

Abstract
The mitogen-activated protein kinase (MAPK)/extracellular signal-regulated kinase (ERK) cascade plays a central role in intracellular signaling by many extracellular stimuli. One target of the ERK cascade is peroxisome proliferator-activated receptor γ (PPARγ), a nuclear receptor that promotes differentiation and apoptosis. It was previously demonstrated that PPARγ activity is attenuated upon mitogenic stimulation due to phosphorylation of its Ser84 by ERKs. Here we show that stimulation by tetradecanoyl phorbol acetate (TPA) attenuates PPARγ's activity in a MEK-dependent manner, even when Ser84 is mutated to Ala. To elucidate the mechanism of attenuation, we found that PPARγ directly interacts with MEKs, which are the activators of ERKs, but not with ERKs themselves, both in vivo and in vitro. This interaction is facilitated by MEKs' phosphorylation and is mediated by the basic D domain of MEK1 and the AF2 domain of PPARγ. Immunofluorescence microscopy and subcellular fractionation revealed that MEK1 exports PPARγ from the nucleus, and this finding was supported by small interfering RNA knockdown of MEK1 and use of a cell-permeable interaction-blocking peptide, which prevented TPA-induced export of PPARγ from the nucleus. Thus, we show here a novel mode of downregulation of PPARγ by its MEK-dependent redistribution from the nucleus to the cytosol. This unanticipated role for the stimulation-induced nuclear shuttling of MEKs shows that MEKs can regulate additional signaling components besides the ERK cascade.
The peroxisome proliferator-activated receptor γ (PPARγ) is a nuclear receptor that participates in the regulation of a large number of cellular processes including differentiation, immune response, and metabolism (31). The activity of PPARγ is normally induced by binding of specific ligands that activate its genomic transcriptional activity and thus initiate the expression of several effector genes. In addition, as a central signaling component, the activity of PPARγ is well regulated and can be inhibited under various cellular conditions such as stimulation of cells with growth factors and protein kinase C activators (24). Among the molecular mechanisms that prevent the activity of PPARγ upon these conditions is the phosphorylation of Ser84/Ser112 within PPARγ1/PPARγ2 by mitogen-activated protein kinases (MAPKs)/extracellular-signal regulated kinases 1 and 2 (ERK1/2) (14, 22). Stress stimuli (11) and proinflammatory mediators (30) also prevent PPARγ activation, but this is mediated mainly by other MAPK cascades including c-Jun N-terminal kinase and p38MAPK, which seem to induce phosphorylation of Ser84/112 as well. The inhibition of PPARγ by growth factors and gamma interferon are in accordance with the antiproliferative and anti-inflammatory role of PPARγ. Interestingly the MAPK cascade of ERK5 was reported to interact with PPARγ, but unlike the other MAPKs, this direct interaction induces activation rather than inhibition of PPARγ transcriptional activity (4). Finally, it was also shown that PPARγ ligands can trigger an activation of the ERK cascade (19, 37); however, the exact role of this activation is not clear as yet.
One important parameter that participates in the regulation and function of PPARγ as well as MAPK signaling is their subcellular localization (27, 42). Regarding the ERK cascade, it was shown that both ERKs and MEKs are localized in the cytosol of resting cells and that they translocate into the nucleus upon cellular stimulation (46). However, while ERKs remain in the nucleus of the stimulated cells for up to 180 min, MEKs are rapidly exported out of the nucleus due to their nuclear export signals (NES) (23). In addition, slow cytonuclear shuttling of inactive MEKs can occur without extracellular stimulation (18) and may assist export of inactive ERKs from the nucleus (1). Unlike the ERK components, little is known about the regulation of the intracellular distribution of PPARs. PPARγ was shown in several studies to localize mainly in the nucleus (6, 20). However, evidence for a significant cytosolic localization upon ligand binding has been presented as well (35, 39, 42). Further indications for extranuclear localization may be deduced from the reported relationships with cytosolic/membranal proteins such as HSP90 (36) and caveolin-1 (10). Therefore, although PPARγ functions primarily in the nucleus, it can redistribute to different compartments under distinct physiological conditions. However, no NES or a definite “shuttle” protein for PPARγ has been described, and therefore, the mechanism that allows the redistributions is not clear. Interestingly, the cytoplasmic retention sequence/common docking domain (CRS/CD) (32, 38) of ERK exhibits significant sequence similarity to the AF2 motif of PPARγ (9). Since CRS/CD participates in ERK-MEK interaction by docking to MEKs' D domain (38), it is possible that the CRS-like motif in PPARγ may mediate a selective interaction with MEKs.
Here we report that a physical association between MEK1 and PPARγ does exist, and we show that this interaction is important for the subcellular localization and transcriptional activity of PPARγ. Our data support a model in which PPARγ is localized in the nucleus of resting cells. Mitogenic stimulation of the cells causes interaction of active nuclear MEKs with PPARγ, and this is followed by a rapid nuclear export of the complex, which is mediated by the NES of MEKs. This nuclear export reduces the transcriptional activity of PPARγ and allows it to interact with cytosolic and membranal components. Therefore, we show here a novel role for MEKs, beside the regulation of ERKs, which is determination of the subcellular localization of other nuclear proteins to modulate their activities.
MATERIALS AND METHODS
Reagents.
Rosiglitazone was synthesized at Fa. Hoffmann-La Roche AG (Basel, Switzerland). Tetradecanoyl phorbol acetate (TPA), PD98059, U0126, and leptomycin B (LMB) were from Sigma (St. Louis, MO). Secondary antibody (Ab) conjugates were from Jackson Immunoresearch (West Grove, PA). Monoclonal PPARγ (E-8, sc-7273), polyclonal PPARγ (H-100, sc-7196), MEK1 (Sc-6250), MEK1 C terminal (Sc 219) histone H1 and tubulin E-19 Abs were from Santa Cruz Biotechnology (CA), the hemagglutinin (HA) and green fluorescent protein (GFP) Abs were from Roche Diagnostics GmbH (Mannheim, Germany), and pMEK1/2 Ab was from Cell Signaling (Beverly, MA). MEK1(C-ter) and ERK Ab were from Sigma (Rehovot, Israel).
Peptides.
Peptides (derived from the human protein sequences) were synthesized by Roche Diagnostics (Penzberg, Germany). Lyophilizates were dissolved as 50 mM stock solutions in dimethyl sulfoxide (DMSO), aliquoted, and stored at −20°C. Sequences were as follows: D-box (PPARγ), PPARγ (amino acids [aa] 147 to 158) DLNCRIHKKSRN; leucine-rich (ERK), ERK2 (aa 285 to 292) LDLLDKML; leucine-rich (PPARγ), PPARγ (aa 433 to 440) LLQKMTDL; CRS (ERK), ERK2 (aa 298 to 319) KRIEVEQALAHPYLEQYYDPSD; AF2 (PPARγ), PPARγ (aa 455 to 475) KKTETDMSLHPLLQEIYKDLY. In cell-permeable peptides, the efficient YARAAARQARA (21) sequence was used as a leader peptide. The peptides were CRSPerE (YARAAARQARAKRIEVEQALAHPYLEQYYDPSDE) and LRPerP (YARAAARQARAKKTETDMSLHPLLQEIYKDLY).
Plasmids.
