

Abstract
It is well established that the nuclear factor-κB (NF-κB) family of transcription factors participates in the regulation of many aspects of innate and adaptive immunity. The majority of these reports have focused on the role of NF-κB in accessory cell and T or B cell function, but less is known about the role of NF-κB in NK cells. However, several studies have demonstrated that these transcription factors are required for NK cell production of IFN-γ and proliferation. The studies presented here examine the role of two NF-κB members, c-Rel and p50, in NK cell function. In vitro data revealed that in the absence of c-Rel, NK cells have a defect in their ability to secrete IFN-c, but remain unaffected in their capacity to proliferate. In contrast, p50-/- NK cells have enhanced proliferative and IFN-γ responses compared with wild-type NK cells. The latter findings suggest a role for p50 as a negative regulator of NK cell production of IFN-γ and chromatin immunoprecipitation assays demonstrated the association of p50 with the IFN-γ promoter of resting NK cells. Consistent with the in vitro studies, in vivo studies with NF-κB gene-deficient mice infected with Toxoplasma gondii revealed that the absence of p50 leads to enhanced NK cell proliferation and production of IFN-γ. Together, these studies define distinct roles for c-Rel and p50 in the function of NK cells.
Keywords: cytokine gene expression regulation, innate immunity, natural killer cells, Toxoplasma gondii, transcription factors
Introduction
NK cells are part of the innate arm of the immune system and play a prominent role in tumor surveillance (1, 2) as well as resistance to many bacterial, viral and parasitic organisms (3-5). While one of the primary activities of NK cells is cytolysis of appropriate targets, these cells also produce cytokines that have important functions in immunity. In particular, the ability of NK cells to produce IFN-γ provides an innate mechanism of resistance to a variety of intracellular pathogens prior to the development of adaptive T cell responses. In addition, it has been proposed that the early production of IFN-γ by NK cells can influence subsequent T cell responses (6). As a consequence of these activities, NK cells represent an important bridge between innate and adaptive immunity.
Although NK and T cells represent the major sources of IFN-γ, most of our understanding about the events that regulate the production of this cytokine comes from work on T cells. These studies have led to the identification of a number of transcription factors, including T-box factors, signal transducers and activators of transcription (STAT) proteins and nuclear factor-κB (NF-κB) family members involved in T cell production of IFN-γ (7-10). However, these transcription factors are also expressed in NK cells, and regulate the ability of these cells to produce IFN-γ. For example, the ability of IL-12 to activate STAT4 is an important event that leads to T and NK cell IFN-γ production (9, 11). Similarly, T-bet, which is known to promote Th1 cell differentiation, is required for sustained NK cell production of IFN-γ (7, 12). However, there are T-bet- and STAT4-independent pathways that contribute to the production of IFN-γ in T and NK cells (13, 14). One such pathway is provided by the cytokines IL-1, IL-18 and tumornecrosis factor-α (TNF-α) which activate NF-κB (15-18), and which when used in combination with IL-12 enhance NK and T cell functions, including the production of IFN-γ (19-21).
The correlation of NF-κB signaling with a number of cytokine-mediated pathways that promote NK cell activity and cytokine production, provided the impetus to define the role of these transcription factors in NK cell function. The NF-κB family contains five members, NF-κB1 (p50), NF-κB2 (p52), RelA, RelB and c-Rel, that are evolutionarily conserved and which play an important role in many aspects of innate and adaptive immunity (22, 23). Most studies thus far have focused on the role of NF-κB in accessory, B and T cell function; fewer have examined the role of this transcription family in NK cells. However, there are reports that indicate an important role for NF-κB in NK cell activity. Initial studies showed that pharmacological inhibitors of NF-κB reduced spontaneous NK cytotoxicity (24). Subsequent in vitro studies revealed that for NK cells, a number of different signaling pathways including stimulation through CD40L and receptors for IL-2 and IL-18 lead to NF-κB activation and subsequent increases in perforin and IFN-c production, as well as enhanced cytotoxicity (25-28). It has also been reported that the ligation of toll-like receptor 2 (TLR2) or TLR3 on NK cells initiates NF-κB signaling and IFN-γ production (29, 30). Moreover, NK cell expression of a dominant negative form of IκBα, a global inhibitor of NF-κB, reduces their ability to become activated and produce IFN-γ during infection with the intracellular parasite, Toxoplasma gondii (31). However, while global inhibition of NF-γB clearly decreases NK activity, there are few examples that have addressed the role of individual family members in NK cell function. The studies presented here define a role for c-Rel in promoting the optimal production of IFN-γ by NK cells, and indicate that p50 is a negative regulator of proliferation and IFN-γ production.
Methods
Mice, parasites and tissue culture
Mice.
c-Rel KO and p50 KO mice on a C57Bl/6 background were bred and housed within microisolator caging units at the University Laboratory Animal Resource facilities at the University of Pennsylvania (32, 33). Mice were bred as homozygous knockouts for each strain. Wild-type (Wt) C57Bl/6 mice were used as age- and sex-matched controls in all experiments.
Parasites.
For in vivo experiments, female mice were inoculated intra-peritoneally with 20 cysts of the Me-49 strain of T. gondii which had been prepared from the brains of chronically infected CBA/CaJ mice.
Lymphokine-activated killer cells.
Wt lymphokine-activated killer (LAK) cells were derived from the bone marrow of female RAG KO mice on a C57Bl/6 background plated with 4 × 104 U ml-1 of rhuIL-2 (Chiron, Emeryville, CA, USA) for 7 days before harvesting for cell culture. LAK cells from KO mice and Wt controls were generated by purifying NK cells from splenocytes before being cultured in vitro in complete RPMI with 4 × 104 U ml-1 IL-2 for 7 days to generate and expand a LAK cell population. While highly purified populations of LAK cells could be obtained from the spleens of Wt mice, this was not the case with spleens from c-Rel or NF-κB1 KO mice. The basis for this observation remains unclear to us, but precluded the analysis of NF-κB complexes in the KO NK cells.
Detection of IFN-γ (in vitro and ex vivo)
Spleens were harvested and dissociated into single-cell suspensions in complete RPMI 1640 (Life Technologies, Gaithersburg, MD, USA) supplemented with 10% FCS (HyClone Laboratories, Logan, UT, USA), 2% HEPES, 50 lM 2-mercaptoethanol, 0.1 mM non-essential amino acids, 10 U ml-1 penicillin, and 0.1 mg ml-1 streptomycin (Life Technologies). Erythrocytes were depleted using 0.83% ammonium chloride, and cells were washed in complete RPMI 1640 before further analysis. Ex vivo cultures were stimulated with 50 ng ml-1 phorbol myristate acetate (PMA) and 500 ng ml-1 ionomycin in the presence of brefeldin A (10 μl ml-1), for 4 h, before harvesting and staining for flow cytometric analysis of IFN-γ expression. All samples were analyzed using a FACSCalibur flow cytometer (BD Biosciences) and data were analyzed using FlowJo software (Tree Star, San Carlos, CA, USA).
