in deionized water in ratios such that the fraction of heparin as a percentage of the total drug mixture was 0%, 6.25%, 12.5%, 25%, or 37.5%. The dissolved drug was lyophilized back to a powder, ground, and sieved between 40 and 500 μm. This process ensured complete mixing of heparin and BSA and provided uniform drug particle sizes in the subsequent EVAc matrix.
EVAc (0.75 g) was dissolved in 5 ml of dichloromethane (Mallinckrodt) to a concentration of 15% (wt/vol). The powdered drug mixture was added to the EVAc solution, mixed on a Vortex mixer, and poured into 2.5-cm-long upright molds at room temperature. The outer surface of the mold was made from Silastic tubing (inside diameter 0.64 cm; Dow Corning), the inner surface was a stainless steel 18-gauge needle (Baxter), and caps for the ends consisted of concentric Silastic tubes cut into disks that fit around the needle and within the outer tubing. The mold was quickly capped, which kept the needle aligned with the axis of the cylinder, and was then placed immediately in a bed of powdered dry ice to harden. The outer Silastic tube was quickly cut away, and the EVAc matrix was placed back in the dry ice for an additional 15 min. The EVAc device was transferred to a precooled screen at −20°C and left overnight with the needle still in the core.
The EVAc collars were warmed to room temperature, the ends were removed with a razor blade, and the collars were cut into 7- to 11-mm lengths. The needles were removed and the collars were weighed and their length was measured. An impermeable EVAc layer was applied to the external surfaces by reinserting a needle back into the lumen and dipping the collars twice into 20% EVAc (wt/vol) in dichloromethane. Care was taken to prevent the collar from sliding along the needle, wherein the polymer solution might cover any of the inner surface. Collars were air-dried on the needles for at least 3 hr and then placed under vacuum (10−3 mbar; 0.1 Pa) overnight. The excess coating at the end of each collar was removed with a razor blade, and each collar was slit longitudinally to enable placement around an intact artery. Irradiating under an ultraviolet lamp for 30 min with periodic rotation provided further sterilization. The devices were stored at 4°C in individual sterile vials until implantation.
Delivered Dose.
To vary the dose delivered to each animal, the heparin fraction of the drug mixture was adjusted to be between 0 and 0.375, the remainder being BSA. The release kinetics from the collars were appropriately nonlinear (see Results: In Vitro Release), with the rate of release decreasing slightly over time (22, 23). We further altered delivered dose by prereleasing the heparin from the EVAc collars in phosphate-buffered saline (PBS; Sigma) at 37°C for 3–12 days before implantation. The delivered dose was calculated from the 2-week portion of the in vitro release profiles just after the prerelease period.
In Vitro Release Characterization.
In vitro release characteristics were determined from EVAc collars that contained trace amounts of [3H]heparin (NEN–DuPont). Collars were prepared as above, except that [3H]heparin was added as a tracer during initial heparin and BSA mixing (1 μCi/mg of heparin; 1 μCi = 37 kBq). Six [3H]heparin-releasing EVAc collars were made, all with a drug mixture that was 37.5% heparin and 62.5% BSA. Two-milliliter aliquots of PBS at 37°C were placed in glass scintillation vials (VWR). Two ligatures were placed around the circumference of each collar to close the longitudinal slit. The lumen of each collar was primed with a 0.1-ml infusion of PBS taken from each initial aliquot. Each collar was immersed in the remaining PBS in the vial and stored at 37°C under gentle agitation. At each time interval, the collars were transferred with clean forceps to new vials containing 2 ml of fresh PBS at 37°C. Eighteen milliliters of Hionic-Fluor (Packard) was added to each vial, and the amount of [3H]heparin in each was determined by liquid scintillation spectroscopy (Rack Beta, LKB Wallach).
Arterial Injury.