Human pSG5-PPARγ1, -PPARγ1-S84A, 5× serum response element (SRE), and 3× PPRE-TK-luc plasmids were from Fa. Hoffmann-La Roche AG. The dominant-negative constructs pCMX-PPARγΔC5 and pCMX-PPARγΔ459 were a gift from Y. Barak (Salk Institute, La Jolla, CA). pCI-nGFP-C656G-mouse PPARγ was obtained from F. Gonzalez (NIH/NCI, Washington, DC). The various cDNAs of rat ERK2 and human MEK1 were previously described (32, 43, 45). Mutations were introduced by PCR-based site-directed mutagenesis and yielded dominant-negative DN-MEK1-GFP (S218A, S222A), inactive MEK1 (K97A-MEK1), constitutively active CA-MEK1-GFP (S218, 222E), hyperactive MEK1 (Δ32-51+S218, 222E), K3-5A-MEK1-GFP (KKK), and NES-MEK1-GFP (Δ32-51). The mammalian two-hybrid vectors pFA-CMV-AD (NF-κB) and pFR-luc were from Stratagene (Cedar Creek, TX). Fragments of MEK1 (N terminus, residues 1 to 70), inverted rat ERK2 (inverted C terminus, aa 246 to 360), and human steroid receptor coactivating factor 1 (SRC1) (LXXLL motifs, aa 595 to 781) were fused C-terminally to the activation domain of NF-κB. The ligand binding domain (LBD) (aa 174 to 476) of PPARγ1 was fused to the GAL4-DNA binding domain (aa 1 to 147) in pFA-CMV-BD (Stratagene). The glutathione S-transferase (GST)-LBD (aa 174 to 476) of PPARγ1 was in pGEX-4T2 (Amersham, United Kingdom). The His6-tagged SRC1 fragment (aa 595 to 781) was cloned into pET11b (Novagen, San Diego CA). pSuper was a gift from R. Agami (The Netherlands Cancer Institute, Amsterdam, The Netherlands). To generate small interfering RNA (siRNA) of MEK1/2, we inserted the sequences TGGATCAAGTCCTGAAGAA and TGGAGGTTCTCTGGATCAA (MEK1) and CAAGGTTGGCGAACTCAAA and AAAGACGATGACTTCGAAA (MEK2) into pSuper (7). The combination of these constructs was used for the siRNA experiments.
Cell culture.
HEK-293T, HeLa, CHO, and COS7 cells were grown in Dulbecco's modified Eagle medium with penicillin/streptomycin (1,000 U/ml each), 20 mM glutamine, and 10% (vol/vol) fetal calf serum (FCS; all from Gibco/Invitrogen, Carlsbad, CA). MKN45 cells were grown in RPMI 1640 medium with 10% (vol/vol) FCS, 20 mM glutamine, and penicillin/streptomycin (1,000 U/ml; all from Gibco/Life Technologies).
Transient-transfection and reporter gene assays.
Cells were grown to 50% confluence in 6-well plates (for reporter assay), on coverslips (for immunofluorescence), in 24-well plates (for RNA extraction), or in 100-mm dishes (for immunoprecipitation [IP]). The cells were then transiently cotransfected in Opti-MEM (Gibco) medium with 2 μg DNA/well or 5 μg DNA/dish using LipofectAMINE (Gibco), FuGENE-6 (Roche), or DEAE-dextran according to the manufacturer's instructions. Equal transfection efficiency for the reporter assay was monitored by cotransfection with pCMV-SEAP (Tropix, Bedford, MA) or pEGFPC1 (Clontech, Mountain View, CA). Six hours later, complete medium was added, and cells were either left in their dishes or, for the reporter assay, cells were reseeded in 96-well plates for an additional 24 h. After a 16-h starvation, cells were stimulated and were either processed (for IP, staining, RNA) or subjected to reporter activity assay. Luciferase activity was measured in wells by the Steady Glo luciferase assay system (Promega, Madison, WI) and together with SEAP activity (Roche Diagnostics) using a luminometer (Topcount X100; Hewlett-Packard, Palo Alto, CA).
Quantitative real-time RT-PCR.
Total RNA was isolated with the RNeasy system (QIAGEN, Hilden, Germany) and converted to cDNA using random hexamer primers and the First Strand cDNA synthesis kit for reverse transcription (RT)-PCR (avian myeloblastosis virus; Roche Diagnostics). Quantitative real-time PCR was performed in an ABIprism7700 thermocycler using the SYBR green PCR kit (Applied Biosystems, Foster City, CA) as detailed by the manufacturer. The numbers of amplification cycles required for a log-linear phase double-stranded DNA product to cross the background noise line (termed crossing point) were normalized to the numbers of S12 copies in the same sample. Results were calculated as the relative increase of mRNA compared to the DMSO control from duplicate reactions, using the same cDNA preparation. Primer sequences (human) were as follows: acyl coenzyme oxidase (ACO) F, TCTGTTGACCTTGTTCGAGCAA; ACO R, CAAGCACAGAGCCAAGTGTCAC; S12 F, GCATTGCTGCTGGAGGTGTAAT; S12 R, CTGCAACCAACCACTTTACGG.
Coimmunoprecipitation experiments.
Two micrograms of the designed Abs were incubated for 24 h at 4°C with 30 μl of a 50% (wt/vol) slurry of protein A/G-agarose (Santa Cruz). Cytosolic lysates were prepared by being washed twice with ice-cold phosphate-buffered saline (PBS), followed by hypotonic lysis in low-stringency buffer (20 mM HEPES, pH 7.9; 2 mM MgCl2; 2 mM EGTA, Complete protease inhibitor cocktail; 1 mM dithiothreitol; 0.1 mM Na3VO4) for 20 min on ice. Nuclei were pelleted (7,000 rpm, 10 min, 4°C), and aliquots of the cytosolic lysates (∼100 to 200 μl, 400 μg total protein) were incubated at 4°C with bead alone (control) or the Ab-coupled protein A/G-agarose for 2 h. Immune complexes were washed three times with low-stringency buffer plus 150 mM NaCl and 0.1% Triton X-100. The bound proteins were eluted by a 1-min incubation in 100 μl of 100 mM glycine (pH 2.2), neutralization by 10 μl of 1.5 M Tris-HCl (pH 8.8), and boiling in sample buffer.
GST pull-down assay.
Five micrograms of GST control protein or purified recombinant GST-PPARγ-LBD protein was coupled to 15 μl glutathione-agarose (50% slurry) for 2 h at 4°C in GST-binding buffer (20 mM HEPES, pH 7.5; 100 mM NaCl; 0.1 mM EDTA; 2.5 mM MgCl2; 0.01% NP-40 [vol/vol]; 10% [vol/vol] glycerol). Unbound GST proteins were removed by washing the beads twice with PBS and twice with GST-binding buffer. Aliquots of cytosolic lysates (∼100 to 200 μl, 400 μg of total protein) or 5 μg purified His-tagged MEK1 (full length) (Santa Cruz) was then incubated for 2 h at 4°C with 50 μl GST-loaded beads in GST-binding buffer supplemented with protease inhibitor cocktail, 1 mM dithiothreitol, and 0.1 mM Na3VO4. GST-bound complexes were then centrifuged, washed three times with GST-binding buffer, and subjected to Western blotting.
Immunofluorescence microscopy.
Cells were fixed on coverslips in 3% (wt/vol) paraformaldehyde in PBS, followed by a 5-min permeabilization in 0.2% (vol/vol) Triton X-100 in PBS at 23°C. PPARγ was detected by a 1:50 dilution of the primary Ab for 1 h at 23°C, washing, and incubation with 1:250 dilutions of mouse immunoglobulin G-rhodamine X red conjugate for 1 h. Nuclei were visualized upon a 15-min incubation with 0.1 mg/ml 4′,6′-diamidino-2-phenylindole (DAPI). Slides were photographed using a digital camera-connected fluorescence microscope (Axioscop microscope; Zeiss, Thornwood, NY).
Cellular fractionation.
