

Abstract
Background:
Resistin-like molecule (RELM) β is a cysteine-rich cytokine expressed in the gastrointestinal tract and implicated in insulin resistance and gastrointestinal nematode immunity; however, its function primarily remains an enigma.
Objective:
We sought to elucidate the function of RELM-β in the gastrointestinal tract.
Methods:
We generated RELM-β gene-targeted mice and examined colonic epithelial barrier function, gene expression profiles, and susceptibility to acute colonic inflammation.
Results:
We show that RELM-β is constitutively expressed in the colon by goblet cells and enterocytes and has a role in homeostasis, as assessed by alterations in colon mRNA transcripts and epithelial barrier function in the absence of RELM-β. Using acute colonic inflammatory models, we demonstrate that RELM-β has a central role in the regulation of susceptibility to colonic inflammation. Mechanistic studies identify that RELM-β regulates expression of type III regenerating gene (REG) (REG3β and γ), molecules known to influence nuclear factor κB signaling.
Conclusions:
These data define a critical role for RELM-β in the maintenance of colonic barrier function and gastrointestinal innate immunity.
Clinical implications:
These findings identify RELM-β as an important molecule in homeostatic gastrointestinal function and colonic inflammation, and as such, these results have implications for a variety of human inflammatory gastrointestinal conditions, including allergic gastroenteropathies.
Keywords: Allergy, colitis, gastrointestinal, inflammatory, innate, IL-13, mucosal, resistin
As we enter the new millennium, the health of the western world is threatened by an increasing prevalence of immune-mediated diseases. For example, asthma (now affecting 300 million persons), inflammatory bowel disease (IBD; now affecting >1 million persons), and obesity (now affecting nearly 300 million persons) are some of the fastest growing and most pervasive public health problems in developed countries and are recognized to involve inflammatory mechanisms.1-3 Of even greater concern is the co-occurrence of multiple inflammatory disorders in the same individual (eg, obesity and asthma), leading to increased morbidity.4 Although the co-occurrence of multiple inflammatory disorders suggests common underlying disease mechanisms, there is a surprising paucity of data concerning the specific mechanisms that might be operational.
Resistin, also called adipocyte-secreted factor or found in inflammatory zone 3 (FIZZ3), is a novel hormone secreted by adipocytes and has been proposed to link obesity with insulin resistance and type II diabetes.5-9 The resistin family of proteins (resistin, resistin-like molecule [RELM] a, RELM-β, and RELM-β) consists of several approximately 12.5-kd conserved subunits with 10 or 11 cysteine residues that promote the formation of unique disulfide-dependent multimeric assembly units.8,9 Recent investigations with experimental models have demonstrated that resistin mediates insulin resistance by antagonizing insulin action and modulating one or more steps in the insulin-signaling pathway.10,11 RELM-α, designated FIZZ1, was originally found in inflammatory zones in a murine model of experimental asthma, yet its role in allergic inflammation has not been elucidated.8 Subsequent investigations have shown that RELM-α is also expressed in adipose tissue, heart, lung, and tongue, whereas RELM-β (FIZZ2) is expressed in the intestine.8 Recently, RELM-γ has been identified, and its highest levels of expression have been found in hematopoietic tissues.12 Preliminary studies have demonstrated that RELMs are secreted proteins that inhibit adipocyte differentiation and neuronal cell survival,8 suggesting that the primary function of these molecules might not be restricted to regulating insulin resistance. Indeed, recent investigations suggest that at least one member of the resistin family, RELM-β, might have an immunoregulatory function.13 Using a murine model of TH2-associated nematode infection, investigators demonstrated that RELM-β is produced by goblet cells in the intestine and possesses antiparasitic activity through an IL-13-dependent mechanism.13 Despite the growing association of RELM family members with inflammatory conditions, there is a paucity of information concerning the function of this family of cytokines. To determine the definitive role of resistin family members, we generated RELM-β gene-targeted mice. We now report the consequences of RELM-β deficiency on colonic epithelial barrier function and susceptibility to colonic inflammation.
METHODS
Generation of RELM-β gene-targeted mice
RELM-β-/- mice were designed and developed by VelociGene technology.14 In brief, the RELM-β gene was replaced by a reporter-selection cassette, which consists of a β-galactosidase enzyme gene and a neomycin resistance gene. The knockout-reporter construct was created by means of bacterial homologous recombination into a bacterial artificial chromosome encoding RELM-β and was constructed so that the cassette’s β-galactosidase gene is placed in frame with the AUG of RELM-β (Fig 1, A). The construct deletes amino acids 2 through 82 of RELM-β contained in exons 1 through 3 of the gene. The knockout-reporter construct was linearized and electroporated into 129S1/Sv-derived embryonic stem (ES) cells, CJ7 clone, and correctly targeted clones were identified by using Taqman screening with 2 probes in the RELM-β gene as loss-of-allele probes.14 Three correctly targeted clones were identified from 192 colonies. Chimeric mice were generated by microinjecting C57BL/6 embryos with the ES clones. Mice from clone 140B-H3 were backcrossed twice to C57BL/6 mice, and F2 mice were crossed to create homozygous RELM-β gene-targeted mice. Mice were identified as heterozygotes and homozygotes by means of the Taqman assay, with probes for the Neo and LacZ genes and the RELM-β loss-of-allele probes. Mice were genotyped by means of PCR (RELM-β forward primer, cctgagctttctggagagtg; RELM-β reverse primer, cctcctcatcaagaaactttag; and lacZ primer, gtctgtcctagcttcctcactg), producing a wild-type (WT) band of 545 bp and a targeted band of 351 bp. Experiments on RELM-β-/- mice were performed on 6- to 8-week-old mice and background-matched WT control animals derived from littermates. All procedures were performed in accordance with the ethical guidelines in the “Guide for care and use of laboratory animals” of the Institutional Animal Care and Use Committee approved by the Veterinary Services Department of the Cincinnati Children’s Hospital Medical Center.
FIG 1.
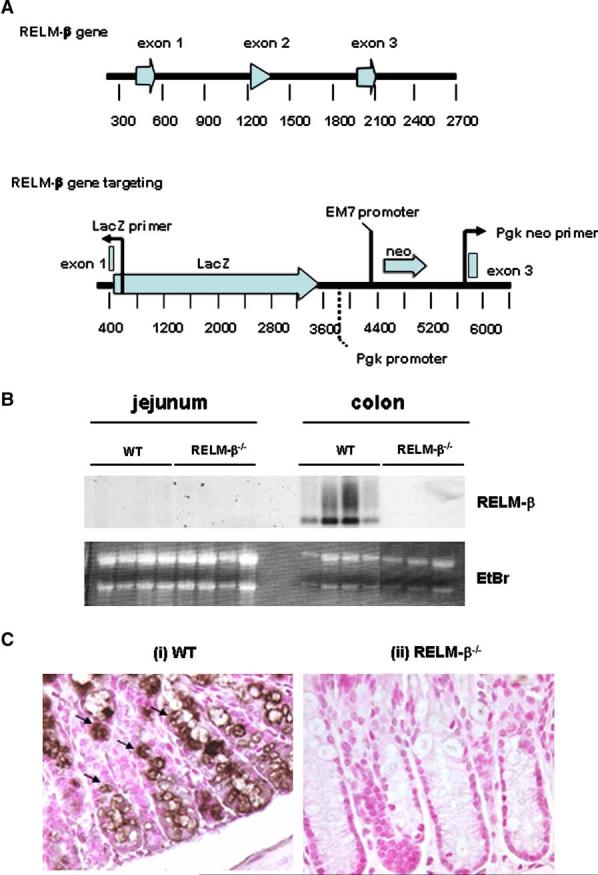
Generation of RELM-β-/- mice. RELM-β-/- mice were developed by using VelociGene technology.14 A, Diagram shows the WT murine RELM-β gene locus and the gene-targeted locus. The RELM-β gene was replaced by a reporter-selection cassette, which consists of a b-galactosidase enzyme gene (LacZ) and a neomycin resistance gene (Neo). B, Northern blot analysis of total jejunum and colon RNA (20 mg) was used to examine RELM-β mRNA expression in WT and RELM-β-/- mice. As an RNA loading control, the position of the 18S and 28S RNA band in the ethidium bromide (EtBr)-stained gels is also shown. Each lane represents a separate animal. C, Immunohistochemically stained sections of colon from WT and RELM-β-/- mice using the RELM-β-specific polyclonal antiserum. Filled arrows depict RELM-β1 cells. Original magnification: C, 3200.