Male Sprague–Dawley rats (325–400 g; Charles River) were anesthetized with an intraperitoneal injection of ketamine (75 mg/mg) and xylazine (5 mg/kg). A midline neck incision exposed the left common and external carotid arteries. A 2 French embolectomy balloon catheter (Baxter) was introduced into the common carotid artery through an arteriotomy in the external carotid artery and withdrawn three times with the balloon distended sufficiently with air to generate slight resistance (24). Upon removal of the catheter, the external carotid artery was ligated and the common carotid artery was dissected free of adventitia and nerves over an approximately 1.5-cm length. Heparin-releasing EVAc collars were placed around the artery by means of the longitudinal slit. The slit was closed with proximal and distal circumferential ligatures to ensure that the collar did not migrate off. Care was taken to ensure that pressure pulsations could be seen at the carotid bifurcation, indicating that the collar did not impede blood flow. Surgical incisions were closed with skin staples, and animals were revived with a 5-ml intraperitoneal injection of normal saline. Thirty-eight rats were injured, 28 were treated with collars containing various amounts of heparin, and 10 were treated with placebo devices devoid of heparin.
Tissue Harvesting.
Two weeks after injury and EVAc collar implantation, animals were anesthetized as above. As in other release device implantation experiments, a thin fibrous layer formed around the outside of the EVAc collar, securing it to the artery. The collar and artery were isolated together and care was taken not to detach the fibrous layer. The animals were perfused clear for 5 min with Ringer’s lactate solution through a left ventricular puncture and fixed under physiologic pressure with 10% buffered formalin (Sigma) for 10 min. The artery and collar were then removed together and immersion-fixed for 24 hr in fresh 10% buffered formalin. The arteries were cut along their midlines, the collar and fibrous layer were teased away, and the arteries were processed for paraffin sectioning.
Histologic cross sections were taken from each half of each artery at approximately 100–300 μm from the cut face. In this way two sections were taken from the center of the artery, where the plane of symmetry allows any potential axial loss of drug to be safely neglected. Sections from each side were stained with ver Hoeff’s elastin stain. The thickness of the neointima, media, and adventitia, as well as the intimal and medial areas, were measured by means of computer-assisted morphometric analysis (14), and results from both sections from the same artery were averaged.
Computational Simulations of Heparin Distribution.
The preceding experiments were simulated through one-dimensional computational models of transmural heparin transport and binding (Appendix) (18). We modeled the rate of drug flux into the adventitia as equivalent to the rate of heparin efflux from the collar as measured in vitro, which depended on the ratio of heparin to BSA in each device and the prerelease period. As the neointimal response to balloon denuding injury formed over the 2-week experiment at an unknown rate, we could not model the neointima as an expanding layer. Instead we formulated the simulations at the two extremes, once without the neointima (model I) and once with the neointima of the final thickness (model II). We used the former to assess the heparin distribution in the artery near the beginning of the experiment and the latter to assess the distribution at the end. The concentration at every location in the arterial wall throughout the experiment was assumed to vary between these extremes. We estimated the time- and space-averaged concentration in the neointima and media throughout the experiment by averaging the space-averaged concentrations from both the beginning (model I) and the end of the experiment (model II).
RESULTS
Effects of Heparin on SMC Growth in Vitro.
A sigmoidal response of sparsely plated, serum-stimulated SMCs in culture to heparin concentration was demonstrated (Fig. 1). These data show that 25–50 μg/ml heparin is needed in the pericellular environment to inhibit proliferation by 60% from control cells that were treated with no drug. No further reduction in proliferation was observed at higher heparin doses.
Figure 1.

The dose response of bovine aortic SMCs to heparin. Cells were plated to 40,000 per well and growth arrested in 0.05% calf serum for 3 days. Heparin was added to 5% calf serum on day 0. Cell numbers were determined after 5 days (±SEM; n = 4).
In Vitro Release.
The heparin-releasing EVAc collars are hollow cylinders whose outer coating constrains drug release to the inner surface that abuts the adjacent artery in vivo. This geometry has been predicted to give zeroth-order release kinetics, where the rate of release is constant and the cumulative release is linear with time (21, 23). The cumulative percent release kinetics of the collars is shown in Fig. 2A. These data were curve-fit (Kaleidograph, Synergy Software, Reading, PA) using a power relationship:
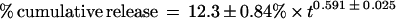 |
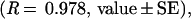 |
where t is time measured in days. Note that the exponent is between the value that is observed with a flat slab (0.5) and the value that is predicted for a hollow cylinder (1.0) (21–23). The amount of heparin released in each time interval as a percentage of the total in the EVAc device decreases over time until day 3 and is thereafter relatively constant and zeroth order (Fig. 2B).
