

Abstract
The product of the von Hippel-Lindau gene (VHL) acts as the substrate-recognition component of an E3 ubiquitin ligase complex that ubiquitylates the catalytic α subunit of hypoxia-inducible factor (HIF) for oxygen-dependent destruction. Although emerging evidence supports the notion that deregulated accumulation of HIF upon the loss of VHL is crucial for the development of clear-cell renal cell carcinoma (CC-RCC), the molecular events downstream of HIF governing renal oncogenesis remain unclear. Here, we show that the expression of a homophilic adhesion molecule, E-cadherin, a major constituent of epithelial cell junctions whose loss is associated with the progression of epithelial cancers, is significantly down-regulated in primary CC-RCC and CC-RCC cell lines devoid of VHL. Reintroduction of wild-type VHL in CC-RCC (VHL−/−) cells markedly reduced the expression of E2 box-dependent E-cadherin-specific transcriptional repressors Snail and SIP1 and concomitantly restored E-cadherin expression. RNA interference-mediated knockdown of HIFα in CC-RCC (VHL−/−) cells likewise increased E-cadherin expression, while functional hypoxia or expression of VHL mutants incapable of promoting HIFα degradation attenuated E-cadherin expression, correlating with the disengagement of RNA polymerase II from the endogenous E-cadherin promoter/gene. These findings reveal a critical HIF-dependent molecular pathway connecting VHL, an established “gatekeeper” of the renal epithelium, with a major epithelial tumor suppressor, E-cadherin.
Individuals who inherit one faulty von Hippel-Lindau gene (VHL) allele are predisposed to VHL disease, which is characterized by the development of cerebellar, spinal, and retinal hemangioblastoma, pheochromocytoma, and clear-cell renal cell carcinoma (CC-RCC) (29). The tumor develops upon the somatic loss of the remaining wild-type VHL allele in a susceptible cell. Importantly, biallelic loss of VHL is associated with the vast majority of sporadic CC-RCCs, establishing VHL as a critical suppressor of renal oncogenesis (29). CC-RCC is resistant to conventional radiation and chemotherapies, and approximately one-quarter of renal cancer patients present with advanced disease, including locally invasive or metastatic CC-RCC (12). Unfortunately, one-third of patients who undergo surgical removal of localized tumors have recurrence of the disease, and the median survival for patients harboring metastatic CC-RCC is 13 months (12). Moreover, the principal cause of morbidity and death of VHL patients is CC-RCC (29). Despite the need to better understand the aggressive nature of CC-RCC, the molecular pathways governing its malignant phenotype remain unresolved.
The most well-characterized function of VHL is as a substrate-recognition component of the SCF (Skp1/Cdc53/F-box protein)-like E3 ubiquitin ligase complex called ECV (elongins/Cul2/VHL) that selectively ubiquitylates oxygen-dependent prolyl-hydroxylated α subunits of hypoxia-inducible factor (HIFα) (24, 26, 43, 49). Under hypoxic conditions, HIFα is stabilized, recruits p300/CBP, and binds to its constitutively expressed partner HIFβ/arylhydrocarbon receptor nuclear translocator (ARNT) (48). The heterodimeric HIF complex binds to a consensus 5′-RCGTG-3′ hypoxia-responsive element in enhancers and promoters to trigger the transcription of numerous hypoxia-inducible genes that promote cellular adaptation to hypoxia, enabling cell survival under compromised oxygen availability (48). Concordantly, tumor cells devoid of VHL have enhanced expression of HIF target genes, such as VEGF (vascular endothelial growth factor; angiogenesis), GLUT-1 (glucose transporter 1; anaerobic metabolism), and EPO (erythropoietin; production of oxygen-carrying red blood cells), irrespective of oxygen tension (29). The overexpression of these and other hypoxia-inducible genes likely explains the angiogenic phenotype of VHL-associated tumors and also supports the notion that constitutive stabilization of HIFα is causally linked to tumorigenesis. In support, Kaelin and colleagues have shown that forced stable expression of HIF-2α in CC-RCC cells ectopically expressing wild-type VHL overrides the tumor suppressor capacity of VHL and restores the tumorigenic potential of CC-RCC cells in an animal xenograft system (32). Conversely, short hairpin RNA (shRNA)-mediated knock-down of HIF-2α is sufficient to suppress the tumorigenic capacity of RCC cells devoid of VHL (31). Notably, all CC-RCC-causing VHL mutants tested to date have shown a failure in either assembling of ECV complex or binding to HIFα (11, 21). However, the critical event(s) downstream of HIF that causes neoplastic transformation of proximal renal tubular epithelial cell (the origin of CC-RCC) is unclear.
Proper regulation of cell-cell adhesion is vital during cell growth, differentiation, and tissue development. E-cadherins, homophilic adhesion molecules, and their associated catenins are the major constituents of cell junctions in polarized epithelial cells (46). Increased expression of E-cadherin is associated with the differentiation of mesenchymal cells into tubular epithelial cells of the adult nephron. Conversely, loss of cell-cell adhesion is frequently associated with tumor progression and metastasis (46). In support of this paradigm, the loss of E-cadherin is associated with the progression of numerous carcinoma types, and forced expression of E-cadherin suppresses tumor development and invasion in various in vitro and in vivo tumor model systems, establishing E-cadherin as a critical tumor suppressor of the epithelium (46). Recently, Krishnamachary et al. and Esteban et al. independently showed that the loss of VHL in CC-RCC cells results in the loss of E-cadherin expression in an HIF-dependent manner (16, 34). Krishnamachary et al. argue that the regulation of E-cadherin expression is exclusively HIF-1-dependent, while Esteban et al. demonstrated dependency on both HIF-1 and HIF-2 (16, 34). Furthermore, Krishnamachary et al. showed that the activation of the HIF-1 pathway upon the loss of VHL transactivates E-cadherin transcriptional repressors TCF3 (also known as E12/E47), ZFHX1A (δEF1 or ZEB1), and SIP1 (Smad-interacting protein-1; also known as ZEB-2 or ZFHX1B), which correlated with the down-regulation of E-cadherin transcription (34).
Here, we examined over 100 primary CC-RCC samples via Affymetrix oligonucleotide arrays and show that the expression of E-cadherin transcripts is significantly down-regulated in CC-RCC. In addition, the analysis of 56 CC-RCC samples on tissue microarrays and immunohistochemistry on CC-RCC nephrectomy specimens from 13 patients revealed a strong positive correlation between the expression of VHL and E-cadherin. Small interfering RNA (siRNA)-mediated knockdown of endogenous VHL or functional hypoxia resulted in dramatic attenuation of E-cadherin expression. Importantly, reintroduction of wild-type VHL, but not a CC-RCC-causing VHL mutant incapable of promoting HIFα degradation, in CC-RCC (VHL−/− HIF-1α−/−) cells fully restored E-cadherin expression, in part, via HIF-dependent regulation of E2 box-dependent transcriptional repressors Snail and SIP1 but not Slug, TCF3, or ZFHX1A, and subsequent engagement of RNA polymerase (Pol) II on endogenous E-cadherin promoter/gene. These findings provide a compelling molecular mechanism governing the transcription of E-cadherin by the VHL-HIF pathway and strengthen the potential involvement of E-cadherin in the development of CC-RCC.
MATERIALS AND METHODS
Cells.
HEK293A embryonic kidney cells, U2OS osteosarcoma, and 786-O (VHL−/−) renal clear-cell carcinoma cell lines were obtained from the American Type Culture Collection (Rockville, MD) and maintained in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% heat-inactivated fetal bovine serum (Sigma) at 37°C in a humidified 5% CO2 atmosphere. 786-O subclones ectopically expressing wild-type hemagglutinin (HA)-VHL (786-VHL) or mutant HA-VHL(C162F) or HA-VHL(L188V) were previously described (21, 39). RCC4 (VHL−/−) renal clear-cell carcinoma subclones stably expressing HA-VHL (RCC4-VHL) or empty plasmid (RCC4-MOCK) were previously described (43). 786-VHL cells stably expressing HIF-2α(P531A) (786-VHL+HIF-2α) or empty control (786-VHL+MOCK) via retrovirus were previously described (32) and were generously provided by William G. Kaelin. 786-O subclones stably expressing pRetroSUPER-empty or pRetroSUPER-HIF2α shRNA were previously described (31).
Antibodies.
Monoclonal anti-HA antibody (12CA5) was obtained from Roche Molecular Biochemicals. Monoclonal anti-VHL antibody (IG32) was as previously described (30). Anti-β-catenin, anti-lamin A/C, and anti-α-tubulin antibodies were obtained from Santa Cruz (Santa Cruz, CA), Abcam (Cambridge, MA), and Sigma-Aldrich (Oakville, Ontario, Canada), respectively. Anti-E-cadherin antibody was obtained from BD Transduction Labs (Mississauga, Canada). Anti-HIF-2α antibody was obtained from Novus Biologicals Inc. (Littleton, CO).
Plasmids.
Mammalian expression plasmid pRc-CMV-HA-VHL(WT) was described previously (39). Expression plasmid encoding Snail was generously provided by Paul Hamel. E-cadherin core promoter (−308/+21)-luciferase reporter plasmids (E-cad prom-luc wild type [WT] or mutE2, which contains inactivating mutations in both E2 boxes) and expression plasmids encoding SIP1 were previously described (13). Plasmids containing shRNA against E-cadherin were generated using the Invitrogen pENTR/U6 system. Inserts containing hairpin DNA were generated using Invitrogen's Block iT siRNA designer, and insert cassettes were bought polyacrylamide gel electrophoresis (PAGE) purified from Invitrogen using the pENTR recommended protocol. The four generated shRNAs were chosen based on their predicted ability of silencing and their placement within the E-cadherin transcript. Each shRNA has 21 mer of complementary sequences starting at positions 1184, 1660, 1973, and 2128 of the E-cadherin gene (accession number NM004360). A CGAA loop is included in all plasmids. The nontargeting or scrambled shRNA was created in a similar manner with a sequence of 5′-GGAGCACGGTATTCGGGTCTACTAA-3′, which bears no sequence similarity with the E-cadherin transcript.
Immunoprecipitation and immunoblotting.
