

Abstract
The importance of quantifying and accounting for stochastic genotyping errors when analyzing microsatellite data is increasingly being recognized. This awareness is motivating the development of data analysis methods that not only take errors into consideration but also recognize the difference between two distinct classes of error, allelic dropout and false alleles. Currently methods to estimate rates of allelic dropout and false alleles depend upon the availability of error-free reference genotypes or reliable pedigree data, which are often not available. We have developed a maximum-likelihood-based method for estimating these error rates from a single replication of a sample of genotypes. Simulations show it to be both accurate and robust to modest violations of its underlying assumptions. We have applied the method to estimating error rates in two microsatellite data sets. It is implemented in a computer program, Pedant, which estimates allelic dropout and false allele error rates with 95% confidence regions from microsatellite genotype data and performs power analysis. Pedant is freely available at http://www.stats.gla.ac.uk/∼paulj/pedant.html.
THE importance of quantifying and accounting for stochastic genotyping errors in microsatellite-based studies is becoming ever more widely recognized. Undetected errors can impair inference across a range of fields, including forensics, genetic epidemiology, kinship analysis, and population genetics (Pompanon et al. 2005). All studies that have looked for genotyping errors have found them at appreciable levels (0.2–15% per locus; Pompanon et al. 2005). Even at low error rates, the frequency of erroneous genotypes increases rapidly with the number of marker loci assayed: from 1% in one locus, to 10% in 10 loci, to a potentially destructive 63% in 100 loci. Error-free microsatellite data sets must therefore be rare and will become rarer as improving laboratory methods allow increasing numbers of samples and markers to be assayed. Thus, although errors are most obviously harmful when genotyping highly error-prone noninvasive samples (Gagneux et al. 1997), they can frustrate analysis of the cleanest data, for example, in mapping genes that contribute to complex disease (Feakes et al. 1999; Walters 2005). The consequences of undetected genotyping errors can be particularly adverse for parentage analysis, especially when using exclusion, where incompatibilities between candidate parents and offspring are used to exclude all but the true parent (Gagneux et al. 1997; Jones and Ardren 2003). Even an error rate as low as 2% in a nine-locus data set can result in false exclusion of >20% of fathers (Hoffman and Amos 2005).
Given that genotyping errors cannot be eliminated with certainty, a more pragmatic approach is to minimize (Piggott et al. 2004), quantify (Bonin et al. 2004; Broquet and Petit 2004; Hoffman and Amos 2005), and integrate them in statistical analysis (Marshall et al. 1998; Sobel et al. 2002; Wang 2004). Most studies quantify error rate as a single quantity, such as error rate per allele or per single-locus genotype. However, stochastic errors (as opposed to systematic errors, for example, null alleles) can be divided into two distinct classes: allelic dropout, where one allele of a heterozygote randomly fails to PCR amplify, and false alleles, where the true allele is misgenotyped because of factors such as PCR or electrophoresis artifacts or human errors in reading and recording data (Broquet and Petit 2004). These two classes of error can bias analyses in fundamentally different ways. For example, a high level of undetected allelic dropout could be misinterpreted as evidence for inbreeding, while false alleles can lead to substantial overestimation of census size (Waits and Leberg 2000; Creel et al. 2003).
The essential difference between the effects of the two classes of error, as far as kinship inference is concerned, is that both homozygotes and heterozygotes potentially contain false alleles, but only homozygotes can be suspected of allelic dropout. Consider a data set with a low false allele rate but a high allelic dropout rate, a common scenario for genotypes from noninvasive samples such as feces or hair (Broquet and Petit 2004). A candidate father with genotype AA could not be excluded with high confidence from paternity of an offspring with genotype CC because of the high probability of allelic dropout, but if the observed genotypes were AB and CD, respectively, the candidate father could be excluded with greater certainty. Thus, knowledge of both error rates allows the likelihood of paternity to be assessed more accurately than is possible when using a single composite error rate. By the same logic, any microsatellite analysis that incorporates error probability would benefit from differentiating between allelic dropout and false alleles.
This approach is being increasingly applied to analysis methods such as sibship reconstruction (Sieberts et al. 2002; Wang 2004), parentage assignment (Hadfield et al. 2006), and establishment of genotypic identity between two DNA samples (Kalinowski et al. 2006). By analyzing simulated data from 100 individuals at eight microsatellite loci, Wang (2004, Figure 2 therein) showed that sibships can be reconstructed accurately when the probabilities of allelic dropout and false alleles are as high as 20% each per single-locus genotype and only 3% of multilocus genotypes are expected to be error free.
Figure 2.—


The effect of sample size (n), false allele error rate (ɛ2), and expected heterozygosity (He) on relative bias in estimating an allelic dropout rate (ɛ1) of 0.01. We analyzed 5000 simulated data sets for each parameter combination. (A) He = 0.5; (B) He = 0.85.
The utility of such methods depends on the ability to quantify the separate error rates. Errors can be quantified separately either through Mendelian inconsistencies between parent–offspring pairs or by comparing error-prone genotypes with reference genotypes, which are assumed to be error free. Reference genotypes can be obtained either from high-quality template DNA (but see Jeffery et al. 2001) or by repeated PCRs (Ewen et al. 2000; Broquet and Petit 2004; Pompanon et al. 2005). However, in many studies neither pedigree data nor reference samples will be available, and the production of reference data by multiple genotyping can be time consuming and expensive (Navidi et al. 1992; Taberlet et al. 1996; Smith et al. 2000; Miller et al. 2002). There is therefore a need to develop a method for estimating allelic dropout and false allele error rates that does not depend on pedigree data, high-quality reference samples, or multiple genotyping.
We describe a method for estimating maximum-likelihood rates of allelic dropout and false allele error from microsatellite genotype data. The method compares duplicate genotypes and estimates error rates on the basis of the frequency and nature of mismatches. It is already considered good practice to duplicate 5–10% of genotypes to monitor overall genotyping error (Bonin et al. 2004; Hoffman and Amos 2005; Pompanon et al. 2005), but it has not been possible to use these data to assess both allelic dropout and false alleles separately. The method presented here therefore extracts valuable information from data that researchers will often have already obtained. Both duplicate genotypes are assumed to be error prone, so reference data are not required. We demonstrate the effectiveness of the method using simulated and real data.
METHODS
Notation and terminology:
Genotyping error rates are conventionally expressed per genotype rather than per allele, reflecting the fact that they are usually calculated in terms of the observed number of erroneous genotypes (judged against reference genotypes) divided by the number of genotypes in which an error could have been observed (Broquet and Petit 2004). The per-genotype allelic dropout rate (p) is calculated as a proportion of observed heterozygotes (because allelic dropout can be observed only in heterozygotes) whereas the false allele rate (f) is calculated as a proportion of all genotypes. Per-genotype error rates are convenient to use and simple to calculate when reference genotypes are available but in the method presented here, which involves modeling the processes through which errors affect individual alleles, per-allele rates are more useful. In practice, conversion between per-allele and per-genotype rates is straightforward (see appendix).
Using Wang's (2004) notation for per-allele error rates, ɛ1 is the population allelic dropout rate and ɛ2 is the population false allele rate (defined below). Additionally, we define the sample error rates, 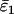 and
and 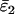 , which are the error rates in a single sample of replicate genotypes. For example, a sample of 200 duplicate genotypes contains 800 scored alleles. If 4 of these have dropped out then
, which are the error rates in a single sample of replicate genotypes. For example, a sample of 200 duplicate genotypes contains 800 scored alleles. If 4 of these have dropped out then 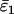 = 0.005, regardless of whether the dropouts occurred visibly in heterozygotes or invisibly in homozygotes. Finally, the error rate estimates are
= 0.005, regardless of whether the dropouts occurred visibly in heterozygotes or invisibly in homozygotes. Finally, the error rate estimates are 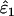 and
and 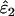 .
.
Where ambiguity between true and error-prone genotypes is possible, we refer to the recorded error-prone genotype as the “observed genotype” and the unknown true genotype (which would be observed in the absence of errors) as the “underlying genotype.” An underlying genotype that has been ascertained by a process that is assumed to be error free is referred to as a “reference genotype.” In reality even reference genotypes can contain errors. Finally, unless stated otherwise, the term “genotype” refers to a single-locus genotype.
General approach:
When reference genotypes are available, quantifying allelic dropout and false alleles is a simple matter of comparing the reference and observed genotypes and counting frequencies of the two error classes. For example, a mismatch between a reference genotype AB and an observed genotype AA indicates an allelic dropout, whereas a mismatch between a reference genotype AA and an observed genotype AB indicates the appearance of a false allele (B) in the observed genotype (hereafter all replicate genotypes are shown in the form AA.AB).
Although errors can also be counted from a single set of duplicate genotypes in the absence of reference data, classifying them is more problematic because both replicate genotypes are error prone and allelic dropout and false alleles can produce equivalent mismatches (e.g., the mismatch AA.AB could have been produced by allelic dropout in AB or the occurrence of a false allele in AA). However, information about the magnitudes of ɛ1 and ɛ2 can be derived from the frequencies of different categories of mismatch. When ɛ1 is high, AA.AB mismatches will be common, while a high frequency of AB.AC mismatches indicates high ɛ2.
A simple way to circumvent the ambiguity of AA.AB mismatches is to estimate ɛ2 and ɛ1 consecutively. AB.AC mismatches are unambiguously attributable to false allele errors in heterozygotes and can be used to estimate ɛ2 across the entire sample. This estimate of ɛ2 can in turn be used to estimate the proportion of AA.AB observations expected to result from a false allele in a homozygote. The remainder of AA.AB duplicates can then be attributed to allelic dropout and used to estimate ɛ1. However, because both replicate genotypes are error prone, the probability of both replicates incurring errors (a double error) is not negligible. If the total probability of an error of either class occurring in a single genotype (p + f) is a realistically high 0.2 (Broquet and Petit 2004), then 5.2% of replicates will be hit by at least two errors, and these will account for 27% of errors. Ignoring these classes will result in substantial underestimates of high error rates (we verified this result using simulations—data not shown), and including them in the simple sequential approach described above becomes impossible because of ambiguity in the origins of some categories of replicate genotype. For example, if double errors are considered, the apparently error-free AA.AA category could arise from an underlying genotype of AB by two dropouts, and AA.BB could originate either from AB by two dropouts or from AA by acquisition of a false allele (B) followed by a dropout. Given these complications, rather than calculating ɛ2 and ɛ1 sequentially, it is preferable to estimate them simultaneously from all the available data using maximum likelihood (ML). This method has the added advantage of allowing simple calculation of confidence limits.