Culture conditions.
Prior to use, LAK cell cultures were rested for 3 h (rested LAK) after plating at 4 × 105 cells per well in a final volume of 200 ll. In vitro cultures of rested LAK cells were left untreated or were stimulated with 1000 U ml -1 of rhuIL-2 plus 10 ng ml -1 rmuIL-12 (Genzyme, Cambridge, MA, USA) or 10 ng ml-1 IL-18 (BD PharMingen, San Diego, CA, USA), and incubated at 37°C in 5% CO2 for 4 h. Cells were then harvested and surface stained with fluorochrome conjugated antibodies (BD Pharmingen) against CD3 and NK1.1 before intracellular staining for IFN-γ as previously described (31).
Proliferation assays (carboxyfluorescein succinimidyl ester and 5′-bromo-2′-deoxyuridine)
In vitro proliferative capacity. Naive splenocytes from all strains were isolated and labeled with 2.5 lM carboxyfluorescein succinimidyl ester (CFSE) before plating at a concentration of 4 × 105 cells per well, in triplicate in a final volume of 200 ll. Cells were cultured with 0-1000 U ml -1 of rhuIL-2 and incubated for 4 days at 37°C in 5% CO2. Cells were harvested and surface stained as above for flow cytometric collection and analysis of proliferation.
In vivo proliferation.
Days 1 and 2 after intra-peritoneal infection with T. gondii, mice were injected intra-peritoneally with 0.2 ml 5′-bromo-2′-deoxyuridine (BrdU) (Sigma-Aldrich, St. Louis, MO, USA) at 4 mg ml -1 stock concentration. Peritoneal exudate cells (PECs) and splenocytes were isolated from infected mice and uninfected controls. Surface staining was done as described above before fixation with 4% PFA (Sigma) overnight. Cells were then re-suspended in 0.2 ml of 1% PFA/0.05% Tween-20 (Sigma) in PBS and incubated for 30 min at room temperature (RT), followed by 30 min at 4°C. Cells were centrifuged and re-suspended in 1 ml DNase solution [0.15 M NaCl, 4.2 mM MgCl2, 150 K units ml -1 DNase I(Sigma)] and incubated at RT for 30 min. Cells were centrifuged and supernatant expelled before staining with anti-BrdU or isotype control (PharMingen) and incubated a final 30 min at RT, prior to washing 23 in FACS buffer and analysis by flow cytometry.
Western blots
Rested LAK cells were cultured in media alone or combinations of IL-2, IL-12 and IL-18 as described previously. Cells were then harvested, and nuclear and cytoplasmic extracts were isolated as previously described (34). Nuclear extracts were resolved on a 10% SDS/PAGE gel and then transferred to a nitrocellulose membrane (BioRad, Hercules, CA, USA) using a Trans-Blot Semi-Dry cell (BioRad) according to the manufacturer’s instructions. Nitrocellulose filters were then incubated with wash buffer (25 mM Tris, pH 7.5, 500 mM NaCl, 0.1% (vol/vol) Tween-20) containing 5% milk protein overnight at 4°C to block non-specific protein binding. Primary antibodies against p50 (sc-114), c-Rel (sc-70) or p65 (sc-372-G) (all from Santa Cruz Biotechnology, Inc., Santa Cruz, CA, USA) were diluted 1:1000 in wash buffer containing 0.5% milk protein and applied to the filter for 2 h at RT. Following washing, the blots were incubated with HRP-conjugated anti-rabbit IgG or anti-goat IgG (Pierce, Rockford, IL, USA) (diluted 1:5000 in wash buffer containing 0.5% milk protein) for 45 min at RT. Bands were visualized by the enhanced chemilumines-cence system (Amersham Pharmacia Biotech, Piscataway, NJ, USA).
Electorphoretic mobility shift assays
LAK cells were generated as above and rested prior to in vitro stimulation. Cells (5 × 106) were stimulated for 1 or 3 h either individually or in combination with IL-2, IL-12 and IL-18. Nuclear and cytoplasmic extracts were isolated as previously described and frozen at -80°C until used in electorphoretic mobility shift assays (EMSAs). In brief, double-stranded oligodeoxynucleotides corresponding to the palindromic jB site (5′-GGGAATTCCC-3′) were labeled by filling the overlapping ends with the Klenow fragment of DNA polymerase I and [32P]dCTP. Following removal of unincorporated nucleotides, labeled oligonucleotide (50 000 counts per minute) was incubated with 6 lg protein extracts and 2 lg poly(dI-dC) in buffer containing 20 mM HEPES (pH 7.9), 100 mM NaCl, 5 mM MgCl2, 1mM dithiothreitol, 0.7 mM phenylmethylsulphonylfluoride and 17% glycerol in a final volume of 22 ll for 15 min at 20°C. Supershift analysis was performed by pre-incubating samples with antibodies to p50, c-Rel or p65 (Santa Cruz) prior to incubation with labeled oligonucleotide. Complexes were separated on 5.5% polyacrylamide gels run on 0.25% Tris-borate-EDTA buffer, dried and exposed to Kodak film at -80°C.
Chromatin immunoprecipitation assays
For each condition, 2 × 106 LAK cells were rested for 3-4 h prior to stimulation with cytokines for 1 h. Histones were then cross-linked by adding formaldehyde (Fisher Scientific, Pittsburgh, PA, USA) at 1% of total volume directly to the cells and incubating for 10 min at 37°C. Chromatin immunoprecipitation (ChIP) assays were performed according to the manufacturer’s kit protocol (Upstate, Charlottesville, VA, USA). Briefly, cells were washed in ice-cold PBS containing the inhibitors aprotinin, leupeptin and NPGB (all from Sigma) and pelleted before a brief incubation in SDS lysis buffer (Upstate) plus inhibitors and sonication to fragment DNA. Precipitation of DNA was performed using antibodies for p50 (Santa Cruz) or rabbit IgG as a control (Upstate) and eluted before histone-DNA cross-linkages were reversed by heating at 65°C for 4 h. Finally, DNA was recovered by phenol/chloroform extraction and PCR was performed using primers specific to exon 4 of the IFN-c promoter region as previously described (35); sense: 5′-CGT AAT CCC GAG GAG CCT TC-3′; antisense: 5′-CTT TCA ATG ACT GTG CCG TGG-3′.