Ussing chambers
Four 1-cm segments of mucosa were stripped of muscle and mounted in U2500 Dual Channel Ussing chambers (Warner Instruments, Hamden, Conn) that exposed 0.30 cm2 of tissue to 10 mL of Krebs buffer. Agar-salt bridges and electrodes were used to measure the potential difference. Every 50 seconds the tissues were short circuited at 1 V (EC 800 Epithelial Cell voltage Clamp; Warner Instruments), and the short-circuit current was monitored continuously. In addition, every 50 seconds the clamp voltage was adjusted to 1 V for 10 seconds to allow calculation of tissue resistance using Ohm’s law. After the preparation had stabilized for 10 minutes and baseline potential difference and resistance had been established, fluorescein isothiocyanate (FITC)-dextran (2.2 mg/mL; molecular mass, 4.4 kd; Sigma-Aldrich, St Louis, Mo) was added to the mucosal reservoir. Medium (0.25 mL of 10 mL) was removed from the serosal reservoir and replaced with fresh medium every 20 minutes over a period of 180 minutes for measurement of FITC-dextran.
Solutions and drugs
Krebs buffer contained 4.70 mM KCl, 2.52 mM CaCl2, 118.5 mM NaCl, 1.18 mM NaH2PO4, 1.64 mM MgSO4, and 24.88 mM NaHCO3 on each side. The tissues were allowed to equilibrate for 15 minutes in Krebs buffer containing 5.5 mM glucose. All reagents were obtained from Sigma-Aldrich unless stated otherwise.
Microarray analysis
To evaluate the effect of the loss of RELM-β on the colonic mucosa, we used Affymetrix GeneChip (Santa Clara, Calif) micro-arrays (MOE430_2, a whole-genome expression chip encoding 45,101 genes) to obtain gene expression profiles from 5 independent WT and 6 independent gene-targeted animals. GeneChip CEL data were subjected to normalization and quantification by using the Robust MultiArray Analysis algorithm developed by Irizarry et al,15 as implemented in GeneSpring 7.0 (Sun Microsystems, Santa Clara, Calif). Gene expression levels for each gene were normalized relative to the median value across the 6 microarrays.
Induction of acute colonic inflammation
Dextran sodium sulfate-induced acute colonic inflammation
Dextran sodium sulfate (DSS; ICN Biomedical Inc, Costa Mesa, Calif) used for the induction of acute colonic inflammation was supplied as the sodium salt with an average molecular weight of 41 kd. It was used as a supplement in the drinking water of the mice for 8 days as 2.5% (wt/vol) solution.
Trinitrobenzene sulfonate-induced acute colonic inflammation
Mice were lightly anesthetized with isoflurane and then administered trinitrobenzene sulfonate (TNBS) (3.75 mg/150 μL dissolved in ethanol [50%]/saline [50%]) or vehicle (ethanol [50%]/saline [50%]) intrarectally through a catheter equipped with a 1-mL syringe. The catheter was advanced into the rectum until the tip was 4 cm proximal to the anal verge, at which time the haptenating agent was administered in a total volume of 150 μL. Mice were held in a vertical position for 30 seconds after the intrarectal injection to ensure distribution of the haptenating agent within the entire colon and cecum.
Disease activity index
The disease activity index (DAI) was derived by scoring 3 major clinical signs (weight loss, diarrhea, and rectal bleeding).16 The clinical features were scored separately and then correlated with a histologic score. DAI = (Body weight loss)+ (Diarrhea score) + (Rectal bleeding score).
Body weight.
A change in body weight was calculated as the difference between the expected and actual weight. The formula for predicted body weight was derived by means of simple regression by using the body weight data for the control group. The following formula was used: Y = a + kx, where Y is body weight change (loss or gain), k is daily increase in body weight, x is day, and a is starting body weight.
Diarrhea.
The appearance of diarrhea was defined as mucus-fecal material adherent to anal fur. The presence or absence of diarrhea was scored as either 1 or 0, respectively. The presence or absence of diarrhea was confirmed by means of examination of the colon after completion of the experiment. Mice were killed, and the colon was excised from the animal. Diarrhea was defined by the absence of fecal pellet formation in the colon and the presence of continuous fluid fecal material in the colon.
Rectal bleeding.
The appearance of rectal bleeding was defined as diarrhea containing visible blood, mucus, or both or gross rectal bleeding and scored as described for diarrhea.
Assessment of intestinal inflammation
Assessment of body weight and evaluation of stool consistency (diarrhea) and rectal bleeding were performed on a daily basis. Body weight was expressed as percentage body weight change from baseline. Diarrhea and rectal bleeding were defined as described above. The presence-absence of diarrhea and rectal bleeding was given a score of 0 or 1 and the diarrhea-rectal bleeding score (0-2) is the accumulation of these values.
Intestinal histopathologic examination
Animals were killed on day 7, and the colon was excised. The length of the colon was measured with digmatic calipers (Mitutoyo, Kawasaji, Japan). Tissue specimens were then fixed in 4% paraformaldehyde and stained with hematoxylin and eosin and Masson trichrome by using standard histologic techniques. The percentage colon length with mucosal ulceration was determined by performing morphometric analysis of the colon with the ImageProPlus 4.5 software package (Media Cybernetics, Inc, Silver Spring, Md). In brief, digital images of longitudinal sections (1-2 cm in length) of hematoxylin and eosin-stained colons were produced.
Assessment of inflammation in TNBS colitis
The colons were divided longitudinally into 2 parts, one of which was used for histologic assessment. The longitudinally divided colons were fixed in 4% formalin and embedded in paraffin for routine histology. One investigator, who was blinded for treatment allocation of the mice, scored the following parameters, as previously reported17,18: (1) percentage of area involved, (2) edema, (3) erosionulceration, (4) crypt loss, and (5) infiltration of mononuclear and polymorphonuclear cells (neutrophils and eosinophils) into the lamina propria and submucosa. The percentage of area involved and the crypt loss were scored on a scale ranging from 0 to 4 as follows: 0, normal; 1, less than 10%; 2, 10%; 3, 10% to 50%; and 4, greater than 50%. Erosions were defined as 0 if the epithelium was intact, 1 for involvement of the lamina propria, 2 for ulcerations involving the submucosa, and 3 when ulcerations were transmural. The severity of the other parameters was scored on a scale of 0 to 3 as follows: 0, absent; 1, weak; 2, moderate; 3, severe.