The cellular fractionation procedure was performed essentially as previously described (29). HeLa cells were grown in 10-cm plates to subconfluence and then serum starved for 16 h (0.1% FCS). Subsequently, the cells were washed with PBS and scraped into ice-cold buffer H (50 mM β-glycerophosphate, pH 7.3, 1.5 mM EGTA, 1 mM EDTA, 1 mM dithiothreitol, 0.1 mM sodium vanadate, 1 mM benzamidine, 10 μg/ml aprotinin, 10 μg/ml leupeptin, and 2 μg/ml pepstatin A). The cells were then spun down (12,000 × g, 5 min), resuspended in 0.1% NP-40, and spun down again. The supernatant, containing the cytosolic fraction, was boiled in sample buffer. The pellet, containing the nuclei, was resuspended in an extraction buffer (420 mM NaCl, 50 mM β-glycerophosphate, 0.5 mM Na3VO4, 1.5 mM MgCl2, 0.2 mM EDTA, 1 mM dithiothreitol, 25% glycerol), and disrupted by sonication (twice for 10 s). The extract was then cleared by centrifugation (12,000 × g, 5 min), and the supernatant was boiled in sample buffer as the nuclear fraction.
RESULTS
MEK-dependent but S84-independent attenuation of PPARγ transcriptional activity upon TPA stimulation.
It has previously been demonstrated that PPARγ1 activity is attenuated upon mitogenic stimulation due to phosphorylation of its Ser-84 by ERKs (22). To further study the effect of the ERK cascade on PPARγ's activity, we examined the transcriptional activity of PPARγ1 in the presence of exogenous MEK constructs or their inhibitor. These experiments were performed in stimulated and nonstimulated HEK-293 cells cotransfected with a 3× PPRE-luciferase plasmid as a reporter of PPARγ's genomic activity. Thus, PPARγ-dependent transcription driven by wild type (WT) PPARγ1 was elevated by rosiglitazone (2.5-fold) (Fig. 1A), while TPA had an inhibitory effect on the basal as well as rosiglitazone-stimulated PPARγ activity. Addition of PD98059, which is a specific inhibitor of MEK1/2 and MEK5, did not change much the basal or ligand-driven activity of PPARγ but completely prevented the inhibitory effect of TPA. Since ERK5 is not activated much by TPA under the examined conditions and because ERK5 has a stimulatory role in PPARγ activation (4), it is likely that the inhibitory effect is fully mediated by MEK1 and MEK2.
FIG. 1.

Ser-84-independent attenuation of PPARγ activity upon TPA stimulation. HEK-293 cells were transiently cotransfected with mutants of PPARγ and MEK1-GFP together with a 3× PPRE-luciferase reporter plasmid. Twenty-four hours after transfection, cells were starved for 16 h and then treated with rosiglitazone (Rosi, 10 μM), TPA (100 nM), or 25 μM PD098059 for another 24 h before harvesting. Luciferase activity was detected as described in Materials and Methods and is expressed as n-fold increase ± standard deviation corrected to SEAP activity and compared to vehicle-treated control cells (n = 3). Effects of different stimuli (A and B) and different MEK1-GFP mutants (C) on WT PPARγ or S84A-PPARγ-driven PPRE transactivation upon stimulation with the indicated reagents (A and B) or rosiglitazone (C) are presented. (D) The effect of the indicated MEK1 constructs on 5× SRE-luciferase activity is presented as a control for the downstream activity of the MEKs.
We then used a PPARγ construct in which ERK's phosphorylation site was mutated to alanine (PPARγ1-S84A). The basal as well as stimulated activities of this construct were higher than those of the WT PPARγ, indicating that indeed the inhibitory effect of Ser-84 phosphorylation was eliminated. However, to our surprise, the activity of this construct was still sensitive to TPA treatment, which caused a very significant inhibitory effect on both rosiglitazone-stimulated and nonstimulated PPARγ activity (Fig. 1A). In similarity to the WT PPARγ, the inhibitory effect was eliminated when PD98059 was added to the cells, indicating that MEKs play a role also in this ERK-independent PPARγ inhibition. To verify the results with the PD98059, we used a dominant-negative (DN) (DN-S218, 222A-MEK1) construct to reduce MEK activation. Cotransfection of WT MEK1 with WT PPARγ caused a small elevation in PPARγ transcriptional activity (Fig. 1A versus Fig. 1B) but had no effect on the inhibitory action of TPA (Fig. 1B). On the other hand, the DN MEK1 significantly enhanced basal and rosiglitazone-stimulated WT as well as S84A-PPARγ activity and eliminated the inhibitory activity of TPA, corroborating the PD98059 results. Taken together, these results suggest that the inhibitory effect of TPA is dependent on MEKs but independent of the ERK-mediated Ser-84 phosphorylation. Interestingly, MEK1 lacking its NES (NES-MEK) enhanced PPARγ activity compared with the WT or constitutively active (CA) MEK1 (CA-S218, 222E-MEK1) on both WT and S84A PPARγ (Fig. 1C). This is despite lack of activity on the SRE, which is a known MEK/ERK target (Fig. 1D). Therefore, it is likely that the S84-independent effect of MEK1 is mediated not only by MEK1 activity but also by its localization.
The PPARγ target gene ACO is regulated by MEK localization.
To establish a physiological role for the inhibition of PPARγ by MEK localization, we examined expression of ACO, which is a known target gene of PPARγ (41). For this study, we used MKN45 cells that normally express high levels of endogenous PPARγ and therefore serve as a good model for the study of the downstream effect of this protein (33). These cells were first used to examine the effect of TPA and rosiglitazone on the expression of ACO and other target genes of PPARγ by quantitative RT-PCR. As expected, addition of rosiglitazone to the cells for 24 h increased the expression of the ACO's mRNA by 2.7-fold (Fig. 2). Addition of TPA alone did not influence the expression levels of ACO, but it did inhibit the effect of rosiglitazone in correlation with the effect detected with the PPRE reporter shown above. Similar effects were detected also with other gene targets of PPARγ such as PEPCK and PTEN (data not shown). Interestingly, the effects on some of the genes ware observed already 1 h after stimulation (data not shown), indicating that the effects of TPA are direct and immediate. To verify that the effects on ACO were related to MEK localization, we transfected the MKN45 cells with WT MEK1, DN MEK1, and NES-MEK1, followed by their activation with rosiglitazone, TPA, or both. Quantitative RT-PCR on RNA extracts from these cells revealed that both DN MEK1 and NES-MEK1 enhanced the effects of rosiglitazone on ACO expression. These results correlate with the PPRE reporter above and support a role of MEKs activation and subcellular localization in the downstream activity of PPARγ.
FIG. 2.
