

Abstract
Gene duplication can occur on two scales: whole-genome duplications (WGD) and smaller-scale duplications (SSD) involving individual genes or genomic segments. Duplication may result in functionally redundant genes or diverge in function through neofunctionalization or subfunctionalization. The effect of duplication scale on functional evolution has not yet been explored, probably due to the lack of global knowledge of protein function and different times of duplication events. To address this question, we used integrated Bayesian analysis of diverse functional genomic data to accurately evaluate the extent of functional similarity and divergence between paralogs on a global scale. We found that paralogs resulting from the whole-genome duplication are more likely to share interaction partners and biological functions than smaller-scale duplicates, independent of sequence similarity. In addition, WGD paralogs show lower frequency of essential genes and higher synthetic lethality rate, but instead diverge more in expression pattern and upstream regulatory region. Thus, our analysis demonstrates that WGD paralogs generally have similar compensatory functions but diverging expression patterns, suggesting a potential of distinct evolutionary scenarios for paralogs that arose through different duplication mechanisms. Furthermore, by identifying these functional disparities between the two types of duplicates, we reconcile previous disputes on the relationship between sequence divergence and expression divergence or essentiality.
GENE duplication is a major source of new genes and is thus a central factor influencing genome evolution (Ohno 1970; Wolfe and Li 2003). Such duplication can occur on two scales: the duplication of the whole genome (WGD) and smaller-scale duplications (SSD), which occur continuously and involve individual genes or genomic segments (see review in Sankoff 2001). Duplicated genes can be retained due to different selection mechanisms and can thus undergo different evolutionary fates. Paralogs may be selected for increased dosage or as a repository for gene conversion against deleterious changes in either copy and result in functional redundancy (Nadeau and Sankoff 1997; Nowak et al. 1997; Gu 2003; Gu et al. 2003). Alternatively, the paralogs may diverge either for generation of new gene functions (neofunctionalization) (Taylor and Raes 2004) or for subdividing multiple functions (subfunctionalization) through complementary degeneration (Force et al. 1999; Stoltzfus 1999; Lynch and Force 2000). However, the relative importance of these mechanisms in preserving WGD vs. SSD duplicates, indicated by the resultant functional conservation/divergence between paralogs, has not been investigated.
Functional studies focusing on either WGD or SSD or the combination of the two sets have established some insights with respect to different attributes of duplicate genes. For example, on the basis of 41 WGD pairs, Baudot et al. (2004) examined the function of duplicate pairs through interaction-network analysis, but found no simple relationship between the sequence identity and functional similarity. An earlier study (Brun et al. 2003) reached a similar result on the basis of a limited combination of WGD and SSD pairs. Also, on the basis of WGD paralogs, Seoighe and Wolfe (1999) found that increased levels of gene expression were a significant factor in determining which genes were retained as duplicates. However, as the selection stages through which the duplicates must pass to become a persistent part of the genome are somewhat different for the WGD and SSD sets (Davis and Petrov 2005), the two modes of duplication may generate genes with different molecular attributes. Thus results based on combined analysis of both types of duplicates ignore properties unique to a particular set. Furthermore, individual analysis of either group does not necessarily generalize to the other. In fact, due to such differences in data sets, two previous studies (Wagner 2000a; Gu et al. 2003) drew inconsistent conclusions on the relationship between the sequence divergence of duplicate genes and the fitness effect of a null mutation. In addition, in many ways the relationship between gene duplication and evolution of transcriptional regulation has been controversial (see review in Li et al. 2005): Wagner (2000b) suggested that the regulatory sequences and mRNA expression patterns of duplicate gene pairs evolve independently of the coding sequence, whereas others have found a highly significant relationship with sequence divergence or age (Gu et al. 2002, 2005; Zhang et al. 2004).
The above studies were based on either only WGD duplicates or a combination of both duplication groups. An open question is whether WGD and SSD duplicates undergo different evolutionary scenarios, thus explaining the disparities among these studies. Despite the unambiguous identification of WGD blocks in Saccharomyces cerevisiae (Dietrich et al. 2004; Kellis et al. 2004a), few previous studies have focused on the global differences between the WGD and SSD duplicates. To our knowledge, the only study to consider the global differences between the two sets was Davis and Petrov (2005), which showed that the two sets differ significantly in overall molecular functional enrichment, but are similar with respect to codon bias and evolutionary rate. Yet no study has focused on the differences between WGD and SSD gene sets with respect to evolutionary fates and consequent functional conservation/divergence between paralogs.
To address this problem, an important and as yet unresolved question is to discriminate the functions of the paralogs and evaluate their subtle differences. The divergence between paralogs has been intensively studied at the sequence level in S. cerevisiae (e.g., Lynch and Conery 2000; Kondrashov et al. 2002), mostly through examining the number of synonymous (dS) vs. nonsynonymous (dN) substitutions. Unfortunately, an attempt to calculate the nonsynonymous vs. synonymous substitution for WGD paralogs shows that most of the dS's are saturated (supplemental Figure S1 at http://www.genetics.org/supplemental/). This hinders an overall perspective of the functional divergence between the WGD paralogs vs. SSD paralogs. The evolutionary rate relative to the orthologs has been shown to be similarly biased (Davis and Petrov 2005). However, the rates calculated by referring to orthologs do not represent the divergence between the paralogs, which is affected by gene conversion. One alternative is to compare the function of paralogs through their gene ontology (GO) (Ashburner et al. 2000) annotations. Unfortunately, subtle differences between paralogs prevented the successful function discrimination based on GO annotations (Baudot et al. 2004), which themselves are sometimes “inferred from sequence or structural similarity” (ISS).