Murine colonoscopy
Mice were anesthetized with triple sedative (ketamine, 35 mg/kg; xylazine, 5 mg/kg; and acepromazine, 1 mg/kg), and colonoscopies were performed with a small-animal rigid telescope (30°, 1.9 mm × 10 cm) connected to a Cold light Fountain Xenon nova (Model 20 131520) and an Endovision TRICAM SL (Model 20) PAL/NTSC camera (Karl Storz Veterinary Endoscopy, Goleta, Calif).
Detection of RELM-β1 cells by means of immunohistochemistry
The colon segment of the gastrointestinal tract was immunostained with antiserum against mouse RELM-β, as previously described.13 Briefly, 5-μm sections were quenched with H2O2, blocked with normal goat serum, and stained with a rabbit anti-murine RELM-β antiserum. The slides were then washed and incubated with biotinylated goat anti-rabbit antibody and avidin-peroxidase complex (Vectastain ABC Peroxidase Elite kit; Vector Laboratories, Burlingame, Calif). The slides were developed by using nickel diaminobenzidine enhanced cobalt chloride to form a black precipitate and counterstained with nuclear fast red.
Northern blot analysis
RNA was extracted from the lung tissue with Trizol reagent (Gibco-BRL, Grand-Island, NY), according to the manufacturer’s protocol. Twenty micrograms of total RNA was used for Northern blot analysis, as previously described.19
Real-time PCR analysis
The RNA samples (500 ng) were subjected to reverse transcription analysis with Superscript II reverse transcriptase (Invitrogen, Carlsbad, Calif), according to the manufacturer’s instructions. Regenerating gene (REG)3β and REG3γ were quantified by means of real-time PCR with the LightCycler instrument and LightCycler FastStart DNA Master SYBR Green I as a ready-to-use reaction mix (Roche Diagnostics Corp, Indianapolis, Ind). Results were then normalized to GAPDH amplified from the same cDNA mix and expressed as fold induction compared with the control values. cDNAs were amplified by using the following primers: murine REG3β (151 bp), tcccaggcttatggctccta and gcaggccagttctgcatca; murine REG3γ (251 bp), catcaactgggagacgaatcc and cagaaatcctgaggctcttgaca; and GAPDH (400 bp), tggaaatcccatcaccatct and gtcttctgggtggcagtgat.
Statistical analysis
Data are expressed as means ± SEM. Statistical significance comparing different sets of mice was determined by using the Student t test. In experiments comparing multiple experimental groups, statistical differences between groups were analyzed by using the 1-way ANOVA nonparametric Kruskal-Wallis test. P values of less than .05 were considered significant. All analyses were performed with Prism 4.0 software.
RESULTS
RELM-β-/- mice were generated by means of homologous recombination with VelociGene technology (Fig 1, A).14 These mice were fully fertile and produced offspring at predicted Mendelian inheritance patterns, with no gross abnormalities observed. The specific ablation of the RELM-β gene was demonstrated by the absence of RELM-β mRNA and protein expression in the colon (Fig 1, B and C). Notably, RELM-β-/- mice still had expression of RELM-β mRNA, as demonstrated by means of Northern blot analysis in the allergen-challenged lung (data not shown). To test the effect of RELM-β deletion on the composition and homeostasis of the peripheral immune system, the development and activation status of B- and T-cell subsets were analyzed by means of flow cytometry. There was no significant difference in the level and phenotype of lymphocytes in the spleen and mesenteric lymph nodes in RELM-β-/- mice. Furthermore, there was also no difference in the complete blood counts and differentials (data not shown).
Analysis of RELM-β mRNA expression in various gastrointestinal compartments at baseline revealed a high level of RELM-β expression in the large intestine (Fig 1, B, and data not shown). Immunohistologic staining demonstrated that RELM-β expression was predominantly restricted to goblet cells and enterocytes (Fig 1, C). Notably, the colons of RELM-β-/- mice were morphologically normal and contained similar levels of goblet cells, and their mucin gene products compared with those of WT control mice (data not shown).
We were next interested in examining whether RELM-β deficiency affected epithelial cells in the colon. Initially we examined colonic epithelial cell populations (goblet cells and endocrine cells) in the colons of WT and RELM-β-/- mice and found no difference in the numbers of epithelial cell populations between the groups (results not shown). We next examined intestinal epithelial cell resistance in RELM-β-/- and WT mice. Resistance, a measure of tissue permeability, was significantly decreased in RELM-β-/- mice compared with WT mice (Fig 2, A). To confirm altered epithelial cell barrier function in RELM-β-/- mice, we examined intestinal permeability by analyzing FITC-dextran transport in colonic segments ex vivo. Compared with control mice, RELM-β-/- colons had increased intestinal permeability to FITC-dextran (Fig 2, B). For example, the FITC-dextran permeability of the colonic layer of RELM-β-/- mice was significantly increased by 3 hours compared with colonic layers from WT animals (0.75 ± 0.36 mg/mL vs 2.6 ± 0.4 mg/mL × 10-12 at 180 minutes, P< .01). On average, we observed a 266% increase in FITC-dextran permeability of the colonic layer of RELM-β-/- mice compared with that seen in WT animals. Collectively, these studies demonstrate a role for RELM-β in epithelial cell barrier function.
FIG 2.

RELM-β deficiency alters transepithelial barrier permeability. A, Transepithelial resistance of colonic segments of muscle-free intestinal mucosa from WT and RELM-β-/- mice. B, FITC-dextran (molecular weight 4400) permeability in muscle-free intestinal mucosal specimens from the colon from WT and RELM-β-/- mice. Values are presented as means 6 SEM (n 5 3-8 mice per group). Statistical significance of differences was determined by using an unpaired Student t test. *P < .05 compared with WT mice.
The colonic epithelium acts as a selective barrier to luminal antigens and pathogens. Clinical and experimental studies suggest that disruption of epithelial barrier function can predispose to chronic intestinal inflammatory processes. Indeed, disruption of barrier-protective genes (CD73, multidrug resistance gene 1, intestinal trefoil factor, and hypoxia inducible factor 1) alters barrier function and increases susceptibility to chemically induced colitis.20-23 The localized expression of RELM-β within the colonic epithelium and the demonstration of altered epithelial barrier function in RELM-β-/- mice led us to hypothesize that RELM-β might have a role in colonic inflammatory susceptibility. In particular, we hypothesized that increased permeability would increase susceptibility to colonic inflammation in RELM-β-/- mice. We thus used 2 models of acute inflammatory colonic injury, a DSS-induced T cell-independent model that reproduces features of ulcerative colitis24 and a second model of TNBS-induced colitis that captures features of Crohn’s disease,25-27 and examined the effect of RELM-β deficiency on colonic injury.