There are seven possible categories of duplicate genotype: (1) AA.AA, (2) AB.AB, (3) AA.AB, (4) AA.BB, (5) AB.AC, (6) AB.CC, and (7) AB.CD. Categories that include two allelic dropouts in a single genotype are not counted because double dropouts are indistinguishable from other causes of PCR failure and are therefore unreliable indicators of ɛ1.
Like the simple sequential method described above for estimating ɛ1 and ɛ2 without reference data, the basis of the ML method is the sensitivity of categories 1–7 to ɛ1 and ɛ2. Assuming that the underlying proportion of heterozygotes is known, the expected frequencies of all categories and the likelihood of the observed frequencies can be calculated for any ɛ1 and ɛ2, allowing the ML estimates, 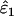 and
and 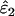 , to be obtained.
, to be obtained.
Assumptions and model:
We base our assumptions and error model on those proposed by Wang (2004), with minor modifications. We make the following assumptions:
The genotypes are diploid and codominant.
The sampled population is in Hardy–Weinberg equilibrium. This assumption is usually desirable because it allows expected heterozygosity (
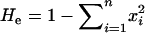 , where xi is the frequency of the ith of n alleles) to be used to gauge the probability that an underlying genotype is heterozygous. In an error-free data set this probability is the observed heterozygosity (Ho), but direct estimates of Ho will be biased downward by allelic dropout and upward by false alleles. Neither dropout nor false alleles should significantly bias estimation of He, assuming that all alleles are equally likely to drop out and that false alleles generally do not create new allelic states. A known degree of nonrandom mating can be accounted for by estimating Ho from He and FIS, the inbreeding coefficient, where Ho = He(1 − FIS). In practice, unbiased FIS estimates are unlikely to be available when errors are frequent, but in cases where the sample contains more than one population, heterozygote deficiency due to spatial genetic structure could be quantified and corrected for by replacing FIS with FST, which is relatively insensitive to genotyping error (Taberlet et al. 1999). We investigate the effect of undetected deviation from random mating on error rate estimation using simulations.
, where xi is the frequency of the ith of n alleles) to be used to gauge the probability that an underlying genotype is heterozygous. In an error-free data set this probability is the observed heterozygosity (Ho), but direct estimates of Ho will be biased downward by allelic dropout and upward by false alleles. Neither dropout nor false alleles should significantly bias estimation of He, assuming that all alleles are equally likely to drop out and that false alleles generally do not create new allelic states. A known degree of nonrandom mating can be accounted for by estimating Ho from He and FIS, the inbreeding coefficient, where Ho = He(1 − FIS). In practice, unbiased FIS estimates are unlikely to be available when errors are frequent, but in cases where the sample contains more than one population, heterozygote deficiency due to spatial genetic structure could be quantified and corrected for by replacing FIS with FST, which is relatively insensitive to genotyping error (Taberlet et al. 1999). We investigate the effect of undetected deviation from random mating on error rate estimation using simulations.Each sample is equally likely to incur an error. In reality errors will preferentially affect low-quality samples (Pompanon et al. 2005). The effect of nonindependence among errors on both simulated and real data is explored and discussed below. Moreover, false alleles might not affect homozygotes and heterozygotes with equal probability. For example, allele-calling errors are probably more likely in heterozygotes, whereas PCR artifacts are more likely to be recorded in homozygous genotypes. The confounding effect of such opposing biases could be overcome by modeling false alleles as a product of two or more processes. However, to do so would require the introduction of at least one additional parameter at the cost of reduced statistical power, increased mathematical complexity, and longer computing time.
Both alleles of a heterozygote are equally likely to drop out. In many instances of allelic dropout, short alleles are preferentially amplified over long alleles (short allele dominance; see Wattier et al. 1998; Ewen et al. 2000; Jeffery et al. 2001). High rates of short allele dominance will cause underestimation of allelic dropout rates by any method dependent on replication of error-prone genotypes.
A false allele always takes an allelic state not already present in the duplicate genotype. For example, an underlying genotype AB can be duplicate genotyped as AB.CC only by the occurrence in the second genotype of a false allele (C) in one allele followed by a dropout of the other allele, not by the occurrence of two identical false alleles. Likewise, AB.AB can arise only by an error-free read of an underlying AB, not by the same false allele (B) occurring in both genotypes from an underlying AA. In real data, most false alleles are recorded as existing true alleles (P. C. D. Johnson, personal observation), so that in practice two identical false alleles could occur in a duplicate genotype in either the AB.CC or the AB.AB case. This could lead to underestimation of high ɛ2 at loci with few alleles. However, given the high number of alleles present at most microsatellite loci and the relative rarity of double occurrences of false alleles, this rule is unlikely to lead to significant underestimation of ɛ2. A consequence of this assumption is that we do not include the number of alleles in a data set as a parameter in our model, which greatly simplifies the calculation of the expected duplicate genotype frequencies.
Following Wang's (2004) error model, we define two classes of error. Class 1 consists of allelic dropouts only: each allele drops out with probability ɛ1. Class 2 includes all stochastic errors that lead to a false allele being recorded, such as those caused by PCR and electrophoresis artifacts, allele miscalling either by software or by human error, and data entry. This class comprises all stochastic errors outside class 1. A false allele is recorded with probability ɛ2. Systematic errors, which might be caused by null alleles, contaminant DNA, or systematic miscalling of an allele, are excluded from both classes. Thus in the production of a single diploid genotype, the probabilities of an error of class i occurring in neither allele, one allele, and both alleles are (1 − ɛi)2, 2ɛi(1 − ɛi), and ɛi 2, respectively.
Our error model differs from Wang's (2004) with respect to his assumption that dropouts always precede false allele errors when they occur together in a single allele, resulting in a heterozygous observed genotype (e.g., AB drops out to AA, which acquires a false allele to become AC). We reverse the order, so that dropouts overrule false alleles, leading to a homozygous observed genotype. For example, a dropout and a false allele would coincide in the second allele of an underlying AA as follows: first AA acquires a false allele to be recorded as AB and then the false allele drops out to give AA. Neither model perfectly fits reality: Wang's order correctly models PCR artifacts that are mistaken for alleles, whereas ours fits any false allele that is able to drop out (e.g., miscalling or data entry errors). We chose to make dropouts dominant, first, because miscalling and data entry errors appear to be more common than PCR artifacts, at least in high-quality data (Paetkau 2003; Bonin et al. 2004; Hoffman and Amos 2005; Pompanon et al. 2005), and, second, because it simplifies the calculation of the expected frequencies of the seven categories. In practice, this difference between the two models is slight, as the probability of both errors coinciding in at least one allele in a duplicate genotype is small even at very high error rates (0.05 when ɛ1 = 0.17 and ɛ2 = 0.08).
We make a further simplifying assumption that at all realistic values of ɛ2 the frequency of replicate genotypes affected by three or four false allele errors is negligible. This frequency is 0.2% when the false allele error rate is as high as 0.16 per single-locus genotype (equivalent to ɛ2 = 0.083), the highest recorded in a literature review by Broquet and Petit (2004), so this assumption seems justified.
Under the assumptions above, the expected frequency of each of the seven replicate genotype categories is a function of three parameters: the error rates to be estimated, ɛ1 and ɛ2, and the known expected heterozygosity, He. The likelihood of any combination of ɛ1 and ɛ2 can then be calculated, given He and the counts of the seven categories (see appendix for derivation of equations). Calculating log likelihood across a sufficiently large number of error rate combinations allows a log-likelihood surface to be constructed and the ML estimates 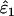 and
and 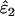 to be located (Figure 1).
to be located (Figure 1).
Figure 1.—

Maximum-likelihood (ML) estimates (+) and confidence regions for the population allelic dropout and false allele rates, ɛ1 and ɛ2 (⋄). ML error rates were estimated from 100 simulated duplicate genotypes where He = 0.85, ɛ1 = 0.05, and ɛ2 = 0.01. The seven duplicate genotype category counts were (10, 67, 17, 2, 2, 0, 0) with two double dropouts uncounted. The sample error rates, 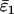 and
and 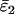 , are also shown (□). Confidence regions were calculated by descending
, are also shown (□). Confidence regions were calculated by descending 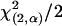 log-likelihood units from the ML estimate, where α = 1 − confidence (Wilks interval).
log-likelihood units from the ML estimate, where α = 1 − confidence (Wilks interval).
A computer program, Pedant, that implements the above method was written in the programming language Delphi version 7.0 (Borland Software). Pedant automates the categorization of the replicate genotypes and finds the ML error rates using a simulated annealing algorithm (see appendix for details). The advantage of simulated annealing is that it reduces the danger of the search getting stuck on a local maximum when searching likelihood space with multiple maxima. In practice, most, if not all, real data sets will produce unimodal surfaces resembling Figure 1. However, it is possible to create artificial data sets that produce two peaks, so it seems prudent to allow for this possibility in the search algorithm, particularly considering that the cost in computation time is small (typically <1 sec/locus using 20,000 search iterations on a 3-GHz Celeron PC).
Simulations:
We tested the method by estimating errors from data with known error rates simulated under the assumptions of the error model. We then repeated the simulation analysis using data that violated the stronger assumptions of the error model. For each data set we generated n underlying genotypes, which were heterozygous with probability He, and then simulated two observed genotypes each with error probabilities ɛ1 and ɛ2. Error rates were estimated from data simulated with low (ɛ1 = 0.01, ɛ2 = 0.0015), intermediate (ɛ1 = 0.09, ɛ2 = 0.04), and high (ɛ1 = 0.17, ɛ2 = 0.08) error rates in all nine possible combinations. These values of ɛ1 and ɛ2 were chosen to reflect a realistic range of genotyping error rates, from levels typical of high-quality samples (Ewen et al. 2000) to rates representing data from low-quality noninvasive samples (Broquet and Petit 2004). Other parameters tested were low (He = 0.5) and high (He = 0.85) expected heterozygosities and small (n = 50), intermediate (n = 100), and large (n = 200) sample sizes. Sampling error in He was simulated as a function of He and n, assuming a broken-stick distribution of allele frequencies (see appendix). All 54 possible combinations of ɛ1, ɛ2, He, and n were simulated. Because generally only a fraction (e.g., 10%) of genotypes are replicated to calculate error rates, sampling error in He was calculated for 10n samples.