Here we took advantage of diverse functional genomic and high-throughput data and carried out genomewide analyses of the divergence of biological function between whole-genome paralogs (506 pairs) and smaller-scale duplicates (1193 pairs including 1862 genes). Using a Bayesian methodology to integrate diverse functional genomic data from >6500 publications, we accurately predicted specific function for each gene. This enabled us to demonstrate that WGD paralogs, independent of sequence divergence level, are in general more likely to share physical protein–protein interaction partners and functional relationships. In addition, WGD paralogs show lower essentiality and higher synthetic lethality frequency. However, such functional compensation between paralogs is not followed by complete redundancy, as the more diverse expression patterns and upstream regulatory regions between WGD paralogs suggest their role in modulating expression level. Moreover, the propensity to have similar, compensatory functions but to diverge in expression patterns is unique to WGD paralogs, in comparison to SSD paralogs, which suggests a potential of distinct evolutionary scenarios for paralogs that arose through different duplication mechanisms
METHODS
Identifying WGD and SSD paralogs:
To define a set of paralogous pairs in the yeast genome, we first constructed a set of alignments among S. cerevisiae proteins. Protein sequences for all ORFs in S. cerevisiae (except dubious ORFs and pseudogenes) were downloaded from the Saccharomyces Genome Database (SGD) (Cherry et al. 1998). For each of these ORFs, we then used protein BLAST (Altschul et al. 1990) with E = 0.01 to find all protein hits within the S. cerevisiae genome. We then used these alignments to identify suboptimal matches (the best match is self-alignment) on the basis of the Kellis et al. (2004b) method. This approach takes into account the fact that similarity between query protein x and target protein y can be split into multiple BLAST hits. Intuitively, the BLAST hits between x and y are weighted by the amino acid percentage of identity and length aligned and thereby grouped into a single match. Compared to global alignment, this method includes duplicate pairs that have internal inversion in one of the members. The detailed procedure is as follows.
The weight for each hit is assigned as
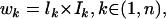 |
where lk is the length and Ik is the overall amino acid identity of hit k, and n is the total number of hits for protein x and target protein y.
To group all BLAST hits into a single match, the nonoverlapping portions of these hits were added to obtain the maximized identity number between x and y. For each paralogous pair (x, y), the wk is ranked and its correspondent start and ending sites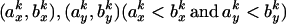 were recorded. The top ranked wk was added to the total weight W(x,y) and only those wj whose corresponding start and ending sites satisfying
were recorded. The top ranked wk was added to the total weight W(x,y) and only those wj whose corresponding start and ending sites satisfying 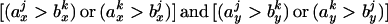 were retained. The above process was repeated until all the hits were added into W(x,y). Hits that overlapped with another hit of higher weight were discarded during this iteration. The summed W(x,y) gives the maximized identity number between protein x and y. Percentage of identity is calculated as
were retained. The above process was repeated until all the hits were added into W(x,y). Hits that overlapped with another hit of higher weight were discarded during this iteration. The summed W(x,y) gives the maximized identity number between protein x and y. Percentage of identity is calculated as
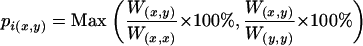 |
Suboptimal matches [pi(x,y), x ≠ y], were used to construct the paralogous pairs. WGD duplicates (528 pairs) were classified depending on their inclusion in the WGD duplicate blocks characterized by genomewide comparisons of S. cerevisiae to Kluyveromyces waltii (Kellis et al. 2004a; Byrne and Wolfe 2005), which diverged just prior to the polyploidization event. SSD duplicate pairs are defined as paralogous pairs not included in the WGD list. Due to the different methods used for identifying paralogous pairs for WGD (both synteny and sequence similarity) or SSD (sequence similarity only), measurements for SSD pairs may contain more fluctuations, thereby further necessitating statistical analyses that we performed in this study.
In this way we identified 2604 pairs (including 528 WGDs and 2076 potential SSDs) according to their percentage of identity. To reduce data fluctuation, we constructed 23 groups with a sliding window of 400 pairs in size (as a sum of WGD and SSD duplicates) and 100 pairs per window slide. Using nonoverlapping bins demonstrates the same trends in each of the attributes we studied (supplemental Figure S10 at http://www.genetics.org/supplemental/). However, these overlapping bins and a unified grouping of the two sets of paralogs ensured that enough WGD and SSD pairs were included in each bin for statistical analysis and enabled us to compare WGD and SSD paralogs of similar sequence identity (Figure 1). The percentage of identity assigned to each group in the figures is the median value of each group. Because there are no or few WGD paralogous pairs falling into the last several bins (Figure 1), and pairs at the very low-alignment bins are not likely to be true paralogs, the last nine bins (<20%, with <25 WGD pairs) were excluded from further analysis. Thus 506 WGD pairs and 1193 unique SSD pairs were used.
Figure 1.—

The number of duplicate pairs at each sequence divergence level. The duplicates were grouped into 23 bins with a sliding window of 400 pairs in size and 100 pairs per window slide. Such grouping is used in the following analysis that included percentage of identity. Thus adjacent bins may include the same pairs so as to smooth the pattern and identify the general trends of different attributes of the WGD and SSD sets.
We also examined the effect of the large number of ribosomal WGD paralogs on our results. We identified ribosomal genes as those annotated with the protein biosynthesis term in the gene ontology (see supplemental files for this list of genes at http://www.genetics.org/supplemental/). For this analysis, any gene pair in which one or both paralogs were ribosomal genes was excluded from the analysis. Furthermore, to control for results being biased by duplicates from large gene families, we repeated several analyses using reciprocal best hits.
Prediction of shared protein–protein interaction partners and functional relationships:
We predicted protein–protein interaction partners and functionally related proteins using a Bayesian data integration method described by us in Myers et al. (2005) to incorporate diverse genomic data sources. This method integrated different types of data (for example, gene expression, interaction data, high-throughput data, or single experiments), using a Bayesian network trained using the expectation-maximization learning algorithm (Dempster et al. 1977) with known functionally relevant GO biological process annotations (Myers et al. 2006) as the gold standard. Intuitively, for each gene i–gene j pair, the network asked the following question: What is the probability, on the basis of the experimental evidence presented, that products of gene i and gene j have a functional relationship (i.e., are involved in the same biological process)? The trained network integrated data sets by weighing relative accuracy and coverage of each experimental method; thus data sets that were more accurate in predicting known GO annotations were given higher weight in predicting interaction partners and functional relationships. The weighted data sets were then used to predict the confidence of a relationship between two proteins. This Bayesian data integration step thus reduced the heterogeneous input data to protein pairs with a score indicating the likelihood that they functionally (or physically) interact, allowing different types of data to be combined with each other.