WT and RELM-β-/- mice were treated with DSS, and we examined colonic RELM-β expression throughout the time course (Fig 3). Notably, although there was some variability between mice, levels of RELM-β mRNA increased after DSS exposure (Fig 3, A). Surprisingly, RELM-β-/- mice had marked protection from most disease features, including weight loss, colonic shortening, diarrhea-rectal bleeding, and protection from mortality (Fig 3, B-F). For example, on day 7, the DAI reached 7.37 ± 2.4 in WT mice compared with 1.53 ± 1.53 in RELM-β-/- mice (mean ± SEM; n = 4-5 mice per group; P < .05), respectively, after DSS treatment. Histologic assessment of the degree of tissue inflammation revealed that RELM-β-/- mice had less epithelial damage and submucosal inflammation compared with WT mice (Fig 4, A-F). The protection from experimental disease occurred at all time points in this acute model of colonic inflammation. Additionally, we assessed the degree of mucosal damage by performing colonoscopy in WT and RELM-β-/- mice (Fig 4, G-L; see this article’s Fig E1 and Videos 1-3 in the Online Repository at www. jacionline.org). Colonoscopy of WT control-treated mice revealed a healthy, translucent colonic mucosa with visible vasculature (Fig 4, G). In contrast, the colonic mucosa of DSS-treated WT mice appeared friable, with readily detectable erythema, strictures, and mucosal injury, as characterized by ulcerations, rectal bleeding, and mucus (Fig 4, H-J). Notably, these lesions were barely detectable in RELM-β-/- mice (Fig 4, K and L). Control-treated RELM-β-/- mice exhibit markedly reduced endoscopic, pathologic, and clinical signs (diarrhea, rectal bleeding, or weight loss) and gross morphologic changes to the gastrointestinal tract normally associated with spontaneous intestinal inflammation (Fig 4, C, and results not shown). Because a DSS-induced colonic inflammation model has been previously shown to occur independent of an adaptive immune response,28 our data highlight the involvement of RELM-β in innate immunity.
FIG 3.

DSS treatment induces experimental colitis in WT and RELM-β-/- mice. A, Northern blot analysis of RELM-β mRNA expression in the colons of control (top panel) and DSS-treated (middle panel) WT mice and kinetics (days 0-8) of RELM-β mRNA expression in the colons of WT mice and IL-13-/- mice (bottom panel) on day 7. Total RNA is shown by ethidium bromide (EtBr) staining. DAI (B), weight change (C), diarrhea-rectal bleeding score (D), and colon length (E; day 7) of control- and DSS-treated WT and RELM-β-/- mice. F, The percentage survival during the course of DSS treatment in WT and RELM-β-/- mice. Data in Fig 3, B through F, represent the mean 6 SEM of 4 to 5 mice per group from triplicate experiments. Statistical significance of differences was determined by using the Kruskal-Wallis test (*P < .05).
FIG 4.
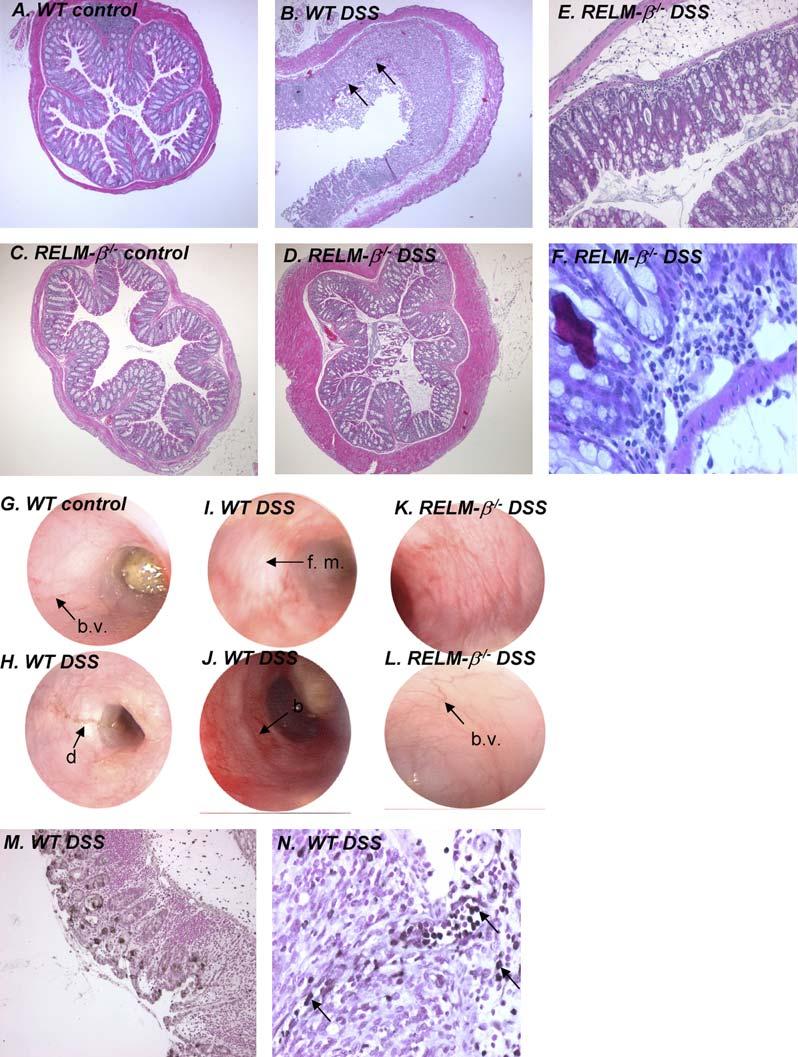
Histopathology and colonoscopy in the colons of DSS-treated WT and RELM-β-/- mice. A-F, Representative photomicrographs of colons from control and DSS-treated WT and RELM-β-/- mice. G-L, Representative colonoscopy photographs of colons of control and DSS-treated WT and RELM-β-/- mice. M and N, Representative photomicrographs of immunohistochemically stained colon sections from DSS-treated WT mice using the RELM-β-specific polyclonal antiserum. Black arrows depict RELM-1 inflammatory cells. Black arrows in Fig 4, B, depict ulceration of the epithelial cell layer: black arrows in Fig 4, G and L, depict normal colonic vasculature (b.v., blood vessel); black arrows in Fig 4, I, H, and J, depict mucus and diarrhea (d), friable mucosa (f. m.), and rectal bleeding (b). Fig 4, H, Narrowing of colon, with evidence of a stricture. Original magnification: A-D, 350; E and M, 3100; F and N, 3200.
To identify the RELM-β-expressing cell population in the inflamed colon, we performed immunohistochemistry with anti-murine RELM-β antiserum. In DSS-treated WT mice we identified RELM-β expression in gastrointestinal epithelial cells and also in infiltrating inflammatory cells (Fig 4, M and N). The RELM-β1 staining of the epithelium was predominantly restricted to goblet cells (Fig 4, M). Thus the variable expression of RELM-β mRNA in DSS-treated mice (Fig 3, A) likely reflects the loss of RELM-β+ epithelial cells. The RELM-β+ inflammatory cells were localized to the muscularis mucosa and were also present within the ulcerated epithelial mucosa (Fig 4, N). We observed no positive staining in the colonic epithelium from RELM-β-/- mice (results not shown).