The performance of the method in estimating the population error rates was assessed by analyzing error rate estimates from 5000 simulated data sets. Performance was gauged by the mean square error (MSE) between the estimated (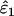 or
or 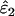 ) and the population error rate (ɛ1 or ɛ2). The MSE can be split into two components, bias and standard error (SE), where MSE = bias2 + SE2. We calculated bias, relative bias, and standard error for each
) and the population error rate (ɛ1 or ɛ2). The MSE can be split into two components, bias and standard error (SE), where MSE = bias2 + SE2. We calculated bias, relative bias, and standard error for each 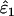 and
and 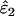 . Bias was calculated as the mean estimated error rate minus the population error rate, and relative bias as bias divided by population error rate.
. Bias was calculated as the mean estimated error rate minus the population error rate, and relative bias as bias divided by population error rate.
MSE depends not only on sampling error in 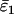 and
and 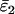 , but also on the number of hidden and ambiguous errors in the sample. The sample will have incurred on average 4nɛ1 dropouts and 4nɛ2 false alleles, so for any ɛ1 and ɛ2, how well the sample represents the population is therefore a function of n and not useful in assessing the performance of the method. Of greater interest is how well the method recovers the sample error rates in spite of the hidden and ambiguous errors, that is, what proportion of the error in the ML estimates is intrinsic to the method (intrinsic error) and not due to sampling error. Intrinsic error was calculated as (mean square error between the estimate and the sample error rate) divided by MSE. The closer that bias, MSE and intrinsic error were to zero, the better the method was judged to be performing.
, but also on the number of hidden and ambiguous errors in the sample. The sample will have incurred on average 4nɛ1 dropouts and 4nɛ2 false alleles, so for any ɛ1 and ɛ2, how well the sample represents the population is therefore a function of n and not useful in assessing the performance of the method. Of greater interest is how well the method recovers the sample error rates in spite of the hidden and ambiguous errors, that is, what proportion of the error in the ML estimates is intrinsic to the method (intrinsic error) and not due to sampling error. Intrinsic error was calculated as (mean square error between the estimate and the sample error rate) divided by MSE. The closer that bias, MSE and intrinsic error were to zero, the better the method was judged to be performing.
The robustness of the method was tested by rerunning the performance analysis on simulated data sets that deviated from some assumptions of the error model in the following ways.
Deviation from Hardy–Weinberg equilibrium:
The effect of undetected nonrandom mating within the sampled population was tested by simulating data at two levels of heterozygote deficiency, defined by FIS-values of 0.0625 and 0.125. These FIS-values equate to inbreeding among first cousins and half-siblings, respectively, and were chosen to represent moderate and high levels of inbreeding within wild vertebrate populations (Slate et al. 2004). Heterozygote deficiency might also result from cryptic genetic structure. We concentrated on investigating heterozygote deficiency (FIS > 0), first, because extreme heterozygote excess (FIS < −0.125) is likely to be rare in nature (Figure 2 of Chesser 1991) and, second, because preliminary simulation analysis suggested that the most severe biases are typically about three times smaller when FIS is negative than when it is positive.
Increased sampling error in He:
The method does not take into account sampling error in He. The effect of increasing sampling error in He to its maximum (when no additional samples are available for estimating He) was tested by decreasing the number of samples from which He was estimated from 10n to n.
Sample quality variation:
We investigated the validity of the assumption that each sample is equally likely to incur an error. This assumption is unlikely to hold true for most data sets: when sample quality varies, errors preferentially occur in low-quality samples (Gagneux et al. 1997; Wandeler et al. 2003; Bonin et al. 2004; Pompanon et al. 2005). To test the effect of variable sample quality on error estimates, we simulated data using a nonuniform distribution of error rates across samples (see appendix).
Dominance of dropouts over false alleles:
The effect of the assumption that dropouts always hide false alleles was assessed by reducing the probability of a dropout hiding a false allele from 1 to 0.5 in the simulated data.
Reference data (e.g., consensus genotypes or pedigree data) are generally required to estimate allelic dropout and false allele error rates. Therefore, we tested the ML method against a reference data-dependent method. We compared the ML estimates with estimates that could have been obtained conventionally had one of the two duplicate genotypes been a reference genotype rather than an error-prone genotype. Thus the reference data method has the advantage that visible errors are unambiguous, but the disadvantage of increased sampling error. The MSEs of the ML and reference data (RD) methods (MSEML and MSERD) were compared for both error rates. To allow this comparison the ML estimates were converted from per-allele to per-genotype rates (see appendix).
Application to real microsatellite data:
The method was tested on microsatellite genotypes from two error-prone sources of DNA: red fox teeth that had been autoclaved to denature rabies virus and fecal samples from Ethiopian wolves. Individual and consensus genotypes were kindly provided by P. Wandeler (foxes) and D. A. Randall (wolves).
For the fox samples, 149–182 consensus genotypes were established from up to nine repeated PCRs at 16 loci: V142, V374, V402, V468, V502, V602, V622 (Wandeler and Funk 2006), AHT-130 (Holmes et al. 1995), CXX-156, CXX-250, CXX-279 (Ostrander et al. 1993), CXX-434, CXX-466, CXX-606, CXX-608 (Ostrander et al. 1995), and c2088 (Holmes et al. 1995; Wandeler 2004). Prior information from genetic data regarding the validity of assuming Hardy–Weinberg equilibrium in fox populations was not available, although using these data Wandeler (2004) found modest heterozygote deficits within populations (FIS = 0.01–0.02) as well as low levels of interpopulation structure (FST = 0.035). The assumption of Hardy–Weinberg equilibrium was therefore unjustified in this data set. We did not correct for either source of heterozygote deficit because FIS would not normally be accurately quantifiable prior to error estimation, and the same may apply to low levels of FST (Bonin et al. 2004). Simulation of error estimation under this total level of heterozygote deficit (FIT = 0.05) predicted small biases of 3% in 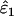 and −8% in
and −8% in 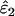 .
.
For the wolf data set, 72–121 consensus genotypes were based on up to 19 repeated genotypes from 17 loci: c377 (Ostrander et al. 1993), FH2001, FH2054, FH2119, FH2137, FH2138, FH2140, FH2159, FH2174, FH2226, FH2293, FH2320, FH2422, FH2472, FH2537 (Breen et al. 2001), Pez17, and Pez19 (Neff et al. 1999). Ethiopian wolf microsatellites would not be expected to show significant deviation from Hardy–Weinberg equilibrium on the basis of prior information from other markers (Gottelli et al. 1994), and this has been confirmed using the present data (Randall 2006).
The first two sets of repeat genotypes were analyzed using the ML method to give 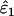 and
and 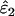 with their 95% confidence limits, the first set of genotypes providing the estimate of He. Error rate estimates and confidence limits were converted to per-genotype error rates for comparison with the sample error rates. The ML estimates were judged by their proximity to the true error rates in the sample, which were counted with reference to the consensus genotypes (with the exception of dropouts in homozygotes, which are invisible even when reference data are available). Because population error rates were not known, relative bias was calculated as (ML estimate)/(sample error rate when greater than zero). These data sets provide a thorough challenge for the ML method because, in addition to covering a wide range of He-values from 0.30 to 0.90 (mean He-values: 0.79 in the foxes, 0.63 in the wolves), they also violate one of the stronger assumptions of the method in having a nonuniform distribution of error rates among samples.
with their 95% confidence limits, the first set of genotypes providing the estimate of He. Error rate estimates and confidence limits were converted to per-genotype error rates for comparison with the sample error rates. The ML estimates were judged by their proximity to the true error rates in the sample, which were counted with reference to the consensus genotypes (with the exception of dropouts in homozygotes, which are invisible even when reference data are available). Because population error rates were not known, relative bias was calculated as (ML estimate)/(sample error rate when greater than zero). These data sets provide a thorough challenge for the ML method because, in addition to covering a wide range of He-values from 0.30 to 0.90 (mean He-values: 0.79 in the foxes, 0.63 in the wolves), they also violate one of the stronger assumptions of the method in having a nonuniform distribution of error rates among samples.
RESULTS
Simulations:
The ML method performed well across the full range of error rates, with rare exceptions, which are detailed below (Table 1). It performed best when He and n were high and error rates were intermediate or high, that is, when the number of errors and the proportion of informative genotypes were high.
TABLE 1.