Results
Translocation and activation of NF-jB after cytokine stimulation of NK cells
The stimulation of resting or activated NK cells with combinations of cytokines such as IL-2 + IL-12 or IL-18 + IL-12 leads to proliferation and the production of IFN-γ by these cells. To characterize which NF-κB family members are associated with these events, resting LAK cells were incubated with these stimuli and nuclear extracts prepared at different times after stimulation to assess overall levels of nuclear NF-κB and the levels of DNA-binding activity in these samples. Western blots revealed the presence of low levels of p50, p65 and c-Rel in the nucleus of resting LAK cells, however, after stimulation with either IL-2 or IL-12 alone or the combination of IL-2 + IL-12 or IL-12 + IL-18 for 3 h there was an accumulation of these transcription factors in the nucleus (Fig. 1A). In contrast, only a slight increase in nuclear localization of c-Rel was found when rested LAK cells were stimulated with IL-18 alone (Fig. 1A). When EMSAs were performed to measure DNA-binding capacity, significant levels of activated NF-κB were observed in resting LAK cells, and supershifts revealed that the predominant family member that bound DNA was p50 (Fig. 1B). As expected, there was a significant increase in the amount of activated NF-κB in the nucleus of cytokine-stimulated cells, however, supershifts of these samples revealed that p50 and c-Rel made up the majority of the activated dimers especially when stimulated with IL-2 + IL-12 (Fig. 1B). Supershifts for p65 showed that while there was some p65 present in activated dimers in response to IL-12 + IL-18, this NF-jB family member was not present in significant levels after stimulation with IL-2 + IL-12. Together, these data suggest that p50 homodimers were the primary source of activated NF-κB in unstimulated cells, whereas c-Rel was specifically activated in response to these stimuli.
Fig. 1.
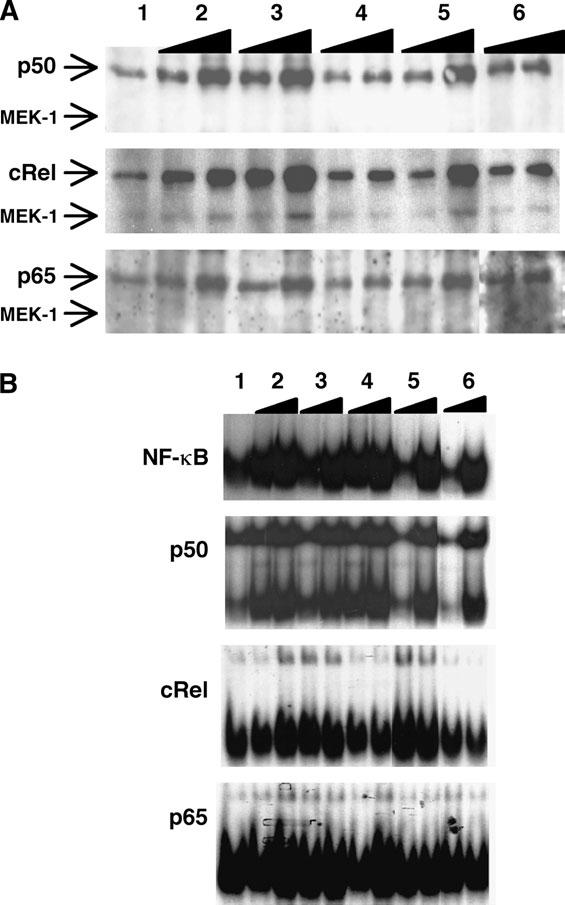
Activated NF-jB dimers translocate into the nucleus of activated NK cells. LAK cells were stimulated in vitro with media alone (1); IL-2 (2); IL-12 (3); IL-18 (4); IL-2 + IL-12 (5); or IL-12 + IL-18 (6) for 1and 3 h and nuclear extracts were prepared as described in Methods. These samples were used in western blots for p50, c-Rel and p65, as well as the cytoplasmic protein MEK-1 to verify purity of extracts (A) or EMSAs (B) that included supershifts for p50, c-Rel and p65.
Role of c-Rel and p50 in NK cell proliferation
Given the prominent activity of p50 and c-Rel in response to different combinations of cytokines, studies were performed to assess the role of these factors in NK cell function. Since stimulation of NK cells with IL-2 leads to activation of NF-κB (26, 36, 37) and proliferation of NK cells (38), experiments were performed to determine how the absence of c-Rel or p50 affected NK cell proliferation in vitro. Whole splenocytes from either Wt or gene knockout mice were labeled with CFSE and cultured with cytokine in vitro for 4 days before analyzing NK cell proliferation by FACS. In the absence of stimulation, no proliferation was observed in any of the groups. However, in response to IL-2, both Wt and c-Rel/NK cells underwent multiple rounds of division compared with media controls, while NK cells deficient in p50 were hyperproliferative at every concentration of IL-2 tested (Fig. 2A and data not shown). Further analysis revealed that the percentage of cells responding to stimulation was similar for Wt or c-Rel -/- NK cells and was ∼20% at the lowest concentration of IL-2 tested (Fig. 2B). Consistent with these data, the overall proliferative capacity for Wt and c-Rel -/- NK cells was also similar, averaging ∼10 daughters per responding cell in both groups. In contrast, over 40% of p50 -/- NK cells responded to low concentrations of IL-2 and progressed through significantly more rounds of division than Wt or c-Rel-deficient cells (Fig. 2A and B). However, at higher concentrations of IL-2, there was little difference in responder frequency. Nevertheless, a comparison of the number of progeny revealed that a significantly higher average number of daughters per responding cell was achieved in the p50-deficient group at all concentrations of IL-2 tested (Fig. 2C). Together, these data reveal that c-Rel is not required for NK cell proliferation, but the absence of p50 leads to heightened sensitivity of NK cells to IL-2 and an increased proliferative capacity.
Fig. 2.
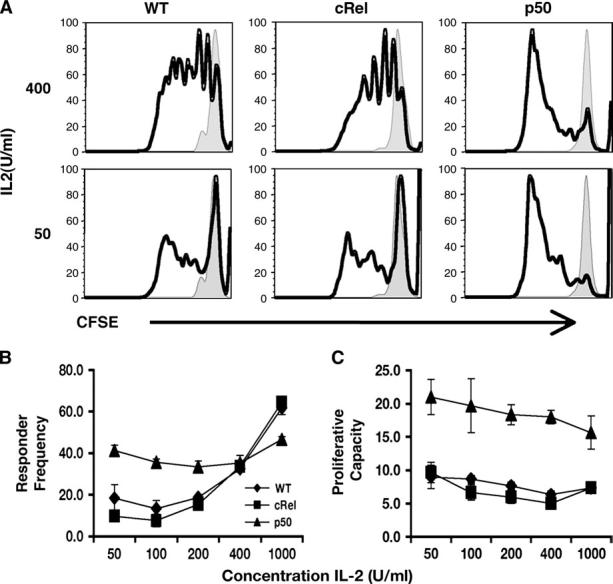
Proliferation of Wt and p50- and c-Rel-deficient NK cells. Splenocytes from Wt or KO mice were labeled with CFSE and stimulated in vitro with IL-2 for 4 days and then used for FACS analysis to identify proliferating NK cells. Representative histograms of live, CD3, NK1.1+ cells (A) of stimulated (bold line) or media controls (shaded); line graph of combined responder frequency (B) and proliferative capacity (C). All differences in values between Wt and p50 are statistically significant except for responder frequency at 400 U ml -1 IL-2.