We considered two types of relationships: general functional relationships, which indicate proteins involved in the same biological process, and physical interactions. The evidence for protein–protein interaction predictions included yeast two-hybrid, copurification, and affinity-precipitation data, etc. (for the full list of evidence, please see supplemental information at http://www.genetics.org/supplemental/). Experimental evidence for a general functional relationship includes all the data supportive of involvement in the same biological process (Myers et al. 2005), including physical and genetic interactions, synthetic data, shared sequence motifs, and curated literature. We considered predictions with Bayesian confidence cutoffs ranging from 0.2 to 0.95 in our experiments (our predictions for physical interaction and functional relationship are available in supplemental information). The percentage of shared interaction partners (or shared functional relationships) between paralogs over the total number of interaction partners (or functional relationships) of the pair was calculated as
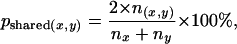 |
where nx and ny represent the number of interactions/functional relationships for x and y proteins, respectively, and n(x,y) represents the number of common interactions/functional relationships between x and y. The cutoffs at which the difference between WGD and SSD groups was most sensitive were used in the functional analyses parsed by percentage of identity, but results are robust to different cutoffs across the whole cutoff range.
Essentiality, synthetic lethality, upstream regulatory region, and expression data retrieval and analysis:
To assess essentiality, results of systematic deletion experiments of S. cerevisiae were retrieved from the Saccharomyces genome deletion project on 11/25/05 (http://www-sequence.stanford.edu/group/yeast_deletion_project/). We used synthetic lethality data retrieved from GRID (Breitkreutz et al. 2003) and MIPS (Tong et al. 2001; Mewes et al. 2004).
For analysis of upstream regulatory regions, we aligned the upstream 1000 bp for each paralogous pair using the same method as in identifying duplicates, with E = 1, as we expected faster divergence of the noncoding sequences. The average percentage of identity between upstream sequences of paralogous pairs was calculated.
For analysis of transcription factor-binding sites, we used Lee et al.'s (2002) data that report confidence values (P-values) for each binding site–gene combination. We calculated the frequency of shared transcription factor-binding sites between paralogs across different confidence values as cutoffs.
For coexpression analysis, we used the microarray data from Brem and Kruglyak (2005). The data were filtered (for genes missing in >50% of measurements) and imputed using KNNimpute (Troyanskaya et al. 2001) to fill in missing values, and any gene replicates were averaged together.
Functional clustering:
To perform a global comparison between the WGD and SSD sets with respect to their gene ontology annotation, we obtained GO annotations (Ashburner et al. 2000) from the SGD (Cherry et al. 1998). The enrichment of each GO term (in percentage) for the WGD and SSD sets was found using a hypergeometric distribution to identify the most enriched GO terms with the lowest Bonferonnni-corrected P-value (see supplemental information at http://www.genetics.org/supplemental/ for full results of this analysis). To summarize the information in Figure 2 and supplemental Figure S2 (http://www.genetics.org/supplemental/), we performed a similar analysis with the GO slim terms and assessed the enrichment in terms of cumulative distribution function.
Figure 2.—

Distribution of GO slim biological process annotations. Differential enrichment of GO slim biological process categories for WGD and SSD genes is shown. The graph represents the enrichment (in cumulative distribution function) of each set of genes in each GO term, in comparison to the genome average, with darker shading representing higher enrichment.
RESULTS
WGD paralogs exhibit a higher propensity than SSD paralogs to share protein–protein interaction partners and functional relationships:
We first addressed the question of whether the WGD and SSD duplicates participate in different biological processes by examining biases in the GO annotations (Ashburner et al. 2000). We found significant differences in enrichment between the WGD and SSD sets with respect to biological process (Figure 2) and cellular component annotations (supplemental Figure S2A at http://www.genetics.org/supplemental/). We also confirmed the enrichment differences in terms of molecular function annotation (Davis and Petrov 2005, supplemental Figure S2B at http://www.genetics.org/supplemental/). With respect to the biological process, for example, while both sets are enriched in genes involved in cell homeostatis, morphogenesis, protein modification, response to stress, transport, and vesicle-mediated transport (P < 0.05), WGD genes are uniquely enriched (P < 0.05 and cumulative distribution function for the SSD set <0.5) in conjugation and protein biosynthesis (Figure 2). On the other hand, SSD genes are uniquely enriched (P < 0.05 and cumulative distribution function for the WGD set <0.5) in DNA metabolism and protein catabolism (Figure 2). These differences in enrichment between the two sets of duplicates are themselves statistically significant (see supplemental information at http://www.genetics.org/supplemental/ for complete enrichment statistics).
In addition to the general gene ontology enrichment of the two sets, we focused on addressing the functional divergence between paralogs, which is informative of the evolutionary fate of duplicates. To assess subtle functional differences between paralogs on a whole-genome scale, we used heterogeneous high-throughput functional genomic data integrated using a Bayesian network (see methods). First, we predicted the physical interaction partners of each paralog. As proteins that have similar functions share interaction partners (Jacq 2001; Brun et al. 2003), this analysis allowed us to assess the degree of functional similarity in paralogous pairs. Second, we predicted functionally related proteins for each paralog on the basis of the Bayesian network. Such functional relationship represents the likelihood that two proteins are involved in the same biological processes. The frequency of shared interaction partners and shared functional relationships was significantly higher for the WGD paralogs compared to the SSD paralogs and this result is robust against different confidence levels used as the cutoff in the Bayesian predictions (Figure 3A, P < 0.01 over all cutoffs and Figure 3B, P < 0.002 over all cutoffs).