Immunoprecipitation and Western blotting were performed as described previously (50). In brief, cells were lysed in EBC buffer (50 mM Tris [pH 8.0], 120 mM NaCl, 0.5% NP-40) supplemented with a cocktail of protease and phosphatase inhibitors (Roche, Laval, Canada). Immunoprecipitates immobilized on protein A-Sepharose beads (Amersham Biosciences, Piscataway, NJ) were washed five times with NETN buffer (20 mM Tris [pH 8.0], 120 mM NaCl, 1 mM ETDA, 0.5% NP-40), eluted by boiling in sodium dodecyl sulfate (SDS)-containing sample buffer, and size-fractionated by SDS-PAGE. Resolved proteins were then electrotransferred onto polyvinylidene difluoride membrane (Bio-Rad Laboratories, Hercules, CA), immunoblotted with the various antibodies, and visualized by chemiluminescence (Amersham Biosciences, Piscataway, NJ).
Hypoxia treatment of cells.
Cells were maintained at 1% O2 for the times indicated in the legend to Fig. 6 in a ThermoForma (Marietta, OH) hypoxia chamber (5% CO2, 10% H2, 85% N2). Cell lysates were prepared in the chamber in hypoxic environment prior to further experimentation.
FIG. 6.

VHL activity is required for E-cadherin transcription. (A) ChIP using anti-RNA Pol II antibody was performed on sheared chromatin from 786-O cell lines (VHL−/−) that had been stably transfected with wild-type VHL (open bar) or mutant VHL(C162F) (solid bar). IP DNA was determined for the promoter and exon 10 of E-cadherin and the promoter of cyclophilin A using real-time PCR, and the value in VHL(WT) cells was arbitrarily set to 1.0. (B) RNA Pol II ChIPs were performed in 786-VHL(WT) cells exposed to 4 or 20 h of hypoxia (1% oxygen). IP DNA for exon 10 of E-cadherin was normalized to the IP DNA for the cyclophilin A promoter (left). Normoxia was arbitrarily set to 1.0. Expression of VEGF was assessed by real-time PCR as internal control for hypoxia treatment (right).
Immunohistochemical staining.
Formalin-fixed paraffin-embedded sections from 13 nephrectomy specimens with renal cell carcinoma of clear-cell type (CC-RCC) were obtained from the files of The Department of Pathology and Laboratory Medicine at The University Health Network (Toronto, Canada). These tissue blocks were used and processed in accordance with a University Health Network Research Ethics Board-approved protocol concerning gene expression in renal cell carcinoma. Tissues were fixed in 10% neutral buffered formalin for 24 to 36 h. Representative sections of tumor with adjacent nontumor renal parenchyma, 3 to 4 mm in thickness, were embedded in paraffin, and 5-μm sections were cut and placed on coated slides for light microscopy. Tumor morphology and classification were assessed using standard hematoxylin and eosin staining. The tumors were classified as CC-RCC according to criteria described in the World Health Organization classification of renal tumors (15). Immunohistochemical staining for E-cadherin and VHL was performed manually using a standard avidin-biotin-peroxidase complex method. Sections were incubated overnight in a humidified chamber with either unlabeled mouse anti-human E-cadherin or mouse anti-human VHL antibodies, each at a 1:2,000 dilution, following microwave pretreatment for antigen retrieval. The sections were then incubated with a biotinylated secondary antibody (horse anti-mouse immunoglobulin G at a dilution of 1:200) and the avidin-peroxidase complex. The color reaction was visualized using diaminobenzidine as the chromogen. The tissue was then lightly counterstained with hematoxylin.
TMAs.
Tissue microarrays (TMAs) consisting of quadruplicate representative 1.0-mm cores from 56 CC-RCCs with various Fuhrman grades (grades 1 to 4) and stages (pT1-pT4) were used to analyze the correlation between E-cadherin and VHL protein expression patterns. Five-micrometer sections from the CC-RCC TMAs were stained with anti-E-cadherin and anti-VHL antibodies as described above in “Immunohistochemical staining.” The slides were then scanned using a ScanScope (Aperio Technologies, Vista, CA) and scored by two observers. In order to be considered for evaluation, at least half of the TMA core was required to be present on the slide with at least 50% of the tissue present being tumor cells. Cores were scored as being either positive or negative. Only tumors with three or more cores in agreement were used for further analysis. Based on these criteria, 70% (39 out of 56) of the tumors on the TMA were suitable for the correlation analysis between E-cadherin and VHL.
Migration assay.
Cells were seeded on uncoated six-well plates, and scraping the cells with a sterile pipette tip generated the wounds. Cells were washed once and maintained in warm DMEM containing 0.5% fetal bovine serum (FBS) for the duration of the migration assay. A Nikon Eclipse TE200 inverted microscope equipped with a heated stage and a Hamamatsu digital camera (model C4742-95) was used to capture the images at the time points indicated in the legend to Fig. 3. Each wound measurement was taken in triplicate, and the experiment was repeated three times. Percent wound closure was determined by the measurement of the surface area of the wound at various time points relative to the starting wound surface area using the AxioVision AC program (version 4.2.0.0).
FIG. 3.

Down-regulation of E-cadherin increases the migration of embryonic kidney cells and invasion of CC-RCC cells. (A) HEK293A cells were transiently transfected with a plasmid encoding the scrambled shRNA or a cocktail of four E-cadherin-specific shRNAs. Equal amounts of the whole-cell lysates were immunoprecipitated with an anti-E-cadherin antibody, resolved by SDS-PAGE, and immunoblotted with an anti-E-cadherin antibody. Equal amounts of the remaining whole-cell lysates were resolved by SDS-PAGE and immunoblotted with an anti-γ-tubulin antibody. E-cadherin signal intensities were quantified using a Kodak Image Station 2000R densitometer and normalized against the corresponding γ-tubulin signals; values are indicated in the parentheses. (B) Wounds were created 48 h posttransfection with the indicated plasmids. Percent wound closure was determined by measuring the migration of cells from the wound edge 25 h postwound scrape. Each wound measurement was taken in triplicate, and the experiment was repeated three times. (C) Line graph representing early migration profile, as indicated by percent wound closure, as measured in the experiment shown in panel B, of HEK293A cells transfected with the indicated shRNA plasmids. (D) 786-VHL cells were transiently transfected with a plasmid encoding the scrambled shRNA or a cocktail of four E-cadherin-specific shRNAs. Equal amounts of the whole-cell lysates were immunoprecipitated with an anti-E-cadherin antibody, resolved by SDS-PAGE, and immunoblotted with an anti-E-cadherin antibody. Equal amounts of the remaining whole cell lysates were resolved by SDS-PAGE and immunoblotted with an anti-hnRNP antibody. E-cadherin signal intensities were quantified using a Kodak Image Station 2000R densitometer and normalized against the corresponding hnRNP signals; results are given in parentheses. (E) 786-VHL cells were transiently transfected with the indicated plasmids as shown in panel D. Cells were counted 72 h posttransfection, and 2.5 × 104 cells were seeded into BD Matrigel Invasion Chambers and incubated for 22 h. The invading cells were stained with 0.1% crystal violet, and images were captured under an inverted light microscope. Cells were counted from photographs of the membrane, and each experiment was repeated twice. The relative change in invasion was determined by counting the number of invading cells transfected with E-cadherin-specific shRNA and normalizing the value against the number of invading cells transfected with the scrambled shRNA (arbitrarily set at 1.0). Anti-E-cad, anti-E-cadherin; IP, immunoprecipitation; IB, immunoblot; Anti-Tub, anti-γ-tubulin; shE-cad, E-cadherin-specific shRNA; shScram, scrambled shRNA; T, time.
Invasion assay.
BioCoat Matrigel Invasion Chambers (BD Biosciences) containing inserts with a matrigel and an underlying membrane with 8-μm pores were used as previously described (35). Briefly, the inserts were hydrated with warm DMEM without FBS in 5% CO2 at 37°C for two hours. A total of 2.5 × 104 cells were seeded into the upper chamber containing DMEM with no FBS, and the lower chamber contained DMEM with 10% FBS. Cells were incubated at 37°C in 5% CO2 for 22 h. Noninvading cells were removed from the upper membrane by scrubbing with a cotton swab. The invading cells were fixed with 95% ethanol, stained with 0.1% crystal violet (Sigma), and visualized under an inverted light microscope. Cells were counted from photographs of the membrane and each experiment was repeated twice.
Subcellular fractionation.
Cells were resuspended in buffer A (10 mM HEPES [pH 7.9], 10 mM KCl, 1.5 mM MgCl2, 0.34 M sucrose, 10% glycerol) supplemented with protease and phosphatase inhibitors (Roche, Laval, Canada) and 1 mM dithiothreitol (DTT) and subsequently lysed with 0.1% Triton X-100. Samples were incubated for 7 min on ice and centrifuged. While the supernatant was recovered (cytoplasmic fraction), the pellet was washed with buffer A and resuspended in buffer B (0.2 mM EGTA [pH 8], 3 mM EDTA [pH 8]) supplemented with protease and phosphatase inhibitors (Roche, Laval, Canada) and 1 mM DTT. After a 30-min incubation period, samples were centrifuged, and the resulting supernatant was isolated (nuclear fraction).
Dual-luciferase assay.
U2OS osteosarcoma cells grown on six-well plates were transfected with a total of 2.5 μg of expression plasmids using Fugene 6 (Roche). E-cad prom-luc WT or mutE2 (0.9 μg per transfection) was used to measure E-cadherin core promoter-mediated transcription, and 0.1 μg of the Renilla luciferase plasmid, pRL-SV40 (Promega), was used as a transfection control. An empty pcDNA3.1 plasmid (Invitrogen) was used to maintain a constant final amount of transfected DNA. Cells were lysed 48 h after transfection, and luciferase assays were performed using the Dual-Luciferase Reporter Assay system (Promega), and relative light units (RLUs) were measured using a Lumat LB9507 luminometer (Berthold Technologies). Firefly luciferase RLUs were normalized against Renilla luciferase RLUs and standardized to the result of transfection with only the E-cad prom-luc WT, which was arbitrarily set to 1.0. Experiments and transfections were performed in triplicate with one representative experiment presented. Error bars represent standard deviations.
Microarray analysis.