Production and regulation of IFN-γ by c-Rel and p50
Although the studies above address the role of p50 and c-Rel in NK cell proliferation, these cells are also a prominent source of IFN-c. To directly address if c-Rel and p50 are involved in NK cell production of this cytokine, Wt, c-Rel -/- or p50 -/- LAK cells were stimulated with IL-2 + IL-12, and FACS analysis used to assess the levels of intracellular IFN-c. In the absence of stimulation,<2% of cells were positive for IFN-γ. After 4 h of stimulation, the percentage of IFN-c+ cells from the different strains showed distinct patterns. Whereas 23% of NK cells from Wt mice were positive for IFN-γ, there was a marked reduction in the percentage of c-Rel-deficient NK cells that were IFN-γ+. In contrast, the absence of p50 resulted in a significant increase in the frequency of IFN-γ+ cells as well as an increased mean fluorescence intensity (MFI) after 4 h, with an average MFI in Wt cells of 113 compared with a MFI of 145 for p50 -/- NK cells (Fig. 3A). Similar effects were also observed after stimulation with other cytokine combinations (Fig. 3B and data not shown). Together, these studies indicate that in NK cells c-Rel promotes, whereas p50 antagonizes the production of IFN-γ.
Fig. 3.
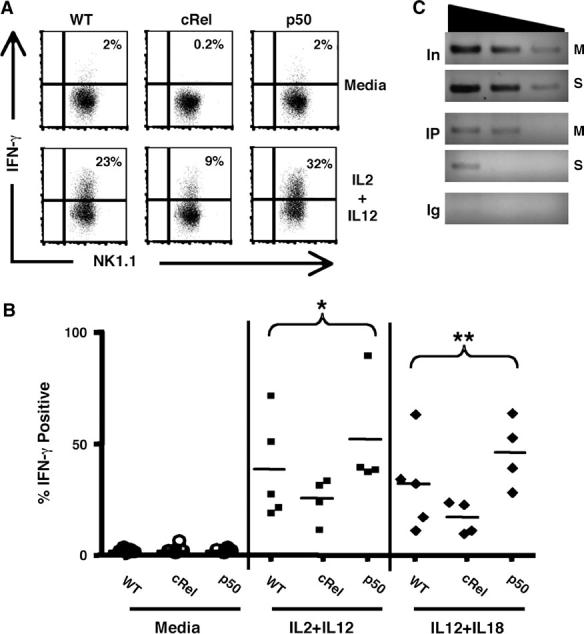
Production of IFN-c by NK cells. Dot plots of LAK cells generated from Wt, p50 or c-Rel KO mice were rested and then re-stimulated in vitro for 4 h (A) and scatter plots of the percentage of IFN-c+ cells after stimulation with IL-2 + IL-12 and IL-12 + IL-18 or media alone in individual mice for all experiments; *P = 0.04, **P = 0.003 (B). LAK cells generated from Wt, mice were rested for 3 h before incubation with media alone (M) or IL-2 + IL-12 (S) for 1 h in vitro (C). PCR of DNA derived from a ChIP assay representing total DNA (In) and DNA precipitated using either an antibody for p50 (IP) or rabbit IgG (Ig) and using primers specific to the exon 4 region of the IFN-c promoter.
Recent studies show that in contrast to naive T cells, the IFN-γ promoter in NK cells exists in a constitutively ‘open’ configuration that would allow the rapid production of IFN-γ (39-41). Since p50 homodimers can act as inhibitors of gene transcription (42) and p50 is readily detected in the nucleus of resting LAK cells (see Fig. 1), one possible explanation for the increase in IFN-γ production in p50 -/- NK cells is that p50 is constitutively associated with the IFN-γ promoter in NK cells. Histone acetylation in exon 4 of the IFN-γ gene was previously associated with IFN-γ production in T and NK cells (35, 39), and binding sites for NF-κB have been previously described in this and other areas of the IFN-κ promoter in T cells (10, 43). Therefore, ChIP assays were performed to determine if p50 was associated with this region of the IFN-γ promoter in resting NK cells. These studies revealed that in resting LAK cells p50 is readily associated with this region, whereas after stimulation with IL-2 + IL-12 there was a marked reduction in the levels detected by PCR (Fig. 3C). Interestingly, we did not detect c-Rel binding to the region of the promoter which has been previously shown to enhance transcription of IFN-γ in human T cells (10) (data not shown). Together with the functional data in Fig. 3(A) and studies in other experimental systems (44), these findings support the hypothesis that in NK cells p50 acts as a constitutive repressor at the IFN-γ locus.
NK cell function in the absence of c-Rel or p50 during infection
The in vitro studies described above indicate distinct roles for c-Rel and p50 in the regulation of NK cell proliferation and production of IFN-γ. In order to determine whether this was relevant to an in vivo situation, Wt and KO mice were infected with the parasite T. gondii, a potent inducer of NK cell proliferation and IFN-γ production (45, 46), and the responses of these cells were evaluated. To assess NK cell proliferation in vivo, mice were injected with the thymidine analog BrdU during challenge, and spleens and PECs were harvested day 3 post-infection and FACS was used to asses the levels of BrdU incorporation. In naive mice, NK cells exhibited a low level of homeostatic proliferation (Fig. 4), similar to previously published observations (47). However, p50 -/- NK cells from uninfected mice had elevated basal levels of BrdU incorporation compared with Wt and c-Rel -/- NK cells (Fig. 4C). Challenge with T. gondii led to an overall increase in levels of BrdU incorporation by splenic NK cells in all strains of mice examined (Fig. 4A and C) and similar levels of proliferation were observed for Wt and c-Rel -/- NK cells. Nevertheless, the highest levels of infection-induced BrdU incorporation were observed with p50 -/- NK cells (Fig. 4B and D). These findings were consistent with the in vitro studies, in which p50-deficient NK cells stimulated with IL-2 exhibited increased proliferative capacity (Fig. 2).
Fig. 4.