RELM-β expression has previously been associated with TH2-immune responses, such as experimental asthma and helminthic infestations.8,13 As such, we were interested in testing the hypothesis that DSS-induced RELM-β expression was dependent on the cytokine IL-13; this cytokine has previously been shown to be important in TH2-associated immunity.29 As such, we subjected WT and IL-13-/- mice to DSS-induced experimental colitis. Notably, the expression of RELM-β was markedly decreased in IL-13-/- mice (Fig 3, A). Indeed, IL-13-/- mice were protected from pathologic responses associated with DSS-induced colitis (DAI index was measured on day 7: 3.905 ± 0.44 vs 2.55 ± 0.13, DSS-treated WT mice (BALB/c) vs IL-13-/- mice; mean ± SEM [n = 3-4]; P< .05).
We next examined the role of RELM-β in the development of TNBS-induced colitis, a rapidly evolving transmural colitis. Administration of TNBS to RELM-β-/- mice was associated with increased disease severity, as assessed on the basis of clinical symptoms (weight loss), survival, and tissue histology (cryptitis and intercryptal inflammation and ulceration), compared with that seen in WT mice (Fig 5). Histologic analysis of colonic tissue from TNBS-treated RELM-β-/- mice revealed extensive ulceration and erosion of the colonic epithelium, crypt loss, and a pronounced cellular infiltrate that was primarily comprised of mononuclear and polymorphonuclear (neutrophils and eosinophils) cells (Table I). Histologic scoring of colonic tissue revealed significantly greater pathology than that observed in TNBS-treated WT mice (Table I).
FIG 5.
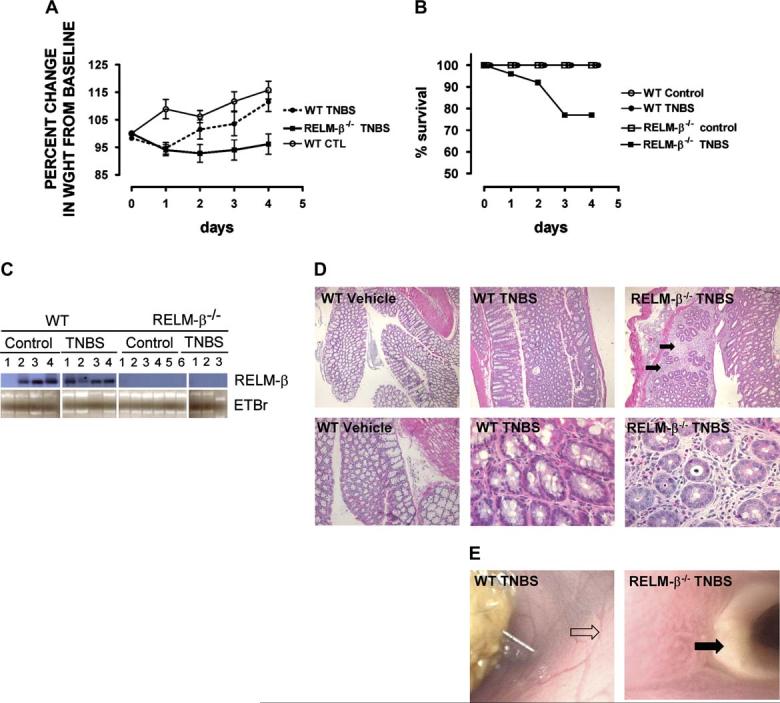
Colon histopathology and colonoscopy in TNBS-treated WT and RELM-β-/- mice. A, Percentage weight change of control-treated and TNBS-treated WT and RELM-β-/- mice. B, Percentage survival during the course of DSS treatment in WT and RELM-β-/- mice. C, Northern blot analysis of RELM-β mRNA expression in the colons of control-treated and TNBS-treated WT and RELM-β-/- mice on day 5. Total RNA is shown by means of ethidium bromide (EtBr) staining. D, Hematoxylin and eosin staining of representative colon transverse sections on day 5 after TNBS administration. E, Representative colonoscopy from TNBS-treated WT and RELM-β-/- mice. The black arrows in Fig 5, D, depict cellular infiltrate. The open arrow in Fig 5, E, depicts normal colonic vasculature, and the black arrow depicts a colonic ulcer. Data in Fig 5, A and B, represent the mean 6 SEM of 4 to 5 mice per group from triplicate experiments. Statistical significance of differences was determined by using the Kruskal-Wallis test. *P < .05.
We observed no significant alteration in colonic morphology between control-treated WT and RELM-β-/- mice (Table I). The very modest disease induced in WT mice might be due to the relative resistance of the C57Bl/6 strain to this model of colitis.27 The protection from DSS-induced injury and the enhanced susceptibility to TNBS-induced colitis might be related to different induction patterns of RELM-β in each model. Indeed, in contrast to DSS exposure, TNBS treatment of WT mice did not enhance colonic RELM-β expression compared with that seen in control mice (Fig 5, C).
To elucidate the molecular basis of altered colonic function in RELM-β-/- mice, we took an empiric approach involving genome-wide expression profile analysis with Affymetrix oligonucleotide chips and probing colon RNA from WT and RELM-β-/- mice. Using criteria of a 2.0-fold change and a Welch t test with a P value cutoff of .05, we identified 32 genes altered in the RELM-β-/- mice (Fig 6, A). Of these transcripts, 8 were upregulated, and 24 were downregulated. Importantly, RELM-β was the most decreased gene, validating the approach, and there was no evidence of compensatory increases in RELM-α expression; in fact, expression of the latter gene was modestly reduced. Functional classification of the altered transcripts revealed a significant predominance of the C-type lectin gene superfamily (approximately 15%). Several families of functional molecules emerged. In particular, the most dysregulated gene family members of the type III subclass of the REG gene family (REG3β and REG3γ) were downregulated 2.7- and 2.2-fold in the RELM-β-/- mice, respectively (Fig 6, B). The type III subclass of the REG gene family (REG3 α, β, γ, and δ) are orthologues of the human pancreatitis-associated protein.30 These proteins are synthesized by Paneth cells in the small intestine and by crypt epithelium in the colon. Given that our analysis used a whole genome-wide approach and that 2 related immunity genes were similarly downregulated, we prioritized confirming these observations by performing real-time PCR analysis. Indeed, we were able to demonstrate a mean 2-fold decrease in REG3β and REG3γ mRNA in the colons of RELM-β-/- mice compared with WT mice (Fig 6, C and D). These studies demonstrate that RELM-β has a role in the regulation of REG3β and REG3γ expression in the colon.
FIG 6.

Gene profile analysis of the colon in the absence of RELM-β. A, Microarray analysis of the transcripts expressed in colonic samples. RNA from each mouse was subjected to chip analysis with Affymetrix mouse Genome MOE430_2 GeneChips. The RELM-β-/- group is composed of 5 mice, and the WT group is composed of 6 mice. The 32 genes differentially expressed (2-fold cut off and P < .05) in the RELM-β-/- mice compared with the WT mice have been ordered (fold change); upregulated genes are shown in red, and downregulated genes are shown in blue. The magnitude of the gene changes is proportional to the darkness of the color. Each column represents an individual mouse, and each line represents a separate gene. B, Quantitative analysis of REG3β and REG3γ expression levels in the colons of WT and RELM-β-/- mice using Affymetrix mouse Genome U430 Plus 2.0 GeneChips. C and D, Quantitative analysis of REG3β (Fig 6, C) and REG3γ (Fig 6, D) mRNA levels in the colons of WT and RELM-β-/- mice by using real-time PCR analysis. REG3 expression was normalized to GADPH expression in each individual sample. Results are expressed as fold change over WT mice. The black line represents the mean value in each group.