Figure 2.

The cumulative percent release (A) and the percent release rate (B) of heparin in vitro from EVAc collars. The collars were loaded with a drug mixture that was 37.5% heparin and 62.5% BSA with a trace amount of [3H]heparin. They were coated with an impermeable layer on their outer surface so that drug release was exclusively toward the core (±SEM; n = 6). The precoated weights of these collars ranged between 83.3 and 111.1 mg, 12.5% of which was heparin.
In Vivo Delivery.
The average intima-to-media area ratio (±SD) of the 10 arteries treated with placebo EVAc collars was 0.86 ± 0.17, a value similar to the response to arterial injury that did not involve treatment with any implant (25). Increasing amounts of delivered heparin reduced the neointimal response to injury, which was maximally suppressed by the release of 2 mg over the arterial length (Fig. 3).
Figure 3.

The average intima-to-media area ratios were determined from two histologic sections from the center of each artery and plotted against the calculated mass of released heparin.
An example of the predicted transmural concentration profiles is shown for an artery of intermediate neointimal thickness in Fig. 4. The concentration profile near the beginning of the experiment was computed at 2 hr, which was sufficient for heparin to fully migrate across the arterial wall to the lumen, establishing quasisteady transport as predicted by the computational simulations. The precipitous drop in concentration between the media and the adventitia reflects differential fractional available spaces for heparin distribution. The drug levels at the outer edge of the adventitia fall slightly over the course of the experiment. Concurrently, the concentration difference across the arterial wall is spread over a greater distance by the addition of the neointima, tending to raise heparin levels in the media. The heparin concentration throughout the release is considered bounded by these two profiles (shaded region, Fig. 4). Each intima-to-media area ratio is shown both with the range of space-averaged concentrations and with the corresponding estimated time- and space-averaged heparin concentrations (Fig. 5). These data suggest that approximately 0.3 mg/ml heparin is needed in the arterial wall to maximally inhibit the hyperproliferative response to injury.
Figure 4.

Transmural concentration profiles generated by simulations for a representative artery with an intermediate neointimal thickness. The EVAc collar was prereleased for 7 days, the heparin fraction of the drug mixture was 0.375, and the resulting intima-to-media area ratio was 0.31. The profile at 2 hr was generated without a neointimal layer (model I, ■), and the profile at 2 weeks was generated with a neointimal layer of the final thickness measured in each artery (model II, ▵). The intimal and medial concentrations are estimated to be within the shaded region throughout the experiment.
Figure 5.

Intima-to-media area ratios versus the time- and space-averaged concentrations (♦). The range of space averaged concentrations in the intima and media over the 2-week experiment is shown by the lines. The time- and space-averaged concentrations were estimated as the average between these two limits. An estimated sigmoidal fit illustrates the dose response.
DISCUSSION
Interest in local drug delivery waxes and wanes to a great degree, reflective of the excitement and potential of this technology on the one hand and the frustration of variable efficacy on the other. A part of this variability may arise from inconstant tissue levels that may be achieved after delivery. Although it is relatively easy to calculate how much drug has been delivered, until now it has been difficult to reliably quantify the amount of drug that deposits within an artery and more importantly to correlate drug effect with tissue drug concentration. As a result it has never been clear whether the behavior of local release modalities arises from the activity of the compounds released or the efficiency of drug delivery. We now report on the correlation of arterial wall heparin concentration with inhibitory effect on neointimal formation after controlled vascular injury. This quantitative demonstration of tissue concentration after local application may set the standard for how pharmacokinetics are considered and pharmacodynamics are measured in the future.