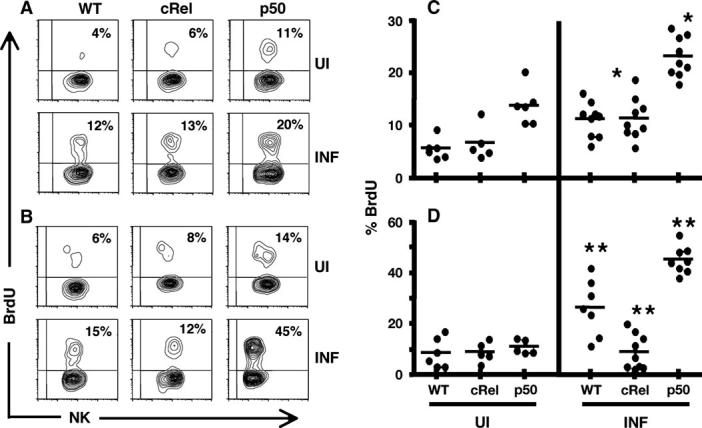
Assessment of in vivo proliferation of NK cells in response to infection. NK cells derived from the spleens (A and C) and peritoneal exudate cells (B and D) of Wt and KO mice harvested on day 3 post-infection with Toxoplasma gondii, to analyze incorporation of BrdU. Percentages represent percentage of live, CD3 NK cells positive for BrdU. Contour plots showing one representative sample from each (A and B) and scatter plots showing values for individual mice from all experiments (C and D). *, ** denote average values which are significantly different with a P-value <0.005.
In order to compare the ability of NK cells to produce IFN-γ following infection in the absence of c-Rel or p50, splenocytes from uninfected and infected mice were cultured directly ex vivo with PMA and ionomycin in the presence of brefeldin A for 4 h before analyzing by FACS. In the absence of ex vivo stimulation there was no detectable IFN-c, however, stimulation with PMA and ionomycin led to the visualization of intracellular IFN-c in NK cell populations. In naive mice, when overall percentages of IFN-γ+ NK cells were compared, no significant differences were observed between the three strains (Fig. 5). After infection, there was a trend towards an increase in the percentage of IFN-γ+ NK cells in Wt and p50 KO mice and lowest levels were seen with c-Rel-deficient mice. However, these differences did not reach statistical significance in any of the strains tested. Nevertheless, when the infection-induced expansion of these cells was taken into account, there was typically a 2- to 4-fold increase in the total numbers of IFN-γ+ NK cells (data not shown).
Fig. 5.

IFN-c production of NK cells after infection with Toxoplasma gondii. Scatter diagram showing the percentage of IFN-c+ splenic NK cells from Wt and KO mice directly ex vivo, day 3 after infection. Values represent percentages of live CD3, NK1.1+ cells that were IFN-γ+. UN = uninfected controls and INF = infected with T. gondii.
Discussion
The generic activation of NF-jB is now a hallmark of many innate immune responses, but based on structural similarity and expression patterns it was initially thought that a significant amount of redundancy existed within this family. Recently, a number of studies have helped to define specific roles for individual NF-κB members in innate immunity (22, 48-51). However, few studies have directly assessed the role of these transcription factors in NK cells. The studies presented here indicate a role for c-Rel in the optimal production of IFN-γ by NK cells, and this is consistent with previous studies that identified a role for c-Rel in T cell production of this cytokine (52-54). However, the fact that c-Rel -/- NK cells were still capable of significant production of IFN-γ in vitro indicates the presence of c-Rel-independent pathways that can promote IFN-γ production. Moreover, it is important to recognize that following challenge of c-Rel -/- mice with T. gondii, there was no obvious defect in NK cell production of IFN-γ. This effect may be a function of the use of PMA and ionomycin, a stimulus for IFN-γ production that bypasses receptor-mediated processes. Another possible explanation is that this infection generates an environment that is characterized by high levels of multiple cytokines and co-stimulatory molecules (TNF, IL-1, IL-2, IL-12, IL-15, IL-18, CD80, CD86, B7RP-1) that enhance NK cell production of IFN-γ and the availability of these multiple stimuli may override a requirement for c-Rel. In addition, other transcription factors such as RelB, STAT4, T-bet and Eomes that are implicated in the ability of NK cells to produce IFN-c may be sufficient for normal NK responses in this inflammatory environment. Nevertheless, the finding that c-Rel can regulate NK cell production of IFN-γ highlights a role for c-Rel in the innate events associated with resistance to multiple intracellular pathogens, in addition to its important role in accessory cell production of IL-12 explored in previous reports (49, 50, 55).
The finding that the absence of p50 led to increased production of IFN-c was a surprise in part because p50 -/- thymocytes have been shown to be defective in their ability to produce IFN-γ (56) and the absence of p50 does not lead to increased T cell production of this cytokine. We have also identified an important intrinsic role for p50 in promoting T cell production of IFN-γ in response to infection with T. gondii (S. Shapira and C. A. Hunter, unpublished observations). Taken together, these data indicate distinct roles for p50 in the production of IFN-γ in closely related immune cells. While there are many possible explanations for these findings, recent reports have highlighted that the rapid response of NK cells to infection, that precede adaptive T cell-mediated immunity, is associated with the presence of constitutively modified chromatin that allows for the proliferation-independent up-regulation of activating receptors (57), cytokine transcription (39, 40) and perforin expression (58). This level of ‘readiness’ implies that there must be a degree of regulation required to keep NK cells quiescent until needed, as well as to down-regulate these responses, that is distinct from cells of the adaptive response. The association of p50 with the IFN-γ promoter in resting NK cells implicates this NF-κB family member in the direct regulation of cytokine transcription and suggests that NK cells, but not T cells, are regulated at the level of transcription by inhibitory complexes, such as p50 homodimers. This idea is consistent with previous work in which p50 homodimers associated with DNA were found in complexes with HDAC, an inhibitor of DNA acetylation (44). In addition, more recent work with macrophages suggests that p50 homodimers may promote BCL-3 induction and association with HDAC as a possible mechanism for transcriptional inhibition (59).
The studies presented here also indicate that p50 plays an important role in the regulation of proliferation at two levels. First, the response threshold for activation is lowered as reflected in the higher constitutive level of homeostatic proliferation, and the increased responder frequency in p50 -/- NK cells after stimulation with low levels of IL-2. The second effect observed in the p50 -/- increased NK cells is the proliferative capacity, or number of progeny, derived from each responding cell. At present, the basis for these effects is unclear, but one possible explanation is provided by studies which established that the ability of IL-2 to activate STAT5 is necessary for cell cycle progression in T cells (60) and that at some level NF-κB signaling is required for this effect (37). Therefore, it is possible that the increased proliferation in p50-deficient NK cells is due to enhanced STAT5 activity in the absence of p50 homodimers. However, it is difficult to determine from these data if this enhanced expansion is simply due to a decreased threshold to initiate proliferation, or is accompanied by an increased rate of division or alterations in survival and future studies are required to address this issue.
