

Abstract
In Arabidopsis recombinant inbred line (RIL) populations are widely used for quantitative trait locus (QTL) analyses. However, mapping analyses with this type of population can be limited because of the masking effects of major QTL and epistatic interactions of multiple QTL. An alternative type of immortal experimental population commonly used in plant species are sets of introgression lines. Here we introduce the development of a genomewide coverage near-isogenic line (NIL) population of Arabidopsis thaliana, by introgressing genomic regions from the Cape Verde Islands (Cvi) accession into the Landsberg erecta (Ler) genetic background. We have empirically compared the QTL mapping power of this new population with an already existing RIL population derived from the same parents. For that, we analyzed and mapped QTL affecting six developmental traits with different heritability. Overall, in the NIL population smaller-effect QTL than in the RIL population could be detected although the localization resolution was lower. Furthermore, we estimated the effect of population size and of the number of replicates on the detection power of QTL affecting the developmental traits. In general, population size is more important than the number of replicates to increase the mapping power of RILs, whereas for NILs several replicates are absolutely required. These analyses are expected to facilitate experimental design for QTL mapping using these two common types of segregating populations.
QUANTITATIVE traits are characterized by continuous variation. The establishment of the genetic basis of quantitative traits is commonly referred to as quantitative trait locus (QTL) mapping and has been hampered due to their multigenic inheritance and the often strong interaction with the environment. The principle of QTL mapping in segregating populations is based on the genotyping of progeny derived from a cross of distinct genotypes for the trait under study. Phenotypic values for the quantitative trait are then compared with the molecular marker genotypes of the progeny to search for particular genomic regions showing statistically significant associations with the trait variation, which are then called QTL (Broman 2001; Slate 2005). Over the past few decades, the field has benefited enormously from the progress made in molecular marker technology. The ease by which such markers can be developed has enabled the generation of dense genetic maps and the performance of QTL mapping studies of the most complex traits (Borevitz and Nordborg 2003).
QTL analyses make use of the natural variation present within species (Alonso-Blanco and Koornneef 2000; Maloof 2003) and have been successfully applied to various types of segregating populations. In plants, the use of “immortal” mapping populations consisting of homozygous individuals is preferred because it allows performance of replications and multiple analyses of the same population. Homozygous populations can be obtained by repeated selfing, like for recombinant inbred lines (RILs), but also by induced chromosomal doubling of haploids, such as for doubled haploids (DHs) (Han et al. 1997; Rae et al. 1999; Von Korff et al. 2004). Depending on the species one can in principle also obtain immortality by vegetative propagation, although this is often more laborious. RILs are advantageous over DHs because of their higher recombination frequency in the population, resulting from multiple meiotic events that occurred during repeated selfing (Jansen 2003).
Another type of immortal population consists of introgression lines (ILs) (Eshed and Zamir 1995), which are obtained through repeated backcrossing and extensive genotyping. These are also referred to as near-isogenic lines (NILs) (Monforte and Tanksley 2000) or backcross inbred lines (BILs) (Jeuken and Lindhout 2004; Blanco et al. 2006). Such populations consist of lines containing a single fragment or a small number of genomic introgression fragments from a donor parent into an otherwise homogeneous genetic background. Although no essential differences exist between these populations, we use the term near-isogenic lines for the materials described here. A special case of ILs are chromosomal substitution strains (CSSs) (Nadeau et al. 2000; Koumproglou et al. 2002), where the introgressions span complete chromosomes.
All immortal populations except those that can be propagated only vegetatively share the advantage that they can easily be maintained through seeds, which allows the analysis of different environmental influences and the study of multiple, even invasive or destructive, traits. Statistical power of such analyses is increased because replicate measurements of genetically identical individuals can be done.
In plants, RILs and NILs are the most common types of experimental populations used for the analysis of quantitative traits. In both cases the accuracy of QTL localization, referred to as mapping resolution, depends on population size. For RILs, recombination frequency within existing lines is fixed and can therefore be increased within the population only by adding more lines (i.e., more independent recombination events). Alternatively, recombination frequency can be increased by intercrossing lines before fixation by inbreeding as homozygous lines (Zou et al. 2005). In NIL populations resolution can be improved by minimizing the introgression size of each NIL. Consequently, to maintain genomewide coverage a larger number of lines are needed. Despite the similarities between these two types of mapping populations, large differences exist in the genetic makeup of the respective individuals and the resulting mapping approach. In general, recombination frequency in RIL populations is higher than that in equally sized NIL populations, which allows the analysis of less individuals. Each RIL contains several introgression fragments and, on average, each genomic region is represented by an equal number of both parental genotypes in the population. Therefore, replication of individual lines is often not necessary because the effect of each genomic region on phenotypic traits is tested by comparing the two genotypic RIL classes (each comprising approximately half the number of lines in the population). In addition, the multiple introgressions per RIL allow detection of genetic interactions between loci (epistasis). However, epistasis together with unequal recombination frequencies throughout the genome and segregation distortions caused by lethality or reduced fitness of particular genotypes may bias the power to detect QTL. Furthermore, the wide variation of morphological and developmental traits present in most RIL populations may hamper the analysis of traits requiring the same growth and developmental stage of the individual lines. When many traits segregate simultaneously, this often affects the expression of other traits due to genetic interactions. Moreover, large-effect QTL may mask the detection of QTL with a small additive effect.
In contrast to RILs, NILs contain only a single introgression per line, which increases the power to detect small-effect QTL. However, the presence of a single introgression segment does not allow testing for genetic interactions and thereby the detection of QTL expressed in specific genetic backgrounds (epistasis). In addition, because most of the genetic background is identical for all lines, NILs show more limited developmental and growth variation, increasing the homogeneity of growth stage within experiments. Nevertheless, lethality and sterility might sometimes hinder the obtaining of specific single introgression lines.
The choice of one mapping population over another depends on the plant species and the specific parents of interest. In cases where different cultivars or wild accessions are studied preference is often given to RILs. However, when different species or when wild and cultivated germ plasm are combined (Eshed and Zamir 1995; Jeuken and Lindhout 2004; Von Korff et al. 2004; Blair et al. 2006; Yoon et al. 2006) NILs are preferred. For instance, in tomato the high sterility in the offspring of crosses between cultivated and wild species made the use of NIL populations (Eshed and Zamir 1995) preferable because genomewide coverage cannot be obtained with RIL populations due to sterility, etc. Furthermore, the analysis of agronomical important traits (such as fruit characters) cannot be performed when many genes conferring reduced fertility segregate. In Arabidopsis, the easiness to generate fertile RIL populations with complete genome coverage, due to its fast generation time, has led to their extensive use in mapping quantitative traits.
NILs have been developed in various studies using Arabidopsis to confirm and fine map QTL previously identified in RILs (Alonso-Blanco et al. 1998b, 2003; Swarup et al. 1999; Bentsink et al. 2003; Edwards et al. 2005; Juenger et al. 2005a; Teng et al. 2005) for which also heterogeneous inbred families (HIFs) (Tuinstra et al. 1997) have been used (Loudet et al. 2005; Reymond et al. 2006). A set of chromosomal substitutions of the Landsberg erecta (Ler) accession into Columbia (Col) has been developed to serve as starting material for making smaller introgressions (Koumproglou et al. 2002). In mice CSSs are widely used for mapping purposes and have proven to be a valuable complement to other population types (Stylianou et al. 2006). However, no genomewide set of NILs that allows mapping to subparts of the chromosome has been described in Arabidopsis and, to our knowledge, no empirical comparative study has been performed between the two population types within a single species.
In this study we aim to compare a RIL population with a NIL population in terms of QTL detection power and localization resolution. For that, we generated a new genomewide population of NILs using the same Ler and Cape Verde Islands (Cvi) parental accessions as used earlier to generate a RIL population (Alonso-Blanco et al. 1998a). The two experimental populations were grown simultaneously in the same experimental setup, including multiple replicates. QTL mapping analyses were performed on six different traits and the results of these analyses were compared in both populations.