The growth-inhibitory response of cultured smooth muscle cells to exogenous heparin was demonstrated almost two decades ago (1–6). Such in vitro preparations provide a well controlled cellular environment and a convenient platform to determine the dose response of cells to exogenous compounds. While cell culture experiments are instrumental in describing dose-dependent pharmacologic effects, they represent only a portion of the events and conditions observed in living organisms. For example, the stimulation for proliferation in the injured arterial wall is likely far more complex than that offered by serum in tissue culture. Culture media may not represent true extracellular conditions after injury, and monolayers of cells in isolation are not subject to physical forces and flows, or physiologic contact with serum, neighboring cells, and tissues. Processing steps in harvesting SMCs that include enzymatic degradation can alter cells and their responses, and harvested SMCs might have even undergone a phenotypic change from contractile to synthetic phenotype.
In contrast to cell culture, the cellular environment in arterial tissues in vivo is not well described, and exogenous drug levels are not easily controlled. Unlike that in a cell culture dish, the concentration of exogenous drug in the extracellular milieu is not static or easy to assess. Drugs administered to arteries in vivo are subject to complex local and systemic pharmacokinetics, such as elimination from tissues by diffusional and convective mechanisms and rapidly changing blood levels (14, 17).
An optimal experimental preparation for pharmacologic analysis would combine the advantages of cell culture and in vivo animal models, by both providing controlled and well described drug levels while simultaneously maintaining the natural environment of the tissue. We have demonstrated the feasibility and utility of such a model system by measuring the in vivo dose response of injured arteries to heparin. Drug was applied at a near constant rate directly to arteries, and the observed biologic effect was correlated with the determination of heparin concentrations within the target tissue. This technique effectively sets target tissue levels for local delivery systems to achieve, rather than focusing on the dose of drug administered.
Heparin Delivery in Vivo.
The EVAc collars released heparin solely to the adventitial surface of a segment of injured artery, in a circumferential and axisymmetric fashion. The amount of released heparin necessary to minimize the intimal response was approximately 2.0 mg (Fig. 3). Delivery methods less efficient at transferring drug to the arterial wall may need to administer more drug to achieve the same biologic response. To circumvent these confounding issues of delivery efficiency, and to make these results relevant and extrapolatable to all methods of administration, we examined the dose requirements in terms of the heparin concentration in the arterial tissue.
Accurate determination of the arterial heparin concentrations required fabrication and use of specialized drug delivery devices. The EVAc collars used in this study were designed to release heparin exclusively to the perivascular surface and allow for accurate prediction of drug distribution and deposition throughout the blood vessel wall. As the neointimal layer developed at an unknown rate, we had to consider two permutations of the simulations to predict bounds for the actual arterial concentrations. In one case we simulated conditions at the beginning of the experiment when the neointimal layer had not yet formed (model I). In the other we simulated conditions at the end of the experiment when the neointimal layer had developed to the final thickness measured in histologic sections (model II). Sample transmural concentration profiles are shown at 2 hr and 2 weeks (Fig. 4) for an artery of intermediate neointimal thickness. The computations predict that 2 hr is sufficient for heparin to traverse the arterial wall and for quasisteady transport to be established. The actual concentration at each location falls between these two predicted extremes (shaded region in Fig. 4).
The ranges of space-averaged concentrations between 2 hr and 2 weeks are shown with the corresponding intima-to-media area ratios (Fig. 5). The space- and time-averaged concentrations in the arterial wall were estimated by averaging these two extreme space-averaged concentrations. Although there is scatter, these data suggest that a tissue heparin concentration of approximately 0.3 mg/ml is needed to maximally inhibit the proliferative response to injury. This value may be an underestimate of the concentration in the extracellular space. Our models predict tissue concentration, the mass of drug in a volume of tissue normalized by that volume, which includes both intracellular and extracellular spaces. Heparin remains principally extracellular (18), as hydrophobic cell membranes and slow endocytotic processes limit access to cell interiors. The extracellular concentration may therefore be greater than the tissue concentration.