Figure 3.—

Frequency of shared interaction partners and functional relationships predicted by a Bayesian network at various confidence levels. We predicted interaction partners and functionally related proteins for each paralog on the basis of a Bayesian analysis of diverse genomic data. Then we calculated the percentage of shared interaction partners/functional relationships between paralogs over the total number of interaction partners/functional relationships of the pair. (A) A Bayesian network integrating evidence for physical interactions was used to predict interaction partners. (B) A Bayesian network integrating diverse genomic data was used to predict broad functional relationships. The WGD group shows a substantially higher percentage of shared interaction partners and functional relationships across all the Bayesian confidence levels. Fluctuations in the WGD graph are most likely due to variations in availability of different experimental data sets that served as input to the Bayesian analysis.
Propensity to share protein-interaction partners and functional relationships is intrinsic to WGD paralogs and independent of sequence divergence level:
Interpretation of the above result is complicated by the different times of the two duplication events: the WGD duplicates arose at one specific time point while the SSD paralogs are heterogeneous in their time of divergence. The saturation of most dS prevented us from grouping the paralogs according to number of synonymous substitutions. Thus, we grouped paralogous pairs according to the divergence level of the ORF amino acid sequence (see methods). Although sequence divergence does not directly represent the time since duplication, as shown by the varying divergences of the WGD set, this stratification allowed us to identify trends that were robust across scenarios covering a wide range of divergence levels. This analysis also avoids a major complication due to the prospect that duplicate pairs undergo decelerated evolution after initial acceleration (Jordan et al. 2004).
Our results show that the WGD paralogs are more similar to each other in protein–protein interactions and functional relationships over different levels of sequence similarity (Figure 4A, P < 0.05 when percentage of identity >25% and Figure 4B, P < 0.05 when percentage of identity >17%). The robustness of the result against the sequence divergence level indicates that such tendencies are possibly due to the differences in selection pressure after duplication (Davis and Petrov 2005). In addition, the relationship between functional relationships and sequence similarity appears linear in the SSD set, whereas in the WGD set, above 45% identity, there is no obvious decrease as the paralogous sequences diverge (Figure 4B). Although such trends do not necessarily indicate a biological relationship, this result does suggest that using either or a mixture of the two sets (for example, as in the Brun et al. 2003 study that did not identify this relationship) may establish a biased result and miss characteristics specific to only WGD or SSD paralogs (see discussion).
Figure 4.—

Propensity of sharing interaction partners and functional relationships between paralogs across sequence divergence levels. (A) Sharing of interaction partners between paralogs predicted from the Bayesian network on the basis of evidence of protein–protein interactions. (B) Sharing of functional relationships between paralogs predicted from the Bayesian network predicting functional relationships. For the same level of sequence divergence, the WGD paralogs are more likely to share protein–protein interaction partners and functional relationships. The linear relationship between sequence divergence and shared functional relationships is evident in SSD duplicates (R2 = 0.8688). In the WGD set, above 45% sequence identity such linear relationship is not observable.
A previous study suggested that when compared to orthologous nonduplicated genes, the evolutionary rate difference between WGD and SSD duplicates results mainly from the overenrichment of ribosomal genes in the WGD set (Davis and Petrov 2005). To assess this hypothesis, we removed pairs with ribosomal genes in both sets and repeated the analysis on functional divergence. We observed a similar pattern, which suggests that the trend to be functionally similar between WGD paralogs is general rather than an effect of enrichment in ribosomal genes (see supplemental Figure S3, A and B, at http://www.genetics.org/supplemental/). To further control for the differences in functional enrichment between the two sets of paralogs, we randomly selected subsets of SSD pairs with the same GO slim molecular function annotation distribution as the WGD set and repeated our analysis. The above differences remain even after this normalization of functional coverage between the two groups of paralogs (supplemental Figure S4 at http://www.genetics.org/supplemental/). We also repeated this analysis with reciprocal best hits (supplemental Figure S3, C and D, at http://www.genetics.org/supplemental/). The results remain robust, suggesting that the above differences between WGD and SSD sets were not caused by potential biases toward larger gene families in the SSD set.
Another concern in this analysis is that the difference observed above may be an artifact caused by the differences in numbers of interaction partners between WGD and SSD genes. However, this is not likely, because the frequency of shared interaction partners does not appear to depend on the total number of interaction partners (supplemental Figure S5 at http://www.genetics.org/supplemental/). In addition, for the SSD set, lower sequence similarity between paralogs correlates to a higher number of interactions, perhaps indicating functional divergence caused by generation of new interaction partners.
Compensation between WGD paralogs:
Because the WGD paralogs tend to be similar in function as suggested by shared interactions and functional relationships, we reasoned that they should show a high frequency of compensation. This would predict that, first, WGD paralogs should be more dispensable since their partner can take over their functions. Second, a high frequency of compensation should result in a higher rate of synthetic lethality of WGD paralogs.
The overall proportion of essential genes is much less (p < 10−300) in the WGD set (4.15%) than in the SSD set (18.2%) (the genome average is 18.9%), although SSD paralogs with lower sequence similarity are more likely to be essential genes (Figure 5A). Interestingly, the relationship between sequence similarity and frequency of essential genes is not evident in the WGD set. We found similar results after the removal of ribosomal gene pairs (supplemental Figure S6A at http://www.genetics.org/supplemental/). The relationship between sequence similarity and fitness effect is different between the two duplicate sets, which indicates that contradictory conclusions could be drawn if examining either or a mixture of the two sets (see discussion).
Figure 5.—

Patterns of essentiality and synthetic lethality of the duplicates across sequence divergence levels. (A) The percentage of essential genes in the WGD and SSD sets. Frequency of essentiality of the SSD duplicate genes increases as the paralogous sequences diverge. In contrast, the essentiality rate stays low and at a relatively constant level for WGD genes. The SSD duplicates generally show a higher proportion of essential genes except in high sequence similarity (>60%) bins, which include 11% of the SSD set only. (B) The percentage of synthetic lethal pairs in WGD and SSD sets. The synthetic lethality rate is generally higher in WGD paralogs, which suggests compensation and functional conservation between paralogs. The synthetic lethality proportion decreases as the paralogous sequences diverge in the WGD set, whereas such a trend is not observable in the SSD set.