Lastly, it is interesting to note that the decreased NK activity observed after global inhibition of NF-κB or in the absence of c-Rel or RelB is consistent with the phenotype of patients that have mutations in NF-κB essential modulator (NEMO), a molecule with a central role in the activation of NF-κB. This primary immune defect is associated with impaired NK cell cytotoxicity, a phenotype which correlates with increased susceptibility to certain viral pathogens (36) and these patients highlight the important role of NF-κB in the activation of NK cells. In contrast, there is no clear evidence that a lack of p50 activity is associated with immune hyperactivity in humandisease. However, the same patients with defects in NEMO also develop colitis (61) and a single nucleotide polymorphism in p50 is associated with human colitis (62). Moreover mice deficient in p50 have an increased susceptibility to infection induced colitis (63-65). Together these findings suggest a prominent role for the inhibitory effects of p50 in immune homeostasis and additional studies will be required to assess whether this impacts on immune-mediated disease in humans.
Acknowledgements
We would like to thank the members of the Pathobiology Department for their help and support during the writing of the manuscript. This work is supported by the National Institutes of Health (NIH) Grant AI 46288, Parasitology Training Grant AI07532 and the State of Pennsylvania (C.A.H.); NIH AI50827 (N.M.) and NIH AI061570 (D.A.).
Abbreviations
- BrdU
5′-bromo-2′-deoxyuridine
- CFSE
carboxyfluorescein succinimidyl ester
- ChIP
chromatin immunoprecipitation
- EMSA
electorphoretic mobility shift assay
- LAK
lymphokine-activated killer
- MFI
mean fluorescence intensity
- NEMO
NF-κB essential modulator
- NF-κB
nuclear factor-κB
- NIH
National Institutes of Health
- PMA
phorbol myristate acetate
- RT
room temperature
- STAT
signal transducers and activators of transcription
- TLR
toll-like receptor
- TNF
tumor necrosis factor
- Wt
wild type
References
- 1.Miller JS. Biology of natural killer cells in cancer and infection. Cancer Investig. 2002;20:405. doi: 10.1081/cnv-120001185. [DOI] [PubMed] [Google Scholar]
- 2.Fauriat C, Marcenaro E, Sivori S, et al. Natural killer cell-triggering receptors in patients with acute leukaemia. Leuk. Lymphoma. 2003;44:1683. doi: 10.1080/1042819031000104006. [DOI] [PubMed] [Google Scholar]
- 3.Bancroft GJ, Sheehan KC, Schreiber RD, Unanue ER. Tumor necrosis factor is involved in the T cell-independent pathway of macrophage activation in scid mice. J. Immunol. 1989;143:127. [PubMed] [Google Scholar]
- 4.Gazzinelli RT, Hieny S, Wynn TA, Wolf S, Sher A. Interleukin 12 is required for the T-lymphocyte-independent induction of interferon c by an intracellular parasite and induces resistance in T-cell-deficient hosts. Proc. Natl Acad. Sci. USA. 1993;90:6115. doi: 10.1073/pnas.90.13.6115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Biron CA, Brossay L. NK cells and NKT cells in innate defense against viral infections. Curr. Opin. Immunol. 2001;13:458. doi: 10.1016/s0952-7915(00)00241-7. [DOI] [PubMed] [Google Scholar]
- 6.Murphy KM, Reiner SL. The lineage decisions of helper T cells. Nat. Rev. Immunol. 2002;2:933. doi: 10.1038/nri954. [DOI] [PubMed] [Google Scholar]
- 7.Szabo SJ, Kim ST, Costa GL, Zhang X, Fathman CG, Glimcher LH. A novel transcription factor, T-bet, directs Th1 lineage commitment. Cell. 2000;100:655. doi: 10.1016/s0092-8674(00)80702-3. [DOI] [PubMed] [Google Scholar]
- 8.Pearce EL, Mullen AC, Martins GA, et al. Control of effector CD8+ T cell function by the transcription factor Eomeso-dermin. Science. 2003;302:1041. doi: 10.1126/science.1090148. [DOI] [PubMed] [Google Scholar]
- 9.Kaplan MH, Sun YL, Hoey T, Grusby MJ. Impaired IL-12 responses and enhanced development of Th2 cells in Stat4-deficient mice. Nature. 1996;382:174. doi: 10.1038/382174a0. [DOI] [PubMed] [Google Scholar]
- 10.Sica A, Dorman L, Viggiano V, et al. Interaction of NF-jB and NFAT with the interferon-gamma promoter. J. Biol. Chem. 1997;272:30412. doi: 10.1074/jbc.272.48.30412. [DOI] [PubMed] [Google Scholar]
- 11.Thierfelder WE, van Deursen JM, Yamamoto K, et al. Requirement for Stat4 in interleukin-12-mediated responses of natural killer and T cells. Nature. 1996;382:171. doi: 10.1038/382171a0. [DOI] [PubMed] [Google Scholar]
- 12.Townsend MJ, Weinmann AS, Matsuda JL, et al. T-bet regulates the terminal maturation and homeostasis of NK and Va14i NKT cells. Immunity. 2004;20:477. doi: 10.1016/s1074-7613(04)00076-7. [DOI] [PubMed] [Google Scholar]
- 13.Kaplan MH, Wurster AL, Grusby MJ. A signal transducer and activator of transcription (Stat)4-independent pathway for the development of T helper type 1 cells. J. Exp. Med. 1998;188:1191. doi: 10.1084/jem.188.6.1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Okamura H, Tsutsi H, Komatsu T, et al. Cloning of a new cytokine that induces IFN-c production by T cells. Nature. 1995;378:88. doi: 10.1038/378088a0. [DOI] [PubMed] [Google Scholar]
- 15.Robinson D, Shibuya K, Mui A, et al. IGIF does not drive Th1 development but synergizes with IL-12 for interferon-gamma production and activates IRAK and NFkappaB. Immunity. 1997;7:571. doi: 10.1016/s1074-7613(00)80378-7. [DOI] [PubMed] [Google Scholar]
- 16.Matsumoto H, Suzuki K, Tsuyuguchi K, et al. Interleukin-12 gene expression in human monocyte-derived macrophages stimulated with Mycobacterium bovis BCG: cytokine regulation and effect of NK cells. Infect. Immun. 1997;65:4405. doi: 10.1128/iai.65.11.4405-4410.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kojima H, Aizawa Y, Yanai Y, et al. An essential role for NF-jB in IL-18-induced IFN-c expression in KG-1 cells. J. Immunol. 1999;162:5063. [PubMed] [Google Scholar]