Analysis of the performance of the ML method in estimating allelic dropout and false allele error rates from simulated data
|
n = 50
|
n = 100
|
n = 200
|
|||||
|---|---|---|---|---|---|---|---|
| ɛ1, ɛ2 | He | Mean 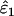 , , 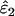
|
IE (%) | Mean 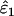 , , 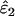
|
IE (%) | Mean 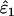 , , 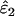
|
IE (%) |
| 0.01, 0.0015 | 0.5 | 0.0108 (0.0111), 0.0013 (0.0033) | 63, 52 | 0.0104 (0.0079), 0.0014 (0.0026) | 62, 51 | 0.0101 (0.0057), 0.0014 (0.0019) | 64, 51 |
| 0.85 | 0.0103 (0.0082), 0.0015 (0.0030) | 21, 16 | 0.0101 (0.0056), 0.0015 (0.0022) | 21, 16 | 0.0099 (0.0039), 0.0015 (0.0015) | 22, 17 | |
| 0.01, 0.04 | 0.5 | 0.0162 (0.0195), 0.0376 (0.0164) | 92, 30 | 0.0122 (0.0135), 0.0392 (0.0119) | 92, 27 | 0.0110 (0.0101), 0.0398 (0.0084) | 92, 29 |
| 0.85 | 0.0108 (0.0096), 0.0398 (0.0149) | 52, 15 | 0.0100 (0.0069), 0.0404 (0.0108) | 53, 15 | 0.0100 (0.0050), 0.0402 (0.0076) | 51, 15 | |
| 0.01, 0.08 | 0.5 | 0.0151 (0.0200), 0.0793 (0.0224) | 94, 20 | 0.0125 (0.0145), 0.0802 (0.0156) | 91, 20 | 0.0102 (0.0102), 0.0809 (0.0111) | 92, 21 |
| 0.85 | 0.0108 (0.0103), 0.0807 (0.0212) | 66, 14 | 0.0102 (0.0075), 0.0808 (0.0150) | 59, 14 | 0.0100 (0.0056), 0.0813 (0.0107) | 62, 16 | |
| 0.09, 0.0015 | 0.5 | 0.0915 (0.0356), 0.0016 (0.0044) | 70, 61 | 0.0913 (0.0247), 0.0014 (0.0029) | 67, 61 | 0.0904 (0.0170), 0.0015 (0.0021) | 67, 58 |
| 0.85 | 0.0913 (0.0262), 0.0015 (0.0033) | 42, 30 | 0.0907 (0.0183), 0.0016 (0.0024) | 40, 30 | 0.0905 (0.0129), 0.0015 (0.0016) | 39, 32 | |
| 0.09, 0.04 | 0.5 | 0.0933 (0.0411), 0.0403 (0.0195) | 74, 48 | 0.0908 (0.0283), 0.0401 (0.0136) | 75, 48 | 0.0905 (0.0200), 0.0404 (0.0095) | 74, 47 |
| 0.85 | 0.0916 (0.0269), 0.0405 (0.0167) | 42, 30 | 0.0906 (0.0189), 0.0399 (0.0116) | 42, 30 | 0.0900 (0.0130), 0.0402 (0.0084) | 41, 28 | |
| 0.09, 0.08 | 0.5 | 0.0924 (0.0408), 0.0815 (0.0261) | 76, 38 | 0.0903 (0.0284), 0.0813 (0.0180) | 75, 39 | 0.0907 (0.0199), 0.0811 (0.0127) | 77, 39 |
| 0.85 | 0.0911 (0.0276), 0.0817 (0.0239) | 43, 29 | 0.0903 (0.0191), 0.0814 (0.0165) | 42, 30 | 0.0907 (0.0134), 0.0812 (0.0116) | 42, 30 | |
| 0.17, 0.0015 | 0.5 | 0.1747 (0.0534), 0.0014 (0.0046) | 74, 69 | 0.1727 (0.0368), 0.0015 (0.0033) | 74, 68 | 0.1712 (0.0259), 0.0015 (0.0024) | 74, 65 |
| 0.85 | 0.1721 (0.0383), 0.0015 (0.0036) | 52, 46 | 0.1709 (0.0273), 0.0015 (0.0026) | 51, 42 | 0.1706 (0.0188), 0.0015 (0.0018) | 52, 41 | |
| 0.17, 0.04 | 0.5 | 0.1735 (0.0549), 0.0402 (0.0214) | 77, 57 | 0.1728 (0.0380), 0.0399 (0.0152) | 77, 55 | 0.1709 (0.0265), 0.0401 (0.0105) | 76, 55 |
| 0.85 | 0.1723 (0.0381), 0.0404 (0.0186) | 52, 42 | 0.1709 (0.0274), 0.0403 (0.0131) | 52, 41 | 0.1706 (0.0192), 0.0401 (0.0093) | 51, 42 | |
| 0.17, 0.08 | 0.5 | 0.1740 (0.0547), 0.0819 (0.0287) | 75, 50 | 0.1716 (0.0383), 0.0811 (0.0202) | 73, 50 | 0.1707 (0.0264), 0.0814 (0.0140) | 75, 49 |
| 0.85 | 0.1721 (0.0380), 0.0815 (0.0261) | 51, 42 | 0.1715 (0.0267), 0.0813 (0.0184) | 52, 41 | 0.1699 (0.0190), 0.0815 (0.0131) | 52, 41 | |
Mean estimated error rates (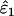 ,
, 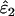 ) with SE (in parentheses) and intrinsic error (IE, the proportion of the total estimation error that is due to the ML method and not to sampling error) were calculated by analyzing 5000 data sets simulated using each of the 54 possible combinations of nine population error rates (ɛ1, ɛ2), three sample sizes (n), and two expected heterozygosities (He = 0.5 and He = 0.85).
) with SE (in parentheses) and intrinsic error (IE, the proportion of the total estimation error that is due to the ML method and not to sampling error) were calculated by analyzing 5000 data sets simulated using each of the 54 possible combinations of nine population error rates (ɛ1, ɛ2), three sample sizes (n), and two expected heterozygosities (He = 0.5 and He = 0.85).
Bias:
When He = 0.85 and n = 200, relative bias was insignificant for both 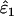 and
and  across all error rate combinations (range −1.8–0.8%). However, relative bias increased with decreasing He and n (Figure 2) and was generally greater in
across all error rate combinations (range −1.8–0.8%). However, relative bias increased with decreasing He and n (Figure 2) and was generally greater in 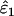 than in
than in 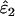 . Although the magnitude of bias in
. Although the magnitude of bias in 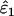 was always low (range 0.0001–0.0062), relative bias in
was always low (range 0.0001–0.0062), relative bias in 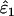 became high when He, n, and ɛ1 were low (i.e., when the number of visible dropouts was low) and ɛ2 was high, reaching a maximum of 62% at ɛ1 = 0.01, ɛ2 = 0.04, He = 0.5, and n = 50 (Figure 2). It appears that when the number of visible dropouts is low and the numbers of false alleles are high, some false alleles in AA.AB-type mismatches are “mistaken” for allelic dropouts. Although a bias of 62% in estimating ɛ1 of 0.01 may seem severe, high bias occurred only in circumstances where the data contained very few visible dropouts (ɛ1 = 0.01, He = 0.5, n = 50) and consequently was characterized by high sampling error, so that even the highest biases were overwhelmed by estimation error. The highest contribution of bias to MSE in
became high when He, n, and ɛ1 were low (i.e., when the number of visible dropouts was low) and ɛ2 was high, reaching a maximum of 62% at ɛ1 = 0.01, ɛ2 = 0.04, He = 0.5, and n = 50 (Figure 2). It appears that when the number of visible dropouts is low and the numbers of false alleles are high, some false alleles in AA.AB-type mismatches are “mistaken” for allelic dropouts. Although a bias of 62% in estimating ɛ1 of 0.01 may seem severe, high bias occurred only in circumstances where the data contained very few visible dropouts (ɛ1 = 0.01, He = 0.5, n = 50) and consequently was characterized by high sampling error, so that even the highest biases were overwhelmed by estimation error. The highest contribution of bias to MSE in 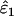 (bias2/MSE) was 9.1%, which occurred when bias was 62%. Bias in
(bias2/MSE) was 9.1%, which occurred when bias was 62%. Bias in 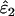 was always low, ranging from −15 to 2.6%. The largest bias, of −15%, was in estimating ɛ2 of 0.0015 at He = 0.5 and n = 50 and was responsible for only 0.4% of MSE.
was always low, ranging from −15 to 2.6%. The largest bias, of −15%, was in estimating ɛ2 of 0.0015 at He = 0.5 and n = 50 and was responsible for only 0.4% of MSE.
Variance:
Like bias, the variance in the error rate estimates was lowest, relative to the population error rates, when the amount of information in the data was highest, that is, when error rates, He, and n were all high (see standard errors in Table 1).
Intrinsic error:
Unsurprisingly, the standard error of the estimates was smallest at high n. More significantly, intrinsic error did not vary with n, but rather was sensitive to the two error rates and He (Table 1, Figure 3). Like bias, intrinsic error was higher in 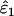 (21–94%) than in
(21–94%) than in 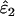 (14–69%) and highest when estimating low ɛ1 at low He and high ɛ2. At He = 0.85 intrinsic error in
(14–69%) and highest when estimating low ɛ1 at low He and high ɛ2. At He = 0.85 intrinsic error in 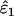 and
and 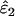 averaged 46 and 29%, respectively, across the range of simulated error rates and sample sizes, compared with 77 and 47% at He = 0.5.
averaged 46 and 29%, respectively, across the range of simulated error rates and sample sizes, compared with 77 and 47% at He = 0.5.
Figure 3.—


The degree of error in ML estimating the allelic dropout rate (ɛ1) that is due to uncertainty inherent in the method (intrinsic error) rather than sampling error in the production of genotyping errors. The two plots summarize the analysis of 5000 data sets of intermediate sample size (n = 100) simulated for each of nine error coordinates at low (A: He = 0.5) and high heterozygosity (B: He = 0.85).
The simulation analysis above was carried out using data simulated under the assumptions of the error model. We now show the effect of deviation from some of these assumptions.
Deviation from Hardy–Weinberg equilibrium:
The only substantial effect of moderate (FIS = 0.0625) and high (FIS = 0.125) heterozygote deficiency was to bias 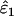 and
and 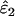 . The relationship between FIS and relative bias was consistently linear across the range of parameter values tested. Relative bias in
. The relationship between FIS and relative bias was consistently linear across the range of parameter values tested. Relative bias in 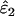 was generally low, being ∼ −FIS. The effect of FIS on relative bias in
was generally low, being ∼ −FIS. The effect of FIS on relative bias in 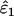 was also generally tolerable (5–25%) but became severe when allelic dropouts were very infrequent relative to false alleles (Figure 4). The most extreme bias occurred when ɛ1 was estimated as 0.028 at ɛ1 = 0.01, ɛ2 = 0.08, He = 0.5, FIS = 0.125, and n = 50 (bias 176%). However, even this considerable bias contributed only 29% to MSE. Moreover, it is very rare for false alleles to outnumber allelic dropouts to such an extent (Ewen et al. 2000; Broquet and Petit 2004; see also Table 2). In summary, we suggest that levels of bias caused by moderate deviation from Hardy–Weinberg equilibrium will generally be acceptable. Specific judgments will depend on specific parameter values, as is clear from Figure 4, as well as the degree of precision demanded by the downstream analysis.
was also generally tolerable (5–25%) but became severe when allelic dropouts were very infrequent relative to false alleles (Figure 4). The most extreme bias occurred when ɛ1 was estimated as 0.028 at ɛ1 = 0.01, ɛ2 = 0.08, He = 0.5, FIS = 0.125, and n = 50 (bias 176%). However, even this considerable bias contributed only 29% to MSE. Moreover, it is very rare for false alleles to outnumber allelic dropouts to such an extent (Ewen et al. 2000; Broquet and Petit 2004; see also Table 2). In summary, we suggest that levels of bias caused by moderate deviation from Hardy–Weinberg equilibrium will generally be acceptable. Specific judgments will depend on specific parameter values, as is clear from Figure 4, as well as the degree of precision demanded by the downstream analysis.