We have established a large gene expression profiling database of renal tumors, some of which have previously been published (18, 63). For this study, we selected a total of 105 renal tumors of clear-cell type and 12 normal kidney tissue samples. The Affymetrix HGU133 Plus 2.0 GeneChip oligonucleotide arrays were used for all 117 cases. The HGU133 Plus 2.0 arrays contain 54,675 probe sets, representing approximately 47,000 transcripts and variants. The manufacturer's recommended protocol (1) was followed for expression profiling. Briefly, for oligonucleotide expression profiling, 5 to 20 μg of total RNA was used to prepare antisense biotinylated RNA. A subset of cases was spiked with external poly(A) RNA positive controls (Affymetrix, Santa Clara, CA). Synthesis of cDNA was performed with the use of T7-oligo(dT) primer. In vitro transcription was performed using a Bioarray Transcript Labeling Kit (Enzo, New York). The biotinylated cRNA was subsequently fragmented, and 15 μg was hybridized to each array at 45°C for 16 h. Scanning was performed in a GeneChip 3000 scanner. Quality assessment was performed in a GeneChip Operating System 1.4 (Affymetrix) using global scaling to a target signal of 500. Quality assessment was also performed using denaturing gel electrophoresis. Median background was 73, the median scaling factor was 3.06, and the median GADPH (glyceraldehyde-3-phosphate dehydrogenase) 3′/5′ ratio was 1.03, indicative of a high overall array and RNA quality.
Statistical analyses were performed in the statistical environment R 2.2, utilizing packages from the Bioconductor project. The MAS 5 algorithm was used to perform preprocessing of the CEL files, including background adjustment, quartile normalization, and summarization. The means and the standard errors for E-cadherin gene expressions were calculated for each of the group of samples. A two-tailed Student's t test was used to determine statistically significant differences between various groups.
siRNA-mediated VHL knockdown.
siGENOME SMARTpool targeted to VHL was used (Dharmacon, Austin, TX). A nontargeting scrambled siRNA duplex was used as a negative control (5′-CCAUUCCGAUCCUGAUCCG-3′). HEK293A (VHL+/+) cells grown on six-well tissue culture plates were transfected with scrambled and VHL siRNA at a final concentration of 200 nM. Briefly, 8 μl of Oligofectamine (Invitrogen) was incubated with 48 μl of Opti-MEM I (Gibco/Invitrogen) for 8 min. The Oligofectamine mixture was added to the siRNA diluted in 175 μl of Opti-MEM I and incubated for 20 min before adding the resulting mixture to 800 μl of Opti-MEM I into the wells. After 3 h, 300 μl of DMEM containing 30% heat-inactivated FBS (Sigma) was added to the plates. RNA was extracted 48 h after transfection using an RNeasy kit (QIAGEN, Mississauga, Ontario, Canada) treated with RNA-free DNase (Ambion, Texas), and first-strand cDNA synthesis was performed.
Quantitative real-time PCR.
For first-strand cDNA synthesis, 1 μl of oligo(dT)23 primer (Sigma) was incubated with 5 μg of RNA and distilled H2O (total reaction volume of 20 μl) for 10 min at 70°C in a thermal cycler (MJ Research, Boston, MA). The mixture was cooled to 4°C, at which time 4 μl of 5× first-strand reaction buffer, 2 μl of 0.1 M DTT, 1 μl of a 10 mM concentration of each deoxynucleoside triphosphate, and 1 μl of Superscript II reverse transcriptase (Invitrogen) were added. cDNA synthesis was performed for 1.5 h at 42°C, followed by 15 min at 70°C in the thermal cycler. Human genomic DNA standards (human genomic DNA was obtained from Roche, Mannheim, Germany) or cDNA equivalent to 20 ng of total RNA were added to the quantitative PCR (qPCR) reaction mixture in a final volume of 10 μl containing 1× PCR buffer (without MgCl2), 3 mM MgCl2, 0.25 units of Platinum Taq DNA polymerase, a 0.2 mM concentration of each deoxynucleoside triphosphate, 0.3 μl of SYBR Green I, 0.2 μl of ROX reference dye, and a 0.5 μM concentration of each primer (Invitrogen). Amplification conditions were as follows: 95°C (3 min), 40 cycles of 95°C (10 s), 65°C (15 s), 72°C (20 s), and 95°C (15 s). qPCR was performed using the ABI Prism 7900HT Sequence Detection System (Applied Biosystems, Foster City, CA). Gene-specific oligonucleotide primers designed using Primer Express (Applied Biosystems) were as follows: Snail primer set (5′-TTCAACTGCAAATACTGCAACAAG-3′ and 5′-CGTGTGGCTTCGGATGTG-3′), SIP1 primer set (5′-CCACACTTCGCGGCTTCTT-3′ and 5′-CGATCTGCGAAGTCTTGTTTGT-3′), E-cadherin primer set (5′-GTCATCCAACGGGAATGCA-3′ and 5′-TGATCGGTTACCGTGATCAAAA-3′), GLUT-1 primer set (5′-CACCACCTCACTCCTGT-TACTT-3′ and 5′-CAAGCATTTCAAAACCATGTTTCTA-3′), VEGF primer set (5′-CTCTCTCCCTCATCGGTGACA-3′ and 5′-GGAGGGCAGAGCTGAGTGTTAG-3′), and U1AsnRNP1 primer set (5′-CAACGACAGCCGAGACATGTA-3′ and 5′-AGCCTCCATCAAATACCCATTC-3′). SYBR Green I fluoresces during each cycle of the qPCR by an amount proportional to the quantity of amplified cDNA (the amplicon) present at that time. The point at which the fluorescent signal is statistically significant above background is defined as the cycle threshold (CT). Expression levels of the various transcripts were determined by taking the average CT value for each cDNA sample performed in triplicate and measured against a standard plot of CT values from amplification of serially diluted human genomic DNA standards. Since the CT value is inversely proportional to the log of the initial copy number, the copy number of an experimental mRNA can be obtained from linear regression of the standard curve. A measure of the relative difference in copy number was determined for each mRNA. Values were normalized to expression of U1AsnRNP1 mRNA, expressed relative to scrambled siRNA samples (arbitrarily set to 1.0), and represented as the mean value of three independent experiments performed in triplicate ± standard deviations.
ChIP.
Chromatin immunoprecipitation (ChIP) was performed as published previously using the Upstate ChIP assay kit (17). Five micrograms of anti-RNA polymerase II (N-20) antibody (Santa Cruz) was added to sheared, formaldehyde cross-linked chromatin preparations from 1 × 106 cells, and immunoprecipitation was performed overnight at 4°C. A control immunoprecipitation without the addition of antibody was also performed in parallel. An 18-μl aliquot (of a total of 1,800 μl) of chromatin was removed prior to immunoprecipitation to serve as an input control. The cross-links were reversed by addition of 2 μl of 5 M NaCl, and the sample was diluted 1 in 10 before real-time PCR was performed. Immune complexes were collected with protein A-agarose beads, and, after extensive washing, immune complexes were released, formaldehyde cross-links were reversed, and DNA was purified by phenol-chloroform extraction. Following ethanol precipitation, DNA was resuspended in 30 μl of water. Real-time PCR was performed on 2 μl of anti-Pol II immunoprecipitated DNA, 2 μl of no-antibody control, and 2 μl of the diluted input sample. Real-time PCR was performed in triplicate using SYBR green chemistry. Copies of the target gene were determined using genomic DNA as a standard curve (where 1 ng of genomic DNA is equivalent to 300 copies of a single copy gene). Immunoprecipitated DNA (IP DNA) was determined by subtracting the number of copies of the no-antibody control from the anti-Pol II immunoprecipitated DNA and dividing by the number of copies in the diluted input sample. Primers were designed to amplify the human E-cadherin promoter (forward, 5′-CCACGCACCCCCTCTCAGT-3′; reverse, 5′-GAGCGGGCTGGAGTCTGAAC-3′), human E-cadherin exon 10 (forward, 5′-CCGTGGATGTGCTGGATGTGA-3′; reverse, 5′-TGGGCAGTGTAGGATGTGATTTC-3′), and the human cyclophilin A promoter (forward, 5′-CCTCATGTGTCGTCCCCATCA-3′; reverse, 5′-CGCCCGTTTTATACCACGTTCG-3′).
RESULTS
Expression of E-cadherin is down-regulated in CC-RCC and correlates with VHL status.
We have established a large gene expression profiling database of renal tumors, some of which have previously been published (18, 63). For this study, we selected a total of 105 human renal tumors of clear-cell type and 12 normal kidney tissue samples. Using the Affymetrix HGU133 Plus 2.0 GeneChip oligonucleotide arrays for all 117 cases, we found that the expression of E-cadherin transcripts was significantly down-regulated in CC-RCC (Fig. 1A). This is consistent with immunohistochemical studies that showed reduced E-cadherin staining in the vast majority of CC-RCC tumor samples and cell lines tested (45, 58). However, the molecular mechanism that accounts for the frequent loss of E-cadherin in CC-RCC is unknown.
FIG. 1.

Expression of E-cadherin is down-regulated in CC-RCC and correlates with VHL status. (A) 105 CC-RCC tumor samples and 12 normal kidney tissue samples were analyzed using Affymetrix HGU133 Plus 2.0 GeneChip oligonucleotide arrays. Mean E-cadherin expression and standard error were calculated, and a two-tailed Student's t test was used to determine statistical significance between the two groups. (B) Immunohistochemical staining of a representative CC-RCC with anti-E-cadherin and anti-VHL antibodies. Note the negative staining of the tumor cells (upper right in each image) with each marker, in contrast to the positive staining shown by core tubule epithelium in the adjacent nontumor renal cortex (lower left in each image) (original magnification, ×50). (C) TMAs containing 56 CC-RCC were immunostained with anti-VHL and anti-E-cad antibodies. The slides were then scanned using Aperio ScanScope and scored blind by two independent observers as being either positive or negative. Tumor cores meeting the quality standard criteria (see Materials and Methods) were considered for the correlation analysis (bar graph). Representative cores of CC-RCC on TMAs stained with anti-VHL and anti-E-cadherin antibodies are shown where the top row depicts a representative core showing negative staining for VHL and E-cadherin and the bottom row depicts a representative core showing positive staining for VHL and E-cadherin. Tumor morphology and classification were assessed using standard hematoxylin and eosin staining.