Second, we predict that the frequency of synthetic lethality between paralogs should be higher in the WGD set than in the SSD set, because deletion of both of the paralogs is expected to abolish their common function. Indeed, the average synthetic lethality frequency of the WGD set is 14% compared to 3.7% of the SSD set (p < 10−300). Synthetic lethality is low across varying sequence divergence levels for the SSD set, while it is generally high at moderate to high sequence similarity for the WGD duplicates (Figure 5B). Interestingly, the decreasing frequency of compensation between duplicate genes as sequences diverge is observable in the WGD set but not in the SSD set. To control for potential functional bias in synthetic lethality results for WGD and SSD pairs, we randomly selected SSD pairs with the same GO slim biological process distribution as the WGD genes and repeated the analysis. As shown in supplemental Figure S6C (http://www.genetics.org/supplemental/), our results are robust to the functional composition differences in the two sets. In addition, to control for the potential complicating factor that the above differences were caused by a larger number of multigene families in the SSD set, we repeated the analysis with reciprocal best hits. The result (supplemental Figure S6, D and E) is robust against this modification.
Expression pattern is more diverged between the WGD paralogs:
Population genetics predicts that a duplicate copy that is entirely redundant cannot be maintained in the genome for a long time, except in cases of concerted evolution for which a larger amount of gene product is beneficial (Zhang 2003). As WGD genes are more similar in function compared to the SSD set, we suspect that their partition of function is achieved at the expression level. We approached this possibility by examining three lines of evidence. First, from a sequence perspective, we found that the upstream regulatory regions are more diverged between WGD paralogs for the same level of ORF sequence divergence (Figure 6A, P = 0.01). Such a result is consistent after removal of ribosomal genes (Figure 6B, P < 0.005). Second, we used the Lee et al. (2002) data measuring transcription factor binding by ChIP–chip and found that over different ORF sequence divergence levels, there are less transcription factor-binding sites in common between WGD paralogs (Figure 6C), with a similar result found after removal of ribosomal genes (Figure 6D). This result is robust against different cutoff levels (supplemental Figure S7 at http://www.genetics.org/supplemental/, P < 1.0−30 over all cutoffs) used for identifying transcription factor-binding sites from the ChIP–chip data. These together suggest that at the same level of sequence similarity, regulatory sequences of the WGD paralogs diverge more than those of the SSD paralogs.
Figure 6.—

Divergence of the upstream regulatory region and transcription factor-binding sites between paralogs across sequence divergence. (A) Alignment of the upstream 1000 kb between pairs. The nonoverlapping percentage of identity of the upstream 1000 kb (E = 1) was calculated and the average was taken. The upstream regions between the background duplicate pair generally align better, especially at high percentage of identity groups. Such a result is in accordance with the expression pattern of which WGD pairs diverge more. (B) Alignment of the upstream 1000 kb between pairs after removal of ribosomal genes. (C) Frequency of shared transcription factor-binding sites. WGD paralogs are significantly weaker in sharing transcription factor-binding sites. (D) Frequency of shared transcription factor-binding sites between paralogs after removal of ribosomal genes.
Third, we analyzed the similarity of gene expression using a microarray data set in which transcripts were treated as quantitative traits (Brem and Kruglyak 2005). This experiment was uniquely designed to find genetically segregating determinants of gene expression level rather than physiological ones. High correlation in gene expression would thus indicate shared regulators of gene expression whereas lower correlation would imply that different regulators are utilized. The expression correlation result is in congruence with the sharing of transcription factors (Figure 7A, P < 0.02 for bins of >25% sequence similarity). The lower correlation between WGD paralogs is even more pronounced after the removal of the ribosomal genes (Figure 7B, P = 0.024). Our result suggests that coding sequence divergence and expression divergence between duplicate genes are correlated in the SSD set but not in the WGD set, which reconciles previous disputes (Wagner 2000b; Gu et al. 2002, 2005; Zhang et al. 2004) on this relationship (see discussion).
Figure 7.—

Divergence of expression pattern between paralogs across sequence divergence. (A) The correlation of expression patterns between paralogs. Expression patterns between the WGD paralogs are highly diverged especially after the removal of ribosomal genes, indicating their role in finely modulating expression levels. The expression correlation coefficient between two random genes in the data set is on average 0.003. (B) The correlation of expression patterns between paralogs after removal of ribosomal genes.
DISCUSSION
Our genome-scale analysis provided systematic discrimination between WGD and SSD sets with respect to the tendency toward functional conservation/divergence between paralogs. Independent of sequence divergence, the WGD paralogs in general are more likely to share physical protein-interaction partners and functional relationships on the basis of integrated functional genomic evidence. This major tendency toward similar function is supported by the higher rate of synthetic lethality in the WGD paralogous pairs. However, similarity and compensation in function does not lead to complete redundancy, because compared to SSD paralogs, WGD paralogs show more diverged expression patterns and upstream regulatory regions, implying their role in fine tuning expression level. As is discussed later, the results of our global study reconcile several previously inconsistent observations that were due to selection of varying sets of duplicates and suggest different tendencies of evolutionary fate and consequences between whole-genome and smaller-scale duplicates.
It is of interest to explore the biological reasons for the functional differences between the whole-genome and smaller-scale duplicates. The much higher propensity of WGD paralogs to be synthetically lethal suggests that retention of two copies after the whole-genome duplication offers robustness to the genome. Such a result is readily in agreement with our global functional analysis. Besides synthetic lethality, genetic robustness can also be supported by paralogs that could compensate for each other in expression. The WGD paralogs NHP6A and NHP6B, which share 89% sequence identity, are an example of this phenomenon. Deletion of NHP6A leads to a threefold increase in NHP6B synthesis while an extra copy of NHP6A reduces NHP6B expression twofold (Kolodrubetz et al. 2001). Changes in the NHP6B gene copy number cause similar but more moderate changes in NHP6A expression (Kolodrubetz et al. 2001). However, such enhanced expression of one gene in response to deletion of its paralog may not be a general pattern in either the WGD or the SSD sets on the basis of our analysis of genomewide expression data in deletion strains (Hughes et al. 2000) (supplemental Figure S8 at http://www.genetics.org/supplemental/). It should also be emphasized that the observed patterns may be subject to alternative explanations. It is also possible that retaining functionally similar genes could be caused by dosage effect (Zhang 2003; Kondrashov and Kondrashov 2006).