DISCUSSION
The resistin family of proteins is composed of cysteinerich cytokines implicated in insulin and gastrointestinal nematode resistance. The precise role of these molecules in mucosal inflammation has not yet been fully elucidated. In the current study we have examined the role of RELM-β in the gastrointestinal tract. We demonstrate that RELM-β is predominantly expressed by goblet cells and epithelial cells within the colonic epithelium and is involved in the maintenance of colonic epithelial cell barrier function. Gene array analysis revealed that RELM-β has a role in regulating intestinal homeostasis because a number of transcripts normally expressed in the colon were altered in the absence of RELM-β. Notably, the altered genes were rich in enterocyte products, especially innate immune mediators, such as the type III REG gene family members. Using acute colonic inflammatory models, we show an important role for RELM-β in colonic injury susceptibility. RELM-β deficiency was associated with reduced susceptibility to DSS-induced colitis but increased susceptibility to TNBS-induced colitis. Thus our initial hypothesis that RELM-β deficiency would simply enhance susceptibility to colonic inflammation was an oversimplification. Although differentially affecting both models, these results underscore the critical participation of RELM-β in regulating colonic inflammation. Prior studies have reported that distinct mechanisms are involved in TNBS-versus DSS-induced colitis.27,31 We show that the pathogenic role of RELM-β is associated in part with dys-regulation of type III REG gene expression, providing some mechanistic insight that helps explains the RELM-β-mediated effects.
To elucidate whether the increased colonic permeability in RELM-β-/- mice could be explained by altered tight junction protein function, we performed electron microscopy on colonic epithelial cells from WT and RELM-β-/- mice (results not shown). We observed normal colonic epithelial architecture and desmosome formations between epithelial cells in the RELM-β-/- mice, suggesting that altered permeability might not be due to altered tight junction protein function. This is consistent with gene chip analysis because we did not observe any significant change in the expression of tight junction proteins in RELM-β-/- mice compared with WT mice (results not shown). Notably, a number of experimental studies have demonstrated altered intestinal permeability (transepithelial resistance and permeability) without any clinical phenotype (diarrhea and epithelial cell ulceration and erosions), suggesting that the increased permeability might be unrelated to the clinical phenotype observed after TNBS treatment. Interestingly, recent investigations have demonstrated a genetic link between mutations in CARD15 with increased intestinal permeability in families with IBD,32 yet approximately 25% of first-degree relatives of patients with IBD show increased intestinal permeability in the absence of clinical symptoms.33,34
RELM-β deficiency was associated with decreased expression of REG3β and REG3γ mRNA in the colon. Although the overall fold change in type III REG proteins (REG3β and REG3γ) was modest, we demonstrated a similar 2.5-fold reduction in levels using 2 independent experimental systems. Notably, we did not observe any change in other REG protein family members, highlighting the specificity to these proteins. Furthermore, the modest change in genome transcripts indicates no major alteration (either directly or indirectly through compensatory mechanisms) as a result of RELM-β deficiency. The increased permeability observed in RELM-β-/- mice was associated with decreased expression of REG3β and REG3γ mRNA in the colon. The function of these proteins in the gastrointestinal tract has only preliminarily been elucidated; recent evidence suggests that they might be involved in epithelial cell repair and Schwann cell regeneration.30 The involvement of REG3β and REG3γ in RELM-β-mediated maintenance of colonic epithelial cell barrier function requires further analysis.
We demonstrate that RELM-β deficiency is associated with decreased susceptibility to DSS-induced colitis and increased susceptibility to TNBS-induced colitis. These 2 models have previously been shown to involve distinct disease mechanisms, such as involving both T cell-dependent and T cell-independent pathways.27 Collectively, these studies suggest that RELM-β might act a critical checkpoint in the regulation of innate immunity. Notably, REG3β has been shown to downregulate TNF-α-induced nuclear factor κB (NF-κB) activation in monocytic and epithelial cells, reduce proinflammatory cytokine mRNA levels, and regulate innate immune responses.30,35 Both in human IBD and in murine models of IBD, the inflammation is likely to depend, at least in part, on the activation and nuclear translocation of NF-κB family members.36 It is tempting to speculate that RELM-β-mediated dysregulation of REG3β and REG3γ expression might regulate susceptibility to NF-κB-dependent inflammation and disease pathology. The analysis of NF-κB activation in RELM-β-deficient mice now deserves future attention.
Notably, we demonstrate that colonic expression of RELM-β was dependent on IL-13. This is consistent with our previous investigations using gastrointestinal nematode models associated with TH2 immunity.13 In these investigations we demonstrated that gastrointestinal nematode infestation associated with TH2 inflammation upregulates RELM-β expression. Furthermore, we demonstrated that gastrointestinal nematode-induced expression of RELM-β was dependent on the IL-4Rα pathway. To confirm a role for IL-13, we showed that administration of recombinant IL-13 induces RELM-β expression in the intestine.
Structural analysis of RELM family members, resistin, and RELM-β reveals a multimeric structure comprised of protomers consisting of carboxy-terminal, disulfide-rich, β-sandwich “head” domain and an amino-terminal α-helical domain.9 It is notable that the 3-dimensional structure of RELM-β requires regulated disulfide-dependent assembly similar to that used by IgM.9 As a secreted product produced by intestinal epithelial cells, RELM-β can function as a regulatory molecule involved in luminal molecule recognition and transportation across the epithelial barrier. This view is supported by our finding that in the absence of RELM-β, there is decreased expression of immunologic effector molecules in the colon. Prior studies have associated RELM-β with innate immunity because expression is induced by bacterial colonization in the gastrointestinal tract through a Cdx2-dependent mechanism.37 Furthermore, RELM-β has been shown to directly bind to parasitic nematodes and possesses antichemotactic activity against the parasite.13 The selective decreased expression of type III REG proteins in the absence of RELM-β draws attention to the interaction of RELM-β and REG3, directly or indirectly. These data, taken together with our present findings that RELM-β is critical in acute models of inflammation, draw attention to the central role of RELM-β in innate intestinal immunity.
Although elegant studies in mice have demonstrated a role for resistin and RELM in regulating glucose metabolism, the exact homolog of murine RELM-β is controversial. We have not observed differences in baseline glucose levels in RELM-β-/- mice (data not shown). Taken together, murine RELM-β behaves more as a cytokine involved in immune regulation rather than a metabolic hormone. Our results highlight the importance of determining the exact biology of the RELM family members in the human system. Our finding defines a new biology for the RELM family of proteins by identifying a regulatory role for RELM-β in colonic barrier function and colonic inflammatory injury. These results have potential clinical applications because altered intestinal inflammation is seen in a variety of human diseases, including IBD and allergic gastroenteropathy. The finding that RELM-β is downstream from IL-13 signaling after DSS exposure highlights the potential involvement of RELM-β in TH2-associated inflammatory responses. Notably, the chromosome position of human RELM-β (3q13.1) has recently been linked to allergic inflammation, tempting speculation that perhaps RELM-β is responsible.38 Although not directly examined in this study, these results imply a similar immunomodulatory role for RELM-β in TH2-associated human lung disease (eg, asthma), especially in view of the overexpression of RELM-β in experimental asthma models.39,40
TABLE I.