To date, VHL is the most frequently mutated gene in CC-RCC, and biallelic inactivation of the VHL locus is associated with the development of greater than 80% of sporadic CC-RCC. Thus, we asked whether the expression of E-cadherin is associated with the VHL status. Hematoxylin and eosin staining of representative sections from nephrectomy specimens from 13 patients confirmed the characteristic morphological features of CC-RCC including nests of cells with abundant, optically clear cytoplasm and delicate cell membranes surrounded by a network of small, thin-walled blood vessels (data not shown). Each section studied by immunohistochemistry contained normal renal parenchyma including core convoluted tubules within the renal cortex (Fig. 1B, left lower half of the micrograph) adjacent to CC-RCC (right upper half of the micrograph). Membranous anti-E-Cadherin staining and cytoplasmic/membranous anti-VHL staining shown by core convoluted tubules were used as internal positive controls on each slide. Cells in this representative tumor sample showed correlative staining for E-cadherin and VHL, where negative staining for VHL observed in CC-RCC corresponded with markedly reduced staining of E-cadherin (Fig. 1B, compare upper and lower panels).
To further validate the positive correlation between VHL and E-cadherin expression, TMAs consisting of 56 CC-RCC cores in quadruplicate were generated. Thirty-nine of the CC-RCC samples met the quality standard criteria (see Materials and Methods) and were analyzed for E-cadherin and VHL protein expression patterns. While only 33% (5/15) of the tumors that stained negative for VHL (15/39) stained positive for E-cadherin, the majority (67% or 16/24) of tumors that stained positive for VHL (24/39) also stained positive for E-cadherin (Fig. 1C). However, a positive stain for VHL does not formally indicate the presence of a wild-type VHL, as, for example, a subtle point mutation will likely produce a positive staining signal. Thus, additional mutational analysis will be required to generate a more precise E-cadherin:VHL correlation index.
Knockdown of endogenous VHL results in a dramatic attenuation of E-cadherin expression.
Reconstitution of 786-O (VHL−/− HIF-1α−/−) or RCC4 (VHL−/−) renal carcinoma cells with HA-VHL dramatically restored the expression of E-cadherin protein and mRNA, as measured by Western blotting and quantitative real-time PCR, respectively (Fig. 2A and B). In addition, siRNA-mediated knockdown of endogenous VHL in HEK293A embryonic kidney cells resulted in marked down-regulation of E-cadherin expression (Fig. 2C). Microarray (Fig. 1A) and real-time PCR data strongly suggest that E-cadherin regulation by VHL is at the pretranslational level. The cytoplasmic domain of E-cadherin is in a complex with β-catenin, implicating a potential “outside-in” signaling where a loss of E-cadherin would release β-catenin to associate with the leukocyte enhancer factor /T-cell factor to regulate the transcription of cell cycle-related genes (e.g., cyclin D1) or invasion-related genes (e.g., metalloproteinase matrilysin and fibronectin) (46). Interestingly, an increased level of cyclin D1 has been observed in RCC cells devoid of VHL at high cell density (4), and cells expressing tumor-causing VHL mutants fail to assemble proper extracellular fibronectin matrices (50, 62). However, both the overall expression and subcellular localization of β-catenin remained unaffected by VHL (Fig. 2A and see Fig. S1 in the supplemental material), suggesting that β-catenin-mediated transcription is likely not involved in potential outside-in signaling via the loss of E-cadherin in the context of CC-RCC.
FIG. 2.

Loss of VHL results in down-regulation of E-cadherin. (A) VHL−/− 786-O and RCC4 cells stably expressing wild-type VHL or empty plasmid (MOCK) were lysed, equal amounts of total cellular lysates were separated by SDS-PAGE, and immunoblotted with the indicated antibodies. Anti-α-tubulin immunoblotting was performed as an internal loading control. (B) Expression of E-cadherin was measured by quantitative real-time PCR in 786-MOCK and 786-VHL cells and normalized to U1AsnRNP1 mRNA level. The E-cadherin level in 786-VHL cells was arbitrarily set to 1.0. Error bars represent standard deviations of the relative increases in expression between the indicated cell types over three independent experiments. (C) Endogenous VHL in HEK293A cells was knocked down using VHL-specific siRNA or scrambled nontargeting control siRNA. RNA was then extracted for cDNA synthesis and endogenous transcript levels of VHL, E-cadherin, and U1AsnRNP1 measured. Error bars represent standard deviations of the relative increases between the expression of the indicated mRNA relative to its expression using control siRNA (arbitrarily set to 1.0) over three independent experiments.
shRNA-mediated down-regulation of E-cadherin increases the invasive potential of CC-RCC.
The role of E-cadherin in modulating the migration and invasion properties of epithelial cells is well established. However, it is not known whether E-cadherin has similar biological effects in the context of kidney epithelial cells or CC-RCC. Although E-cadherin expression can be predictably determined by manipulating the status of VHL, altering the expression level of VHL has other consequences that can influence the motility and invasion properties of CC-RCC (see Discussion). Thus, we used an shRNA approach to specifically down-regulate the endogenous expression level of E-cadherin in HEK293A embryonic kidney epithelial cells, which resulted in a significant enhancement of migration as measured by percent wound closure (61.2% ± 4.8%) compared to cells expressing scrambled shRNA (43.0% ± 3.0%) (Fig. 3A and B). The change in motility was noticeable from the early time points, suggesting that the effect of modulating the expression of E-cadherin is not only potent but also immediate (Fig. 3C). shRNA-mediated down-regulation of E-cadherin consistently increased the motility of 786-VHL cells in a similar wound assay but was not statistically significant (data not shown). However, the invasion potential was increased (2.4 ± 0.2)-fold in comparison to 786-VHL cells expressing the scrambled shRNA, as measured on the standard matrigel invasion chambers (Fig. 3D and E). It should be noted that the changes in motility and invasion are likely underestimated due to the incomplete knockdown of E-cadherin (Fig. 3A and D). Nevertheless, these results suggest that the diminution of E-cadherin expression would promote the invasive property of CC-RCC.
VHL regulates E-cadherin expression via an HIF-dependent mechanism.
We next asked whether the regulation of E-cadherin expression by VHL is mediated through the activity of HIF. RCC4-VHL and 786-VHL cells were maintained under normoxic (21% oxygen) (Fig. 4A) or hypoxic (1%) (see Fig. S2 in the supplemental material) conditions for 16 h and analyzed by Western blotting. The expression of E-cadherin was dramatically reduced under hypoxia while preserving the expression status of VHL (Fig. 4A). The effect of hypoxic treatment was confirmed by the increase in HIF-2α expression (Fig. 4A). Although this result suggests that hypoxia-induced stabilization of HIF results in repression of E-cadherin expression, it is formally possible that VHL, independent of HIF, regulates the expression of E-cadherin in an oxygen-dependent manner. Therefore, we examined various VHL mutants that have retained or lost the ability to regulate HIF. Certain non-CC-RCC-associated VHL mutants have been shown to retain the ability to regulate HIF activity (11, 21, 62). For example, The L188V mutation allows proper oxygen-dependent degradation of HIFα and is associated with a subclass of VHL disease (type 2C), which is clinically characterized by the exclusive development of pheochromocytoma (21). In contrast, invariably all CC-RCC-associated VHL mutations tested to date, such as C162F, result in a complete loss of the ability to mediate the destruction of HIFα via the ubiquitin-proteasome pathway (11, 21). 786-O cells ectopically expressing VHL(C162F) showed negligible expression of E-cadherin, while those expressing VHL(L188V) showed higher detectable levels of E-cadherin, albeit at a lower level than observed in cells expressing VHL(WT) (Fig. 4B). In addition, 786-O (VHL−/− HIF-1α−/−) cells stably expressing wild-type VHL (786-VHL) infected with retroviruses that express functional and stable HIF-2α(P531A) (escapes VHL recognition) demonstrated a reduced level of E-cadherin relative to 786-VHL cells infected with “empty” retrovirus (Fig. 4C, compare lanes 1 and 2). Notably, the level of E-cadherin was inversely proportional to the level of HIF-2α (Fig. 4C). Conversely, 786-O subclones infected with retroviruses that express HIF-2α-specific shRNA demonstrated a markedly increased level of E-cadherin relative to 786-O cells infected with empty retrovirus (Fig. 4D). In addition, the activity of the exogenous E-cadherin promoter-driven luciferase reporter was much higher in 786-O cells reconstituted with wild-type VHL (786-WT; low HIF activity) than in 786-MOCK (high HIF activity) cells (Fig. 4E). Taken together, these results strongly suggest that HIF negatively regulates E-cadherin expression, at least in part, at the level of transcription.
FIG. 4.

VHL regulation of E-cadherin is HIF mediated. (A) RCC4-VHL cells were maintained under normoxia (N; 21% O2) or hypoxia (H; 1% O2) for 16 h, lysed, resolved on SDS-PAGE, and immunoblotted with anti-HIF-2α, anti-E-cadherin, and anti-HA antibodies. (B) 786-O cells stably expressing HA-VHL(WT), HA-VHL(C162F), or HA-VHL(L188V) were lysed, resolved on SDS-PAGE, and immunoblotted with the indicated antibodies, where α-tubulin served as an internal loading control. (C) 786-MOCK and 786-VHL cells infected with “empty” retrovirus (786-VHL+EMPTY) or retrovirus expressing constitutively stable and functional HIF-2α(P531A) were lysed, resolved by SDS-PAGE, and immunoblotted with the indicated antibodies, where α-tubulin served as an internal loading control. Signal intensities were quantified using a Kodak Image Station 2000R densitometer and normalized against the corresponding α-tubulin signals; results are given in parentheses. Experiments were performed three times with one representative experiment presented. (D) 786-O (VHL−/−) subclones stably expressing pRetroSUPER-empty or pRetroSUPER-HIF2α shRNA were lysed, and comparable amounts of whole-cell extracts were immunoprecipitated and immunoblotted with an anti-E-cadherin antibody. Equal amounts of the whole-cell extracts were also resolved on SDS-PAGE and immunoblotted with anti-HIF-2α and anti-actin antibodies. Experiments were performed three times with one representative experiment presented. (E) Dual-luciferase assays were performed in 786-MOCK and 786-VHL cells transfected with the firefly luciferase construct (E-cad prom-luc) driven by the human E-cadherin promoter sequence. Cytomegalovirus-driven Renilla luciferase was used as an internal transfection control, and the firefly luciferase RLUs were normalized against Renilla luciferase RLUs. Experiments and transfections were performed in triplicate with one representative experiment presented. Error bars represent standard deviations. IP, immunoprecipitation; IB, immunoblot.
VHL down-regulates E-cadherin-specific transcriptional repressors Snail and SIP1.