Another reason for keeping two similar genes in the genome is suggested by the lower level of coexpression and sharing of upstream regulatory regions between the WGD paralogs. This suggests that WGD pairs, while showing a lower frequency of essentiality, could be beneficial under other environmental and experimental conditions. For example, Ser/Thr kinases DBF2 and DBF20 are 77% identical in sequence, highly functionally related (0.99 confidence of functional relationship as predicted by the Bayesian network), and synthetically lethal. However, their expression correlation coefficient is negative on the basis of the data set of Brem and Kruglyak (2005). Functionally related WGD paralogous pairs with negative expression correlations are not uncommon: 74% of paralogs with negative correlation are predicted to be functionally related by our Bayesian method (at a confidence level higher than the baseline prior), compared to the genome average of 1.6%. This suggests a possible role of the WGD duplicates in expression modulation. Another example of this phenomenon is the WGD sulfate permease paralogs SUL1 and SUL2, which share 65% identity, are functionally related on the basis of the integrated genomic data, and show phenotypic enhancement (Cherest et al. 1997). However, their Pearson correlation coefficient is only 0.163, compared to 0.288 of the SSD group at the same sequence divergence level. Additionally, expression of SUL2, but not SUL1, is under the control of SUL3 (Cherest et al. 1997). Our result suggests that divergence at the expression level is more significant in the WGD duplicates than in the SSD set.
We also found that the role of sequence divergence in functional attributes of duplicate genes is often different for the WGD and SSD sets. This difference can cause studies based on different subsets of WGD and SSD data to arrive at different conclusions. In fact, our global analyses reconciled two previous disputes on the essentiality and expression divergence of paralogs in relation to sequence divergence. First, we investigated the relationship between sequence similarity of duplicate genes and the fitness effect of a null mutation. The conclusion that no relationship could be established (Wagner 2000a) was based on 45 duplicate genes having a paralog in a syntenic block on another yeast chromosome, namely, duplicates from WGD. The opposite conclusion that a high correlation can be established (Gu et al. 2003) was based on a genomewide group of 1147 duplicated pairs, corresponding to the combination of the WGD and SSD sets used in our study. Our result (Figure 5A) confirms, but qualifies, both conclusions by showing that the correlation between sequence similarity and essentiality, which is absent in the WGD set as suggested by Wagner (2000a), can be observed in the SSD set as in Gu et al. (2003). Second, we reconcile a previous dispute on whether coding sequence divergence and expression divergence between duplicate genes are coupled. A previous study based on 376 WGD paralogous pairs (after removal of ribosomal genes) found no significant correlation between protein-sequence divergence and regulatory region/expression divergence (Wagner 2000b), which is in agreement with our result based on the WGD set (Figure 6, A and B). On the other hand, a positive correlation between expression divergence and nonsynonymous divergence (Gu et al. 2002, 2005) or the age of duplicates (Zhang et al. 2004) was established on the basis of the paralogs identified genomewide, corresponding to a combination of WGD and SSD duplicates in our study. Our global comparative result on WGD and SSD sets reconciles these different results (Figure 6, A and B, Figure 7) and suggests that the correlation between expression or upstream regulatory region divergence and coding sequence divergence is evident in the SSD set, but relatively weak in the WGD set.
Similarly, caution should be taken when drawing conclusions about the relationship between physical interaction, functional relationship, or synthetic lethality and sequence divergence. For example, while the conclusion that no simple relationship could be established between sequence identity and functional similarity (Baudot et al. 2004) is reasonable for the set of WGD paralogs, we found an almost linear relationship in the SSD set (Figure 4B). Using a mixture of the two sets (for example, Brun et al. 2003 used 10 WGD and 10 SSD duplicates) often fails to establish such a relationship, probably due to the interference from WGD duplicates. The decoupling of sequence divergence and functional divergence in the WGD set is especially worth considering. A recent study (Fares et al. 2006) suggests that positive selection was indeed detected in one or both of the WGD paralogs when compared to nonduplicated orthologous sequences. However, we found that such positive selection on the sequence level cannot be connected to divergence between paralogs on the functional level. Most of the positively selected paralogs that Fares et al. identified, despite their large sequence divergence, are highly functionally related and, in several cases, synthetically lethal. For example, both of the SUMO ligases SIZ1 and SIZ2I are positively selected (Fares et al. 2006) and the in-paralog divergence is large (32% identity). Nevertheless, they are predicted to share a function relationship (0.99 confidence predicted by the Bayesian network) and are synthetically lethal (see supplemental information at http://www.genetics.org/supplemental/).
It should be emphasized that the above results represent behavior of the majority of the WGD duplicates in comparison to the SSD duplicates. Most of the WGD duplicates have moderate or high sequence similarity (Figure 1) and follow the general trends proposed by our analysis. However, at high sequence divergence, the WGD paralogs diverge in function quickly, as indicated by shared interaction partners and functional relationships (Figure 4, A and B). The frequency of synthetic lethal pairs also drops substantially (Figure 5B), indicating that paralogous pairs lose functional compensation as their sequences diverge. Another interesting subset of duplicates was identified by Kellis et al. (2004a), who suggested a group of “fast” WGD pairs showing accelerated protein evolution. The relatively small number of fast WGD pairs prevented us from a thorough analysis by partitioning according to sequence divergence. Nevertheless, we found that the general enrichment of biological processes is different for the fast set from the rest of the WGD paralogs (supplemental Figure S9 at http://www.genetics.org/supplemental/). Thus, while this study provides a global comparison of functional divergence for duplicates that arose from the whole-genome duplication or small-scale duplications, specific subgroups of duplicates may exhibit different behaviors.