MATERIALS AND METHODS
Mapping populations:
Two types of mapping populations were used to analyze six developmental traits. The first population consists of a set of 161 RILs derived from a cross between the accessions Cvi and Ler. The F10 generation has been extensively genotyped (Alonso-Blanco et al. 1998a) and is available from the Arabidopsis Biological Resource Center. All lines were advanced to the F13 generation and residual heterozygous regions, estimated at 0.71% in the F10 generation, were genotyped again with molecular PCR markers to confirm that they were practically 100% homozygous.
The second population consists of a set of 92 NILs. NILs were generated by selecting appropriate Ler/Cvi RILs and repeated backcrossing with Ler as recurrent female parent. A number of these lines have been described previously (Alonso-Blanco et al. 1998b, 2003; Swarup et al. 1999; Bentsink et al. 2003; Edwards et al. 2005; Juenger et al. 2005a; Teng et al. 2005). The progeny of backcrosses was genotyped with PCR markers and lines containing a homozygous Cvi introgression into an otherwise Ler background were selected. The set of selected lines was then extensively genotyped by AFLP analysis using the same restriction enzymes and primer combinations as those used for the genotyping of the RILs (Alonso-Blanco et al. 1998a). The NILs will be made available through the Arabidopsis stock centers.
In both populations each line is almost completely homozygous and therefore individuals of the same line are genetically identical, which allows the pooling of replicated individuals and repeated measurements to obtain a more precise estimate of phenotypic values. For the RIL and NIL population 16 and 24 genetically identical plants were grown per line, respectively. Additionally, 96 replicates were grown for each parental accession Ler and Cvi. All plants were grown in a single experiment with four completely randomized blocks containing 4, 6, and 24 replicates per RIL, NIL, and parent, respectively.
Plant growing conditions:
Seeds were sown in petri dishes on water-soaked filter paper and incubated for 5 days in a cold room at 4° in the dark to promote uniform germination. Subsequently, petri dishes were transferred to a climate chamber (24°, 16 hr light per day) for 2 days before planting. Germinated seedlings were transferred to clay pots, placed in peat, containing a sandy soil mixture. A single plant per pot was grown under long-day light conditions in an air-conditioned greenhouse from July until October. Plants were fertilized every 2 weeks using a liquid fertilizer.
Quantitative traits:
A total of six developmental traits, which were known to vary within the populations for the number of QTL and heritability, were measured on all individuals. We quantified flowering time (FT); main inflorescence length at first silique (SL); total length of the main inflorescence (TL); basal branch number (BB), which is the number of side shoots growing out from the rosette; main inflorescence branch number (IB), which is the number of elongated axillary (secondary) inflorescences along the main inflorescence; and total number of side shoots (TB) (basal plus main inflorescence). Flowering time was recorded as the number of days from the date of planting until the opening of the first flower. All other traits were measured at maturity.
Quantitative genetic analyses:
For both populations and for each trait, total phenotypic variance was partitioned into sources attributable to genotype (VG; i.e., the line effect) and error (VE), using a random-effects analysis of variance (ANOVA, SPSS version 11.0) according to the model 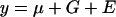 . Variance components were used to estimate broad sense heritability according to the formula
. Variance components were used to estimate broad sense heritability according to the formula 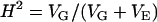 , where VG is the among-genotype variance component and VE is the residual (error) variance component. Genetic correlations (rG) were estimated as
, where VG is the among-genotype variance component and VE is the residual (error) variance component. Genetic correlations (rG) were estimated as 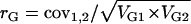 , where cov1,2 is the covariance of trait means and VG1 and VG2 are the among-genotype variance components for those traits. The coefficient of genetic variation (CVG) was estimated for each trait as
, where cov1,2 is the covariance of trait means and VG1 and VG2 are the among-genotype variance components for those traits. The coefficient of genetic variation (CVG) was estimated for each trait as 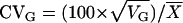 , where VG is the among-genotype variance component and
, where VG is the among-genotype variance component and 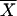 is the trait mean of the genotypes.
is the trait mean of the genotypes.
QTL analyses in the RIL population:
To map QTL using the RIL population, a set of 144 markers equally spaced over the Arabidopsis genetic map was selected from the RIL Ler/Cvi map (Alonso-Blanco et al. 1998a). These markers spanned 485 cM, with an average distance between consecutive markers of 3.5 cM and the largest genetic distance being 11 cM. The phenotypic values recorded, except basal branch number, were transformed (log10(x + 1)) to improve the normality of the distributions and the values of 16 plants per RIL were used to calculate the means of each line for all traits. These means were used to perform the QTL analyses unless otherwise stated. The computer program MapQTL version 5.0 (Van Ooijen 2004) was used to identify and locate QTL linked to the molecular markers, using both interval mapping and multiple QTL mapping (MQM). In a first step, putative QTL were identified using interval mapping. Thereafter, a marker closely linked to each putative QTL was selected as a cofactor and the selected markers were used as genetic background controls in the approximate MQM of MapQTL. LOD statistics were calculated at 0.5-cM intervals. Tests of 1000 permutations were used to obtain an estimate of the number of type 1 errors (false positives). The genomewide LOD score, which 95% of the permutations did not exceed, ranged from 2.6 to 2.8 and chromosomewide LOD thresholds varied between 1.8 and 2.1 depending on trait and linkage group. The genomewide LOD score was then used as the significance threshold to declare the presence of a QTL in MQM mapping, while the chromosomewide thresholds were used to detect putative small-effect QTL. In the final MQM model the genetic effect (μB − μA) and percentage of explained variance were estimated for each QTL and 2-LOD support intervals were established as an ∼95% confidence level (Van Ooijen 1992), using restricted MQM mapping.
Epistatic interactions between QTL were estimated using factorial analysis of variance. For each trait, the mean phenotypic values were used as a dependent variable and cofactors, corresponding to the detected QTL, were used as fixed factors. The general linear model module of the statistical package SPSS version 11.0 was used to perform a full factorial analysis of variance or analysis of main effects only. Differences in R2-values, calculated from the type III sum of squares, were assigned to epistatic interaction effects of detected QTL. Additionally we performed a complete pairwise search (P < 0.001, determined by Monte Carlo simulations) for conditional and coadaptive epistatic interactions for each trait, using the computer program EPISTAT (Chase et al. 1997).
The effect of replication on statistical power was analyzed by performing MQM mapping on means of trait values from 1, 2, 4, 8, 12, and 16 replicate plants, respectively. Analyses were performed on 10 independent, stochastically sampled, data sets for each replication size and trait using automated cofactor selection (P < 0.02). Total explained variance, LOD score of the largest-effect QTL, and number of significant QTL were recorded for each analysis.
The effect of population size on statistical power was analyzed by performing MQM mapping on increasing population sizes. Analyses were performed on 10 independent, stochastically sampled, data sets for each population size. Subpopulations of increasing size, with a step size of 20 lines, were analyzed for each trait using automated cofactor selection (P < 0.02). Total explained variance, LOD score of the largest-effect QTL, and number of significant QTL were recorded for each analysis.
Statistical analyses of NILs:
Differences in mean trait values of Ler and NILs were analyzed by univariate analysis of variance, using the general linear model module of the statistical package SPSS version 11.0. Dunnett's pairwise multiple comparison t-test was used as a post hoc test to determine significant differences. For each analysis, trait values were used as a dependent variable and NILs were used as a fixed factor. Tests were performed two sided with a Bonferroni-corrected significance threshold level of 0.05 and Ler as a control category. To increase statistical power, similar analyses were conducted for bins (see results). For this, trait values of all introgression lines assigned to a certain bin were pooled and compared to values of the Ler parental line. Because each NIL can be a member of more than one bin the significance threshold was lowered to 0.001 to correct for multiple testing. The genetic effect of Cvi bins significantly differing from Ler was calculated as μB − μA, where μA and μB are the mean trait values of Ler and the Cvi bin, respectively. Explained variance was estimated from the partial η2 of the univariate analysis of variance, where η2 is the proportion of total variance attributable to factors in the analysis. The total percentage of explained variance was then estimated by using trait values as a dependent variable and NILs as a fixed factor, where all NILs were included as subjects. The percentage of explained variance of individual QTL was estimated as a fraction of the total variation in the population (including all lines), using a single bin as a fixed factor and as a fraction of the total variation in a comparison of a single bin with Ler only.