Histologic colitis score of colons of TNBS-treated WT and RELM-β-/-mice
| WT mice |
RELM-β-/-mice |
|||
|---|---|---|---|---|
| Control | TNBS | Control | TNBS | |
| Area involved (%) | 0 ± 0 | 0.3 ± 0.7 | 0.1 ± 0.4 | 2.3 ± 2.1* |
| Crypt loss (%) | 0 ± 0 | 0.2 ± 0.4 | 0.1 ± 0.4 | 1.8 ± 1.5* |
| Erosion-ulceration | 0 ± 0 | 0.2 ± 0.4 | 0.1 ± 0.4 | 3.3 ± 3.2* |
| Cellular infiltration | ||||
| Lamina propria | 0 ± 0 | 0.2 ± 0.4 | 0.0 ± 0.0 | 1.5 ± 1.3* |
| Submucosa | 0 ± 0 | 0.1 ± 0.3 | 0.0 ± 0.0 | 1.8 ± 1.5* |
| Edema | 0 ± 0 | 0.2 ± 0.4 | 0.1 ± 0.4 | 1.8 ± 1.5* |
Data are presented as mean histologic colitis scores (points) ± SD (n = 4-8 mice per group). The Kruskal-Wallis test was performed to analyze differences between treatment groups (*P < .05).
Acknowledgments
We thank Drs Nives Zimmermann, Fred Finkelman, Gurjit K. Hershey, Thomas Korfhagen, Patricia Fulkerson, Dominique Brandt, Bruce Aronow, and Gary Ross for helpful discussions and review of this manuscript and Andrea Lippelman for editorial assistance.
Abbreviations used
- DAI
Disease activity index
- DSS
Dextran sodium sulfate
- ES
Embryonic stem
- FITC
Fluorescein isothiocyanate
- FIZZ
Found in inflammatory zone
- IBD
Inflammatory bowel disease
- NF-κB
Nuclear factor κB
- RELM
Resistin-like molecule
- REG
Regenerating gene
- TNBS
Trinitrobenzene sulfonate
- WT
Wild-type
Footnotes
Supported in part by the DDRDC Pilot and Feasibility Grant (NIH R24 DK64403; S.P.H.), R01 AI42242 (M.E.R.), AI45898 (M.E.R.), AI53479 (M.E.R.), and the Burroughs Wellcome Fund (M.E.R.) RO1 AI61570 (D.A.) and the Crohn’s and Colitis Foundation of America’s William and Shelby Modell Family Foundation Research Award (D.A.), and T32 AI060515 (L.S.).
Disclosure of potential conflict of interest: A. Mishra has received grant support from the National Institutes of Health. A. J. Murphy owns stock in and is employed by Regeneron Pharmaceuticals, Inc. M. E. Rothenberg owns stock in Ception Therapeutics, is the inventor of a patent application filed by CCHMC concerning RELM-β, has received grant support from Cambridge Antibody Technology, and is on the speakers’ bureau for Merck. The rest of the authors have declared that they have no conflict of interest.
Marc E. Rothenberg, MD, PhD, Division of Allergy and Immunology, Cincinnati Children’s Hospital Medical Center, 3333 Burnet Ave, ML7028, Cincinnati OH 45229. E-mail: Rothenberg@cchmc.org.
REFERENCES
- 1.Macdonald TT, Monteleone G. Immunity, inflammation, and allergy in the gut. Science. 2005;307:1920–5. doi: 10.1126/science.1106442. [DOI] [PubMed] [Google Scholar]
- 2.Umetsu DT, McIntire JJ, Akbari O, Macaubas C, DeKruyff RH. Asthma: an epidemic of dysregulated immunity. Nat Immunol. 2002;3:715–20. doi: 10.1038/ni0802-715. [DOI] [PubMed] [Google Scholar]
- 3.Abelson P, Kennedy D. The obesity epidemic. Science. 2004;304:1413. doi: 10.1126/science.304.5676.1413. [DOI] [PubMed] [Google Scholar]
- 4.Weiss ST, Shore S. Obesity and asthma: directions for research. Am J Respir Crit Care Med. 2004;169:963–8. doi: 10.1164/rccm.200303-403WS. [DOI] [PubMed] [Google Scholar]
- 5.Steppan CM, Bailey ST, Bhat S, Brown EJ, Banerjee RR, Wright CM, et al. The hormone resistin links obesity to diabetes. Nature. 2001;409:307–12. doi: 10.1038/35053000. [DOI] [PubMed] [Google Scholar]
- 6.Steppan CM, Brown EJ, Wright CM, Bhat S, Banerjee RR, Dai CY, et al. A family of tissue-specific resistin-like molecules. Proc Natl Acad Sci U S A. 2001;98:502–6. doi: 10.1073/pnas.98.2.502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Flier JS. The missing link with obesity? Nature. 2001;409:292–3. doi: 10.1038/35053251. [DOI] [PubMed] [Google Scholar]
- 8.Holcomb IN, Kabakoff RC, Chan B, Baker TW, Gurney A, Henzel W, et al. FIZZ1, a novel cysteine-rich secreted protein associated with pulmonary inflammation, defines a new gene family. EMBO J. 2000;19:4046–55. doi: 10.1093/emboj/19.15.4046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Patel SD, Rajala MW, Rossetti L, Scherer PE, Shapiro L. Disulfide-dependent multimeric assembly of resistin family hormones. Science. 2004;304:1154–8. doi: 10.1126/science.1093466. [DOI] [PubMed] [Google Scholar]
- 10.Banerjee RR, Rangwala SM, Shapiro JS, Rich AS, Rhoades B, Qi Y, et al. Regulation of fasted blood glucose by resistin. Science. 2004;303:1195–8. doi: 10.1126/science.1092341. [DOI] [PubMed] [Google Scholar]
- 11.Rajala MW, Obici S, Scherer PE, Rossetti L. Adipose-derived resistin and gut-derived resistin-like molecule-beta selectively impair insulin action on glucose production. J Clin Invest. 2003;111:225–30. doi: 10.1172/JCI16521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Schinke T, Haberland M, Jamshidi A, Nollau P, Rueger JM, Amling M. Cloning and functional characterization of resistin-like molecule gamma. Biochem Biophys Res Commun. 2004;314:356–62. doi: 10.1016/j.bbrc.2003.12.100. [DOI] [PubMed] [Google Scholar]
- 13.Artis D, Wang ML, Keilbaugh SA, He W, Brenes M, Swain GP, et al. RELMbeta/FIZZ2 is a goblet cell-specific immune-effector molecule in the gastrointestinal tract. Proc Natl Acad Sci U S A. 2004;101:13596–600. doi: 10.1073/pnas.0404034101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Marcus RC, Matthews GA, Gale NW, Yancopoulos GD, Mason CA. Axon guidance in the mouse optic chiasm: retinal neurite inhibition by ephrin “A”-expressing hypothalamic cells in vitro. Dev Biol. 2000;221:132–47. doi: 10.1006/dbio.2000.9660. [DOI] [PubMed] [Google Scholar]
- 15.Irizarry RA, Hobbs B, Collin F, Beazer-Barclay YD, Antonellis KJ, Scherf U, et al. Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics. 2003;4:249–64. doi: 10.1093/biostatistics/4.2.249. [DOI] [PubMed] [Google Scholar]
- 16.Stevceva L, Pavli P, Husband A, Matthaei KI, Young IG, Doe WF. Eosinophilia is attentuated in experimental colitis induced in IL-5 deficient mice. Genes Immun. 2000;1:213–8. doi: 10.1038/sj.gene.6363654. [DOI] [PubMed] [Google Scholar]