Figure 4.—

The effect of heterozygote deficiency, gauged by the inbreeding coefficient FIS, on relative bias in estimating allelic dropout rate (ɛ1) averaged over 5000 simulated data sets. Low (ɛ1 = 0.01, solid line) and intermediate (ɛ1 = 0.09, dashed line) allelic dropout rates were estimated at three false allele error rates (•, ɛ2 = 0.0015; ○, ɛ2 = 0.04; ▾, ɛ2 = 0.08). Other parameter values were He = 0.85 and n = 100.
TABLE 2.
Analysis of the performance of the ML method in estimating allelic dropout and false allele error rates from real data
| Locus | Sample p | ML p (95% C.I.) | Sample f | ML f (95% C.I.) |
|---|---|---|---|---|
| V142 | 0.138 | 0.116 (0.070, 0.176) | 0.028 | 0.011 (0.002, 0.033) |
| V374 | 0.464 | 0.429 (0.341, 0.517) | 0.042 | 0.013 (0.002, 0.035) |
| V402 | 0.000 | 0.000 (0.000, 0.011) | 0.000 | 0.000 (0.000, 0.009) |
| V468 | 0.027 | 0.019 (0.005, 0.049) | 0.003 | 0.000 (0.000, 0.010) |
| V502 | 0.016 | 0.005 (0.000, 0.031) | 0.000 | 0.000 (0.000, 0.011) |
| V602 | 0.084 | 0.069 (0.037, 0.114) | 0.024 | 0.000 (0.000, 0.010) |
| V622 | 0.132 | 0.126 (0.078, 0.196) | 0.036 | 0.008 (0.002, 0.036) |
| AHT-130 | 0.169 | 0.138 (0.090, 0.204) | 0.041 | 0.027 (0.011, 0.059) |
| CXX-156 | 0.175 | 0.161 (0.105, 0.231) | 0.076 | 0.044 (0.018, 0.075) |
| CXX-250 | 0.160 | 0.163 (0.107, 0.230) | 0.049 | 0.045 (0.018, 0.078) |
| CXX-279 | 0.120 | 0.089 (0.050, 0.141) | 0.031 | 0.012 (0.002, 0.036) |
| CXX-434 | 0.302 | 0.304 (0.224, 0.390) | 0.075 | 0.040 (0.013, 0.073) |
| CXX-466 | 0.277 | 0.244 (0.175, 0.323) | 0.045 | 0.042 (0.015, 0.073) |
| CXX-606 | 0.195 | 0.190 (0.127, 0.266) | 0.019 | 0.015 (0.002, 0.042) |
| CXX-608 | 0.213 | 0.224 (0.159, 0.302) | 0.037 | 0.009 (0.002, 0.039) |
| c2088 | 0.211 | 0.239 (0.171, 0.317) | 0.051 | 0.019 (0.004, 0.045) |
| c377 | 0.086 | 0.087 (0.031, 0.167) | 0.072 | 0.049 (0.016, 0.095) |
| FH2001 | 0.086 | 0.101 (0.048, 0.180) | 0.017 | 0.000 (0.000, 0.023) |
| FH2054 | 0.100 | 0.094 (0.042, 0.169) | 0.061 | 0.035 (0.009, 0.078) |
| FH2119 | 0.167 | 0.154 (0.071, 0.275) | 0.014 | 0.010 (0.000, 0.051) |
| FH2137 | 0.088 | 0.048 (0.000, 0.163) | 0.029 | 0.038 (0.010, 0.076) |
| FH2138 | 0.315 | 0.294 (0.194, 0.408) | 0.080 | 0.056 (0.015, 0.109) |
| FH2140 | 0.086 | 0.079 (0.031, 0.151) | 0.050 | 0.044 (0.014, 0.088) |
| FH2159 | 0.100 | 0.070 (0.016, 0.159) | 0.018 | 0.027 (0.005, 0.066) |
| FH2174 | 0.227 | 0.213 (0.128, 0.317) | 0.019 | 0.008 (0.000, 0.040) |
| FH2226 | 0.097 | 0.081 (0.028, 0.160) | 0.058 | 0.060 (0.023, 0.107) |
| FH2293 | 0.222 | 0.235 (0.141, 0.346) | 0.104 | 0.070 (0.024, 0.125) |
| FH2320 | 0.295 | 0.247 (0.141, 0.371) | 0.093 | 0.092 (0.033, 0.148) |
| FH2422 | 0.140 | 0.090 (0.041, 0.161) | 0.026 | 0.041 (0.013, 0.079) |
| FH2472 | 0.207 | 0.218 (0.136, 0.317) | 0.104 | 0.087 (0.032, 0.150) |
| FH2537 | 0.200 | 0.202 (0.126, 0.295) | 0.024 | 0.016 (0.001, 0.055) |
| Pez17 | 0.143 | 0.123 (0.049, 0.228) | 0.035 | 0.038 (0.009, 0.079) |
| Pez19 | 0.167 | 0.076 (0.027, 0.154) | 0.023 | 0.009 (0.000, 0.040) |
| Mean | 0.164 | 0.149 (0.116, 0.183) | 0.042 | 0.029 (0.021, 0.038) |
Sample and ML estimates of error rates p (allelic dropouts per heterozygote) and f (false alleles per genotype) in duplicate genotypes from 16 red fox and 17 Ethiopian wolf microsatellites (top and bottom, respectively). Confidence limits for p and f estimates (in parentheses) were converted from the horizontal and vertical dimensions of the 95% confidence region for ɛ1, ɛ2 (see Figure 1). Confidence limits for the mean are calculated from the normal distribution.
Increased sampling error in He:
The effect on 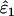 and
and 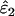 of increasing sampling error in He to its maximum possible level was slight. Bias was unaffected, but overall estimation error (MSE) rose at low He and n. The greatest increase in MSE was 17%, when estimating ɛ2 at He = 0.5, n = 50, ɛ1 = 0.17, and ɛ2 = 0.0015. All other increases in MSE in
of increasing sampling error in He to its maximum possible level was slight. Bias was unaffected, but overall estimation error (MSE) rose at low He and n. The greatest increase in MSE was 17%, when estimating ɛ2 at He = 0.5, n = 50, ɛ1 = 0.17, and ɛ2 = 0.0015. All other increases in MSE in 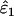 and
and 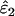 were <10%.
were <10%.
Sample quality variation:
Skewing the distribution of errors across samples had no effect on the variance of the error estimates but did cause considerable underestimation of both error rates at intermediate and high ɛ1. When ɛ1 = 0.17, bias ranged from −13 to −29% in 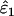 and from −35 to −43% in
and from −35 to −43% in 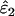 across the 54 simulated parameter sets. Bias in
across the 54 simulated parameter sets. Bias in 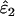 was also substantial at intermediate ɛ1 (range −17 to −28%).
was also substantial at intermediate ɛ1 (range −17 to −28%).
Dominance of dropouts over false alleles:
As might be expected, reducing the proportion of false alleles that are hidden by dropouts from 1 to 0.5 caused underestimation of ɛ1 and overestimation of ɛ2. Bias was greatest when there was the highest probability of both types of error striking the same allele, that is, when both ɛ1 and ɛ2 were high. The degree of bias was small, even at high error rates. When ɛ1 = 0.17 and ɛ2 = 0.08, relative bias was −7 to −8% in 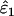 (previous range 0.0–0.1%) and 21% in
(previous range 0.0–0.1%) and 21% in 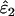 (previous range 0.0–1.8%) across all He and n. Across all other parameter combinations the maximum biases were −4% in
(previous range 0.0–1.8%) across all He and n. Across all other parameter combinations the maximum biases were −4% in 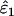 and 11% in
and 11% in 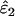 .
.
Performance against reference data method:
For a large majority of parameter combinations, the ML method outperformed the reference data method in estimating ɛ1 and ɛ2. Averaged across 108 comparisons (two estimated error rates × 54 parameter combinations), MSEML was 20% lower than MSERD. MSEML was significantly smaller than MSERD in 90 comparisons, while MSERD was significantly smaller than MSEML in 11 comparisons. In the remaining 7 comparisons there was no significant difference [two-tailed F-test for equality of variances, F(0.025, 4999, 4999) = 1.057]. The 11 competitions in which the reference data method was superior (MSEML/MSERD range 1.13–2.15) shared two common features: low heterozygosity (He = 0.5) and wide disparity between ɛ1 and ɛ2, suggesting that a high rate of one error can interfere with estimating a much lower rate of the other when the data are relatively uninformative. At high heterozygosity (He = 0.85) the RD method was never superior, with MSEML/MSERD ranging from 0.51 to 0.99.
Application to real microsatellite data:
The mean sample error rates across the 16 red fox microsatellite loci were, respectively, 0.17 allelic dropouts per heterozygote per locus (p) and 0.035 false alleles per genotype per locus (f). The mean ML estimates were 0.16 and 0.018, respectively. For the 17 Ethiopian wolf microsatellites, the mean sample error rates were p = 0.16 and f = 0.049, while the mean ML estimates were 0.14 and 0.040, respectively. Thus, with the exception of underestimating f in the fox genotypes, the ML method performed well in estimating the mean cross-locus error rates.
For individual loci, ML estimates of p were generally close to the sample error rates, although they tended to be slightly underestimated in both the fox (mean bias −12%) and wolf data sets (mean bias −13%; Table 2, Figure 5). The bias in the fox data was of opposite sign to the value predicted by the heterozygote deficit (3%), suggesting that other factors have greater influence on bias in real data. Almost half of the bias in analyzing the wolf data set was due to two loci, Pez19 and FH2137. Underestimation of p in FH2137 was explained by a lack of information in the data due to a low He of 0.30 (sample p = 0.087, ML p = 0.048), while short allele dominance severely affected locus Pez19 (sample p = 0.17, ML p = 0.076), although it was not a significant factor in any other locus. The linear relationship between sample and ML p was tight ([ML p] = 0.97 × [sample p] − 0.0097, R2 = 0.94), and the slope of the line was not significantly different from 1 (t31 = 0.69, P = 0.50).
Figure 5.—

Performance of the ML method in estimating allelic dropout rate per heterozygote (A) and false allele rate per genotype (B) from real data. Two duplicated microsatellite data sets were analyzed: 149–182 red fox teeth genotyped at 16 loci (▴) and 72–121 Ethiopian wolf fecal samples genotyped at 17 loci (○). The dotted line shows equality between ML and sample error rates. The sample error rates in the duplicate genotypes were counted by referring to the consensus genotypes. Error bars show 95% confidence intervals for the ML estimates, calculated as described in Table 2.