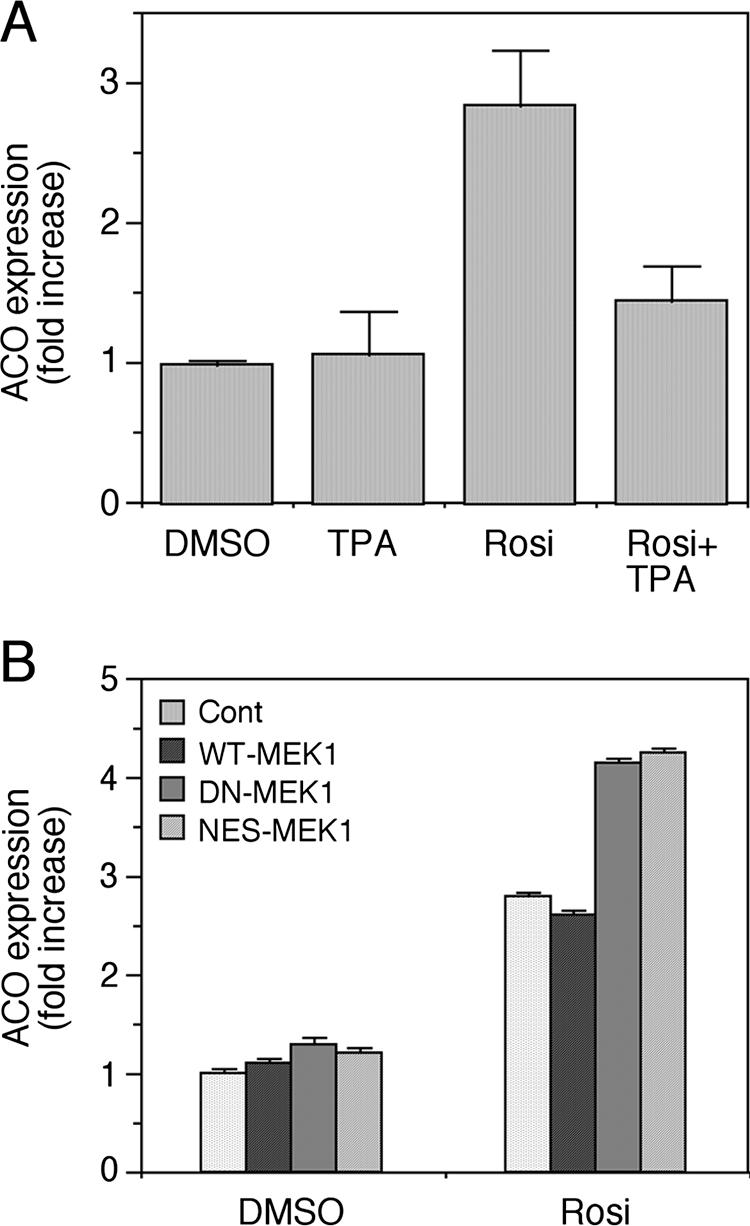
Regulation of the PPARγ target gene ACO by MEK1. (A) MKN45 cells were starved for 16 h and stimulated with TPA (100 nM), rosiglitazone (Rosi, 1 μM), or a combination of both for 24 h. Total RNA of these cells was extracted, and quantitative RT-PCR was performed with ACO primers as indicated in Materials and Methods. Results are expressed as mean n-fold increase ± standard deviation of mRNA (normalized to S12 RNA) compared to vehicle control (n = 3 independent experiments). (B) To study the effect of MEKs on rosiglitazone-induced expression of ACO, MKN45 cells were transiently transfected with empty vector, WT MEK1, DN MEK1, and NES-MEK1. Two days later, the cells were starved (0.1% FCS) for 16 h and were stimulated with either rosiglitazone (Rosi, 1 μM) or DMSO as a control, both for 24 h. The quantitative RT-PCR was performed on total RNA as indicated in Materials and Methods. Results are expressed as mean n-fold increase of mRNA (normalized to S12) compared to vehicle control of cells transfected with empty vector of three samples.
MEK1 but not ERK1 interacts with PPARγ in vitro and in vivo.
Because of the activatory effect of NES-MEK1 and because ERK does not seem to phosphorylate other residues on PPARγ besides Ser-84 itself, we examined the possibility that the above effects are mediated by a direct protein-protein interaction with ERKs or MEKs. For this purpose, HEK-293 cells were transiently cotransfected with a PPARγ1 together with either GFP-ERK2, MEK1-GFP, or a control construct of GFP alone. Cell extracts were then subjected to coimmunoprecipitation (CoIP) with PPARγ Ab, leading to precipitation of MEK1-GFP but not of GFP-ERK2 or GFP alone (Fig. 3A, upper panel). The amount of the PPARγ precipitated was similar in all three treatments (data not shown). Then the experiment was performed in the reverse order by using GFP Ab for the CoIP. Also under these conditions, PPARγ failed to interact with GFP alone or with GFP-ERK2 but significantly precipitated with GFP-MEK1 (Fig. 3A, second panel), although the amount of immunoprecipitated GFP constructs was comparable between the treatments. The amount of PPARγ and GFP constructs expressed in the different cells was very similar, as judged by blotting with Abs to these proteins. Importantly, CoIP in HEK-293 cells was also observed between the endogenous PPARγ and MEKs, even after extensive washes of the precipitants (Fig. 3B). However, as for the overexpressed proteins, ERKs did not seem to be involved in this PPARγ complexation, as ERKs were not detected in the CoIP, even under conditions were MEK1 was readily precipitated by PPARγ (data not shown).
FIG. 3.

PPARγ association with MEK1. (A) CoIP. HEK-293 cells were transiently cotransfected with PPARγ1 and GFP control vector, GFP-ERK2, or MEK1-GFP expression plasmids. Cytosolic extracts were incubated with the indicated Abs conjugated to protein A/G-agarose beads or with beads alone, and then CoIP was performed. The amount of MEK1-GFP and GFP-ERK2 was determined by Western blotting (IB) with GFP Ab (first panel). Reverse CoIP with GFP Ab was analyzed by Western blotting with PPARγ Ab (second panel). The equal amount of GFP-ERK2 or MEK1-GFP was determined by IP and Western blotting with GFP Ab (third panel). The amounts of PPARγ and GFP proteins in the extracts were determined by Western blotting with PPARγ and GFP Ab (fourth and fifth panels). (B) CoIP. Cytosolic extracts from HEK-293 cells were incubated with PPARγ Ab or Ab against the C terminus of MEK1 [MEK1 (C)] conjugated to protein A/G-agarose beads or beads alone. IP was performed as described, and the amount of precipitated proteins was determined using Ab to the N terminus [MEK1 (N)] or C terminus of MEK1 and to PPARγ as indicated. The amounts of MEK1 and PPARγ in the extracts were determined by Western blotting with the appropriate Abs (fourth and fifth panels). Usp, unspecified band. (C) Cellular GST pull down. HEK-293 cells were transfected as for panel A, and their cytosolic extracts were incubated with GST-PPARγ. (D) In vitro GST pull down. Recombinant HIS-MEK1(FL) was incubated with GST-PPARγ or GST alone in the presence of 0.1% bovine serum albumin to avoid nonspecific interactions. (E and F) Interaction of MEK1 and PPARγ detected by mammalian two-hybrid analyses. Cells were cotransfected with a GAL4-UAS-reporter plasmid and GAL4-PPARγ-LBD together with pCMV-AD or fusions, in which the N terminus of MEK1(N), the central LXXLL domain of SRC1, or the inverted C-terminal domain of ERK2(C) were inserted downstream of the NF-κB activation domain. Graphs represent n-fold increases ± standard deviations of luciferase activity in the presence of elevating concentrations of rosiglitazone (n = 3). (E) Basal interactions in HEK-293 cells. (F) Ligand-driven interactions in HEK-293.
To further verify the direct interaction between MEK1 and PPARγ, we used the GST pull-down method. For this purpose, human recombinant GST-tagged PPARγ-LBD, which contains the important protein interaction domains of PPARγ (aa 174 to 476) was used to precipitate proteins from cytosolic extracts of HEK-293 cells that had been transfected with control GFP, MEK1-GFP, or GFP-ERK2 constructs. Similar to the CoIP, significant precipitation of MEK1-GFP but none of GFP or GFP-ERK2 from the different extracts (Fig. 3C) was detected, despite the approximately equal amounts of GFP constructs present in the extracts that were loaded on the GST-PPARγ beads (Fig. 3C, right panel). We found that GST-PPARγ-LBD is able to precipitate the full-length human recombinant His-MEK1. As seen in Fig. 3D, there was some unspecific interaction of the purified His-MEK1 with GST alone, but the amount of the 50-kDa His-MEK1 band was much increased when pulled with GST-PPARγ-LBD. These data further support a direct interaction between MEK1 and PPARγ. However, since the interaction with the cytosolic extract appears to be stronger than the interaction with the recombinant His-MEK1, as judged by a comparison between Fig. 3C and D, it is also possible that the interaction is facilitated by other cytosolic proteins, i.e., putative scaffold or adaptor proteins.