The range of space-averaged concentrations is broad in some experiments and narrow in others. Two factors influenced the evolution of the arterial heparin concentration over the 2-week experiment, an increasing neointimal thickness and a subtle decay in heparin release rate. The neointima adds resistance to heparin efflux into the lumen, tending to raise the concentration of drug in both the media and the adventitia (Fig. 4), to a greater extent for thicker neointimal layers. At the same time, the in vitro kinetics show that the rate of release decays with time (Fig. 2B), tending to decrease heparin levels in the artery over the 2-week experiment. Longer prerelease periods, however, shift the 2-week experimental interval to lower but steadier rates of release. The range of space-averaged concentrations in the arterial wall may be broad for experiments with short prerelease periods and thin neointimal layers, where the drop in concentration from decaying release rates is not balanced by a larger intimal resistance to heparin transport.
Summary.
To our knowledge, this is the first known quantification of a dose response from local release platforms that examines target tissue concentration, rather than administered dose. By combining local drug delivery with computational models of vascular pharmacokinetics we have determined the concentration of heparin in arterial wall required to inhibit intimal proliferation. This concentration is the required dose needed to be achieved within the target tissue, independent of the route of drug delivery. The tissue concentration needed to inhibit proliferation in injured arteries is an order of magnitude higher than the concentration required to retard the growth of vascular SMCs in culture (Fig. 1) (1, 2, 4, 5). That significantly less drug is required in vitro than in vivo is likely a result of differential stimulation for growth, as well as differential mechanical and chemical environments. This distinction illustrates that dosing in culture reflects a cellular response alone, whereas dosing in vivo reflects a tissue response. The impact of many more complex events in vivo highlights the need for studying pharmacodynamics in a generalizable fashion in the natural tissue environment. These techniques demonstrate a new paradigm for considering local drug delivery strategies, and the data enable one to finally define goals for local pharmacologic therapies.
Acknowledgments
We thank Carmen Berg, Kristy Hong, Danielle Bornstein, and Cindy Richmond for their help in the laboratory. This study was supported in part by grants from the National Institutes of Health (GM/HL 49039), the Burroughs-Welcome Fund in Experimental Therapeutics, and the Whitaker Foundation in Biomedical Engineering. E.R.E. is an Established Investigator of the American Heart Association.
ABBREVIATIONS
- SMC
smooth muscle cell
- EVAc
ethylene–vinyl acetate copolymer
Appendix
The arterial distribution of drug was simulated for each experiment by using a one-dimensional computational model of transmural heparin transport and binding, based on a modified forward-difference algorithm that was programmed in Matlab (Mathworks, Natick, MA). This algorithm was derived in detail elsewhere (18) and is summarized below with boundary conditions that reflect the perivascular delivery of heparin from the EVAc collars. Briefly, the average thickness of each distinct concentric tissue layer was divided into consecutive elements, 4 for the neointima, 10 for the media, and 3 for the adventitia. The number of elements in thinner tissue layers was reduced to minimize computation time. Drug in each element was considered to be homogeneous and distributed between soluble and reversibly bound phases. For each step in time, the diffusive transport of soluble heparin into and out of each element was computed, and then the reversibly bound and soluble phases were redistributed according to local equilibrium within each element (18). Convective motion was neglected because the EVAc collars dampened potential hydraulic flows (17). The transendothelial resistance to heparin transport was excluded because regeneration after injury is slow and incomplete (26, 27).
The physicochemical parameters that characterize the binding and distribution of heparin in arterial media and adventitia have been measured in previous studies and are summarized in Table 1 (14, 15). They are effective diffusivity, the fractional available space for drug distribution, the average binding site density, and the dissociation constant of the average binding site. As a first-order approximation, these values for the neointimal layer were assumed to be identical to those of the arterial media. The neointimal response to balloon denuding injury formed at an unknown rate. We therefore formulated the simulations twice, once without and once with a neointimal layer.
Table 1.
Physical constants for heparin in arterial tissues
| Tissue | Effective diffusivity D, μm2/s | Fractional space ɛ′ | Binding site density BT, μM | Average dissociation constant K′, μM |
|---|---|---|---|---|
| Intima | 7.7 | 0.61 | 2.5 | 5.0 |
| Media | 7.7 | 0.61 | 2.5 | 5.0 |
| Adventitia | 12 | 0.85 | 0.0022 | 0.0081 |
Model I: Without the Neointima.