To determine the effect of replicated measurements we calculated the power of detecting significant differences between Ler and NILs using various replicate numbers. For each trait we calculated the minimal relative difference in mean trait values that could still be significantly detected. Calculations were performed using a normal distribution two-sample equal variance power calculator from the UCLA department of statistics (http://calculators.stat.ucla.edu/). We first calculated for each trait the mean phenotypic value of 96 Ler replicate plants (μA) and for each line the standard deviation of 24 replicate plants. The mean line standard deviation of each trait was taken as a measure of variation (σ) in all subsequent calculations. The significance level, the probability of falsely rejecting the null hypothesis (H0: μA = μB) when it is true, was set to 0.05 and power, the probability of correctly rejecting the null hypothesis when the alternative (H1: μA ≠ μB) is true, was set to 0.95. The sample size of Ler (NA) was always identical to the sample size of NILs (NB) and ranged from 2 to 24 individuals. For each trait and sample size the mean trait value (μB) for NILs was then calculated as the minimum value to meet the alternative hypothesis (H1: μA ≠ μB) in a two-sided test. These minimum values were then converted in a fold-difference value compared to the Ler value, calculated as (|μB − μA| + μA)/μA, to obtain a relative estimate independent of trait measurement units.
The effect of replication on statistical power was also analyzed by performing bin mapping using 2, 4, 8, 12, and 16 replicate plants, respectively. Analyses were performed on 10 independent, stochastically sampled, data sets for each replication size and trait and the number of significant QTL was recorded for each analysis.
RESULTS
Construction of a genomewide near-isogenic line population:
We constructed a population of 92 introgression lines carrying between one and four Cvi introgression fragments in a Ler genetic background. Lines were genotyped using 349 AFLP and 95 PCR markers to determine the number, position, and size of the introgressions (see materials and methods). This set of lines was selected to provide together an almost complete genomewide coverage (Figure 1). Forty lines contained a single introgression while 52 lines carried several Cvi fragments. From those, 32, 19, and 1 line bore two, three, and four introgressions, respectively. The genetic length of the introgression fragments was estimated using the map positions of the introgressed markers in the genetic map constructed from the existing RIL population derived from the same Ler and Cvi parental accessions (Alonso-Blanco et al. 1998a). The average genetic sizes of the main, second, third, and fourth introgression fragments were 31.7, 11.1, 6.7, and 5.2 cM, respectively. Thus, lines with multiple Cvi fragments carried a main large introgression and several much smaller Cvi fragments. Additionally, we selected a core set of 25 lines that together covered >90% of the genome (supplemental Table 1 at http://www.genetics.org/supplemental/).
Figure 1.—

Graphical genotype of the Ler/Cvi NIL population. Bars represent introgressions. Solid bars represent the genetic position of Cvi introgressions in individual NILs. Shaded bars represent crossover regions between markers used for the genotyping of the lines. Numbers at the top indicate the five linkage groups.
Genetic analyses of developmental traits:
Six traits were measured and analyzed in the RIL and NIL populations (Table 1). Although plants were grown in four replicated blocks, block effects were negligible and were therefore not used as a factor in subsequent analyses. In both populations, among-genotype variance was highly significant (P < 0.0001) for all traits. In the RIL population, broad sense heritability estimates ranged from 0.34 (basal branch number) to 0.92 (total plant length) (Table 1). Statistical parameters of most traits were similar to those described by Alonso-Blanco et al. (1998b, 1999) and Juenger et al. (2005b). However, Ungerer et al. (2002) reported much lower average values for plant height and branch number although time to flower was similar. Moreover, among-genotype variance estimates were lower and within-genotype variance estimates higher, resulting in lower heritability values compared to our analyses.
TABLE 1.
Descriptive statistics for six developmental traits analyzed in two mapping populations and their parents
| Trait | 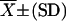 |
[VG]a | [VE]b | [H2]c | [CVG]d |
|---|---|---|---|---|---|
| Parents | |||||
| FT (days) | 24.30 (1.03)e | 8.74 | 3.57 | 0.71 | 10.85 |
| 30.21 (2.47)f | |||||
| SL (cm) | 9.58 (0.98)e | 3.27 | 3.14 | 0.51 | 15.87 |
| 13.21 (2.30)f | |||||
| TL (cm) | 23.59 (1.92)e | 26.81 | 10.53 | 0.72 | 17.99 |
| 33.95 (4.17)f | |||||
| IB | 2.21 (0.46)e | 0.02 | 0.33 | 0.05 | 5.53 |
| 2.49 (0.67)f | |||||
| BB | 1.54 (0.68)e | 0.00 | 0.65 | 0.00 | 0.00 |
| 1.48 (0.91)f | |||||
| TB | 3.75 (0.77)e | 0.01 | 0.82 | 0.01 | 1.88 |
| 3.97 (1.02)f | |||||
| RIL population | |||||
| FT (days) | 26.06 (6.03) | 32.59 | 3.82 | 0.90 | 21.91 |
| SL (cm) | 9.89 (3.39) | 9.70 | 1.80 | 0.83 | 31.49 |
| TL (cm) | 26.13 (9.22) | 78.53 | 6.52 | 0.92 | 33.91 |
| IB | 2.34 (1.22) | 0.99 | 0.50 | 0.67 | 42.66 |
| BB | 1.43 (0.93) | 0.30 | 0.57 | 0.34 | 37.98 |
| TB | 3.77 (1.27) | 0.78 | 0.84 | 0.48 | 23.36 |
| NIL population | |||||
| FT (days) | 23.68 (3.60) | 10.78 | 2.21 | 0.83 | 13.87 |
| SL (cm) | 9.81 (2.18) | 3.17 | 1.58 | 0.65 | 18.15 |
| TL (cm) | 24.50 (5.95) | 31.24 | 4.10 | 0.87 | 22.82 |
| IB | 2.26 (0.88) | 0.51 | 0.27 | 0.65 | 31.42 |
| BB | 1.56 (0.84) | 0.18 | 0.53 | 0.24 | 26.92 |
| TB | 3.82 (1.06) | 0.48 | 0.64 | 0.42 | 18.25 |
FT, flowering time; SL, length until first silique; TL, total plant length; IB, main inflorescence branch number; BB, basal branch number; TB, total branch number.
Among-genotype variance component from ANOVA: tests whether genetic differences exist among genotypes for specified traits (P < 0.0001).
Residual variance component from ANOVA.
Measure of total phenotypic variance attributable to genetic differences among genotypes (broad sense heritability) calculated as VG/(VG + VE).
Coefficient of genetic variation calculated as 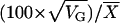 .
.
Landsberg erecta parent.
Cape Verde Islands parent.
For the NIL population, mean trait values were closer to those measured for Ler due to the genetic structure of the population, consisting of lines carrying only small Cvi introgressions in a Ler background. Furthermore, variance components from ANOVA were lower in the NIL population but heritability estimates differed only slightly compared to the RIL population (Table 1).
Strong and similar genetic correlations were observed between traits in the two Ler/Cvi populations, indicating partial genetic coregulation (Table 2). Flowering time shows the highest correlation with the number of main inflorescence branches but is negatively correlated with basal branch number. Flowering time is also, but to a lesser degree, correlated with plant height. Correlations were also found between plant height and branching with again positive values with the number of main inflorescence branches and negative correlations with basal branch number. These results contrasted with those from Ungerer et al. (2002), who found negative correlations between flowering time, plant height, and branching in all pairwise comparisons, which is probably due to the different environmental setups in the two laboratories.
TABLE 2.
Genetic correlations among developmental traits analyzed in two mapping populations
| Trait | FT | SL | TL | IB | BB | TB |
|---|---|---|---|---|---|---|
| FT | 0.63* | 0.38* | 0.97* | −0.49* | 0.80* | |
| SL | 0.39* | 0.90* | 0.52* | −0.39* | 0.35* | |
| TL | 0.21* | 0.88* | 0.18* | −0.32* | 0.00 | |
| IB | 0.91* | 0.31* | 0.09* | −0.54* | 0.95* | |
| BB | −0.26* | −0.28* | −0.26* | −0.35* | 0.12* | |
| TB | 0.77* | 0.15* | −0.07 | 0.85* | 0.31* |
The top right and the bottom left halves of the table represent values calculated for the RIL and the NIL populations, respectively. FT, flowering time; SL, length until first silique; TL, total plant length; IB, main inflorescence branch number; BB, basal branch number; TB, total branch number. *Significant at P < 0.001.