- 17.Ten Hove T, Drillenburg P, Wijnholds J, te Velde AA, van Deventer SJ. Differential susceptibility of multidrug resistance protein-1 deficient mice to DSS and TNBS-induced colitis. Dig Dis Sci. 2002;47:2056–63. doi: 10.1023/a:1019629013945. [DOI] [PubMed] [Google Scholar]
- 18.Camoglio L, te Velde AA, de Boer A, ten Kate FJ, Kopf M, van Deventer SJ. Hapten-induced colitis associated with maintained Th1 and inflammatory responses in IFN-gamma receptor-deficient mice. Eur J Immunol. 2000;30:1486–95. doi: 10.1002/(SICI)1521-4141(200005)30:5<1486::AID-IMMU1486>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- 19.Rothenberg ME, Luster AD, Lilly CM, Drazen JM, Leder P. Constitutive and allergen-induced expression of eotaxin mRNA in the guinea pig lung. J Exp Med. 1995;181:1211–6. doi: 10.1084/jem.181.3.1211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Karhausen J, Furuta GT, Tomaszewski JE, Johnson RS, Colgan SP, Haase VH. Epithelial hypoxia-inducible factor-1 is protective in murine experimental colitis. J Clin Invest. 2004;114:1098–106. doi: 10.1172/JCI21086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Synnestvedt K, Furuta GT, Comerford KM, Louis N, Karhausen J, Eltzschig HK, et al. Ecto-5'-nucleotidase (CD73) regulation by hypoxia-inducible factor-1 mediates permeability changes in intestinal epithelia. J Clin Invest. 2002;110:993–1002. doi: 10.1172/JCI15337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Panwala CM, Jones JC, Viney JL. A novel model of inflammatory bowel disease: mice deficient for the multiple drug resistance gene, mdr1a, spontaneously develop colitis. J Immunol. 1998;161:5733–44. [PubMed] [Google Scholar]
- 23.Mashimo H, Wu DC, Podolsky DK, Fishman MC. Impaired defense of intestinal mucosa in mice lacking intestinal trefoil factor. Science. 1996;274:262–5. doi: 10.1126/science.274.5285.262. [DOI] [PubMed] [Google Scholar]
- 24.Forbes E, Murase T, Yang M, Matthaei KI, Lee JJ, Lee NA, et al. Immunopathogenesis of experimental ulcerative colitis is mediated by eosinophil peroxidase. J Immunol. 2004;172:5664–75. doi: 10.4049/jimmunol.172.9.5664. [DOI] [PubMed] [Google Scholar]
- 25.Hendrickson BA, Gokhale R, Cho JH. Clinical aspects and pathophysiology of inflammatory bowel disease. Clin Microbiol Rev. 2002;15:79–94. doi: 10.1128/CMR.15.1.79-94.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bouma G, Strober W. The immunological and genetic basis of inflammatory bowel disease. Nat Rev. 2003;3:521–33. doi: 10.1038/nri1132. [DOI] [PubMed] [Google Scholar]
- 27.Strober W, Fuss IJ, Blumberg RS. The immunology of mucosal models of inflammation. Annu Rev Immunol. 2002;20:495–549. doi: 10.1146/annurev.immunol.20.100301.064816. [DOI] [PubMed] [Google Scholar]
- 28.Dieleman LA, Ridwan BU, Tennyson GS, Beagley KW, Bucy RP, Elson CO. Dextran sulfate sodium-induced colitis occurs in severe combined immunodeficient mice. Gastroenterol. 1994;107:1643–52. doi: 10.1016/0016-5085(94)90803-6. [DOI] [PubMed] [Google Scholar]
- 29.Elias JA, Lee CG, Zheng T, Ma B, Homer RJ, Zhu Z. New insights into the pathogenesis of asthma. J Clin Invest. 2003;111:291–7. doi: 10.1172/JCI17748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Okamoto H. The REG gene family and REG proteins: with special attention to the regeneration of pancreatic B-cells. J Hepatobiliary Pancreat Surg. 1999;6:254–62. doi: 10.1007/s005340050115. [DOI] [PubMed] [Google Scholar]
- 31.Blumberg RS, Saubermann LJ, Strober W. Animal models of mucosal inflammation and their relation to human inflammatory bowel disease. Curr Opin Immunol. 1999;11:648–56. doi: 10.1016/s0952-7915(99)00032-1. [DOI] [PubMed] [Google Scholar]
- 32.Buhner S, Buning C, Genschel J, Kling K, Herrmann D, Dignass A, et al. Genetic basis for increased intestinal permeability in families with Crohn’s disease: role of CARD15 3020insC mutation? Gut. 2006;55:342–7. doi: 10.1136/gut.2005.065557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Peeters M, Geypens B, Claus D, Nevens H, Ghoos Y, Verbeke G, et al. Clustering of increased small intestinal permeability in families with Crohn’s disease. Gastroenterol. 1997;113:802–7. doi: 10.1016/s0016-5085(97)70174-4. [DOI] [PubMed] [Google Scholar]
- 34.Wyatt J, Vogelsang H, Hubl W, Waldhoer T, Lochs H. Intestinal permeability and the prediction of relapse in Crohn’s disease. Lancet. 1993;341:1437–9. doi: 10.1016/0140-6736(93)90882-h. [DOI] [PubMed] [Google Scholar]
- 35.Gironella M, Iovanna JL, Sans M, Gil F, Penalva M, Closa D, et al. Antiinflammatory effects of pancreatitis associated protein in inflammatory bowel disease. Gut. 2005;54:1244–53. doi: 10.1136/gut.2004.056309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fichtner-Feigl S, Fuss IJ, Preiss JC, Strober W, Kitani A. Treatment of murine Th1- and Th2-mediated inflammatory bowel disease with NF-kappa B decoy oligonucleotides. J Clin Invest. 2005;115:3057–71. doi: 10.1172/JCI24792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wang ML, Shin ME, Knight PA, Artis D, Silberg DG, Suh E, et al. Regulation of RELM/FIZZ isoform expression by Cdx2 in response to innate and adaptive immune stimulation in the intestine. Am J Physiol Gastrointest Liver Physiol. 2005;288:G1074–83. doi: 10.1152/ajpgi.00442.2004. [DOI] [PubMed] [Google Scholar]
- 38.Brasch-Andersen C, Haagerup A, Borglum AD, Vestbo J, Kruse TA. Highly significant linkage to chromosome 3q13.31 for rhinitis and related allergic diseases. J Med Gen. 2006;43:10–7. doi: 10.1136/jmg.2005.035519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zimmermann N, King NE, Laporte J, Yang M, Mishra A, Pope SM, et al. Dissection of experimental asthma with DNA microarray analysis identifies arginase in asthma pathogenesis. J Clin Invest. 2003;111:1863–74. doi: 10.1172/JCI17912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zimmermann N, Hershey GK, Foster PS, Rothenberg ME. Chemokines in asthma: cooperative interaction between chemokines and IL-13. J Allergy Clin Immunol. 2003;111:227–42. doi: 10.1067/mai.2003.139. [DOI] [PubMed] [Google Scholar]