Mutational analyses have shown that biallelic somatic inactivating mutations of E-cadherin are rare (7, 8), and emerging evidence suggests that the loss or reduction in E-cadherin expression in cancer cells primarily occurs at the level of transcription (6, 19, 28, 65). The two major regulators of E-cadherin transcription are the zinc finger transcriptional repressors Snail and SIP1, which bind evolutionarily conserved E2 boxes located within the E-cadherin core promoter resulting in the inhibition of E-cadherin transcription (5, 10, 13). Moreover, hypoxic treatment of ovarian carcinoma cells was shown to attenuate the expression of E-cadherin via the upregulation of Snail (23).
Quantitative real-time PCR analysis showed a significant attenuation of both Snail and SIP1 in CC-RCC 786-O cells restored with VHL (Fig. 5A). However, negligible changes in the expression were noted for the other known E-cadherin transcriptional repressors Slug, TCF3, and ZFHX1A (data not shown). As expected, restoration of VHL reduced the expression of HIF target genes, VEGF and GLUT-1, and increased E-cadherin expression (Fig. 5A; compare with results shown in Fig. 2). These results suggest the possibility that VHL may increase the expression of E-cadherin by down-regulating the transcriptional repressors Snail and SIP1. In a complementary experiment, we tested the ability of VHL in the transactivation of E-cadherin promoter-driven luciferase reporter (Fig. 5B). As expected, E-cadherin promoter containing both E2 boxes had lower basal transcriptional activity relative to E-cadherin promoter with point mutations in the E2 boxes that abrogate Snail/SIP1 binding (Fig. 5B). Importantly, the addition of VHL markedly increased the wild-type E-cadherin promoter-driven luciferase transcription, but had insignificant effect on the E2 mutant E-cadherin promoter (Fig. 5B). As predicted, the increase in VHL-mediated transactivity of wild-type E-cadherin promoter-luciferase was dampened by the addition of SIP1 or/and Snail in a dosage-dependent manner (Fig. 5C). Coexpression analysis indicated that Snail or SIP1 had negligible effect on the steady-state level of VHL (see Fig. S3 in the supplemental material; also data not shown). These results demonstrate that the E2 boxes are functionally important in upregulating E-cadherin transcription by VHL, in part, via the down-regulation of SIP1 and/or Snail. However, neither SIP1 nor Snail individually or in combination achieved a complete inhibition of E-cadherin promoter-driven reporter activity. This suggests that there exist other yet-to-be-defined VHL/HIF-mediated E-cadherin-specific transcriptional repressors or that full repression requires the concerted actions of multiple repressors and involves, in addition to the E2 boxes, other elements within the E-cadherin promoter.
FIG. 5.

VHL-mediated transcription of E-cadherin is attenuated by Snail and SIP1 via the conserved E2 boxes. (A) Expression levels of E-cadherin, Snail, SIP1, VEGF, and GLUT-1 were measured by quantitative real-time PCR in 786-MOCK and 786-VHL cells and normalized to U1AsnRNP1 mRNA expression. Solid bars represent expression of the indicated mRNA in 786-MOCK cells relative to its expression in 786-VHL cells, which was arbitrarily set to 1.0. (B) Dual-luciferase assays were performed in U2OS cells transfected with the indicated expression plasmids. The firefly luciferase construct (E-cad prom-luc) was driven by the human E-cadherin promoter sequence (WT) or the promoter with point mutations in both E2 boxes (mutE2). Cytomegalovirus-driven Renilla luciferase was used as an internal transfection control, and the firefly luciferase RLUs were normalized against Renilla luciferase RLUs. Experiments and transfections were performed in triplicate with one representative experiment presented. Error bars represent standard deviations. (C) The experiment was performed as described in panel B with increasing concentrations of Snail and SIP1 individually mixed into HA-VHL transfection reactions at a ratio of 1:2, 2:2, and 4:2 (Snail or SIP1:HA-VHL) or equal quantities of SIP1 and Snail combined into HA-VHL transfection reactions at a ratio of 1:1 (Snail and SIP1:HA-VHL). Relative increases in induction (n-fold) were standardized to the E-cad prom-luc activity in the absence of exogenous VHL.
Wild-type, but not CC-RCC-causing mutant VHL, induces transcriptional activation of E-cadherin.
The principal mechanism by which transcriptional repressors attenuate the rate of transcription is by blocking the engagement of RNA Pol II and associated factors to the promoter. Notably, Snail has been shown to repress E-cadherin expression through the binding of Sin3A/histone deacetylase 1 and 2 complex (52), which is thought to impair recruitment of Pol II and transcriptional initiation via repressive changes in chromatin structure. Repressors can also act at a postinitiation step of transcription including inhibition of phosphorylation of the Pol II holoenzyme, which represents a key step in promoter escape and cessation in elongation (40). Therefore, we asked whether the engagement of Pol II on the endogenous E-cadherin promoter/gene is influenced by VHL. ChIP assays at the E-cadherin locus were performed using antibodies recognizing the N terminus of Pol II in 786-O cells expressing wild-type or an HIF regulation-defective mutant VHL(C162F) (Fig. 6A). The total amount of Pol II at the E-cadherin promoter and exon 10 was dramatically decreased in the absence of VHL functional activity (Fig. 6A). This was in contrast to the promoter of the housekeeping gene, cyclophilin A, where binding of Pol II was similar in wild-type and mutant VHL cells (Fig. 6A). This suggests that VHL's functional activity to negatively regulate HIF is necessary for transcriptional activation of E-cadherin.
To further establish a functional role of VHL/HIF in E-cadherin gene transcription, 786-O cells stably expressing wild-type VHL were exposed to hypoxia. Assessment of Pol II binding to genomic regions corresponding to coding regions of E-cadherin (exon 10) revealed that hypoxia decreased E-cadherin transcription (Fig. 6B). A similar decrease in Pol II binding was demonstrated at the promoter of E-cadherin (data not shown). As expected, hypoxia elicited a time-dependent increase in VEGF mRNA expression (Fig. 6B). Taken together, these results suggest that VHL activity, specifically E3 ligase function to negatively regulate HIF, is required for the transcription of the E-cadherin gene. Conversely, cellular hypoxia or loss of HIF-associated function of VHL results in the activation of HIF and disengagement of Pol II from the E-cadherin promoter, resulting in the down-regulation of E-cadherin transcription. However, it is not formally known whether HIF-mediated engagement of Pol II on E-cadherin promoter is strictly SIP1/Snail-dependent.
E-cadherin expression is cell density dependent.
E-cadherin expression in CC-RCC cells is also cell density dependent as measured by Western blotting and quantitative real-time PCR (see Fig. S4A and B in the supplemental material). Interestingly, the expression of VHL is strictly regulated by cell density where the steady-state amount of VHL in human renal proximal tubule epithelial cells was shown to increase more than 100-fold in dense cultures relative to sparse cultures (3). In addition, other components of the VHL E3 ligase complex showed a similar cell density-dependent regulation (44). Importantly, HIF-2α level was elevated in sparsely growing cells with low levels of VHL and significantly reduced or undetectable in confluent cells containing abundant VHL (44). Moreover, the ability of VHL to shuttle between the nucleus and the cytoplasm is also regulated by cell density (37), which in turn may influence the ability of VHL to regulate HIF activity (20). Thus, cell density-dependent expression of E-cadherin may be due to a corresponding cell density-dependent regulation of VHL stability/function.
DISCUSSION
VHL is a direct oxygen-dependent negative regulator of HIFα via the ubiquitin-proteasome pathway (29). The loss of VHL or VHL mutations associated with the development of CC-RCC invariably results in the accumulation/hyperactivation of HIF due to a failure in VHL's ability to either bind or ubiquitylate HIFα. Here, we propose that HIF—stabilized by hypoxia in the presence of wild-type VHL or upon mutation/loss of VHL—activates the transcriptional repressors SIP1 and Snail (likely via the HIF-engagement of the hypoxia-responsive element 5′-GCGTG-3′ found in the Snail promoter at position −86 to −82; SIP1 promoter/enhancer has not been defined), preventing Pol II engagement on the E-cadherin promoter and resulting in the down-regulation of E-cadherin expression (Fig. 7). There is, however, an additional pathway to consider (described below).
FIG. 7.
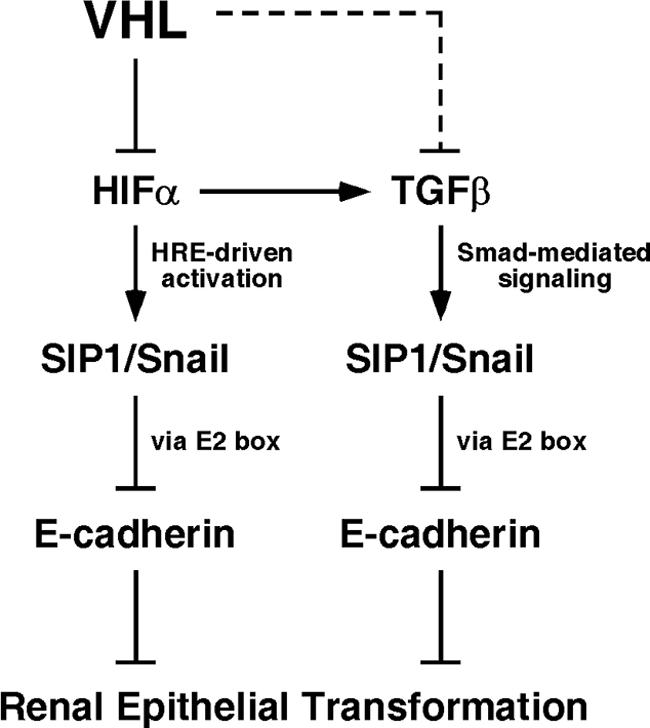
VHL gatekeeper's pathway in renal epithelium. See text for details.