We envision several directions that can further address the differences between whole-genome and small-scale duplications. First, our present study focuses on the behavior of the duplicated genes as the sequences of the paralogs diverge. Several recent studies identified asymmetric evolution based on WGD duplicates compared to nonduplicated orthologs (Kellis et al. 2004a,b; Fares et al. 2006; Kim and Yi 2006, etc.). The relative strength of the asymmetric evolution in WGD vs. SSD duplicates would be interesting to investigate. Second, our results suggest functional divergence at low alignment. However, we do not discriminate between neofunctionalization and subfunctionalization. Studies on individual pairs using interaction data and/or moving-window analysis could be informative. In addition, a recent study reports network partition in the WGD genes (Conant and Wolfe 2006). Investigation of the presence and partition of such a network in the SSD set could provide insight into whether whole-genome duplication is more likely to result in duplication of an entire pathway or expression network. Finally, the present study compares the WGD set and the SSD set by stratifying according to sequence similarity. Estimating the age of the duplicates will further clarify the divergence variation caused by age or a specific duplicate set. The growing amounts of large-scale genomic data can enable comprehensive comparison between whole-genome duplication and small-scale duplication in a variety of organisms.
Acknowledgments
We thank Chad Myers and Matthew Hibbs for insightful discussions, helping with the data, the processing of expression data, and correction of the manuscript. We appreciate the constructive suggestions from the two anonymous reviewers. This research was partially supported by National Institutes of Health grant R01 GM071966 and National Science Foundation grant IIS-0513552 to O.G.T. O.G.T. is an Alfred P. Sloan Research Fellow.
References
- Altschul, S. F., W. Gish, W. Miller, E. W. Myers and D. J. Lipman, 1990. Basic local alignment search tool. J. Mol. Biol. 215: 403–410. [DOI] [PubMed] [Google Scholar]
- Ashburner, M., X. A. Ball, J. A. Blake, D. Botstein, H. Butler et al., 2000. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet. 25: 25–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baudot, A., B. Jacq and C. Brun, 2004. A scale of functional divergence for yeast duplicated genes revealed from analysis of the protein-protein interaction network. Genome Biol. 5: R76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breitkreutz, B. J., C. Stark and M. Tyers, 2003. The GRID: the General Repository for Interaction Datasets. Genome Biol. 4(3): R23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brem, R. B., and L. Kruglyak, 2005. The landscape of genetic complexity across 5,700 gene expression traits in yeast. Proc. Natl. Acad. Sci. USA 102(5): 1572–1577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brun, C., A. Guénoche and B. Jacq, 2003. Approach of the functional evolution of duplicated genes in Saccharomyces cerevisiae using a new classification method based on protein-protein interaction data. J. Struct. Funct. Genomics 3: 213–224. [PubMed] [Google Scholar]
- Byrne, K. P., and K. H. Wolfe, 2005. The Yeast Gene Order Browser: combining curated homology and syntenic context reveals gene fate in polyploidy species. Genome Res. 15(10): 1456–1461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cherest, H., J. C. Davidian, D. Thomas, V. Benes, W. Ansorge et al., 1997. Molecular characterization of two high affinity sulfate transporters in Saccharomyces cerevisiae. Genetics 145: 627–635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cherry, J. M., C. Adler, C. Ball, S. A. Chervitz, S. S. Dwight et al., 1998. SGD: Saccharomyces genome database. Nucleic Acids Res. 26(1): 73–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conant, G. C., and K. H. Wolfe, 2006. Functional partitioning of yeast co-expression networks after genome duplication. PLoS Biol. 4(4): e109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis, J. C., and D. A. Petrov, 2005. Do disparate mechanisms of duplication add similar genes to the genome? Trends Genet. 21: 548–551. [DOI] [PubMed] [Google Scholar]
- Dempster, A. P., N. M. Laird and D. B. Rubin, 1977. Maximum likelihood from incomplete data via EM algorithm. J. R. Stat. Soc. Methodol. 39: 1–38. [Google Scholar]
- Dietrich, F. S., S. Voegeli, S. Brachat, A. Lerch, K. Gates et al., 2004. The Ashbya gossypii genome as a tool for mapping the ancient Saccharomyces cerevisiae genome. Science 304(5668): 304–307. [DOI] [PubMed] [Google Scholar]
- Fares, M. A., K. P. Byrne and K. H. Wolfe, 2006. Rate asymmetry after genome duplication causes substantial long-branch attraction artifacts in the phylogeny of Saccharomyces species. Mol. Biol. Evol. 23(2): 245–253. [DOI] [PubMed] [Google Scholar]
- Force, A., M. Lynch, F. B. Pickett, A. Amores, Y. L. Yan et al., 1999. Preservation of duplicate genes by complementary, degenerative mutations. Genetics 151: 1531–1545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu, X., 2003. Evolution of duplicate genes versus genetic robustness against null mutations. Trends Genet. 19: 354–356. [DOI] [PubMed] [Google Scholar]
- Gu, X., Z. Zhang and W. Huang, 2005. Rapid evolution of expression and regulatory divergences after yeast gene duplication. Proc. Natl. Acad. Sci. USA 102(3): 707–712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu, Z., D. Nicolae, H. H. Lu and W. H. Li, 2002. Rapid divergence in expression between duplicate genes inferred from microarray data. Trends Genet. 18(12): 609–613. [DOI] [PubMed] [Google Scholar]