The deposition and distribution of heparin throughout the media (med) and adventitia (adv) were simulated. The following differential equation describes the diffusive movement of heparin (18).
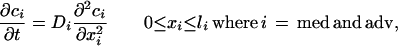 |
A1 |
where c is the concentration of soluble drug, t is time, D is effective diffusivity, x is the position in each layer oriented from the lumen toward the EVAc collar, and l is the thickness of each tissue layer. The following four equations describe the boundary conditions (18):
 |
A2 |
where ɛ′ is the fractional available space for heparin to distribute (Table 1). The boundary layer resistance to heparin transport into the lumen flow (Rbl) was estimated from laminar flow and mass transport theory to be approximately 0.17 s/μm (14), which is small compared with the resistance offered by the arterial media. The concentration of heparin in plasma (cplasma) was considered to be zero throughout the simulations. The following two boundary conditions match the heparin fluxes and concentrations in the accessible spaces across the medial–adventitial interface:
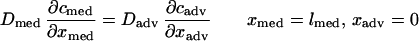 |
A3 |
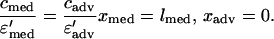 |
A4 |
At the outer edge of the adventitia, the flux of drug was matched to the heparin release from the EVAc collars, as estimated from the in vitro release measurements:
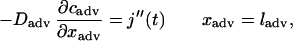 |
A5 |
where j" is the flux, or mass transfer per unit area, of delivered heparin from the inner surface of the collar. Note that the EVAc collars do not release heparin at a perfectly constant rate (Fig. 2B), and thus j" varies with time. The entire artery was assumed to initially contain no heparin:
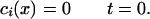 |
A6 |
Model II: With the Neointima.
The addition of the neointima (ni) requires modification of the boundary conditions. Eq. A1 governs transport in all three layers. At the interface between the lumen and neointimal layer:
 |
A7 |
The heparin fluxes and the concentrations in the accessible spaces are matched across the neointimal–medial interface:
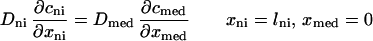 |
A8 |
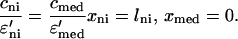 |
A9 |
Eqs. A3–A5 describe the remaining three boundary conditions and Eq. A6 describes the initial conditions.
Footnotes
This paper was submitted directly (Track II) to the Proceedings Office.
Kiesz, R. S., Buszman, P., Deutsch, E., Martin, J. L., Rozek, M. M., Gaszewska, E., Rewicki, M., Seweryniak, P., Kusmider, M. & Tendera, M. (1998) Circulation 98, I-433 (abstr.); Tanguay, J.-F., Wilensky, R. L., Weismann, N. J., Bartorelli, A. L., Mehran, R., Williams, D. O., Bucher, T. A., Wu, H., Popma, J. J. & Kaplan, A. V. (1998) Circulation 98, I-435 (abstr.).