Increased transforming-growth factor β (TGF-β) signaling and expression of Snail and SIP1, as well as the loss of E-cadherin expression, have all been correlated with the epithelial to mesenchymal transition process that occurs during normal development and acquisition of invasive phenotype in epithelial cancers (5, 10, 13, 64). Smad-mediated signaling by TGF-β has been shown to induce the expression of the repressors Snail and SIP1 (13, 27, 60). HIF-1 has been shown to upregulate the expression of the members of TGF-β family in a transcription-dependent manner under hypoxic conditions. HIF-1 and Smad proteins cooperate in regulating the expression of several hypoxia and TGF-β-regulated genes, including the expression of TGF-β2 in human umbilical vein endothelial cells (67). In addition, HIF-1 has been shown to directly bind to the TGF-β3 promoter and upregulate its expression under hypoxia during placental and epithelial development processes (47, 57). Therefore, increased transcriptional activity of HIF by the functional loss of VHL in CC-RCC may result in an upregulation of TGF-β signaling, resulting in a Smad-mediated induction of SIP1 and Snail and subsequent loss of E-cadherin (Fig. 7). Notably, VHL has also been shown to repress the expression of TGF-β1 by regulating its mRNA stability (Fig. 7, dashed line) (2). Whether this process is HIF mediated is currently unknown.
In the current work, we demonstrated that the loss of VHL leads to the dramatic down-regulation of E-cadherin in CC-RCC. VHL-dependent transactivation of E-cadherin was dependent on the conserved E2 boxes known to recruit transcriptional repressors Snail and SIP1 to the promoter of E-cadherin. Reintroduction of VHL in CC-RCC cells devoid of VHL showed a reduction in the expression of both Snail and SIP1, thereby explaining, at least in part, the resulting restoration of E-cadherin expression. Transcriptional repressors principally block transcription by inhibiting the engagement of Pol II to the promoter. In support, Snail has been shown to repress E-cadherin expression through the binding of histone deacetylase, promoting repressive changes in chromatin structure and thereby impairing the recruitment of Pol II and transcription (52). Consistent with this view, wild-type VHL enhanced the recruitment of Pol II to the E-cadherin promoter/gene, whereas hypoxia or a tumor-causing VHL mutant with a failure in targeting HIFα for ubiquitin-mediated destruction dramatically decreased the association of Pol II with the E-cadherin gene. Thus, VHL or functional hypoxia directly affects Pol II engagement on E-cadherin DNA likely via HIF-dependent regulation of E-cadherin-specific transcriptional repressors, revealing a previously unrecognized regulation of a major epithelial tumor suppressor E-cadherin.
During the preparation of the present report, Krishnamachary et al. and Esteban et al. showed that the loss of VHL in CC-RCC cells results in the loss of E-cadherin expression in a HIF-dependent manner (16, 34). Krishnamachary et al. argue that the regulation of E-cadherin expression is exclusively HIF-1 dependent, as 786-O (HIF-1α −/−) cells failed to express E-cadherin after VHL rescue (16, 34). However, our data clearly demonstrate that HIF-2, in the absence of HIF-1α, can restore E-cadherin expression upon reintroduction of VHL in 786-O cells. Although the reason for this discrepancy is unclear, the relative contribution of HIF-1 versus HIF-2 in the development of CC-RCC is an emerging area of research. For example, the introduction of a HIF-1α mutant that escapes VHL recognition into CC-RCC cells reconstituted with wild-type VHL does not produce a tumorigenic phenotype in SCID mice (41). However, the treatment of these VHL-restored CC-RCC cells with an HIF-1α-oxygen-dependent degradation domain peptide that can block VHL binding to HIFα substrates restored the tumorigenic phenotype (41). This finding suggests that although HIF-1α is dispensable, other HIFα subunits (or possibly other ECV substrates) are associated with the tumor suppressor function of VHL. In support of this notion, Kondo et al. demonstrated that, unlike HIF-1α, the nondegradable HIF-2α was able to restore the tumor phenotype in CC-RCC cells expressing wild-type VHL (32), suggesting that HIF-2α is the relevant oncogenic player in the development of CC-RCC. Furthermore, Rankin et al. used the PEPCK (phosphoenolpyruvate carboxykinase) promoter to generate transgenic mice in which Cre-recombinase is expressed in the renal proximal tubules and hepatocytes (54). Conditional inactivation of VHL in PEPCK-Cre mouse resulted in glomerular and tubular renal cysts, increased serum erythropoietin levels, and polycythemia (54). The inactivation of ARNT, but not HIF-1α, rescued the conditional VHL knockout mouse from developing renal cysts (53, 54), further supporting the notion that another partner of ARNT such as HIF-2α, but not HIF-1α, plays a critical role in the transformation of renal proximal tubules.
We show here that the depletion of E-cadherin promotes the invasive potential of VHL-reconstituted CC-RCC cells, which is in keeping with the canonical function of E-cadherin. Krishnamachary et al. recently provided evidence that loss of VHL promotes epithelial-mesenchymal transformation, which is consistent with a loss of adherens junction due to reduced E-cadherin expression (34). There are, however, other notable VHL-dependent events bearing on CC-RCC progression to consider. CXCR4 is a chemokine receptor that aids in the metastasis of tumor cells to organs abundant in CXCR4-specific ligand, stromal cell-derived factor-1α (SDF-1α). Staller et al. showed that the expression of chemokine receptor CXCR4 increases upon the loss of VHL, suggesting a potential mechanism of CC-RCC metastasis (61). In addition, Zagzag et al. recently demonstrated that CC-RCC and hemangioblastoma cells devoid of VHL overexpress not only CXCR4 but also its ligand SDF-1α (66). These findings suggest that loss-of-function of VHL can establish an autocrine signaling pathway providing selective survival advantage and increased tendency for metastasis. The impact of the individual events (i.e., VHL-mediated E-cadherin versus CXCR4/SDF-1α regulation) in CC-RCC progression is not yet established, but nevertheless remains an important question to address.
In addition, VHL binds directly to fibronectin and promotes proper assembly of fibronectin extracellular matrix (ECM) (21, 50). All disease-associated VHL mutants examined to date exhibit decreased or undetectable binding to fibronectin (21, 50), underscoring the importance of VHL-mediated fibronectin assembly in the pathogenesis of VHL disease. In fact, alterations in the fibronectin component of the ECM have been correlated with cellular transformation (22). Conversely, multimeric forms of fibronectin or the overproduction of fibronectin receptors, such as α5β1 integrin, have been shown to promote differentiation and suppress the proliferative and metastatic potentials of transformed cells in various model systems (22, 51, 56). Lieubeau-Teillet et al. demonstrated that CC-RCC cells lacking VHL grow as tightly packed amorphous spheroids in a three-dimensional growth assay, indicative of an undifferentiated phenotype (38). In contrast, CC-RCC cells reconstituted with VHL form loose aggregates, which, on microscopic and ultrastructural examination, exhibit evidence of epithelial differentiation, such as trabecular and tubular structures (38). Davidowitz et al. demonstrated that VHL expressing CC-RCC cells grown on ECM differentiate into organized epithelial monolayers, whereas VHL-deficient cells are branched and disorganized and fail to arrest under high cell density (14). Koochekpour et al. showed that CC-RCC cells lacking functional VHL produce increased levels of matrix metalloproteinases and attenuated levels of tissue inhibitors of metalloproteinases and demonstrate increased invasiveness on growth factor-reduced matrigel (an artificial ECM) in response to hepatocyte growth factor/scatter factor (33). The lack of functional VHL has also been associated with overproduction of carbonic anhydrases 9 and 12, which are involved in the acidification of the microenvironment, favoring the growth and invasive properties of tumor cells (25, 42, 59). Thus, in addition to regulating E-cadherin expression, VHL is seemingly required for multiple additional events governing epithelial transformation from cellular differentiation to invasiveness.
There are other examples of cancer-causing mutations that often increase the expression of HIFα, which provides a mechanistic explanation for hypervascular tumors including CC-RCC that develop in the absence of VHL mutations. Mutations in the TSC2 tumor suppressor gene increase the level of HIFα via the mammalian target of rapamycin-dependent and -independent mechanisms that may involve chromatin remodeling (9). Loss of PTEN, which has been observed in the brain tumor glioblastoma multiforme, results in increased HIF-1α levels via the activation of the Akt/protein kinase B signaling cascade (68). The increased expression of HER2 receptor tyrosine kinase in breast cancer and the loss of p53 in various tumors enhance HIF-1-dependent transcription, often correlating with tumor aggressiveness (36, 55). Although these examples support the notion that there are multiple important regulators of HIF to ultimately promote oncogenic transformation, whether non-VHL-associated HIF activation likewise results in the down-regulation of E-cadherin via the activation of SIP1/Snail family of transcriptional repressors is an important question that remains to be resolved.
Supplementary Material
Acknowledgments
This work was supported by the Canadian Cancer Society of the National Cancer Institute of Canada (NCIC grants 16056 and 15252 to M.O.), Canadian Institutes of Health Research (CIHR grants MOP-77718 and MOP-37778 to M.O. and P.A.M., respectively), and the Kidney Foundation of Canada (KFOC grant to M.O.). R.C.R. is a recipient of the Natural Science and Engineering Research Council of Canada (NSERC) Scholarship. M.A.M. is a recipient of the CIHR Canada Graduate Scholarship. M.L.G. is a recipient of NCIC Graduate Studentship. A.M.R. is a recipient of the NSERC Canada Graduate Scholarship. M.S.I. is a Canada Research Chair in Cancer Biology. M.O. is a Canada Research Chair in Molecular Oncology.
Footnotes
Published ahead of print on 23 October 2006.
Supplemental material for this article may be found at http://mcb.asm.org/.
REFERENCES
- 1.Affymetrix. 2003. GeneChip expression analysis technical manual. Affymetrix, Santa Clara, CA.