Bias in the locus-specific estimates of f of −34% was considerably greater than in the estimates of p, most notably in the fox duplicate genotypes where bias was −55%, much greater than the −8% bias predicted due to heterozygote deficiency. Mean bias in the wolf data set was comparatively low at −17%. There were two principal reasons for underestimation of f in the fox data. First, the accumulation of errors in the most error-prone samples, combined with a tendency for the same rare false allele to recur across repeat genotypes, caused some errors to be hidden. For example, in locus V142 a 142/146 genotype was scored as 142/148.142/148 and a 133/140 genotype as 142/142.142/142. In these two duplicate genotypes, four of the nine false alleles in the V142 genotypes went undetected. However, the greatest cause of underestimation in the fox data, and to a lesser extent also in the wolf data, was the tendency of false alleles to occur preferentially in homozygotes, contrary to the third error model assumption. An excess of false alleles in homozygotes leading to extra ambiguous duplicate genotypes (e.g., AA read as AA.AB) will inflate p at the expense of f.
Taking both data sets together, the scatter of the f estimates was wider than that of the p estimates (Figure 5), possibly as a result of the comparatively small numbers of false alleles in the duplicate genotypes (mean 10.8) compared with the number of allelic dropouts (mean 31.3). Nevertheless, the relationship between sample and ML f estimates was close ([ML p] = 0.76 × [sample p] − 0.0028, R2 = 0.76). The slope of the regression line differed significantly from 1 (t31 = 3.1, P = 0.005), reflecting the greater bias in the f estimates compared with the p estimates.
DISCUSSION
The consequences of undetected microsatellite genotyping errors can range from insignificant biases to outright false conclusions (Gagneux et al. 1997; Abecasis et al. 2001; Hoffman and Amos 2005; Pompanon et al. 2005). Errors have become easier to correct or account for due to a combination of greater caution among researchers and the development of data analysis methods that incorporate a single genotyping error rate. Error-tolerant microsatellite data analysis is more accurate when allelic dropout and false allele probabilities are estimated as separate parameters (Wang 2004; Hadfield et al. 2006; Kalinowski et al. 2006). The method described here estimates allelic dropout and false allele error rates without the limiting requirement for reference data.
The results of the simulation analysis indicate that, even when reference samples are available, the ML method will generally estimate error rates more accurately than possible from an equivalent number of PCRs, provided that the data fit the assumptions of the error model reasonably closely. This counterintuitive result is explained by the fact that the cost in added uncertainty due to both duplicates being error prone is outweighed by the reduction in sampling error caused by doubling the sample size of error-prone genotypes. The simulation analysis also suggested that the ML method is generally robust to modest violations of the assumptions of the underlying error model, although varying sample quality does lead to underestimation of ɛ1 and ɛ2 at high ɛ1. This bias occurs because coincidence of dropouts and false alleles leads to undercounting of errors in the most error-prone simulated duplicate genotypes: the number of uncounted double dropouts will be higher than expected, as will the number of false alleles that are hidden by dropouts. Error estimates should therefore be used with caution when both high dropout levels and highly skewed sample quality are suspected. One way of mitigating this problem would be to identify and eliminate the most error-prone samples by comparing data quality across loci.
To what extent does the assumption of Hardy–Weinberg equilibrium (HWE) limit the use of our method? The standard error of the mean error rates estimated from simulated data was unaffected by high FIS. Bias was also tolerably low except in rare cases where ɛ1 is very low relative to ɛ2. Chesser (1991, Figure 2 therein) has shown theoretically that under most realistic circumstances FIS should not be >0.1, although no cross-taxon survey of FIS-values exists to test this prediction. However, empirical estimates show that HWE is a reasonable assumption within populations of humans (Altshuler et al. 2005), which form the basis of most studies that consider genotyping error (Pompanon et al. 2005). The 12 pedigree studies of bird and mammal populations reviewed by Slate et al. (2004) revealed predominantly low levels of FIT (quoted as f; mean 0.042, range 0.002–0.103), and even these low values probably represent publication bias in favor of high FIT. Assuming that FST is zero or positive, FIT will set an upper bound on FIS because 1 − FIT = (1 − FIS)(1 − FST) (Wright 1951), indicating that in many if not most vertebrate populations HWE will not be an unduly restrictive assumption, provided that the populations are correctly defined and no Wahlund effect is present. It is important to note that a high pedigree inbreeding coefficient (equivalent to Wright's FIT), which indicates shared coancestry within a population (Keller and Waller 2002), does not necessarily imply heterozygote deficiency and therefore would not in itself affect our method. Very large heterozygote deficits that would invalidate our method are likely to be more common in nonvertebrate taxa as a result of greater mating system diversity (e.g., self-fertility in plants and molluscs). However, provided that prior information on mating system and genetic structure is used judiciously, our method should be applicable to a large majority of microsatellite genotyping studies, particularly considering the large bias toward humans and wild vertebrates in microsatellite studies.
When presented with real data, the ML method performed well in approximating the sample error rates of both allelic dropout and false alleles, with the exception that it systematically underestimated the false allele rate in the fox data set. This result suggests that in some data sets at least there will be significant deviation from the assumptions of the error model. The major cause of underestimation, the tendency of false alleles to preferentially affect homozygotes, is probably a consequence of the fact that artifactual alleles, as opposed to scoring and data entry errors, form a much higher proportion of false alleles in data derived from low-quality DNA, such as the genotypes from autoclaved fox teeth and wolf feces analyzed here (Bradley and Vigilant 2002; Pompanon et al. 2005). Whereas a scoring or data entry error in a heterozygote will cause an existing allele to be miscalled, an artifactual allele will create a third allele, with two possible consequences: the genotype will be deleted (i.e., recorded as missing data), or two of the three alleles will be recorded as the genotype, which will include the artifactual allele with at most two-thirds probability. Either result will bias false alleles toward homozygotes when artifactual alleles are frequent, raising the possibility that there are two false allele error rates in error-rich data, one specific to homozygotes and the other to heterozygotes. However, in more typical studies when high-quality DNA is available, this bias should be greatly reduced if not absent. Hoffman and Amos (2005) found that in low-error data (0.0038 errors per single-locus genotype), only 7% of errors that our model would class as false alleles were due to artifacts, the remainder being either due to scoring or data input errors (89%) or of unknown origin (4%). Scoring and data input errors should not affect homozygotes preferentially and may even show a bias toward heterozygotes, which present double the opportunities for error. The other significant source of underestimation of both error rates in the simulated and real data, clustering of errors in low-quality samples, should also be much less prevalent when template DNA is of high quality, principally because the probability of duplicates hit by two errors is the square of the per-genotype error rate.
It remains to be seen how well our error model will fit a wider range of microsatellite data sets with differing frequencies and patterns of errors. The error model could easily be adapted to specific circumstances by adjusting the expected frequencies of the seven duplicate genotype categories. Our intention here was to provide a general model that enables parameters to be estimated for use in analyses that incorporate similar error models (e.g., Wang 2004; Hadfield et al. 2006).
Where such error-tolerant analyses are unavailable, error rate estimates are nevertheless helpful in evaluating the robustness of analyses to genotyping error. In the event that an analysis method is judged unacceptably error sensitive, error rates can be reduced by multiple genotyping. A simulation-based method is available to monitor the error sensitivity of a number of population parameter estimates (allele frequencies, He, Ho, probability of identity, and census size) and to select the minimum number of repeat genotypes required to reduce the impact of errors to acceptable levels (Valière et al. 2002).
What are the consequences of under- or overestimating allelic dropout and false allele error rates? Provided there is sufficient information in the data, highly inaccurate estimates can still allow accurate inference of family relationships (SanCristobal and Chevalet 1997; Sieberts et al. 2002; Wang 2004). Using simulated data where ɛ1 = ɛ2 = 0.05, Wang (2004) showed that at least 90% of full-sib families can be fully reconstructed assuming error rates across the range 0.008–0.36 (range estimated from Wang's Figure 4). It does not follow from this result that there would never be a significant benefit in estimating error rates accurately, particularly when error rates are high and the amount of information in the data is low (in which case a reference data-based method might be preferred), or when solving difficult problems such as assigning paternity among candidate fathers that are numerous or closely related. However, it does imply that occasional egregious estimates (e.g., locus Pez19, Table 2) should not significantly disrupt inference in most analyses, so that resources could be better applied to assaying additional individuals or loci than to refining error estimates by creating consensus genotypes from multiple rounds of genotyping.
An exception to the optimistic conclusion that approximate estimates are adequate occurs when the estimate is zero and the true error rate is greater than zero. For example, our method estimated a false allele rate of zero in loci V468, V602, and FH2001 (Table 2) when the sample (and hence population) rate was nonzero. Using these estimates in relationship inference would lead to problems similar to those encountered when using strict exclusion in parentage analysis (Gagneux et al. 1997). However, the probability of error is greater than zero even in the cleanest data set, so assuming zero error estimates is never advisable. An alternative if somewhat arbitrary approach is to use upper confidence limits rather than ML estimates. Although these values will generally be overestimates, particularly when sample size is small, it may often be safer to overestimate than to underestimate error rates whether error rate estimates are greater than zero or not. Indeed, when there is ample information in the data, gross overestimates can allow greater accuracy in reconstruction of full-sib families than relatively modest underestimates (Figure 4 of Wang 2004). Morrissey and Wilson (2005) reached the opposite conclusion on the basis of parentage analysis of simulated and real data using the method of Marshall et al. (1998). Under certain circumstances (mother unknown, few marker loci, skewed allele frequencies, and a requirement of 95% confidence in correct assignment) there was a benefit in underestimating a 1% error rate. Not surprisingly, this benefit rapidly became a cost with increasing numbers of loci. In this study genotyping error was modeled as a single cross-locus quantity, so it is not clear to what extent, if at all, this result applies to methods that model allelic dropout and false allele error rates separately.
In practice, the level of imprecision that can be tolerated in the error rate estimates could be assessed during the planning stage of a study by analyzing simulated data. The robustness of the analysis to inaccuracy in the error estimates could also be gauged after the data have been generated by repeating the analysis using a range of error estimates based on the confidence regions. Once the desired level of precision in the error estimates is known, how many duplicate samples will be needed to achieve it? As we have shown using simulations, for each locus this number will depend on the expected heterozygosity, which will usually be known, and error rates, for which plausible ranges can usually be predicted. The effect of varying sample size and error rates on error estimate precision can be explored using simulations in Pedant.