Mapping quantitative traits in the Ler/Cvi RIL population:
Each trait was subjected to QTL analysis and three to eight QTL were detected for each trait (Figure 2, Table 3). Major QTL for flowering time, plant height, and branching were in concordance with previously reported studies (Alonso-Blanco et al. 1998b, 1999; Ungerer et al. 2002, 2003; Juenger et al. 2005b), although slight differences for minor QTL were also found. Total explained variance for each trait ranged from 38.5% for basal branch number to 86.3% for total plant height. LOD scores for the largest-effect QTL ranged from 5.7 for basal branch number up to 60.7 for total plant height with corresponding explained variances of 11.0 and 64.0%, respectively. The average genetic length of 2-LOD support intervals was 11.6 cM, ranging from 2.3 (length at first silique) to 33.3 cM (total branch number). Opposing-effect QTL were found for all traits, explaining the observed transgressive segregation within the population (data not shown). Genetic interaction among the detected QTL was also tested. The proportion of variance explained by epistatic interactions ranged from 3.1 (basal branch number) to 20.5% (number of main inflorescence branches) and involved two to five of the detected QTL (Table 3). Using a complete pairwise search of all markers (Chase et al. 1997), a number of additional interactions were detected between loci not colocating with major QTL positions (supplemental Figure 1 at http://www.genetics.org/supplemental/).
Figure 2.—

Genomewide QTL profiles of traits analyzed in the RIL population: (A) flowering time, (B) length at first silique, (C) total plant length, (D) number of main inflorescence branches, (E) basal branch number, and (F) total branch number. Solid lines represent the QTL effect calculated as described in materials and methods. Shaded lines represent LOD scores. Shaded dashed lines represent genomewide significance threshold levels for LOD scores determined by permutation testing.
TABLE 3.
QTL detected in the RIL population
| Trait | Chra | LOD score | Support intervalb (cM) | Explained variancec (%) | Effectd | Total explained variancee (%) | Interactionf (%) |
|---|---|---|---|---|---|---|---|
| FT | 1 | 11.9 | 1.5–9.8g | 13.0 | −3.9 | 68.4 | 9.6 |
| 5 | 18.9 | 388.4–394.5g | 22.2 | 5.7 | |||
| 5 | 11.9 | 408.2–413.7g | 13.0 | 4.4 | |||
| SL | 1 | 9.3 | 0.0–9.3 | 6.3 | −1.7 | 79.5 | 15.0 |
| 1 | 4.8 | 103.1–126.0 | 3.1 | −1.3 | |||
| 2 | 39.7 | 173.2–175.5 | 43.2 | 4.5 | |||
| 3 | 2.9 | 234.2–253.6 | 1.9 | 1.0 | |||
| 3 | 5.0 | 281.5–287.8 | 3.2 | −1.2 | |||
| 5 | 15.7 | 387.9–392.4g | 11.8 | 2.9 | |||
| 5 | 10.2 | 403.6–409.7g | 7.2 | 2.0 | |||
| TL | 1 | 6.5 | 0.0–9.8g | 2.8 | −3.1 | 86.3 | 11.5 |
| 1 | 5.0 | 73.9–84.6 | 2.1 | −2.7 | |||
| 1 | 3.3 | 116.3–126.0 | 1.2 | −2.3 | |||
| 2 | 60.7 | 173.2–176.0g | 64.0 | 14.8 | |||
| 3 | 6.0 | 207.3–225.7g | 2.6 | −3.0 | |||
| 4 | 5.2 | 287.8–307.5g | 2.2 | −2.7 | |||
| 5 | 7.8 | 383.1–392.5g | 3.6 | 4.1 | |||
| 5 | 5.1 | 403.6–411.7 | 2.2 | 3.0 | |||
| IB | 1 | 5.0 | 0.0–13.5g | 5.3 | −0.4 | 65.0 | 20.5 |
| 2 | 2.7 | 154.9–171.0g | 2.8 | −0.3 | |||
| 5 | 15.3 | 387.0–391.9g | 19.7 | 0.9 | |||
| 5 | 10.4 | 398.8–411.7g | 12.3 | 0.7 | |||
| 5 | 3.1 | 472.2–485.3 | 3.2 | −0.3 | |||
| BB | 1 | 5.7 | 72.4–91.0g | 11.0 | 0.4 | 38.5 | 3.1 |
| 2 | 3.2 | 167.0–200.2g | 6.2 | −0.3 | |||
| 4 | 4.6 | 360.7–373.5g | 9.1 | 0.4 | |||
| 5 | 5.5 | 385.6–406.1g | 11.3 | −0.5 | |||
| TB | 1 | 15.5 | 5.3–12.4g | 16.1 | −0.8 | 71.1 | 16.2 |
| 1 | 4.9 | 81.7–93.8g | 4.6 | 0.4 | |||
| 2 | 9.5 | 169.0–180.0g | 9.1 | −0.6 | |||
| 5 | 9.7 | 386.5–392.4g | 9.4 | 0.6 | |||
| 5 | 10.9 | 403.3–412.2g | 10.8 | 0.7 | |||
| 5 | 5.2 | 472.2–485.3 | 4.7 | −0.4 |
FT, flowering time; SL, length until first silique; TL, total plant length; IB, main inflorescence branch number; BB, basal branch number; TB, total branch number.
Chromosome number.
2-LOD support interval.
Percentage of total variation explained by individual QTL.
Effect of QTL calculated as μB − μA, where A and B are RILs carrying Ler and Cvi genotypes at the QTL position, respectively. μB and μA were estimated by MapQTL. Effects are given in days (flowering time), centimeters (length at first silique and total length), or numbers (elongated axils, basal branch number, and total branch number).
Percentage of total variance explained by genetic factors estimated by MapQTL.
Percentage of total variation explained by interaction between individual QTL.
QTL showing significant epistatic interactions (P < 0.05) and used to estimate the percentage of explained variance by genetic interactions.
The smallest significant absolute effect detected was 4.4 days for flowering time, 1.0 and 2.3 cm for length at first silique and total plant length, respectively, and 0.3, 0.3, and 0.4 for the number of main inflorescence branches, basal branch number, and total branch number, respectively. Relative effects, expressed as the fold difference between genotypes, calculated as (|μB − μA| + μA)/μA, then equaled 1.15-, 1.09-, 1.09-, 1.13-, 1.59-, and 1.10-fold, respectively (Tables 3 and 5). As expected, the total explained variance of a trait correlated positively with the smallest significantly detectable effect for that particular trait. In general, smaller effects could be detected with increasing total explained variance. When the chromosomewide threshold for significance was used instead of the genomewide threshold, one additional suggestive QTL was detected for main inflorescence branch number and total branch number and two for length at first silique.
TABLE 5.
Comparative summary of QTL mapping parameters in the Ler/Cvi RIL and NIL populations
| Trait | Populationa | QTLb (no.) | Supportc (cM) | Explained varianced (%) | Total explained variance (%) | Effecte | Relative effectf |
|---|---|---|---|---|---|---|---|
| FT | RIL | 3 | 6.6 | 16.1 | 68.4 | 4.7 | 1.15 |
| NIL | 5 | 35.5 (3.6) | 34.0 | 83.2 | 4.4 | 1.03 | |
| SL | RIL | 7 | 10.1 | 11.0 | 79.5 | 2.1 | 1.09 |
| NIL | 9 | 23.3 (5.2) | 38.7 | 66.1 | 2.1 | 1.11 | |
| TL | RIL | 8 | 11.1 | 10.1 | 86.3 | 4.5 | 1.09 |
| NIL | 6 | 31.4 (3.4) | 50.8 | 87.7 | 6.4 | 1.09 | |
| IB | RIL | 5 | 12.1 | 8.7 | 65.0 | 0.5 | 1.13 |
| NIL | 1 | 45.9 (2.7) | 46.3 | 66.1 | 3.8 | 2.71 | |
| BB | RIL | 4 | 21.3 | 9.4 | 38.5 | 0.4 | 1.59 |
| NIL | 5 | 33.1 (5.6) | 16.1 | 26.7 | 0.7 | 1.30 | |
| TB | RIL | 6 | 9.7 | 9.1 | 71.1 | 0.6 | 1.10 |
| NIL | 4 | 40.5 (5.4) | 22.2 | 44.1 | 1.3 | 1.11 |
FT, flowering time; SL, length until first silique; TL, total plant length; IB, main inflorescence branch number; BB, basal branch number; TB, total branch number.