- 2.Ananth, S., B. Knebelmann, W. Gruning, M. Dhanabal, G. Walz, I. Stillman, and V. Sukhatme. 1999. Transforming growth factor beta1 is a target for the von Hippel-Lindau tumor suppressor and a critical growth factor for clear cell renal carcinoma. Cancer Res. 59:2210-2216. [PubMed] [Google Scholar]
- 3.Baba, M., S. Hirai, S. Kawakami, T. Kishida, N. Sakai, S. Kaneko, M. Yao, T. Shuin, Y. Kubota, M. Hosaka, and S. Ohno. 2001. Tumor suppressor protein VHL is induced at high cell density and mediates contact inhibition of cell growth. Oncogene 20:2727-2736. [DOI] [PubMed] [Google Scholar]
- 4.Baba, M., S. Hirai, H. Yamada-Okabe, K. Hamada, H. Tabuchi, K. Kobayashi, K. Kondo, M. Yoshida, A. Yamashita, T. Kishida, N. Nakaigawa, Y. Nagashima, Y. Kubota, M. Yao, and S. Ohno. 2003. Loss of von Hippel-Lindau protein causes cell density dependent deregulation of CyclinD1 expression through hypoxia-inducible factor. Oncogene 22:2728-2738. [DOI] [PubMed] [Google Scholar]
- 5.Batlle, E., E. Sancho, C. Franci, D. Dominguez, M. Monfar, J. Baulida, and A. Garcia De Herreros. 2000. The transcription factor snail is a repressor of E-cadherin gene expression in epithelial tumour cells. Nat. Cell Biol. 2:84-89. [DOI] [PubMed] [Google Scholar]
- 6.Behrens, J., O. Lowrick, L. Klein-Hitpass, and W. Birchmeier. 1991. The E-cadherin promoter: functional analysis of a GC-rich region and an epithelial cell-specific palindromic regulatory element. Proc. Natl. Acad. Sci. USA 88:11495-11499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Berx, G., A. M. Cleton-Jansen, F. Nollet, W. J. de Leeuw, M. van de Vijver, C. Cornelisse, and F. van Roy. 1995. E-cadherin is a tumour/invasion suppressor gene mutated in human lobular breast cancers. EMBO J. 14:6107-6115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Berx, G., A. M. Cleton-Jansen, K. Strumane, W. J. de Leeuw, F. Nollet, F. van Roy, and C. Cornelisse. 1996. E-cadherin is inactivated in a majority of invasive human lobular breast cancers by truncation mutations throughout its extracellular domain. Oncogene 13:1919-1925. [PubMed] [Google Scholar]
- 9.Brugarolas, J. B., F. Vazquez, A. Reddy, W. R. Sellers, and W. G. Kaelin, Jr. 2003. TSC2 regulates VEGF through mTOR-dependent and -independent pathways. Cancer Cell 4:147-158. [DOI] [PubMed] [Google Scholar]
- 10.Cano, A., M. A. Perez-Moreno, I. Rodrigo, A. Locascio, M. J. Blanco, M. G. del Barrio, F. Portillo, and M. A. Nieto. 2000. The transcription factor snail controls epithelial-mesenchymal transitions by repressing E-cadherin expression. Nat. Cell Biol. 2:76-83. [DOI] [PubMed] [Google Scholar]
- 11.Clifford, S. C., M. E. Cockman, A. C. Smallwood, D. R. Mole, E. R. Woodward, P. H. Maxwell, P. J. Ratcliffe, and E. R. Maher. 2001. Contrasting effects on HIF-1α regulation by disease-causing pVHL mutations correlate with patterns of tumourigenesis in von Hippel-Lindau disease. Hum. Mol. Genet. 10:1029-1038. [DOI] [PubMed] [Google Scholar]
- 12.Cohen, H. T., and F. J. McGovern. 2005. Renal-cell carcinoma. N. Engl. J. Med. 353:2477-2490. [DOI] [PubMed] [Google Scholar]
- 13.Comijn, J., G. Berx, P. Vermassen, K. Verschueren, L. van Grunsven, E. Bruyneel, M. Mareel, D. Huylebroeck, and F. van Roy. 2001. The two-handed E box binding zinc finger protein SIP1 downregulates E-cadherin and induces invasion. Mol. Cell 7:1267-1278. [DOI] [PubMed] [Google Scholar]
- 14.Davidowitz, E. J., A. R. Schoenfeld, and R. D. Burk. 2001. VHL induces renal cell differentiation and growth arrest through integration of cell-cell and cell-extracellular matrix signaling. Mol. Cell. Biol. 21:865-874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Eble, J. N., G. Sauter, J. I. Epstein, and I. A. Sesterhenn. 2004. World Health Organization classification of tumors: pathology and genetics of tumors of the urinary system and male genital organs. IRAC Press, Lyon, France.
- 16.Esteban, M. A., M. G. B. Tran, S. K. Harten, P. Hill, M. C. Castellanos, A. Chandra, R. Raval, T. S. O'Brien, and P. H. Maxwell. 2006. Regulation of E-cadherin expression by VHL and hypoxia-inducible factor. Cancer Res. 66:3567-3575. [DOI] [PubMed] [Google Scholar]
- 17.Fish, J. E., C. C. Matouk, A. Rachlis, S. Lin, S. C. Tai, C. D'Abreo, and P. A. Marsden. 2005. The expression of endothelial nitric-oxide synthase is controlled by a cell-specific histone code. J. Biol. Chem. 280:24824-24838. [DOI] [PubMed] [Google Scholar]
- 18.Furge, K. A., K. A. Lucas, M. Takahashi, J. Sugimura, E. J. Kort, H. O. Kanayama, S. Kagawa, P. Hoekstra, J. Curry, X. J. Yang, and B. T. Teh. 2004. Robust classification of renal cell carcinoma based on gene expression data and predicted cytogenetic profiles. Cancer Res. 64:4117-4121. [DOI] [PubMed] [Google Scholar]
- 19.Graff, J. R., J. G. Herman, R. G. Lapidus, H. Chopra, R. Xu, D. F. Jarrard, W. B. Isaacs, P. M. Pitha, N. E. Davidson, and S. B. Baylin. 1995. E-cadherin expression is silenced by DNA hypermethylation in human breast and prostate carcinomas. Cancer Res. 55:5195-5199. [PubMed] [Google Scholar]
- 20.Groulx, I., and S. Lee. 2002. Oxygen-dependent ubiquitination and degradation of hypoxia-inducible factor requires nuclear-cytoplasmic trafficking of the von Hippel-Lindau tumor suppressor protein. Mol. Cell. Biol. 22:5319-5336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hoffman, M. A., M. Ohh, H. Yang, J. M. Klco, M. Ivan, and W. G. Kaelin, Jr. 2001. von Hippel-Lindau protein mutants linked to type 2C VHL disease preserve the ability to downregulate HIF. Hum. Mol. Genet. 10:1019-1027. [DOI] [PubMed] [Google Scholar]
- 22.Hynes, R. 1992. Integrins: versatility, modulation, and signalling in cell adhesion. Cell 69:11-25. [DOI] [PubMed] [Google Scholar]
- 23.Imai, T., A. Horiuchi, C. Wang, K. Oka, S. Ohira, T. Nikaido, and I. Konishi. 2003. Hypoxia attenuates the expression of E-cadherin via up-regulation of SNAIL in ovarian carcinoma cells. Am. J. Pathol. 163:1437-1447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ivan, M., K. Kondo, H. Yang, W. Kim, J. Valiando, M. Ohh, A. Salic, J. M. Asara, W. S. Lane, and W. G. Kaelin, Jr. 2001. HIFα targeted for VHL-mediated destruction by proline hydroxylation: implications for O2 sensing. Science 292:464-468. [DOI] [PubMed] [Google Scholar]
- 25.Ivanov, S., I. Kuzmin, M.-H. Wei, S. Pack, L. Geil, B. Johnson, E. Stanbridge, and M. Lerman. 1998. Down-regulation of transmembrane carbonic anhydrases in renal cell carcinoma cell lines by wild-type von Hippel-Lindau transgenes. Proc. Natl. Acad. Sci. USA 95:12596-12601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jaakkola, P., D. R. Mole, Y. M. Tian, M. I. Wilson, J. Gielbert, S. J. Gaskell, A. Kriegsheim, H. F. Hebestreit, M. Mukherji, C. J. Schofield, P. H. Maxwell, C. W. Pugh, and P. J. Ratcliffe. 2001. Targeting of HIF-α to the von Hippel-Lindau ubiquitylation complex by O2-regulated prolyl hydroxylation. Science 292:468-472. [DOI] [PubMed] [Google Scholar]
- 27.Jamora, C., P. Lee, P. Kocieniewski, M. Azhar, R. Hosokawa, Y. Chai, and E. Fuchs. 2005. A signaling pathway involving TGF-beta2 and snail in hair follicle morphogenesis. PLOS Biol. 3:e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ji, X., A. S. Woodard, D. L. Rimm, and E. R. Fearon. 1997. Transcriptional defects underlie loss of E-cadherin expression in breast cancer. Cell Growth Differ. 8:773-778. [PubMed] [Google Scholar]
- 29.Kaelin, W. G., Jr. 2002. Molecular basis of the VHL hereditary cancer syndrome. Nat. Rev. Cancer 2:673-682. [DOI] [PubMed] [Google Scholar]
- 30.Kibel, A., O. Iliopoulos, J. D. DeCaprio, and W. G. Kaelin. 1995. Binding of the von Hippel-Lindau tumor suppressor protein to elongin B and C. Science 269:1444-1446. [DOI] [PubMed] [Google Scholar]
- 31.Kondo, K., W. Y. Kim, M. Lechpammer, and W. G. Kaelin, Jr. 2003. Inhibition of HIF2alpha is sufficient to suppress pVHL-defective tumor growth. PLOS Biol. 1:e83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kondo, K., J. Klco, E. Nakamura, M. Lechpammer, and W. G. Kaelin, Jr. 2002. Inhibition of HIF is necessary for tumor suppression by the von Hippel-Lindau protein. Cancer Cell 1:237-246. [DOI] [PubMed] [Google Scholar]
- 33.Koochekpour, S., M. Jeffers, P. Wang, C. Gong, G. Taylor, L. Roessler, R. Stearman, J. Vasselli, W. Stetler-Stevenson, W. J. Kaelin, W. Linehan, R. Klausner, J. Gnarra, and G. Vande Woude. 1999. The von Hippel-Lindau tumor suppressor gene inhibits hepatocyte growth factor/scatter factor-induced invasion and branching morphogenesis in renal carcinoma cells. Mol. Cell. Biol. 19:5902-5912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Krishnamachary, B., D. Zagzag, H. Nagasawa, K. Rainey, H. Okuyama, J. H. Baek, and G. L. Semenza. 2006. Hypoxia-inducible factor-1-dependent repression of E-cadherin in von Hippel-Lindau tumor suppressor-null renal cell carcinoma mediated by TCF3, ZFHX1A, and ZFHX1B. Cancer Res. 66:2725-2731. [DOI] [PubMed] [Google Scholar]