In conclusion, we have developed a method for estimating locus-specific rates, with confidence regions, of allelic dropout and false allele genotypic error from duplicate microsatellite genotypes without the requirement for reference data. These error estimates can provide input for microsatellite data analysis methods that handle allelic dropout and false allele error rates separately. The method described here is implemented in a computer program, Pedant, which also uses simulations to perform power analyses. The source code and executable are freely available for download from http://www.stats.gla.ac.uk/∼paulj/pedant.html.
Acknowledgments
We thank Tanita Casci, Ian Ford, Jarrod Hadfield, Lukas Keller, Barbara Mable, Graeme Ruxton, Peter Wandeler, and Jinliang Wang for discussion and advice and two anonymous reviewers for constructive comments on the manuscript. We are particularly grateful to Peter Wandeler and Deborah Randall for sharing their data. P.C.D.J. was supported by a Leverhulme Trust research project grant.
APPENDIX
Likelihood calculation:
To simplify the presentation of the equations for the expected category frequencies, we use per-genotype rather than per-allele error probabilities. For a single genotype at a single locus, we define the probability of no dropouts as p0 = (1 − ɛ1)2 and the probability of one dropout in a given allele as p1 = ɛ1(1 − ɛ1). Double dropouts are not counted. Similarly, the per-genotype probabilities for false alleles are f0 = (1 − ɛ2)2, f1 = ɛ2(1 − ɛ2), and f2 = 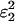 . The expected frequency (P1,2,…,7) of each repeat genotype category can be expressed by summing the probabilities of all the ways in which a repeat genotype can contribute to that category. For example, one way for an observed duplicate genotype to enter category 3 (AA.AB) is via the occurrence in a homozygote of no dropouts and one false allele in any of the four alleles, with probability (
. The expected frequency (P1,2,…,7) of each repeat genotype category can be expressed by summing the probabilities of all the ways in which a repeat genotype can contribute to that category. For example, one way for an observed duplicate genotype to enter category 3 (AA.AB) is via the occurrence in a homozygote of no dropouts and one false allele in any of the four alleles, with probability (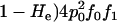 . If all possible states are considered, there are 512 ways of entering the seven categories or the double-dropout category. This number is reduced to 198 by not counting double dropouts and ignoring replicates with more than two false allele errors. The expected frequencies of categories 1–7 are
. If all possible states are considered, there are 512 ways of entering the seven categories or the double-dropout category. This number is reduced to 198 by not counting double dropouts and ignoring replicates with more than two false allele errors. The expected frequencies of categories 1–7 are
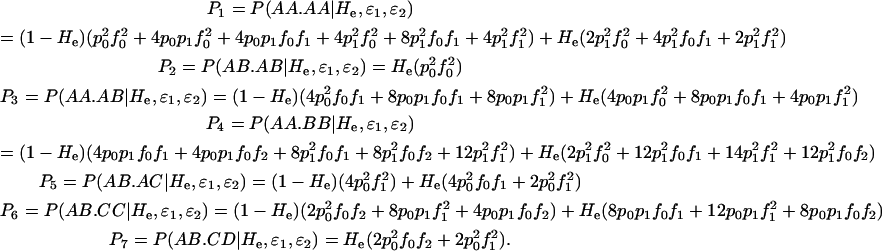 |
Because double dropouts are not counted, these probabilities must be normalized to sum to one, giving the expected frequency of category i,
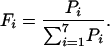 |
The data consist of the seven observed category counts, X1,…, X7, which sum to n, the number of duplicated genotypes. The likelihood, L, of the data given the expected heterozygosity He and error rates ɛ1 and ɛ2 is
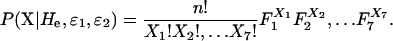 |
Maximum-likelihood search algorithm:
The maximum-likelihood search begins at a random point on the likelihood surface and proceeds by a simulated annealing procedure (Kirkpatrick et al. 1983). New error coordinates are proposed by randomly adding or subtracting a step of size S to or from each error rate. The likelihood of the new coordinates (Lnew) is compared with that of the old (Lold). Uphill steps (Lnew > Lold) are always accepted while downhill steps are accepted with probability (Lnew/Lold)1/T, where T is the annealing temperature, allowing the search to escape from a local maximum. Downhill steps are more likely to be taken when the proposed drop in likelihood is modest and T is high. Because T decreases as the search continues, the probability of a downhill step decreases toward the end of the search. T begins the search at a value of 1000 and decreases multiplicatively every iteration by a factor of 10−11/i, where i is the number of search iterations, so that T approaches 10−8 toward the end of the search regardless of i. During the last 10% of the search only uphill steps are permitted. Like T, S decreases exponentially throughout the search, from a maximum of 0.1 to a minimum of 10−7. The initial and final values of T and S and the shapes of their declines are independent of i, with the practical result that regardless of i approximately the first half of the search is spent searching widely across the likelihood surface for a peak to settle on and the remainder is spent refining the error estimates on that peak. The search jumps to the last 1000 iterations if the likelihood of accepted steps increases by <1% for 1000 consecutive iterations.
An additional search procedure further reduces the probability of the search ending on a local maximum. After i iterations have been completed, the maximum likelihood recorded throughout the search is compared with the final likelihood. If the former is higher, the final coordinates must represent a local maximum, so the search returns to the likelier recorded coordinates, which must be located either on a higher local maximum or on the global maximum. Finally, a further 1000 optimization iterations are completed, again with exponentially decreasing S but with only uphill steps being allowed.
We tested the search algorithm by analyzing artificial data sets that produce bimodal likelihood surfaces with a narrow global maximum and a broad (and therefore more easily located) local maximum. The global maximum was located within 20,000 iterations in 100/100 trials except when the difference in likelihood between the two peaks was low (likelihood ratio <2.2). In these cases 500,000 iterations were required to locate the global maximum in 82/100 trials.
Simulation of sampling error in He:
The point estimate of He used to calculate the expected frequencies is subject to sampling error, which was incorporated into the simulated estimate of He used in estimating the error rates. Estimated He was simulated as a normally distributed random variable with mean He and standard deviation s. Because s is a function of both allele frequency distribution and the number of genotypes from which He was calculated, nH (Nei and Roychoudhury 1974), simulation of s was simplified by assuming a single allele-frequency distribution, the expected frequencies determined from the broken-stick distribution (Legendre and Legendre 1998). Broken-stick frequencies can be used to provide a simple means of simulating realistic microsatellite allele frequencies (Kalinowski et al. 2006). Given broken-stick expected frequencies, for any nH both He and s are discrete functions of the number of alleles. We derived a close approximation of s,
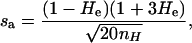 |
by fitting a curve to discrete values of s across a realistic range of He (0.38–0.95, which corresponds to 2–29 alleles) and nH (50–20,000). The approximation sa accounts for 98.1% of the variation in s at nH = 50 and >99.5% at nH ≥ 100. Although all microsatellite allele-frequency distributions will deviate from broken-stick expected frequencies to some extent, comparison of s calculated from data with sa simulated at the same He and nH for data from 54 microsatellite loci (He range, 0.27–0.90; nH range, 172–545) from aphids (Johnson et al. 2000), nematodes (Johnson et al. 2006), red foxes, and Ethiopian wolves (data kindly provided by P. Wandeler and D. A. Randall) suggests that the broken-stick model fits well to reality. The means of the simulated and real standard deviations were not significantly different (mean sa = 0.0124, mean data s = 0.0130, P = 0.24, paired t-test), and the slope of the linear regression line (s = 1.10sa − 0.0007, R2 = 0.62) did not differ significantly from 1 (t52 = 0.87, P = 0.39). Thus a plausible standard deviation of He can be simulated for any combination of He and nH within the limits stated above.
Simulation of nonindependence of error rates among samples:
We simulated the effect of variable sample quality by changing the probability distribution of ranked errors in the simulated data from uniform to a more realistic S shape. Multiplying ɛ1 and ɛ2 in the ith simulated duplicate genotype (i = 1, . . ., n) by
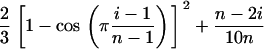 |
skews the distribution of error probability so that 1.7% of error probability is distributed in the first 20% of the simulated genotypes and 48% in the last 20%. This expression was designed to mimic to the most severely skewed error distribution observed among the 16 fox microsatellite loci.