Studying the MEK1-PPARγ interaction using mammalian two-hybrid system.
To study the structural basis for the direct MEK1-PPARγ interaction, we applied a mammalian two-hybrid system (16). In these experiments, the N terminus of MEK1 was fused to the NF-κB transactivation domain of the prey vector pCMV-AD. A construct of the coactivator SRC1 harboring the three central LXXLL motifs essential for the strong binding of this protein to PPARγ and an inverted C-terminal region of ERK2 served as positive and negative controls, respectively. The bait construct GAL4-PPARγ-LBD and the various prey constructs were then transfected into HEK-293 cells together with a GAL4-UAS-luciferase reporter, and luciferase activity was determined after 24 h. Cotransfection of PPARγ and MEK1 constructs to the HEK-293 cells resulted in a significant interaction that was as high as the interaction observed for the SRC1 positive control (Fig. 3E). As expected, the magnitude of interaction was relatively low due to the lack of PPARγ ligands that are required to optimize this reaction. Indeed, rosiglitazone induced a concentration-dependent increase in the luciferase activity driven by the interaction of MEK(N) with PPARγ-LBD of up to 50-fold compared to the control cells that expressed GAL4-PPARγ-LBD with empty pCMV-AD vector (Fig. 3F). The effect of rosiglitazone was comparable to its effect in the SRC1-positive control cells. These results required the presence of serum, as in serum-free medium, there was almost no effect of PPARγ (data not shown). Additionally, we have used fluorescent resonance energy transfer to examine the interaction between MEK1, SRC1, and PPARγ. In this assay, addition of MEK1 was sufficient to abrogate rosiglitazone-induced interaction between the coactivator and the LBD of PPARγ (data not shown), suggesting a competition between SRC1 and MEK1 interactions with PPARγ. Taken together, all these data indicate that MEK1 and PPARγ interact directly, that this interaction is mediated by the N terminus of MEK1 and the PPARγ-LBD, and that it is increased by cellular stimulation.
MEK1-PPARγ interaction is increased upon stimulation with TPA and rosiglitazone.
Since the two-hybrid experiments indicated that the MEK1-PPARγ interaction may be increased upon stimulation, we examined the effect of TPA and rosiglitazone on the CoIP of overexpressed MEK1 and PPARγ in HEK-293 cells. First, we transfected MEK1-HA and GFP-PPARγ into HEK-293 cells, and a CoIP was performed with HA Ab. Similar to the results above (Fig. 3), a significant association of MEK1-HA with the GFP-PPARγ was detected in resting cells, and it was significantly increased upon treatment with TPA and to a lesser extent also by rosiglitazone stimulation (Fig. 4A and B). Similar results were obtained when we performed the reciprocal experiment, namely IP of the overexpressed MEK1-HA followed by the detection of associated PPARγ (data not shown).
FIG. 4.

The interaction between MEK1 and PPARγ is increased upon TPA and rosiglitazone stimulation. (A) HEK-293 cells cotransfected with GFP-PPARγ and MEK1-HA were serum starved (16 h) and then stimulated with 100 nM TPA or 10 μM rosiglitazone for the indicated times. CoIP was carried out with HA Ab, and the amount of coimmunoprecipitated PPARγ was determined by Western blotting (IB) with GFP Ab. The amounts of MEK1-HA and GFP-PPARγ in the extracts were determined by Western blotting with the indicated Abs. (B) Densitometric analyses of CoIP experiments shown in panel A. Values represent n-fold increases ± standard deviations in complex formation compared to vehicle-treated unstimulated cells (n = 3). (C) HEK-293 cells were serum starved (16 h) and then stimulated with 100 nM TPA or 10 μM rosiglitazone for the indicated times. CoIP was carried out on the endogenous proteins using PPARγ Ab. The amount of coimmunoprecipitated MEKs and the amounts of endogenous MEKs and PPARγ in the extracts were determined by Western blotting with the indicated Abs. Con, bead control. (D) Densitometric analyses of CoIP experiments shown in panel C (n = 3). (E) Phosphorylation of MEKs upon rosiglitazone and TPA stimulation of HEK-293 cells for the indicated times was determined by pMEK and general (g) MEK Abs.
Importantly, stimulation of MEK1-PPARγ interaction was detected not only with the overexpressed proteins but also with the endogenous MEK1 and PPARγ proteins (Fig. 4C and D, left panels). Thus, when IP was carried out with PPARγ Ab on extracts from nontransfected HEK-293 cells, a small amount of a 46-kDa protein was detected in the Western blot with MEK Ab. The association was increased upon TPA stimulation, and the level of CoIP correlated with the activation of MEKs detected by pMEK Ab (Fig. 4E). Upon stimulation with rosiglitazone, the association between the endogenous proteins first increased and then decreased to below the basal level (Fig. 4C and D, right panels), which again correlated with the kinetics of MEK phosphorylation. This may indicate that after the initial association, the MEK1-PPARγ complex can dissociate and free the two proteins to other compartments or activities. Combined treatment of TPA and rosiglitazone increased the association to a level between that of rosiglitazone or TPA alone (data not shown). Notably, similar results of coimmunoprecipitation were obtained in other cell line as well (MKN45 and HeLa [data not shown]), supporting the general role of this interaction. Thus, our results indicate that the endogenous PPARγ-MEK1 interaction does occur and is sensitive to stimulation with TPA and PPARγ ligands.
The D domain of MEK1 and the AF2 domain of PPARγ mediate their interaction.
We next undertook the task of a more specific identification of the regions in PPARγ and MEK that are responsible for the association between the two proteins. Likely regions for such an interaction could be the AF2 in PPARγ that has a sequence similarity to the CRS/CD of ERK (9) and the CRS/CD-interacting area in MEKs, which is the N-terminal D domain (38). Therefore, we first used deletion mutants of PPARγ lacking either 5 (ΔC5; with functional LXXLL-binding pocket) or 16 (Δ459, lacking the LXXLL-binding pocket) amino acids of the C-terminal AF2 domain of PPARγ known to act as dominant-negative constructs (DN-PPARγ) (6, 20). Thus, WT PPARγ1, and the PPARγ mutants PPARγΔC5 and PPARγΔ459 were each cotransfected with MEK1-GFP into HEK-293 cells. CoIP was carried out using PPARγ Ab, and the amount of associated MEK1 was assessed with GFP Ab. As seen in Fig. 5A and B, the absence of the 5 amino acids from the C terminus reduced the association between the proteins, although the effect of deleting the last 16 amino acids was much more pronounced. Therefore, although the CRS/CD-like motif, which is located mainly in the last 5 amino acids may be important in the interaction, other parts of the C terminus are likely to contribute to PPARγ binding as well.
FIG. 5.