Population type.
Number of QTL detected.
Average length of support interval. In parentheses: average length of largest-effect bin.
Average explained variance for each QTL.
Average absolute effect for each QTL. Effects are given in days (flowering time), centimeters (length at first silique and total length), or numbers (elongated axils, basal branch number, and total branch number).
Smallest relative effect significantly detected, expressed as fold difference compared to Ler, calculated as (|μB − μA| + μA)/μA.
Mapping quantitative traits in the Ler/Cvi NIL population:
To search for QTL in the NIL population, we divided the Arabidopsis genetic map in adjacent genomic fragments that were individually tested. The complete genome was subdivided into 97 regions, defined by the position of the recombination events of the main introgressions of the 92 NILs (supplemental Table 2 at http://www.genetics.org/supplemental/). These regions are referred to as bins and each NIL was then assigned to those adjacent bins spanned by its Cvi introgression fragment. Thus, each bin contains a unique subset of lines with overlapping Cvi introgressions in that particular region, which were used to test the phenotypic effects of that bin. The average genetic length of the bins was 5.0 cM, ranging from 0.1 to 26.3 cM. The number of NILs per bin ranged from 0 to 13 with an average of 5.1 NILs. Because NILs were assigned only to bins when the complete bin was covered by the introgression, 3 bins remained empty [viz. bins 66 (26.3 cM), 73 (3.3 cM), and 77 (5.4 cM)]. On average each NIL was assigned to 5.4 adjacent bins. One NIL (LCN4-2) was not assigned to any bin because its introgression included only a single marker. Two NILs corresponded to complete chromosomal substitutions: line LCN3-8 (chromosome 3) and line LCN1-8 (chromosome 1), the latter carrying the largest introgression assigned to 27 adjacent bins.
To map QTL in the NIL population, all bins were tested individually by comparing the phenotypes of the NILs assigned to each bin with that of Ler. As shown in Figure 3 and Table 4, one to nine QTL were detected for each trait. The total explained variance for each trait ranged from 26.7% for basal branch number up to 87.7% for total plant height. Explained variances for the largest-effect QTL for each trait ranged from 19.3% for basal branch number to 91.9% for total plant height as calculated from a restricted ANOVA using only lines from the most significant bin and Ler. To show the relative effect of Mendelizing QTL with respect to the total population variance we calculated the explained variances also when all lines of the population were subjected to ANOVA analysis using the most significant bin as a fixed factor (Table 4). Relative effects of QTL were much lower in this unrestricted analysis because all other QTL in the population increase residual variation that is not corrected for, as is done in MQM mapping in the RIL population. Moreover, lines partly overlapping the QTL bin are not assigned to that bin but can still contain the QTL Cvi allele, further increasing the residual variation in the population.
Figure 3.—

QTL profiles of traits analyzed in the NIL population: (A) flowering time, (B) length at first silique, (C) total plant length, (D) number of main inflorescence branches, (E) basal branch number, and (F) total branch number. Solid lines represent the QTL effect calculated as described in materials and methods. Shaded lines represent significance scores. Shaded dashed lines represent significance threshold levels applied in this study.
TABLE 4.
QTL detected in the NIL population
| Support intervalb (cM) | Support bin (cM)c | Explained variance (%)
|
Total explained varianceg (%) | ||||
|---|---|---|---|---|---|---|---|
| Trait | Chra | Restrictedd | Unrestrictede | Effectf | |||
| FT | 1 | 0.0–21.6 | 3.9–7.8 | 70.3 | 3.2 | −3.2 | 83.2 |
| 1 | 31.4–40.6 | 33.4–40.7 | 18.0 | 0.5 | −1.0 | ||
| 1 | 73.3–122.0 | 83.6–87 | 7.1 | 0.7 | −0.7 | ||
| 2 | 174.4–204.7 | 200.9–201.8 | 22.3 | 0.6 | 1.5 | ||
| 5 | 388.4–434.2 | 392.3–395 | 52.1 | 42.8 | 15.7 | ||
| SL | 1 | 10.8–27.4 | 17.3–21.7 | 64.0 | 4.8 | −3.1 | 66.1 |
| 1 | 31.4–40.6 | 33.4–40.7 | 17.1 | 0.6 | −1.1 | ||
| 1 | 73.3–125.9 | 122.1–126 | 34.9 | 2.8 | −1.7 | ||
| 2 | 160.8–207.2 | 162–174.5 | 73.4 | 5.3 | 4.9 | ||
| 3 | 270.1–288.4 | 287.1–288.4 | 37.1 | 1.6 | −1.7 | ||
| 4 | 359.5–375.7 | 368.2–375.7 | 32.2 | 1.7 | −1.6 | ||
| 5 | 388.3–418.9 | 392.3–395 | 32.2 | 0.7 | 2.7 | ||
| 5 | 434.2–436.0 | 434.3–436.1 | 29.6 | 3.8 | −1.4 | ||
| 5 | 441.4–459.3 | 454.3–459.4 | 28.2 | 1.1 | −1.1 | ||
| TL | 1 | 0.0–33.3 | 17.3–21.7 | 66.2 | 1.7 | −6.3 | 87.7 |
| 1 | 64.7–125.9 | 122.1–126 | 48.8 | 3.8 | −3.8 | ||
| 2 | 160.8–207.2 | 174.5–178.8 | 91.9 | 10.5 | 18.5 | ||
| 3 | 287.0–288.4 | 287.1–288.4 | 19.0 | 0.4 | −2.1 | ||
| 5 | 389.9–416.1 | 411.7–416.2 | 34.1 | 1.7 | 3.7 | ||
| 5 | 434.2–454.3 | 434.3–436.1 | 45.0 | 1.4 | −3.9 | ||
| IB | 5 | 388.3–434.2 | 392.3–395 | 46.3 | 37.7 | 3.8 | 66.1 |
| BB | 1 | 0.0–15.1 | 3.9–7.8 | 17.7 | 1.8 | −0.6 | 26.7 |
| 1 | 40.6–125.9 | 94.5–101.6 | 17.9 | 9.0 | 0.8 | ||
| 2 | 174.4–189.1 | 179.7–189.2 | 11.4 | 2.4 | −0.5 | ||
| 5 | 388.3–434.2 | 392.3–395 | 14.4 | 1.7 | −0.7 | ||
| 5 | 483.2–487.8 | 483.2–487.8 | 19.3 | 1.1 | −0.8 | ||
| TB | 1 | 0.0–15.9 | 7.8–9.9 | 24.1 | 2.2 | −0.8 | 44.1 |
| 1 | 40.6–125.9 | 94.5–101.6 | 14.0 | 4.1 | 0.8 | ||
| 2 | 174.4–189.1 | 179.7–189.2 | 7.6 | 1.5 | −0.4 | ||
| 5 | 388.3–434.2 | 392.3–395 | 43.2 | 17.4 | 3.1 | ||
FT, flowering time; SL, length until first silique; TL, total plant length; IB, main inflorescence branch number; BB, basal branch number; TB, total branch number.
Chromosome number.
The region spanned by consecutive bins, significantly (P < 0.001) differing from Ler and sharing the same direction of effect, was taken as the support interval.
Position of the bin within the QTL support interval showing the largest effect.
Within the QTL support interval the bin showing the largest effect was compared to Ler in an ANOVA analysis. The among-genotype component of ANOVA was taken as an estimator of explained variance.
All lines in the population were subjected to ANOVA using the bin described in footnote d as a fixed factor. The among-genotype component of ANOVA was taken as an estimator of explained variance.
Effect of QTL calculated as μB − μA, where μA is the mean value of all Ler lines and μB is the mean value of all lines in the bin described in footnote d. Effects are given in days (flowering time), centimeters (length at first silique and total length), or numbers (main inflorescence branch number, basal branch number, and total branch number).
All bins together with Ler were analyzed by ANOVA and the among-genotype component was taken as a measure of totally explained variance.