Conversion of per-allele to per-genotype error rates:
For allelic dropout
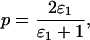 |
where p is the per-heterozygote dropout rate for a single locus (Wang 2004). The conversion of ɛ2 to f is less straightforward because, assuming that dropouts override false alleles, when ɛ1 is high f must be adjusted to account for the proportion of false alleles that will be obscured by allelic dropout. If ɛ1 is zero then f = 1 − f0 = 2ɛ2 − 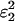 , which can be adjusted to allow for allelic dropout by multiplying by the probability of a false allele not dropping out (ignoring genotypes where both alleles are false), so that
, which can be adjusted to allow for allelic dropout by multiplying by the probability of a false allele not dropping out (ignoring genotypes where both alleles are false), so that
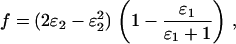 |
which simplifies to
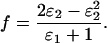 |
References
- Abecasis, G. R., S. S. Cherny and L. R. Cardon, 2001. The impact of genotyping error on family-based analysis of quantitative traits. Eur. J. Hum. Genet. 9: 130–134. [DOI] [PubMed] [Google Scholar]
- Altshuler, D., L. D. Brooks, A. Chakravarti, F. S. Collins, M. J. Daly et al., 2005. A haplotype map of the human genome. Nature 437: 1299–1320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonin, A., E. Bellemain, P. Bronken Eidesen, F. Pompanon, C. Brochman et al., 2004. How to track and assess genotyping errors in population genetics studies. Mol. Ecol. 13: 3261–3273. [DOI] [PubMed] [Google Scholar]
- Bradley, B. J., and L. Vigilant, 2002. False alleles derived from microbial DNA pose a potential source of error in microsatellite genotyping of DNA from faeces. Mol. Ecol. Notes 2: 602–605. [Google Scholar]
- Breen, M., S. Jouquand, C. Renier, C. S. Mellersh, C. Hitte et al., 2001. Chromosome-specific single-locus FISH probes allow anchorage of an 1800-marker integrated radiation-hybrid/linkage map of the domestic dog genome to all chromosomes. J. Genome Res. 11: 1784–1795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broquet, T., and E. Petit, 2004. Quantifying genotyping errors in noninvasive population genetics. Mol. Ecol. 13: 3601–3608. [DOI] [PubMed] [Google Scholar]
- Chesser, R. K., 1991. Influence of gene flow and breeding tactics on gene diversity within populations. Genetics 129: 573–583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Creel, S., G. Spong, J. L. Sands, J. Rotella, J. Zeigle et al., 2003. Population size estimation in Yellowstone wolves with error-prone noninvasive microsatellite genotypes. Mol. Ecol. 12: 2003–2009. [DOI] [PubMed] [Google Scholar]
- Ewen, K. R., M. Bahlo, S. A. Treloar, D. F. Levinson, B. Mowry et al., 2000. Identification and analysis of error types in high-throughput genotyping. Am. J. Hum. Genet. 67: 727–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feakes, R., S. Sawcer, J. Chataway, F. Coraddu, S. Broadley et al., 1999. Exploring the dense mapping of a region of potential linkage in complex disease: an example in multiple sclerosis. Genet. Epidemiol. 17: 51–63. [DOI] [PubMed] [Google Scholar]
- Gagneux, P., C. Boesch and D. S. Woodruff, 1997. Microsatellite scoring errors associated with noninvasive genotyping based on nuclear DNA amplified from shed hair. Mol. Ecol. 6: 861–868. [DOI] [PubMed] [Google Scholar]
- Gottelli, D., C. Sillero-Zubiri, G. D. Applebaum, M. S. Roy, D. J. Girman et al., 1994. Molecular genetics of the most endangered canid: the Ethiopian wolf, Canis simensis. Mol. Ecol. 3: 301–312. [DOI] [PubMed] [Google Scholar]
- Hadfield, J. D., D. S. Richardson and T. Burke, 2006. Towards unbiased parentage assignment: combining genetic, behavioural and spatial data in a Bayesian framework. Mol. Ecol. 15: 3715–3730. [DOI] [PubMed] [Google Scholar]
- Hoffman, J. I., and W. Amos, 2005. Microsatellite genotyping errors: detection approaches, common sources and consequences for paternal exclusion. Mol. Ecol. 14: 599–612. [DOI] [PubMed] [Google Scholar]
- Holmes, N. G., H. F. Dickens, H. L. Parker, M. M. Binns, C. S. Mellersh et al., 1995. Eighteen canine microsatellites. Anim. Genet. 26: 132–133. [DOI] [PubMed] [Google Scholar]
- Jeffery, K. J., L. F. Keller, P. Arcese and M. W. Bruford, 2001. The development of microsatellite loci in the song sparrow, Melospiza melodia (Aves) and genotyping errors associated with good quality DNA. Mol. Ecol. Notes 1: 11–13. [Google Scholar]
- Johnson, P. C. D., K. S. Llewellyn and W. Amos, 2000. Microsatellite loci for studying clonal mixing, population structure and inbreeding in a social aphid, Pemphigus spyrothecae (Hemiptera: Pemphigidae). Mol. Ecol. 9: 1445–1446. [DOI] [PubMed] [Google Scholar]
- Johnson, P. C. D., L. M. I. Webster, A. Adam, R. Buckland, D. A. Dawson et al., 2006. Abundant variation in microsatellites of the parasitic nematode Trichostrongylus tenuis and linkage to a tandem repeat. Mol. Biochem. Parasitol. 148: 210–218. [DOI] [PubMed] [Google Scholar]
- Jones, A. G., and W. R. Ardren, 2003. Methods of parentage analysis in natural populations. Mol. Ecol. 12: 2511–2523. [DOI] [PubMed] [Google Scholar]
- Kalinowski, S. T., M. L. Taper and S. Creel, 2006. Using DNA from non-invasive samples to census populations: an evidential approach tolerant of genotyping errors. Conserv. Genet. 7: 319–329. [Google Scholar]
- Keller, L. F., and D. M. Waller, 2002. Inbreeding effects in wild populations. Trends Ecol. Evol. 17: 230–241. [Google Scholar]
- Kirkpatrick, S., C. D. Gelatt and M. P. Vecchi, 1983. Optimization by simulated annealing. Science 220: 671–680. [DOI] [PubMed] [Google Scholar]
- Legendre, P., and L. Legendre, 1998. Numerical Ecology. Elsevier, Amsterdam.
- Marshall, T. C., J. Slate, L. E. B. Kruuk and J. M. Pemberton, 1998. Statistical confidence for likelihood-based paternity inference in natural populations. Mol. Ecol. 7: 639–655. [DOI] [PubMed] [Google Scholar]
- Miller, C. R., P. Joyce and L. P. Waits, 2002. Assessing allelic dropout and genotype reliability using maximum likelihood. Genetics 160: 357–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrissey, M. B., and A. J. Wilson, 2005. The potential costs of accounting for genotyping errors in molecular parentage analyses. Mol. Ecol. 14: 4111–4121. [DOI] [PubMed] [Google Scholar]
- Navidi, W., N. Arnheim and M. S. Waterman, 1992. A multiple-tubes approach for accurate genotyping of very small DNA samples by using PCR: statistical considerations. Am. J. Hum. Genet. 50: 347–359. [PMC free article] [PubMed] [Google Scholar]
- Neff, M. W., K. W. Broman, C. S. Mellersh, K. Ray, G. M. Acland et al., 1999. A second-generation genetic linkage map of the domestic dog, Canis familiaris. Genetics 151: 803–820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nei, M., and A. K. Roychoudhury, 1974. Sampling variances of heterozygosity and genetic distance. Genetics 76: 379–390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ostrander, E. A., G. F. Sprague and J. Rine, 1993. Identification and characterization of dinucleotide repeat (CA)n markers for genetic mapping in dog. Genomics 16: 207–213. [DOI] [PubMed] [Google Scholar]
- Ostrander, E. A., F. A. Mapa, M. Yee and J. Rine, 1995. One hundred and one new simple sequence repeat-based markers for the canine genome. Mamm. Genome 6: 192–195. [DOI] [PubMed] [Google Scholar]
- Paetkau, D., 2003. An empirical exploration of data quality in DNA-based population inventories. Mol. Ecol. 12: 1375–1387. [DOI] [PubMed] [Google Scholar]
- Piggott, M. P., E. Bellemain, P. Taberlet and A. C. Taylor, 2004. A multiplex pre-amplification method that significantly improves microsatellite amplification and error rates for faecal DNA in limiting conditions. Conserv. Genet. 5: 417–420. [Google Scholar]
- Pompanon, F., A. Bonin, E. Bellemain and P. Taberlet, 2005. Genotyping errors: causes, consequences and solutions. Nat. Rev. Genet. 6: 847–859. [DOI] [PubMed] [Google Scholar]
- Randall, D. A., 2006. Determinants of genetic variation in Ethiopian wolves. Ph.D. Thesis, Department of Zoology, University of Oxford, Oxford.
- SanCristobal, M., and C. Chevalet, 1997. Error tolerant parent identification from a finite set of individuals. Genet. Res. 70: 53–62. [Google Scholar]
- Sieberts, S. K., E. M. Wijsman and E. A. Thompson, 2002. Relationship inference from trios of individuals, in the presence of typing error. Am. J. Hum. Genet. 70: 170–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slate, J., P. David, K. G. Dodds, B. A. Veenvliet, B. C. Glass et al., 2004. Understanding the relationship between the inbreeding coefficient and multilocus heterozygosity: theoretical expectations and empirical data. Heredity 93: 255–265. [DOI] [PubMed] [Google Scholar]
- Smith, K. L., S. C. Alberts, M. K. Bayes, M. W. Bruford, J. Altmann et al., 2000. Cross-species amplification, non-invasive genotyping, and non-Mendelian inheritance of human STRPs in savannah baboons. Am. J. Primatol. 51: 219–227. [DOI] [PubMed] [Google Scholar]
- Sobel, E., J. C. Papp and K. Lange, 2002. Detection and integration of genotyping errors in statistical genetics. Am. J. Hum. Genet. 70: 496–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taberlet, P., S. Griffin, B. Goossens, S. Questiau, V. Manceau et al., 1996. Reliable genotyping of samples with very low DNA quantities using PCR. Nucleic Acids Res. 24: 3189–3194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taberlet, P., L. P. Waits and G. Luikart, 1999. Noninvasive genetic sampling: look before you leap. Trends Ecol. Evol. 14: 323–327. [DOI] [PubMed] [Google Scholar]
- Valière, N., P. Berthier, D. Mouchirod and D. Pontier, 2002. GEMINI: software for testing the effects of genotyping errors and multitubes approach for individual identification. Mol. Ecol. Notes 2: 83–86. [Google Scholar]
- Waits, J. L., and P. L. Leberg, 2000. Biases associated with population estimation using molecular tagging. Anim. Conserv. 3: 191–199. [Google Scholar]
- Walters, K., 2005. The effect of genotyping error in sib-pair genomewide linkage scans depends crucially upon the method of analysis. J. Hum. Genet. 50: 329–337. [DOI] [PubMed] [Google Scholar]
- Wandeler, P., 2004. Spatial and temporal population genetics of Swiss red foxes (Vulpes vulpes) following a rabies epizootic. Ph.D. Thesis, School of Biosciences, University of Cardiff, Cardiff, UK.
- Wandeler, P., and S. M. Funk, 2006. Short microsatellite DNA markers for the red fox (Vulpes vulpes). Mol. Ecol. Notes 6: 98–100. [Google Scholar]
- Wandeler, P., S. Smith, P. A. Morin, R. A. Pettifor and S. M. Funk, 2003. Patterns of nuclear DNA degeneration over time—a case study in historic teeth samples. Mol. Ecol. 12: 1087–1093. [DOI] [PubMed] [Google Scholar]
- Wang, J., 2004. Sibship reconstruction from genetic data with typing errors. Genetics 166: 1963–1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wattier, R., C. R. Engel, P. Saumitou-Laprade and M. Valero, 1998. Short allele dominance as a source of heterozygote deficiency at microsatellite loci: experimental evidence at the dinucleotide locus Gv1CT in Gracilaria gracilis (Rhodophyta). Mol. Ecol. 7: 1569–1573. [Google Scholar]
- Wright, S., 1951. The genetical structure of populations. Ann. Eugen. 15: 323–354. [DOI] [PubMed] [Google Scholar]