Determination of the site of MEK1 and PPARγ interaction. (A and B) HEK-293 cells were transiently cotransfected with WT-PPARγ, PPARγΔC5, and PPARγΔ459 together with MEK1-GFP. Cells were starved and stimulated and CoIP was performed as described for Fig. 3A. Precipitates and original extracts were then analyzed by sodium dodecyl sulfate-polyacrylamide gel electrophoresis and Western blotting (IB) with the indicated Abs. Representative gels (A) and densitometric analyses (B) are shown. Values represent n-fold increases ± standard deviations in complex formation compared to vehicle-treated unstimulated cells (n = 3). (C and D) The same experiments and analyses as described for panels A and B, except that the cells were cotransfected with WT PPARγ together with WT MEK1-GFP, CA MEK1-GFP, and DN MEK1-GFP. (E and F) The same experiments and analyses as described for panels A and B, except that cells were cotransfected with WT PPARγ together with DN MEK1-GFP (DN), CA MEK1-GFP (CA), ΔNES-MEK1-GFP (NES), and Κ3-5A-MEK1-GFP (KKK). (G and H) HEK-293 cells were transiently cotransfected with WT PPARγ together with MEK1-GFP, and IP was carried out as described in the presence of the indicated peptides (400 μM each). The peptides used (see Materials and Methods) were derived form the CRS/CD of ERK (E-CRS), the CRS-like sequence of PPARγ (P-CRS), D box of PPARγ (P-DBox), leucine-rich region of ERK1 (E-LR), and leucine-rich region of PPARγ (P-LR).
We then examined the ability of several MEK1 mutants to undergo CoIP by PPARγ Ab. WT PPARγ 1 was cotransfected with WT, CA, and DN MEK1-GFP into HEK-293 cells. The CoIP revealed a two- to threefold increase in complex formation upon overexpression of CA MEK1 compared to WT or DN MEK1, while the latter was not responsive to TPA (Fig. 5C and D). These data may indicate that the increase in interaction after stimulation demonstrated in Fig. 3 is dependent on the activation of MEKs and not of PPARγ. We then examined two additional mutants in the N terminus of MEK1, which are Κ3-5A-MEK1-GFP, whose basic residues in the D domain had been replaced with alanine residues and therefore has a reduced activity towards ERK (15), and NES-MEK1-GFP, whose NES had been deleted, causing its nuclear localization and slightly elevated activity. When these constructs were subjected to CoIP with PPARγ Ab, the NES-MEK1 showed slightly reduced association compared to CA MEK1 (Fig. 5E and F) and Κ3-5Α-MEK1 showed reduced association compared to DN MEK1 (and also WT MEK1).
Further support for the importance of the D domain of MEK1 and CRS-like AF2 domain of PPARγ came from peptide competition experiments. Thus, a peptide derived from the AF2 domain (P-AF2) and the D box of PPARγ (P-D box, which, in similarity to the D domain of MEK1, contains basic amino acids) inhibited the association between the two proteins by 40 and 70%, respectively (Fig. 5G and H). A peptide derived from the CRS/CD motif of ERK (E-CRS) had a small inhibitory effect (∼25%), probably because of its similarity to the CRS-like AF2 sequence of PPARγ, while the control leucine-rich regions of ERK (E-LR) and PPARγ (P-LR) had no significant effects. These data further support the idea that the N terminus of MEK1 and the CRS/CD-like region within the AF2 of PPARγ are essential for formation of this complex. The results above also suggest that nearby sequences (NES in MEK1, residues within the AF2 domain, and probably also the D box of PPARγ) may contribute to the binding efficiency as well.
PPARγ is retained in the cytosol in a MEK1-dependent manner.
It should be noted that despite the clear interaction between the two proteins, MEK1 failed to phosphorylate PPARγ under any condition examined (data not shown). We therefore investigated whether the interaction serves as a novel mechanism for regulating the subcellular localization of PPARγ. Thus, HeLa cells were transiently cotransfected with MEK1-GFP together with the PPARγ construct in a different plasmid ratio that gave rise to expression ratios of MEK1 to PPARγ of 9:1, 6:1, and 3:1. Forty-eight hours after transfection, the cells were stained with PPARγ Ab. Using this method, overexpressed PPARγ without ectopic addition of MEK1 was detected in the nuclei of the HeLa cells (data not shown). However, increasing expression of MEK1-GFP together with the PPARγ was followed by a considerable extraction of PPARγ from the nucleus toward the cytoplasm (Fig. 6A) in a manner similar to the cytosolic retention of ERKs by MEKs (18, 32). This phenomenon was quantified and found to occur in about 65% of the transfected cells examined. We further evaluated the effect of different mutants on cytosolic localization of PPARγ in COS7 cells. A nuclear staining pattern similar to that of the HeLa cells was visible when PPARγ was overexpressed together with the GFP control vector (Fig. 6B). Coexpression of MEK1-GFP with the PPARγ in these cells partially retained PPARγ in the cytosol and so did constitutively activated MEK1 (EE-MEK1) and the inactive MEK1 (KA-MEK1), which are localized primarily in the cytosol. On the other hand, the hyperactive MEK1 (ΔN-EE-MEK1) did not induce cytosolic retention of PPARγ, most likely because of its nuclear localization. In addition, PPARγΔ459, which lacks the interaction site with MEK1, could not be retained in the cytosol of COS7 cells, supporting the role of this region in the MEK1-regulated localization of PPARγ.
FIG. 6.

Cytosolic retention of PPARγ upon overexpression of MEK1-GFP. Cells were grown on coverslips and transiently cotransfected with PPARγ constructs and the indicated constructs of MEKs fused to GFP as described in Materials and Methods. Thirty-two hours after transfection, the cells were serum starved for 16 h, fixed, and stained for PPARγ and DAPI, which were visualized using a fluorescence microscope (Nikon, Japan) at ×400 magnifications. (A) HeLa cells cotransfected with WT PPARγ and WT MEK1-GFP in the indicated ratios. (B) COS7 cells cotransfected with PPARγ together with either GFP alone, WT MEK1 (WT), hyperactivated MEK1 (ΔNEE), constitutively active MEK1 (EE), or ATP binding site-deficient mutant of MEK1 (KA). In addition, WT MEK1 was cotransfected together with PPARγΔ459. Staining was performed as described above. The nuclei were detected with DAPI, and the fluorescence was visualized as described above.
TPA induces nuclear export of PPARγ dependent on its interaction with MEKs.
We then examined the subcellular distribution of endogenous PPARγ. In our hands, PPARγ was detected in the nuclei of various cell lines, including COS7, HeLa, and MCF7 cells (data not shown). The best quality staining was seen in CHO cells, and those were used to follow the subcellular localization under varying conditions (Fig. 7A and data not shown). Interestingly, PPARγ was exported from the nucleus toward the cytosol upon stimulation with TPA for 15 min. This relocalization was even more pronounced 30 min after stimulation (Fig. 7A) and could be detected up to 60 min after stimulation (data not shown). The addition of rosiglitazone also resulted in a nuclear export of PPARγ, but this was considerably weaker than the export caused by TPA (data not shown). Importantly, TPA-induced relocalization of PPARγ was prevented by the MEK inhibitor U0126, indicating that MEKs are involved in mediating this action.
FIG. 7.

MEKs induce the nuclear export of PPARγ upon TPA stimulation. (A) CHO cells were grown on coverslips as above. Following serum-starvation, CHO cells were treated with TPA (250 nM, 15 and 30 min), or with U0126 (5 μM) 15 min prior to TPA (250 nM, 15 min). The cells were stained with PPARγ Ab or PPARγ Ab and DAPI, and the localization was assessed by fluorescence microscopy as above. (B) HeLa cells were cotransfected with GFP to identify transfected cells together with either combination of 4 siRNA oligonucleotides of MEK1 and 2 (Si-MEK) or with vector (pSuper) alone and then grown on coverslips as described above. Seventy-two hours after transfection, the cells were serum starved (0.1% FCS, 16 h) and then were treated with TPA (250 nM, 30 min) or left untreated. The cells were stained with polyclonal rabbit PPARγ and DAPI and developed with rhodamine-conjugated anti-rabbit secondary Ab. The localization of PPARγ was visualized by a fluorescence microscopy as above. (C) HeLa cells were transfected either with pSuper or with combination of 4 siRNA oligonucleotides of MEK1 and 2. Ninety hours after transfection, the cells were extracted, and the endogenous proteins were subjected to Western blot analysis using C-terminus MEK and actin Abs.