The smallest significant QTL effect detected was 0.7 days for flowering time, 1.1 and 2.1 cm for length at first silique and total plant length, respectively, and 3.8, 0.5, and 0.4 for the number of main inflorescence branches, basal branch number, and total branch number, respectively. Relative effects, expressed as the fold difference between genotypes, calculated as (|μB − μA| + μA)/μA, then equaled 1.03-, 1.11-, 1.09-, 2.71-, 1.30-, and 1.11-fold, respectively (Tables 4 and 5).
For a number of traits several QTL were found that could not be significantly detected in the RIL population. In total 12 of such small-effect QTL were detected for flowering time (3), length at first silique (5), total plant length (2), and basal branch number (2). None of those met the lower chromosomewide significance threshold for suggestive QTL in the RIL population. Although 2 were close to this threshold, 10 of them did not reach LOD scores >1.0 in the RIL population (supplemental Table 3 at http://www.genetics.org/supplemental/).
We defined the support interval in the NIL mapping population as the region spanned by consecutive bins, significantly differing from Ler (P < 0.001) and sharing the same direction of effect. The length of support intervals estimated in this way ranged from 1.4 (total plant length) to 85.3 cM (basal branch number) with an average of 30.9 cM. Alternatively, we also searched for QTL in the NIL population by comparing the phenotype of each NIL individually against Ler (supplemental Figures 2–7 at http://www.genetics.org/supplemental/). In this case, support intervals can be estimated as the length of the overlapping regions between the Cvi introgression fragments of NILs significantly differing from Ler in a particular genomic region. This second method increases the QTL localization resolution, but reduces statistical power. For each bin on average 116 plants could be tested against Ler whereas only 24 plants were available for analysis of individual NILs. Moreover, individual lines may contain multiple opposing-effect QTL, resulting in nonsignificant differences compared to Ler. Therefore, lines spanning the bin support interval were occasionally not significantly different from Ler. Likewise, lines bearing introgressions outside the bin support intervals sometimes differed significantly from Ler, probably due to multiple additive small-effect QTL. Together, the loss of power and the complexity of the traits under study hindered a confident estimation of a NIL support interval. Nevertheless, all QTL detected in the bin analysis could also be detected by analyzing individual NILs. As a compromise between the two methods of support interval estimation we recorded the position of the largest-effect bin within the bin support interval (Table 4). However, it must be noted that bin support intervals may contain multiple QTL of similar direction. The average size of these largest-effect bins was 4.6 cM. Within those bins, at least one individual NIL significantly differing from Ler was always found.
Power in RIL vs.NIL QTL mapping:
The power to detect a QTL at a specific locus basically depends on the difference in mean trait values between A and B genotypes for that particular locus. Although other parameters like trait heritability, genetic interactions, and genetic map quality should not be ignored. Because power increases when variance for mean values decreases, QTL analyses can benefit greatly from multiple measurements. In a RIL population this can be achieved in two ways. First, because segregation of both alleles occurs randomly and each locus is represented equally by the A and the B genotype, provided there is no segregation distortion (Doerge 2002), increasing the number of RILs to be analyzed will increase the number of observations of each genotype at a given genomic position. A further advantage of increasing the RIL population size is that the number of recombination events increases, which can improve resolution. However, when the number of lines is fixed, more accurate trait values of lines can be achieved by measuring replicate individuals of the same line. In addition, accurate trait values based on replicate measurements improve the possibility of detecting smaller-effect QTL.
To test the effect of replicated measurements and population size on the QTL detection power of the two Ler/Cvi populations we analyzed the phenotypic data obtained in these populations by varying both parameters. For the RIL population we performed QTL analyses on different numbers of RILs (population size) and used mean line values obtained with different numbers of replicates (replicate size). The total explained variance in the population, the LOD score of the largest-effect QTL, and the number of detected QTL were then recorded for each trait (Figure 4). When the population size was kept constant (161 lines), the recorded statistics increased when increasing the replicate number from one to four but this increase leveled off rapidly when measuring more replicates (Figure 4, A–C). In contrast, when the number of replicates was kept constant (16 replicated measurements per RIL) and population size was increased, the QTL detection power improved more drastically. However, the total explained variance remained more or less constant over all population sizes (Figure 4D). This phenomenon is commonly known as the Beavis effect and is due to the fact that estimated explained variances of detected QTL are sampled from a truncated distribution because QTL are taken into account only when the test statistics reach a predetermined critical value (Xu 2003). As a result, the expectations of detected QTL effects are biased upward. A second effect of increasing population size is the nearly linear increase of LOD scores, observed for all analyzed QTL (Figure 4E). Significance thresholds determined by permutation tests for each population size were steady around 2.7 LOD for population sizes >30 RILs and increased slightly with smaller population sizes (data not shown). The largest-effect QTL could be significantly detected at all population sizes for all traits except for basal branch number, whose largest-effect QTL could not be significantly detected in population sizes <80 RILs.
Figure 4.—

QTL detection power analysis of the Ler/Cvi RIL population. (A) Effect of replicate number on total explained variance. (B) Effect of replicate number on LOD score of the largest-effect QTL. (C) Effect of replicate number on the number of detected QTL. (D) Effect of population size on total explained variance. (E) Effect of population size on LOD score of the largest-effect QTL. (F) Effect of population size on the number of detected QTL. □, flowering time; ◊, length at first silique; Δ, total plant length; x, main inflorescence branch number; ○, basal branch number; and +, total branch number. Error bars represent SEM of 10 independent analyses.
To evaluate the NIL population, we studied the effect of increasing the number of replicates per line by estimating the relative difference between line mean values that could still be significantly detected with different replicate numbers (see materials and methods). As shown in Figure 5A the power to detect significant phenotypic differences greatly increases when increasing the number of replicate individuals of NILs measured. Furthermore, the lower the heritability of the trait the larger the increase of detection power achieved by increasing the number of replicates per NIL. When a bin analysis was carried out using increasing replicate numbers a similar increase in the number of detected QTL was observed (Figure 5B). Overall, the results presented in Figures 4 and 5 show that the number of replicates used in our analyses (16 individuals for each RIL and 24 individuals for each NIL) approximated the maximum QTL detection power of both Ler/Cvi populations.
Figure 5.—

QTL detection power analysis of the Ler/Cvi NIL population. (A) Effect of replicate number on significantly detectable relative differences, expressed as fold difference between two lines. (B) Effect of replicate number on the number of detected QTL. □, flowering time; ◊, length at first silique; Δ, total plant length; x, main inflorescence branch number; ○, basal branch number; and +, total branch number. Error bars represent SEM of 10 independent analyses.
DISCUSSION
Experimental mapping populations are a basic resource to elucidate the genetic basis of quantitative multigenic traits. In this work, we have developed the first genomewide population of NILs of Arabidopsis thaliana consisting of 92 lines carrying genomic introgression fragments from the parental accession Cvi into the common laboratory genetic background Landsberg erecta. In addition we have empirically compared the mapping power of this population with that of an existing population of recombinant inbred lines, derived from the same parental accessions. RIL and NIL populations have been used extensively in genetic studies (Eshed and Zamir 1995; Rae et al. 1999; Monforte and Tanksley 2000; Koumproglou et al. 2002; Han et al. 2004; Koornneef et al. 2004; Singer et al. 2004; Von Korff et al. 2004) due to the advantages derived from their homozygosity and immortality: they can be used indefinitely; various traits can be analyzed in different experiments and environmental settings; and replicates of the individual lines can be analyzed, enabling a more accurate estimate of the line's phenotypic mean value. However, the main difference between the two populations lies in the nature of their genetic makeup. In a RIL population multiple genomic regions differ between most pairs of RILs and several segregating QTL contribute to phenotypic differences between pairs of lines, making it impossible to assign the observed variation between pairs of lines to a specific genomic region. Therefore, to detect QTL one must perform the simultaneous analysis of a large number of lines. In contrast, in a NIL population, the phenotypic variation observed between pairs of lines can be assigned directly to the distinct genomic regions introgressed in an otherwise similar genetic background. Depending on the desired resolution one can minimize the number of lines by analyzing lines carrying large introgressions or even chromosome substitution strains (Nadeau et al. 2000).