- Gu, Z., L. M. Steinmetz, X. Gu, C. Scharfe, R. W. Davis et al., 2003. Role of duplicate genes in genetic robustness against null mutations. Nature 421: 63–66. [DOI] [PubMed] [Google Scholar]
- Hughes, T. R., M. J. Marton, A. R. Jones, C. J. Roberts, R. Stoughton et al., 2000. Functional discovery via a compendium of expression profiles. Cell 102(1): 109–126. [DOI] [PubMed] [Google Scholar]
- Jacq, B., 2001. Protein function from the perspective of molecular interactions and genetic networks. Brief. Bioinform. 2: 38–50. [DOI] [PubMed] [Google Scholar]
- Jordan, I. K., Y. I. Wolf and E. V. Koonin, 2004. Duplicated genes evolve slower than singletons despite the initial rate increase. BMC Evol. Biol. 4: 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kellis, M., B. W. Birren and E. S. Lander, 2004. a Proof and evolutionary analysis of ancient genome duplication in the yeast Saccharomyces cerevisiae. Nature 428(6983): 617–624. [DOI] [PubMed] [Google Scholar]
- Kellis, M., N. Patterson, B. Birren, B. Berger and E. S. Lander, 2004. b Methods in comparative genomics: genome correspondence, gene identification and regulatory motif discovery. J. Comput. Biol. 11: 319–355. [DOI] [PubMed] [Google Scholar]
- Kim, S. H., and S. V. Yi, 2006. Correlated asymmetry of sequence and functional divergence between duplicate proteins of Saccharomyces cerevisiae. Mol. Biol. Evol. 23(5): 1068–1075. [DOI] [PubMed] [Google Scholar]
- Kolodrubetz, D., M. Kruppa and A. Burgum, 2001. Gene dosage affects the expression of the duplicated NHP6 genes of Saccharomyces cerevisiae. Gene 272: 93–101. [DOI] [PubMed] [Google Scholar]
- Kondrashov, F. A., and A. S. Kondrashov, 2006. Role of selection in fixation of gene duplications. J. Theor. Biol. 239: 141–151. [DOI] [PubMed] [Google Scholar]
- Kondrashov, F. A., I. B. Rogozin, Y. I. Wolf and E. V. Koonin, 2002. Selection in the evolution of gene duplications. Genome Biol. 3(2): RESEARCH0008. [DOI] [PMC free article] [PubMed]
- Lee, T. I., N. J. Rinaldi, F. Robert, D. T. Odom, Z. Bar-Joseph et al., 2002. Transcriptional regulatory networks in Saccharomyces cerevisiae. Science 298: 799–804. [DOI] [PubMed] [Google Scholar]
- Li, W. H., J. Yang and X. Gu, 2005. Expression divergence between duplicate genes. Trends Genet. 21(11): 602–607. [DOI] [PubMed] [Google Scholar]
- Lynch, M., and J. S. Conery, 2000. The evolutionary fate and consequences of duplicate genes. Science 290: 1151–1155. [DOI] [PubMed] [Google Scholar]
- Lynch, M., and A. Force, 2000. The probability of duplicate gene preservation by subfunctionalization. Genetics 154: 459–473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mewes, H. W., C. Amid, R. Arnold, D. Frishman, U. Guldener et al., 2004. MIPS: analysis and annotation of proteins from whole genomes. Nucleic Acids Res. 32: D41–D44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myers, C. L., D. Robson, A. Wible, M. A. Hibbs, C. Chiriac et al., 2005. Discovery of biological networks from diverse functional genomic data. Genome Biol. 6(13): R114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myers, C. L., D. R. Barrett, M. A. Hibbs, C. Huttenhower and O. G. Troyanskaya, 2006. Finding function: evaluation methods for functional genomic data. BMC Genomics 7(1): 187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nadeau, J. H., and D. Sankoff, 1997. Comparable rates of gene loss and functional divergence after genome duplications early in vertebrate evolution. Genetics 147: 1259–1266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nowak, M. A., M. C. Boerlijst, J. Cooke and J. M. Smith, 1997. Evolution of genetic redundancy. Nature 388: 167–171. [DOI] [PubMed] [Google Scholar]
- Ohno, S., 1970. Evolution by Gene Duplication. Springer-Verlag, New York.
- Sankoff, D., 2001. Gene and genome duplication. Curr. Opin. Genet. Dev. 11: 681–684. [DOI] [PubMed] [Google Scholar]
- Seoighe, C., and K. H. Wolfe, 1999. Yeast genome evolution in the post-genome era. Curr. Opin. Microbiol. 2: 548–554. [DOI] [PubMed] [Google Scholar]
- Stoltzfus, A., 1999. On the possibility of constructive neutral evolution. J. Mol. Evol. 49: 169–181. [DOI] [PubMed] [Google Scholar]
- Taylor, J. S., and J. Raes, 2004. Duplication and divergence: the evolution of new genes and old ideas. Annu. Rev. Genet. 38: 615–643. [DOI] [PubMed] [Google Scholar]
- Tong, A. H. Y., M. Evangelista, A. B. Parsons, H. Xu, G. D. Bader et al., 2001. Systematic genetic analysis with ordered arrays of yeast deletion mutants. Science 294: 2364–2368. [DOI] [PubMed] [Google Scholar]
- Troyanskaya, O., M. Cantor, G. Sherlock, P. Brown, T. Hastie et al., 2001. Missing value estimation methods for DNA microarrays. Bioinformatics 17(6): 520–525. [DOI] [PubMed] [Google Scholar]
- Wagner, A., 2000. a Robustness against mutations in genetic networks of yeast. Nat. Genet. 24: 355–361. [DOI] [PubMed] [Google Scholar]
- Wagner, A., 2000. b Decoupled evolution of coding region and mRNA expression patterns after gene duplication: implications for the neutralist-selectionist debate. Proc. Natl. Acad. Sci. USA 97(12): 6579–6584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolfe, K. H., and W. H. Li, 2003. Molecular evolution meets the genomics revolution. Nat. Genet. 33(Suppl.): 255–265. [DOI] [PubMed] [Google Scholar]
- Zhang, J., 2003. Evolution by gene duplication—an update. Trends Ecol. Evol. 18: 292–298. [Google Scholar]
- Zhang, J., J. Gu and X. Gu, 2004. How much expression divergence after yeast gene duplication could be explained by regulatory motif evolution? Trends Genet. 20(9): 403–407. [DOI] [PubMed] [Google Scholar]