- 35.Kurban, G., V. Hudon, E. Duplan, M. Ohh, and A. Pause. 2006. Characterization of a von Hippel Lindau pathway involved in extracellular matrix remodeling, cell invasion, and angiogenesis. Cancer Res. 66:1313-1319. [DOI] [PubMed] [Google Scholar]
- 36.Laughner, E., P. Taghavi, K. Chiles, P. C. Mahon, and G. L. Semenza. 2001. HER2 (neu) signaling increases the rate of hypoxia-inducible factor 1alpha (HIF-1α) synthesis: novel mechanism for HIF-1-mediated vascular endothelial growth factor expression. Mol. Cell. Biol. 21:3995-4004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lee, S., D. Y. T. Chen, J. S. Humphrey, J. R. Gnarra, W. M. Linehan, and R. D. Klausner. 1996. Nuclear/cytoplasmic localization of the von Hippel-Lindau tumor suppressor gene product is determined by cell density. Proc. Natl. Acad. Sci. USA 93:1770-1775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lieubeau-Teillet, B., J. Rak, S. Jothy, O. Iliopoulos, W. Kaelin, and R. Kerbel. 1998. von Hippel-Lindau gene-mediated growth suppression and induction of differentiation in renal cell carcinoma cells grown as multicellular tumor spheroids. Cancer Res. 58:4957-4962. [PubMed] [Google Scholar]
- 39.Lonergan, K. M., O. Iliopoulos, M. Ohh, T. Kamura, R. C. Conaway, J. W. Conaway, and W. G. Kaelin. 1998. Regulation of hypoxia-inducible mRNAs by the von Hippel-Lindau protein requires binding to complexes containing elongins B/C and Cul2. Mol. Cell. Biol. 18:732-741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Luecke, H. F., and K. R. Yamamoto. 2005. The glucocorticoid receptor blocks P-TEFb recruitment by NF-κB to effect promoter-specific transcriptional repression. Genes Dev. 19:1116-1127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Maranchie, J. K., J. R. Vasselli, J. Riss, J. S. Bonifacino, W. M. Linehan, and R. D. Klausner. 2002. The contribution of VHL substrate binding and HIF1-α to the phenotype of VHL loss in renal cell carcinoma. Cancer Cell 1:247-255. [DOI] [PubMed] [Google Scholar]
- 42.Martinez-Zaguilan, R., E. A. Seftor, R. E. Seftor, Y. W. Chu, R. J. Gillies, and M. J. Hendrix. 1996. Acidic pH enhances the invasive behavior of human melanoma cells. Clin. Exp. Metastasis 14:176-186. [DOI] [PubMed] [Google Scholar]
- 43.Maxwell, P., M. Weisner, G.-W. Chang, S. Clifford, E. Vaux, C. Pugh, E. Maher, and P. Ratcliffe. 1999. The von Hippel-Lindau gene product is necessary for oxygen-dependent proteolysis of hypoxia-inducible factor α subunits. Nature 399:271-275. [DOI] [PubMed] [Google Scholar]
- 44.Mohan, S., and R. D. Burk. 2003. von Hippel-Lindau protein complex is regulated by cell density. Oncogene 22:5270-5280. [DOI] [PubMed] [Google Scholar]
- 45.Morell-Quadreny, L., J. Rubio, J. A. Lopez-Guerrero, J. Casanova, D. Ramos, I. Iborra, E. Solsona, and A. Llombart-Bosch. 2003. Disruption of basement membrane, extracellular matrix metalloproteinases and E-cadherin in renal-cell carcinoma. Anticancer Res. 23:5005-5010. [PubMed] [Google Scholar]
- 46.Nelson, W. J., and R. Nusse. 2004. Convergence of Wnt, beta-catenin, and cadherin pathways. Science 303:1483-1487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Nishi, H., T. Nakada, M. Hokamura, Y. Osakabe, O. Itokazu, L. E. Huang, and K. Isaka. 2004. Hypoxia-inducible factor-1 transactivates transforming growth factor-β3 in trophoblast. Endocrinology 145:4113-4118. [DOI] [PubMed] [Google Scholar]
- 48.Ohh, M., and W. G. Kaelin, Jr. 2003. VHL and kidney cancer. Methods Mol. Biol. 222:167-183. [DOI] [PubMed] [Google Scholar]
- 49.Ohh, M., C. W. Park, M. Ivan, M. A. Hoffman, T.-Y. Kim, L. E. Huang, V. Chau, and W. G. Kaelin. 2000. Ubiquitination of HIF requires direct binding to the von Hippel-Lindau protein beta domain. Nat. Cell Biol. 2:423-427. [DOI] [PubMed] [Google Scholar]
- 50.Ohh, M., R. L. Yauch, K. M. Lonergan, J. M. Whaley, A. O. Stemmer-Rachamimov, D. N. Louis, B. J. Gavin, N. Kley, W. G. Kaelin, O. Iliopoulos, and W. G. Kaelin. 1998. The von Hippel-Lindau tumor suppressor protein is required for proper assembly of an extracellular fibronectin matrix. Mol. Cell 1:959-968. [DOI] [PubMed] [Google Scholar]
- 51.Pasqualini, R., S. Bourdoulous, E. Koivunen, V. Woods, Jr., and E. Ruoslahti. 1996. A polymeric form of fibronectin has antimetastatic effects against multiple tumor types. Nat. Med. 2:1197-1203. [DOI] [PubMed] [Google Scholar]
- 52.Peinado, H., E. Ballestar, M. Esteller, and A. Cano. 2004. Snail mediates E-cadherin repression by the recruitment of the Sin3A/histone deacetylase 1 (HDAC1)/HDAC2 complex. Mol. Cell. Biol. 24:306-319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Rankin, E. B., D. F. Higgins, J. A. Walisser, R. S. Johnson, C. A. Bradfield, and V. H. Haase. 2005. Inactivation of the arylhydrocarbon receptor nuclear translocator (Arnt) suppresses von Hippel-Lindau disease-associated vascular tumors in mice. Mol. Cell. Biol. 25:3163-3172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Rankin, E. B., J. E. Tomaszewski, and V. H. Haase. 2006. Renal cyst development in mice with conditional inactivation of the von Hippel-Lindau tumor suppressor. Cancer Res. 66:2576-2583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ravi, R., B. Mookerjee, Z. M. Bhujwalla, C. H. Sutter, D. Artemov, Q. Zeng, L. E. Dillehay, A. Madan, G. L. Semenza, and A. Bedi. 2000. Regulation of tumor angiogenesis by p53-induced degradation of hypoxia-inducible factor 1α. Genes Dev. 14:34-44. [PMC free article] [PubMed] [Google Scholar]
- 56.Ruoslahti, E. 1992. Control of the cell motility and tumour invasion by extracellular matrix interactions. Br. J. Cancer 66:239-242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Scheid, A., R. H. Wenger, L. Schaffer, I. Camenisch, O. Distler, A. Ferenc, H. Cristina, H. E. Ryan, R. S. Johnson, K. F. Wagner, U. G. Stauffer, C. Bauer, M. Gassmann, and M. Meuli. 2002. Physiologically low oxygen concentrations in fetal skin regulate hypoxia-inducible factor 1 and transforming growth factor-β3. FASEB J. 16:411-413. [DOI] [PubMed] [Google Scholar]
- 58.Shimazui, T., L. A. Giroldi, P. P. Bringuier, E. Oosterwijk, and J. A. Schalken. 1996. Complex cadherin expression in renal cell carcinoma. Cancer Res. 56:3234-3237. [PubMed] [Google Scholar]
- 59.Sly, W. S., and P. Y. Hu. 1995. Human carbonic anhydrases and carbonic anhydrase deficiencies. Annu. Rev. Biochem. 64:375-401. [DOI] [PubMed] [Google Scholar]
- 60.Spagnoli, F. M., C. Cicchini, M. Tripodi, and M. C. Weiss. 2000. Inhibition of MMH (Met murine hepatocyte) cell differentiation by TGF(beta) is abrogated by pre-treatment with the heritable differentiation effector FGF1. J. Cell Sci. 113:3639-3647. [DOI] [PubMed] [Google Scholar]
- 61.Staller, P., J. Sulitkova, J. Lisztwan, H. Moch, E. J. Oakeley, and W. Krek. 2003. Chemokine receptor CXCR4 downregulated by von Hippel-Lindau tumour suppressor pVHL. Nature 425:307-311. [DOI] [PubMed] [Google Scholar]
- 62.Stickle, N. H., J. Chung, J. M. Klco, R. P. Hill, W. G. Kaelin, Jr., and M. Ohh. 2004. pVHL modification by NEDD8 is required for fibronectin matrix assembly and suppression of tumor development. Mol. Cell. Biol. 24:3251-3261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Takahashi, M., D. R. Rhodes, K. A. Furge, H. Kanayama, S. Kagawa, B. B. Haab, and B. T. Teh. 2001. Gene expression profiling of clear cell renal cell carcinoma: gene identification and prognostic classification. Proc. Natl. Acad. Sci. USA 98:9754-9759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Vandewalle, C., J. Comijn, B. De Craene, P. Vermassen, E. Bruyneel, H. Andersen, E. Tulchinsky, F. Van Roy, and G. Berx. 2005. SIP1/ZEB2 induces EMT by repressing genes of different epithelial cell-cell junctions. Nucleic Acids Res. 33:6566-6578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Yoshiura, K., Y. Kanai, A. Ochiai, Y. Shimoyama, T. Sugimura, and S. Hirohashi. 1995. Silencing of the E-cadherin invasion-suppressor gene by CpG methylation in human carcinomas. Proc. Natl. Acad. Sci. USA 92:7416-7419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Zagzag, D., B. Krishnamachary, H. Yee, H. Okuyama, L. Chiriboga, M. A. Ali, J. Melamed, and G. L. Semenza. 2005. Stromal cell-derived factor-1alpha and CXCR4 expression in hemangioblastoma and clear cell-renal cell carcinoma: von Hippel-Lindau loss-of-function induces expression of a ligand and its receptor. Cancer Res. 65:6178-6188. [DOI] [PubMed] [Google Scholar]
- 67.Zhang, H., H. O. Akman, E. L. Smith, J. Zhao, J. E. Murphy-Ullrich, and O. A. Batuman. 2003. Cellular response to hypoxia involves signaling via Smad proteins. Blood 101:2253-2260. [DOI] [PubMed] [Google Scholar]
- 68.Zundel, W., C. Schindler, D. Haas-Kogan, A. Koong, F. Kaper, E. Chen, A. Gottschalk, H. Ryan, R. Johnson, A. Jefferson, D. Stokoe, and A. Giaccia. 2000. Loss of PTEN facilitates HIF-1-mediated gene expression. Genes Dev. 14:391-396. [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.