A summary of the differences observed between the RIL and NIL populations derived from Ler and Cvi is shown in Table 5 and in supplemental Figure 8 at http://www.genetics.org/supplemental/. The total number of QTL detected did not differ much between the two populations. However, different loci were detected in both types of populations, showing their complementary properties. For both populations the detection of QTL was highly dependent on the trait under consideration and its genetic architecture (e.g., effect and position of QTL, epistasis). The power of the new NIL population to detect the large-effect loci was close to that of the existing RIL population since most large-effect loci were detected in both populations. However, a few relatively large-effect loci showing significant epistatic interactions could be detected only in the RIL population, but not in the NILs (supplemental Table 3 at http://www.genetics.org/supplemental/). Moreover, localization resolution was higher in the RIL population compared to the bin analysis of the NIL population, allowing separation of linked QTL. This was best illustrated by the two major QTL for flowering time detected in the RIL population on the top of chromosome 5, which not only are linked but also showed strong epistatic interaction. Consequently, these two QTL could not be separated in the NIL population. Nevertheless, the QTL resolution in the NIL population can be increased when analyzing individual lines, although this will be at the cost of mapping power. In total, 14 QTL detected in the RIL population could not be detected in the NIL population, of which 10 showed significant epistatic interaction with other QTL and all others were closely linked to another significant QTL.
In contrast, the average explained variance of single QTL was higher in the NIL population, increasing the power to detect small-effect QTL. This difference can be attributed to the level of transgression, which is stronger in the RIL population, thereby increasing total phenotypic variance. As a result, 13 small-effect QTL could be detected in the NIL population, which were not detected in the RIL population. Nevertheless, some of the small-effect QTL detected in the NILs were close to the significance threshold in the RIL population when using the lower chromosomal LOD thresholds (supplemental Table 3 at http://www.genetics.org/supplemental/). Expectedly, the power to detect small-effect QTL in the NIL population was higher for the more heritable traits (flowering time and plant height) than for those traits with low heritability (branching traits). The different power to detect small-effect QTL of the two populations is due to the effect of the segregation of multiple QTL in the RIL population, which increases the residual variance at each QTL under study.
The analyses of the RIL and NIL populations performed in this work were probably close to the maximum statistical power for the given population sizes since the number of detected QTL leveled off at higher replication sizes (Figures 4 and 5). The power analyses presented here could guide the making of decisions on the number of plants to be analyzed when experiments are costly, laborious, or time consuming and therefore may require the analysis of fewer plants. Overall, for RILs, the effect of population size on mapping power was larger than the effect of replicated measurements of individual lines. Therefore, to reduce the number of plants to be analyzed, it is preferable to first reduce the number of replicates per line, and only thereafter, if required, the number of lines. In our analyses major-effect QTL for most traits could still be significantly detected when only 50 lines were analyzed without replicates (data not shown). However, due to the Beavis effect (Xu 2003) the explained variances obtained with small population sizes were strongly overestimated. In the NIL population, the number of replicated measurements has a larger impact on mapping power and at least five replicated plants should be analyzed to obtain enough statistical power (Figure 5). However, fewer lines can be analyzed as long as genomewide coverage is maintained. In this NIL population this can be achieved using a core set of 25 lines, although localization resolution was diminished. Nevertheless, most QTL detected in the full set could still be detected in the core set (supplemental Figure 9 at http://www.genetics.org/supplemental/). Once a QTL has been identified in a particular region, one can zoom in with a minimal set of lines carrying smaller introgressions defined by crossovers in the support interval of the QTL of interest (Fridman et al. 2004).
The Ler/Cvi NIL population developed in this work provides a useful resource that will facilitate the genetic dissection of quantitative traits in Arabidopsis in various aspects. First, as shown here, it can be analyzed as an alternative segregating population to perform genomewide QTL mapping, with the particular advantage of detecting small-effect QTL. Second, this population can be used to confirm previously detected QTL in the Ler/Cvi RIL population. Third, individual lines of this population can serve as a starting point for the rapid Mendelization of particular QTL and for their fine mapping and cloning (Paran and Zamir 2003). Finally, the single introgression lines of this population may also strongly facilitate the fine mapping of artificially induced mutant alleles in the common laboratory Ler genetic background (or transferred to this accession). The fine mapping of mutant loci affecting quantitative adaptive traits is often hampered by the confounding effects of QTL segregating in the mapping populations derived from crosses between the mutant and another Arabidopsis wild accession. Knowing the approximate genetic location of the mutant locus within a chromosomal arm, specific lines of this NIL population can be selected as carrying a single introgression spanning the map position of the locus of interest. These lines can then be used to derive the required monogenic mapping population, as has been illustrated with the flowering-time locus FVE (Ausin et al. 2004). In conclusion, the elucidation of quantitative traits can benefit from the parallel analysis of both populations.
Acknowledgments
We thank Kieron Edwards for sharing NILs, Johan van Ooijen for helpful assistance in the QTL mapping, and Piet Stam for critical reading of the manuscript. This work was supported by a grant from The Netherlands Organization for Scientific Research, Program Genomics (050-10-029).
References
- Alonso-Blanco, C., and M. Koornneef, 2000. Naturally occurring variation in Arabidopsis: an underexploited resource for plant genetics. Trends Plant Sci. 5: 22–29. [DOI] [PubMed] [Google Scholar]
- Alonso-Blanco, C., A. J. M. Peeters, M. Koornneef, C. Lister, C. Dean et al., 1998. a Development of an AFLP based linkage map of Ler, Col and Cvi Arabidopsis thaliana ecotypes and construction of a Ler/Cvi recombinant inbred line population. Plant J. 14: 259–271. [DOI] [PubMed] [Google Scholar]
- Alonso-Blanco, C., S. E. El-Assal, G. Coupland and M. Koornneef, 1998. b Analysis of natural allelic variation at flowering time loci in the Landsberg erecta and Cape Verde Islands ecotypes of Arabidopsis thaliana. Genetics 149: 749–764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alonso-Blanco, C., H. Blankestijn-De Vries, C. J. Hanhart and M. Koornneef, 1999. Natural allelic variation at seed size loci in relation to other life history traits of Arabidopsis thaliana. Proc. Natl. Acad. Sci. USA 96: 4710–4717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alonso-Blanco, C., L. Bentsink, C. J. Hanhart, H. Blankestijn-De Vries and M. Koornneef, 2003. Analysis of natural allelic variation at seed dormancy loci of Arabidopsis thaliana. Genetics 164: 711–729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ausin, I., C. Alonso-Blanco, J. A. Jarillo, L. Ruiz-Garcia and J. M. Martinez-Zapater, 2004. Regulation of flowering time by FVE, a retinoblastoma-associated protein. Nat. Genet. 36: 162–166. [DOI] [PubMed] [Google Scholar]
- Bentsink, L., K. Yuan, M. Koornneef and D. Vreugdenhil, 2003. The genetics of phytate and phosphate accumulation in seeds and leaves of Arabidopsis thaliana, using natural variation. Theor. Appl. Genet. 106: 1234–1243. [DOI] [PubMed] [Google Scholar]
- Blair, M. W., G. Iriarte and S. Beebe, 2006. QTL analysis of yield traits in an advanced backcross population derived from a cultivated Andean x wild common bean (Phaseolus vulgaris L.) cross. Theor. Appl. Genet. 112: 1149–1163. [DOI] [PubMed] [Google Scholar]
- Blanco, A., R. Simeone and A. Gadaleta, 2006. Detection of QTLs for grain protein content in durum wheat. Theor. Appl. Genet. 112: 1195–1204. [DOI] [PubMed] [Google Scholar]
- Borevitz, J. O., and M. Nordborg, 2003. The impact of genomics on the study of natural variation in Arabidopsis. Plant Physiol. 132: 718–725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broman, K. W., 2001. Review of statistical methods for QTL mapping in experimental crosses. Lab Anim. 30: 44–52. [PubMed] [Google Scholar]
- Chase, K., F. R. Adler and K. G. Lark, 1997. Epistat: a computer program for identifying and testing interactions between pairs of quantitative trait loci. Theor. Appl. Genet. 94: 724–730. [Google Scholar]
- Doerge, R. W., 2002. Mapping and analysis of quantitative trait loci in experimental populations. Nat. Rev. Genet. 3: 43–52. [DOI] [PubMed] [Google Scholar]
- Edwards, K. D., J. R. Lynn, P. Gyula, F. Nagy and A. J. Millar, 2005. Natural allelic variation in the temperature-compensation mechanisms of the Arabidopsis thaliana circadian clock. Genetics 170: 387–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eshed, Y., and D. Zamir, 1995. An introgression line population of Lycopersicon pennellii in the cultivated tomato enables the identification and fine mapping of yield-associated QTL. Genetics 141: 1147–1162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fridman, E., F. Carrari, Y. Liu, A. R. Fernie and D. Zamir, 2004. Zooming in on a quantitative trait for tomato yield using interspecific introgressions. Science 305: 1786–1789. [DOI] [PubMed] [Google Scholar]
- Han, F., S. E. Ullrich, A. Kleinhofs, B. L. Jones, P. M. Hayes et al., 1997. Fine structure mapping of the barley chromosome-1 centromere region containing malting-quality QTLs. Theor. Appl. Genet. 95: 903–910. [Google Scholar]
- Han, F., J. A. Clancy, B. L. Jones, D. M. Wesenberg, A. Kleinhofs et al., 2004. Dissection of a malting quality QTL region on chromosome 1 (7H) of barley. Mol. Breed. 14: 339–347. [Google Scholar]
- Jansen, R. C., 2003. Quantitative trait loci in inbred lines, pp. 445–476 in Handbook of Statistical Genetics, edited by D. J. Balding, M. Bishop and C. Cannings. John Wiley & Sons, Chichester, UK.