References
- 1.Castellot J J, Jr, Addonizio M L, Rosenberg R D, Karnovsky M J. J Cell Biol. 1981;90:372–379. doi: 10.1083/jcb.90.2.372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Castellot J J, Jr, Cochran D L, Karnovsky M J. J Cell Physiol. 1985;124:21–28. doi: 10.1002/jcp.1041240105. [DOI] [PubMed] [Google Scholar]
- 3.Castellot J J, Jr, Favreau L V, Karnovsky M J, Rosenberg R D. J Biol Chem. 1982;257:11256–11260. [PubMed] [Google Scholar]
- 4.Fager G, Hansson G K, Ottoson P, Dahllof B, Bonders G. Exp Cell Res. 1988;176:319–335. doi: 10.1016/0014-4827(88)90334-5. [DOI] [PubMed] [Google Scholar]
- 5.Hoover R L, Rosenberg R, Haering w, Karnovsky M J. Circ Res. 1980;47:578–583. doi: 10.1161/01.res.47.4.578. [DOI] [PubMed] [Google Scholar]
- 6.Benitz W E, Lessler D S, Coulson J D, Bernfield M. J Cell Physiol. 1986;127:1–7. doi: 10.1002/jcp.1041270102. [DOI] [PubMed] [Google Scholar]
- 7.Clowes A W, Karnovsky M J. Nature (London) 1977;265:625–626. doi: 10.1038/265625a0. [DOI] [PubMed] [Google Scholar]
- 8.Clowes A W, Clowes M M. Lab Invest. 1985;52:611–616. [PubMed] [Google Scholar]
- 9.Clowes A W, Clowes M M. Circ Res. 1986;58:839–845. doi: 10.1161/01.res.58.6.839. [DOI] [PubMed] [Google Scholar]
- 10.Gimple L W, Gertz D G, Haber H L, Ragosta M, Powers E R, Roberts W C, Sarembock I J. Circulation. 1992;86:1536–1546. doi: 10.1161/01.cir.86.5.1536. [DOI] [PubMed] [Google Scholar]
- 11.Edelman E R, Adams D A, Karnovsky M J. Proc Natl Acad Sci USA. 1990;87:3773–3777. doi: 10.1073/pnas.87.10.3773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Edelman E R, Karnovsky M J. Circulation. 1994;89:770–776. doi: 10.1161/01.cir.89.2.770. [DOI] [PubMed] [Google Scholar]
- 13.Edelman E R, Lovich M A. Nat Biotechnol. 1998;16:146–147. doi: 10.1038/nbt0298-136. [DOI] [PubMed] [Google Scholar]
- 14.Lovich M A, Edelman E R. Circ Res. 1995;77:1143–1150. doi: 10.1161/01.res.77.6.1143. [DOI] [PubMed] [Google Scholar]
- 15.Lovich M A, Edelman E R. Biophys J. 1996;70:1553–1559. doi: 10.1016/S0006-3495(96)79719-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lovich M A, Brown L, Edelman E R. J Am Coll Cardiol. 1997;29:1645–1650. doi: 10.1016/s0735-1097(97)00123-x. [DOI] [PubMed] [Google Scholar]
- 17.Lovich M A, Philbrook M, Sawyer S, Weselcouch E, Edelman E R. Am J Physiol. 1998;275:H2236–H2242. doi: 10.1152/ajpheart.1998.275.6.H2236. [DOI] [PubMed] [Google Scholar]
- 18.Lovich M A, Edelman E R. Am J Physiol. 1996;271:H2014–H2024. doi: 10.1152/ajpheart.1996.271.5.H2014. [DOI] [PubMed] [Google Scholar]
- 19.Dinbergs I, Brown L, Edelman E R. J Biol Chem. 1996;271:29822–29829. doi: 10.1074/jbc.271.47.29822. [DOI] [PubMed] [Google Scholar]
- 20.Koo E W Y, Gotlieb A I. Am J Pathol. 1989;134:497–503. [PMC free article] [PubMed] [Google Scholar]
- 21.Rhine W D, Hsieh D S, Langer R. J Pharm Sci. 1980;69:265–270. doi: 10.1002/jps.2600690305. [DOI] [PubMed] [Google Scholar]
- 22.Hsieh D S T, Rhine W D, Langer R. J Pharm Sci. 1983;72:17–22. doi: 10.1002/jps.2600720105. [DOI] [PubMed] [Google Scholar]
- 23.Brooke D, Washkuhn F J. J Pharm Sci. 1977;66:159–162. doi: 10.1002/jps.2600660206. [DOI] [PubMed] [Google Scholar]
- 24.Clowes A W, Reidy M A, Clowes M M. Lab Invest. 1983;49:327–333. [PubMed] [Google Scholar]
- 25.Edelman E R, Pukac L A, Karnovsky M J. J Clin Invest. 1993;91:2308–2313. doi: 10.1172/JCI116460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Clowes A W, Clowes M M, Reidy M A. Lab Invest. 1986;54:295–303. [PubMed] [Google Scholar]
- 27.Reidy M A, Clowes A W, Schwartz S M. Lab Invest. 1983;49:569–575. [PubMed] [Google Scholar]