Since TPA induced nuclear export of PPARγ as well as nucleus-cytosol shuttle of MEKs (23), it became important to examine whether these two events are linked and to verify that MEKs regulate PPARγ relocalization upon stimulation. For this purpose, we used siRNA of MEK1 and MEK2 in the pSuper vector to reduce the expression of MEKs. Thus, in HeLa cells transfected with the pSuper vector control, there was no change in the level of MEKs expression, and as in nontransfected cells, PPARγ was localized in the nucleus (Fig. 7B). After stimulation with TPA for 30 min, PPARγ was redistributed and appeared diffused all over the cells. We then used the siRNA constructs to reduce the amount of MEKs by about 70% (Fig. 7C) without affecting their subcellular localization (data not shown). The transfection of this construct did not change the subcellular localization of PPARγ in nonstimulated cells. However, when these cells were stimulated with TPA, PPARγ was exported from the nucleus in cells with high expression levels of MEKs, while in the cells containing reduced MEK levels, the PPARγ was retained in the nucleus. Interestingly, siRNA of ERKs that reduced the expression levels of endogenous ERKs by about 75% did not have any effect on the subcellular localization of PPARγ either before or after TPA stimulation (data not shown).
To verify that the TPA-induced relocalization of PPARγ is mediated by direct MEK-PPARγ interaction, we used cell-permeable versions of the ERK-CRS peptide (CRSPerE) that disturbs the interaction and the PPARγ leucine-rich region peptide (LRPerP) that does not alter it. Both of these peptides do not affect the kinase activity of MEKs or ERKs (data not shown), so any effect of the peptides on the subcellular localization should be due to the protein-protein interaction of MEKs and not to their activity. Thus, application of 1 μg/ml of the CRSPerE peptide (E-CRS) for 45 min significantly reduced the TPA-induced nuclear export of PPARγ, while a similar amount of the LRPerP peptide (P-LR) did not significantly affect this relocalization (Fig. 8). LMB, a potent inhibitor of the nuclear export, prevented the TPA-induced relocalization of PPARγ as well. Since the nuclear export of MEKs is also sensitive to LMB (45), these data are consistent with the role of MEKs in the PPARγ export. The effects of LMB also indicate that the relocation is not the result of a de novo protein synthesis, a parameter that is indicated also by the rapid nature of the effect (within 5 min).
FIG. 8.

Nuclear export of PPARγ is mediated by interaction with MEKs and by exportin. Subconfluent HeLa cells were pretreated with the interfering cell-permeable peptide CRSPerE (E-CRS), the control permeable peptide LRPerP (P-LR; both 1 μg/ml for 45 min), and LMB (5 ng/ml for 60 min). Then, the distinctly treated cells were either left untreated (Basal) or treated with TPA (250 nM, 15 min). After this treatment, the cells were fixed and stained with PPARγ Ab and DAPI, followed by visualization with a fluorescence microscope (Nikon, Japan) at ×400 magnifications. Con, control.
Use of subcellular fractionation to study the TPA- and MEK-dependent subcellular localization of PPARγ.
To further establish the subcellular localization of endogenous PPARγ, we then resorted to a subcellular fractionation technique that may provide a more quantitative detection method. As expected, fractionation of HeLa cells resulted in a clear separation between the nuclear histone H1 and the cytosolic tubulin (Fig. 9A). Notably, in nonstimulated cells, all PPARγ was detected in the nucleus of the cells. However, TPA stimulation induced a rapid appearance of PPARγ in the cytosol and a reduction in its nuclear appearance (Fig. 9A). This effect was dependent on MEKs, since the MEK inhibitor U0126 reversed it, both in the cytosol and in the nuclear fractions (Fig. 9B). We then knocked down the MEK levels in the HeLa cells using siRNA (∼70% reduction) and found that the lack of MEKs in the cells prevents the redistribution of PPARγ upon TPA induction (Fig. 9C). Again, this effect is probably mediated by the protein interaction of MEKs and not their activity, since the CRSPerE (E-CRS), and not the LRPerP (P-LR), peptide prevented this change in distribution (Fig. 9D). Finally, LMB prevented the nuclear export of PPARγ (Fig. 9E). Notably, a small amount of nuclear export of PPARγ was detected upon rosiglitazone stimulation, but rosiglitazone did not affect much the TPA-induced relocation when these two ligands were added together (data not shown). Since siRNA of MEKs, the interaction-modifier peptide, and LMB were all able to prevent the nucleocytosolic redistribution of PPARγ upon mitogenic stimulation, it is likely that the interaction with MEKs and not the activity of these kinases is important for this process.
FIG. 9.

PPARγ nuclear export is mediated by MEKs. Subconfluent, serum-starved (0.1% FCS, 16 h) HeLa cells were treated as follows: (A) stimulated with TPA (250 nM, 15 and 30 min) or left untreated; (B) stimulated with TPA (250 nM, 15 and 30 min) or pretreated with U0126 (5 μM) 15 min prior to addition of TPA (250 nM, 15 min) or vehicle control. (C) HeLa cells were transiently transfected with combination of 4 siRNA vectors of MEK1 and 2 (SiMEK) or with vector (pSuper) control. Four days after transfections, the cells were serum starved for 16 h and treated with TPA (250 nM, 15 min) (+) or left untreated (−). (D) HeLa cells were preincubated for 45 min with interfering cell-permeable peptide CRSPerE (E-CRS) or the control permeable peptide LRPerP (P-LR) or left untreated, followed by TPA stimulation (250 nM, 15 min). (E) HeLa cells were treated with TPA (250 nM, 15 min) or with LMB (5 ng/ml, 60 min) plus TPA (250 nM, 15 min) or left untreated. All treated cells were extracted and subjected to cellular fractionation as described. The lysates were Western blotted with PPARγ Ab and with tubulin Ab and histone H1 (histone) Ab as markers for the cytosolic (Cyt) and nuclear (Nuc) fractions, respectively. (F) The amount of PPARγ in the cytosol was quantitated using densitometry, and the results are shown in a bar graph (n = 3).
DISCUSSION
In the present study, we show for the first time that MEK1 interacts with PPARγ and that this event leads to export of nuclear PPARγ to the cytosol. Our results best fit a model (Fig. 10) in which, in the absence of both ligand and mitogen, i.e., under conditions where both PPARγ and MEK1 are inactive, there is little interaction between the cytosolic MEK1 and nuclear PPARγ. Addition of a mitogen, or to a lesser extent, also a PPARγ ligand, causes the stimulation of MEKs and evokes their translocation into the nucleus, where they interact with free PPARγ and assist its export to the cytosol. This process probably plays a role in terminating the transcriptional activity of PPARγ upon rosiglitazone stimulation and in providing enough PPARγ protein for its extranuclear effects (reference 10 and references therein) but may also lead to stimulation-dependent degradation of this nuclear receptor. Until now, two processes have been proposed for the termination and downregulation of PPARγ signaling, the phosphorylation of Ser-84/112 of PPARγ1/2 by ERKs (2, 22) and the proteasomal degradation of ligand-activated PPARγ (17). Based on the data provided herein, we propose an alternative mechanism of PPARγ downregulation by its interaction with MEKs, which induces its nuclear export and prevents its nuclear activity. This process may be a general mechanism for enabling nonnuclear action of other nuclear receptors such as estrogen (34) and androgen receptors (26).
FIG. 10.