- 18.Yang J, Murphy TL, Ouyang W, Murphy KM. Induction of interferon-gamma production in Th1 CD4+ T cells: evidence for two distinct pathways for promoter activation. Eur. J. Immunol. 1999;29:548. doi: 10.1002/(SICI)1521-4141(199902)29:02<548::AID-IMMU548>3.0.CO;2-Z. [DOI] [PubMed] [Google Scholar]
- 19.Kanakaraj P, Ngo K, Wu Y, et al. Defective interleukin (IL)-18-mediated natural killer and T helper cell type 1 responses in IL-1 receptor-associated kinase (IRAK)-deficient mice. J. Exp. Med. 1999;189:1129. doi: 10.1084/jem.189.7.1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Walker W, Aste-Amezaga M, Kastelein RA, Trinchieri G, Hunter CA. IL-18 and CD28 use distinct molecular mechanisms to enhance NK cell production of IL-12-induced IFN-c. J. Immunol. 1999;162:5894. [PubMed] [Google Scholar]
- 21.Cai G, Kastelein R, Hunter CA. Interleukin-18 (IL-18) enhances innate IL-12-mediated resistance to Toxoplasma gondii. Infect. Immun. 2000;68:6932. doi: 10.1128/iai.68.12.6932-6938.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Caamano J, Hunter CA. NF-jB family of transcription factors: central regulators of innate and adaptive immune functions. Clin. Microbiol. Rev. 2002;15:414. doi: 10.1128/CMR.15.3.414-429.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liou HC. Regulation of the immune system by NF-jB and IjB. J. Biochem. Mol. Biol. 2002;35:537. doi: 10.5483/bmbrep.2002.35.6.537. [DOI] [PubMed] [Google Scholar]
- 24.Valle Blazquez M, Luque I, Collantes E, et al. Cellular redox status influences both cytotoxic and NF-jB activation in natural killer cells. Immunology. 1997;90:455. doi: 10.1111/j.1365-2567.1997.00455.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jyothi MD, Khar A. Regulation of CD40L expression on natural killer cells by interleukin-12 and interferon c: its role in the elicitation of an effective antitumor immune response. Cancer Immunol. Immunother. 2000;49:563. doi: 10.1007/s002620000151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jyothi MD, Khar A. Interleukin-2-induced nitric oxide synthase and nuclear factor-kappaB activity in activated natural killer cells and the production of interferon-gamma. Scand. J. Immunol. 2000;52:148. doi: 10.1046/j.1365-3083.2000.00762.x. [DOI] [PubMed] [Google Scholar]
- 27.Hoshino K, Tsutsui H, Kawai T, et al. Cutting edge: generation of IL-18 receptor-deficient mice: evidence for IL-1 receptor-related protein as an essential IL-18 binding receptor. J. Immunol. 1999;162:5041. [PubMed] [Google Scholar]
- 28.Zhou J, Zhang J, Lichtenheld MG, Meadows GG. A role for NF-jB activation in perforin expression of NK cells upon IL-2 receptor signaling. J. Immunol. 2002;169:1319. doi: 10.4049/jimmunol.169.3.1319. [DOI] [PubMed] [Google Scholar]
- 29.Schmidt KN, Leung B, Kwong M, et al. APC-independent activation of NK cells by the Toll-like receptor 3 agonist double-stranded RNA. J. Immunol. 2004;172:138. doi: 10.4049/jimmunol.172.1.138. [DOI] [PubMed] [Google Scholar]
- 30.Becker I, Salaiza N, Aguirre M, et al. Leishmania lipophosphoglycan (LPG) activates NK cells through toll-like receptor-2. Mol. Biochem. Parasitol. 2003;130:65. doi: 10.1016/s0166-6851(03)00160-9. [DOI] [PubMed] [Google Scholar]
- 31.Tato CM, Villarino A, Caamano JH, Boothby M, Hunter CA. Inhibition of NF-jB activity in T and NK cells results in defective effector cell expansion and production of IFN-c required for resistance to Toxoplasma gondii. J. Immunol. 2003;170:3139. doi: 10.4049/jimmunol.170.6.3139. [DOI] [PubMed] [Google Scholar]
- 32.Liou HC, Jin Z, Tumang J, Andjelic S, Smith KA, Liou ML. c-Rel is crucial for lymphocyte proliferation but dispensable for T cell effector function. Int. Immunol. 1999;11:361. doi: 10.1093/intimm/11.3.361. [DOI] [PubMed] [Google Scholar]
- 33.Sha WC, Liou HC, Tuomanen EI, Baltimore D. Targeted disruption of the p50 subunit of NF-jB leads to multifocal defects in immune responses. Cell. 1995;80:321. doi: 10.1016/0092-8674(95)90415-8. [DOI] [PubMed] [Google Scholar]
- 34.Lernbecher T, Muller U, Wirth T. Distinct NF-kappaB/Rel transcription factors are responsible for tissue-specific and inducible gene activation. Nature. 1993;365:767. doi: 10.1038/365767a0. [DOI] [PubMed] [Google Scholar]
- 35.Fields PE, Kim ST, Flavell RA. Cutting edge: changes in histone acetylation at the IL-4 and IFN-c loci accompany Th1/Th2 differentiation. J. Immunol. 2002;169:647. doi: 10.4049/jimmunol.169.2.647. [DOI] [PubMed] [Google Scholar]
- 36.Orange JS, Brodeur SR, Jain A, et al. Deficient natural killer cell cytotoxicity in patients with IKK-c/NEMO mutations. J. Clin. Invest. 2002;109:1501. doi: 10.1172/JCI14858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mora A, Youn J, Keegan A, Boothby M. NF-jB/Rel participation in the lymphokine-dependent proliferation of T lymphoid cells. J. Immunol. 2001;166:2218. doi: 10.4049/jimmunol.166.4.2218. [DOI] [PubMed] [Google Scholar]
- 38.Biron CA, Young HA, Kasaian MT. Interleukin 2-induced proliferation of murine natural killer cells in vivo. J. Exp. Med. 1990;171:173. doi: 10.1084/jem.171.1.173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Stetson DB, Mohrs M, Reinhardt RL, et al. Constitutive cytokine mRNAs mark natural killer (NK) and NK T cells poised for rapid effector function. J. Exp. Med. 2003;198:1069. doi: 10.1084/jem.20030630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tato CM, Martins GA, High FA, DiCioccio CB, Reiner SL, Hunter CA. Cutting edge: innate production of IFN-c by NK cells is independent of epigenetic modification of the IFN-c promoter. J. Immunol. 2004;173:1514. doi: 10.4049/jimmunol.173.3.1514. [DOI] [PubMed] [Google Scholar]
- 41.Bream JH, Hodge DL, Gonsky R, et al. A distal region in the IFN-c gene is a site of epigenetic remodeling and transcriptional regulation by IL-2. J. Biol. Chem. 2004;279:41249. doi: 10.1074/jbc.M401168200. [DOI] [PubMed] [Google Scholar]