- Jeuken, M. J. W., and P. Lindhout, 2004. The development of lettuce backcross inbred lines (BILs) for exploitation of the Lactuca saligna (wild lettuce) germplasm. Theor. Appl. Genet. 109: 394–401. [DOI] [PubMed] [Google Scholar]
- Juenger, T. E., J. K. Mckay, N. Hausmann, J. J. B. Keurentjes, S. Sen et al., 2005. a Identification and characterization of QTL underlying whole-plant physiology in Arabidopsis thaliana: δ13C, stomatal conductance and transpiration efficiency. Plant Cell Environ. 28: 697–708. [Google Scholar]
- Juenger, T. E., S. Sen, K. A. Stowe and E. L. Simms, 2005. b Epistasis and genotype-environment interaction for quantitative trait loci affecting flowering time in Arabidopsis thaliana. Genetica 123: 87–105. [DOI] [PubMed] [Google Scholar]
- Koornneef, M., C. Alonso-Blanco and D. Vreugdenhil, 2004. Naturally occurring genetic variation in Arabidopsis thaliana. Annu. Rev. Plant Biol. 55: 141–172. [DOI] [PubMed] [Google Scholar]
- Koumproglou, R., T. M. Wilkes, P. Townson, X. Y. Wang, J. Beynon et al., 2002. STAIRS: a new genetic resource for functional genomic studies of Arabidopsis. Plant J. 31: 355–364. [DOI] [PubMed] [Google Scholar]
- Loudet, O., V. Gaudon, A. Trubuil and F. Daniel-Vedele, 2005. Quantitative trait loci controlling root growth and architecture in Arabidopsis thaliana confirmed by heterogeneous inbred family. Theor. Appl. Genet. 110: 742–753. [DOI] [PubMed] [Google Scholar]
- Maloof, J. N., 2003. Genomic approaches to analyzing natural variation in Arabidopsis thaliana. Curr. Opin. Genet. Dev. 13: 576–582. [DOI] [PubMed] [Google Scholar]
- Monforte, A. J., and S. D. Tanksley, 2000. Development of a set of near isogenic and backcross recombinant inbred lines containing most of the Lycopersicon hirsutum genome in a L. esculentum genetic background: a tool for gene mapping and gene discovery. Genome 43: 803–813. [PubMed] [Google Scholar]
- Nadeau, J. H., J. B. Singer, A. Matin and E. S. Lander, 2000. Analysing complex genetic traits with chromosome substitution strains. Nat. Genet. 24: 221–225. [DOI] [PubMed] [Google Scholar]
- Paran, I., and D. Zamir, 2003. Quantitative traits in plants: beyond the QTL. Trends Genet. 19: 303–306. [DOI] [PubMed] [Google Scholar]
- Rae, A. M., E. C. Howell and M. J. Kearsey, 1999. More QTL for flowering time revealed by substitution lines in Brassica oleracea. Heredity 83: 586–596. [DOI] [PubMed] [Google Scholar]
- Reymond, M., S. Svistoonoff, O. Loudet, L. Nussaume and T. Desnos, 2006. Identification of QTL controlling root growth response to phosphate starvation in Arabidopsis thaliana. Plant Cell Environ. 29: 115–125. [DOI] [PubMed] [Google Scholar]
- Singer, J. B., A. E. Hill, L. C. Burrage, K. R. Olszens, J. Song et al., 2004. Genetic dissection of complex traits with chromosome substitution strains of mice. Science 304: 445–448. [DOI] [PubMed] [Google Scholar]
- Slate, J., 2005. Quantitative trait locus mapping in natural populations: progress, caveats and future directions. Mol. Ecol. 14: 363–379. [DOI] [PubMed] [Google Scholar]
- Stylianou, I. M., S. Tsaih, K. Dipetrillo, N. Ishimori, R. Li et al., 2006. Complex genetic architecture revealed by analysis of high-density lipoprotein cholesterol in chromosome substitution strains and F2 crosses. Genetics 174: 999–1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swarup, K., C. Alonso-Blanco, J. R. Lynn, S. D. Michaels, R. M. Amasino et al., 1999. Natural allelic variation identifies new genes in the Arabidopsis circadian system. Plant J. 20: 67–77. [DOI] [PubMed] [Google Scholar]
- Teng, S., J. Keurentjes, L. Bentsink, M. Koornneef and S. Smeekens, 2005. Sucrose-specific induction of anthocyanin biosynthesis in Arabidopsis requires the MYB75/PAP1 gene. Plant Physiol. 139: 1840–1852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuinstra, M. R., G. Ejeta and P. B. Goldsbrough, 1997. Heterogeneous inbred family (HIF) analysis: a method for developing near-isogenic lines that differ at quantitative trait loci. Theor. Appl. Genet. 95: 1005–1011. [Google Scholar]
- Ungerer, M. C., S. S. Halldorsdottir, J. L. Modliszewski, T. F. C. Mackay and M. D. Purugganan, 2002. Quantitative trait loci for inflorescence development in Arabidopsis thaliana. Genetics 160: 1133–1151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ungerer, M. C., S. S. Halldorsdottir, M. D. Purugganan and T. F. C. Mackay, 2003. Genotype-environment interactions at quantitative trait loci affecting inflorescence development in Arabidopsis thaliana. Genetics 165: 353–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Ooijen, J. W., 1992. Accuracy of mapping quantitative trait loci in autogamous species. Theor. Appl. Genet. 84: 803–811. [DOI] [PubMed] [Google Scholar]
- Van Ooijen, J. W., 2004. MapQTL 5, Software for the Mapping of Quantitative Trait Loci in Experimental Populations. Kyazma B.V., Wageningen, The Netherlands.
- Von Korff, M., H. Wang, J. Leon and K. Pillen, 2004. Development of candidate introgression lines using an exotic barley accession (Hordeum vulgare ssp. spontaneum) as donor. Theor. Appl. Genet. 109: 1736–1745. [DOI] [PubMed] [Google Scholar]
- Xu, S., 2003. Theoretical basis of the Beavis effect. Genetics 165: 2259–2268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon, D. B., K. H. Kang, H. J. Kim, H. G. Ju, S. J. Kwon et al., 2006. Mapping quantitative trait loci for yield components and morphological traits in an advanced backcross population between Oryza grandiglumis and the O. sativa japonica cultivar Hwaseongbyeo. Theor. Appl. Genet. 112: 1052–1062. [DOI] [PubMed] [Google Scholar]
- Zou, F., J. A. Gelfond, D. C. Airey, L. Lu, K. F. Manly et al., 2005. Quantitative trait locus analysis using recombinant inbred intercrosses: theoretical and empirical considerations. Genetics 170: 1299–1311. [DOI] [PMC free article] [PubMed] [Google Scholar]