- 42.Beinke S, Ley SC. Functions of NF-jB1 and NF-jB2 in immune cell biology. Biochem. J. 2004;382:393. doi: 10.1042/BJ20040544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sica A, Tan TH, Rice N, Kretzschmar M, Ghosh P, Young HA. The c-rel protooncogene product c-Rel but not NF-jB binds to the intronic region of the human interferon-gamma gene at a site related to an interferon-stimulable response element. Proc. Natl Acad. Sci. USA. 1992;89:1740. doi: 10.1073/pnas.89.5.1740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhong H, May MJ, Jimi E, Ghosh S. The phosphorylation status of nuclear NF-jB determines its association with CBP/p300 or HDAC-1. Mol. Cell. 2002;9:625. doi: 10.1016/s1097-2765(02)00477-x. [DOI] [PubMed] [Google Scholar]
- 45.Hunter CA, Subauste CS, VanCleave VH, Remington JS. Production of gamma interferon by natural killer cells from Toxoplasma gondii-infected SCID mice: regulation by interleukin-10, interleukin-12, and tumor necrosis factora. Infect. Immun. 1994;62:2818. doi: 10.1128/iai.62.7.2818-2824.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hunter CA, Subauste CS, Remington JS. The role of cytokines in toxoplasmosis. Biotherapy. 1994;7:237. doi: 10.1007/BF01878489. [DOI] [PubMed] [Google Scholar]
- 47.Jamieson AM, Isnard P, Dorfman JR, Coles MC, Raulet DH. Turnover and proliferation of NK cells in steady state and lymphopenic conditions. J. Immunol. 2004;172:864. doi: 10.4049/jimmunol.172.2.864. [DOI] [PubMed] [Google Scholar]
- 48.Gadjeva M, Tomczak MF, Zhang M, et al. A role for NF-κB subunits p50 and p65 in the inhibition of lipopolysaccharide-induced shock. J. Immunol. 2004;173:5786. doi: 10.4049/jimmunol.173.9.5786. [DOI] [PubMed] [Google Scholar]
- 49.Mason N, Aliberti J, Caamano JC, Liou HC, Hunter CA. Cutting edge: identification of c-Rel-dependent and -independent pathways of IL-12 production during infectious and inflammatory stimuli. J. Immunol. 2002;168:2590. doi: 10.4049/jimmunol.168.6.2590. [DOI] [PubMed] [Google Scholar]
- 50.Sanjabi S, Hoffmann A, Liou HC, Baltimore D, Smale ST. Selective requirement for c-Rel during IL-12 P40 gene induction in macrophages. Proc. Natl Acad. Sci. USA. 2000;97:12705. doi: 10.1073/pnas.230436397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Speirs K, Lieberman L, Caamano J, Hunter CA, Scott P. Cutting edge: NF-jB2 is a negative regulator of dendritic cell function. J. Immunol. 2004;172:752. doi: 10.4049/jimmunol.172.2.752. [DOI] [PubMed] [Google Scholar]
- 52.Kontgen F, Grumont RJ, Strasser A, et al. Mice lacking the c-rel proto-oncogene exhibit defects in lymphocyte proliferation, humoral immunity, and interleukin-2 expression. Genes Dev. 1995;9:1965. doi: 10.1101/gad.9.16.1965. [DOI] [PubMed] [Google Scholar]
- 53.Hilliard BA, Mason N, Xu L, et al. Critical roles of c-Rel in autoimmune inflammation and helper T cell differentiation. J. Clin. Invest. 2002;110:843. doi: 10.1172/JCI15254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Mason NJ, Liou HC, Hunter CA. T cell-intrinsic expression of c-Rel regulates Th1 cell responses essential for resistance to Toxoplasma gondii. J. Immunol. 2004;172:3704. doi: 10.4049/jimmunol.172.6.3704. [DOI] [PubMed] [Google Scholar]
- 55.Weinmann AS, Mitchell DM, Sanjabi S, et al. Nucleosome remodeling at the IL-12 p40 promoter is a TLR-dependent, Rel-independent event. Nat. Immunol. 2001;2:51. doi: 10.1038/83168. [DOI] [PubMed] [Google Scholar]
- 56.Rodriguez-Galan MC, Bream JH, Farr A, Young HA. Synergistic effect of IL-2, IL-12, and IL-18 on thymocyte apoptosis and Th1/Th2 cytokine expression. J. Immunol. 2005;174:2796. doi: 10.4049/jimmunol.174.5.2796. [DOI] [PubMed] [Google Scholar]
- 57.Tanamachi DM, Moniot DC, Cado D, Liu SD, Hsia JK, Raulet DH. Genomic Ly49A transgenes: basis of variegated Ly49A gene expression and identification of a critical regulatory element. J. Immunol. 2004;172:1074. doi: 10.4049/jimmunol.172.2.1074. [DOI] [PubMed] [Google Scholar]
- 58.Lu Q, Wu A, Ray D, et al. DNA methylation and chromatin structure regulate T cell perforin gene expression. J. Immunol. 2003;170:5124. doi: 10.4049/jimmunol.170.10.5124. [DOI] [PubMed] [Google Scholar]
- 59.Wessells J, Baer M, Young HA, et al. BCL-3 and NF-κB p50 attenuate lipopolysaccharide-induced inflammatory responses in macrophages. J. Biol. Chem. 2004;279:49995. doi: 10.1074/jbc.M404246200. [DOI] [PubMed] [Google Scholar]
- 60.Moriggl R, Sexl V, Piekorz R, Topham D, Ihle JN. Stat5 activation is uniquely associated with cytokine signaling in peripheral T cells. Immunity. 1999;11:225. doi: 10.1016/s1074-7613(00)80097-7. [DOI] [PubMed] [Google Scholar]
- 61.Orange JS, Levy O, Geha RS. Human disease resulting from gene mutations that interfere with appropriate nuclear factor-kappaB activation. Immunol. Rev. 2005;203:21. doi: 10.1111/j.0105-2896.2005.00221.x. [DOI] [PubMed] [Google Scholar]
- 62.Karban AS, Okazaki T, Panhuysen CI, et al. Functional annotation of a novel NFKB1 promoter polymorphism that increases risk for ulcerative colitis. Hum. Mol. Genet. 2004;13:35. doi: 10.1093/hmg/ddh008. [DOI] [PubMed] [Google Scholar]
- 63.Erdman S, Fox JG, Dangler CA, Feldman D, Horwitz BH. Typhlocolitis in NF-jB-deficient mice. J. Immunol. 2001;166:1443. doi: 10.4049/jimmunol.166.3.1443. [DOI] [PubMed] [Google Scholar]
- 64.Artis D, Shapira S, Mason N, et al. Differential requirement for NF-jB family members in control of helminth infection and intestinal inflammation. J. Immunol. 2002;169:4481. doi: 10.4049/jimmunol.169.8.4481. [DOI] [PubMed] [Google Scholar]
- 65.Tomczak MF, Erdman SE, Poutahidis T, et al. NF-κB is required within the innate immune system to inhibit microflora-induced colitis and expression of IL-12 p40. J. Immunol. 2003;171:1484. doi: 10.4049/jimmunol.171.3.1484. [DOI] [PubMed] [Google Scholar]