Model of the role of MEK translocation in the subcellular localization and functional modulation of PPARγ. In resting cells, ERKs and MEKs are bound in the cytosol via their corresponding CRS/CD (C) and D (D) domains, while PPARγ that contains a CRS-like motif (Doc) is localized in the nucleoplasm. Upon addition of TPA, ERKs and MEKs dissociate from each other and translocate into the nucleus where ERKs remain in the nucleus for up to 3 h and phosphorylate PPARγ on Ser84, while active MEKs interact with PPARγ (via their corresponding D and CRS-like domains), and induce the export of PPARγ to the cytosol, where it may participate in nongenomic functions. When rosiglitazone is added, it penetrates to the nucleus and activates PPARγ transcriptional activity. The small amount of MEK/ERK activation that is induced by rosiglitazone is not depicted in this figure. However, after addition of TPA together with rosiglitazone, MEKs, ERKs, and rosiglitazone are translocated to the nucleus. ERKs phosphorylate PPARγ and inhibit its rosiglitazone-induced transcriptional activity. On the other hand, MEKs induce an export of PPARγ as above, and this reduces the amount of active PPARγ in the nucleus and thereby inhibits its activity.
Protein-protein interactions are important for many signaling processes, including that of the ERK signaling cascade. One important docking surface in ERKs is their CRS/CD domain, which allows interaction with regulatory proteins and the retention of ERKs in the cytoplasm (46). Interestingly, the C-terminal AF2 domain of PPARγ possesses sequence similarity to the CRS region, although it lacks at least one acidic residue that was shown to be important for the interaction of ERKs with MEKs (32, 38, 44). Despite the incomplete similarity, we found that MEK1 can interact with PPARγ. This interaction was confirmed using several distinct methods, including (i) CoIP of overexpressed proteins using Abs to PPARγ, (ii) the reciprocal CoIP of using Abs to MEKs, (iii) CoIP of endogenous proteins, (iv) GST pull-down assay, (v) mammalian two-hybrid assay, and (vi) homogenous time-resolved fluorescence/fluorescent resonance energy transfer in which the PPARγ coactivator SRC1 was displaced by MEK1 (Fig. 3 and 4 and data not shown). These methods clearly show that, besides its interaction with ERKs, MEK1 can also interact with PPARγ. This type of result was reproduced in several cell lines (data not shown), supporting the general role of this effect. Because of the very high similarity between MEK1 and MEK2, the results observed here for MEK1 are likely to hold true for MEK2 as well. Moreover, as assumed based on the ERK-MEK interaction, the PPARγ-MEK1 interaction indeed involves the D domain of MEK1 and the CRS-like region of PPARγ (Fig. 5). However, unlike the interaction between ERKs and MEKs, which is inhibited upon activation of the proteins (43), the association between PPARγ and MEK1 seems to increase upon activation of MEK1 (Fig. 4). This result indicates that the MEK1 interaction with the two proteins is not simultaneous. The interaction with ERKs occurs mainly in resting cells when MEK1 is not active, whereas the interaction with PPARγ occurs mainly upon mitogenic stimulation. These differences in the interaction may occur due to additional interacting sequences in the two proteins, including residues in the NES region of MEK1 and additional residues in the AF2 region of PPARγ (Fig. 5). Indeed, several nuclear receptors were reported to interact with regulatory proteins via the AF2 domain, including glucocorticoid receptor (8), estrogen receptor (12), and NF-κB interaction with PPARγ (13). Our results, therefore, are in line with these examples, suggesting that MEKs may regulate PPARγ via the important protein-docking region AF2.
The fact that MEK1 strongly binds to PPARγ raised the question of the localization of this interaction. MEK1 is known to localize primarily in the cytosol of all cells examined (25), while PPARγ is localized primarily in the nucleus of many (5, 6, 20), but not all (35, 39), cells. In our hands, PPARγ was localized primarily in the nucleus of the resting cells, while MEK1 appeared to be localized exclusively in the cytosol. Since the two proteins appear to be localized in different compartments in resting cells, we speculated that they could reach each other after cellular stimulation. One possibility was that, upon stimulation, MEKs translocate to the nucleus and stay there attached to the PPARγ, as previously reported, for the inactivation of MyoD by MEK1 (28). However, this mechanism is unlikely to participate in our system, since in our hands, MEKs were not retained in the nucleus for considerable duration upon stimulation (23, 45). Moreover, in the cell types used, MEK1 seems to translocate to the nucleus upon activation but is rapidly exported from the nucleus due to its N-terminal NES (23, 40, 45). Although this shuttling is robust and rapid, its role(s) is still poorly understood. One suggestion for the role of the translocation is that active MEKs in the nucleus may induce the activation of the nuclear ERK1b/c upon stimulation (3, 47). It was also suggested that nonstimulated MEKs may slowly shuttle into and out of the nucleus and thereby assist a slow and continuous export of inactive ERKs out of the nucleus (1). However, this mechanism is distinct from the MEK-PPARγ interaction, which is enhanced upon stimulation, involves active MEKs, and occurs in a rapid manner. Indeed, we found that overexpression of MEK1 can assist in cytosolic retention of PPARγ (Fig. 6), dependent on the CRS-like motif of the later. Using MEK inhibitors, we also found that, upon stimulation, endogenous PPARγ is exported from the nucleus in a MEK-dependent manner (Fig. 7 and 9). Addition of LMB and a peptide that disturbs the MEK1-PPARγ interaction showed that the relocalization of PPARγ is dependent on the protein-protein interaction of MEK1 and its nuclear shuttling (Fig. 8 and 9) and not on MEKs activity or on de novo protein synthesis. Therefore, our results indicate that MEKs and PPARγ interact during the short stay of active MEK1 in the nucleus, and this interaction allows the nuclear export of PPARγ that leads to the cytosolic localization of the MEK1-PPARγ complexes. This effect demonstrates a novel role for the cytonuclear shuttle of active MEKs and suggests that MEK1, which was previously considered an extremely specific regulator of the ERK cascade, may also play an important role in the regulation of other cellular signaling events.
In summary, we demonstrated here a strong association between MEK1 and PPARγ. The association between the proteins is enhanced upon stimulation and mediates the nuclear export of PPARγ by the cytonuclear shuttling of active MEKs. This represents a novel role for the mitogen-induced shuttling of MEKs, which determines the subcellular localization of PPARγ. MEK-dependent export of PPARγ from the nucleus reduces its transcriptional activity and therefore represents a novel mechanism of downregulation for PPARγ and possibly other nuclear receptors.
Acknowledgments
We thank J. Mizrahi, M. P. Ebert, and R. M. Schmid for their support and discussion and A. Schnoebelen for technical help.
This work was supported by grants from the Israel Academy of Sciences and Humanities, the Binational Israel-USA Foundation (BSF), and the Women Health Center in the Weizmann Institute of Science (to R.S.). E.B. was a recipient of a Minerva Postdoctoral Fellowship. M.L. is an incumbent of the Harold L. Korda Professorial Chair in Biology, and R.S. is an incumbent of the Yale S. Lewine and Ella Miller Lewine Professorial Chair for Cancer Research.
Footnotes
Published ahead of print on 13 November 2006.
REFERENCES
- 1.Adachi, M., M. Fukuda, and E. Nishida. 2000. Nuclear export of MAP kinase (ERK) involves a MAP kinase kinase (MEK)-dependent active transport mechanism. J. Cell Biol. 148:849-856. (Erratum, 149:754.) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Adams, M., M. J. Reginato, D. Shao, M. A. Lazar, and V. K. Chatterjee. 1997. Transcriptional activation by peroxisome proliferator-activated receptor gamma is inhibited by phosphorylation at a consensus mitogen-activated protein kinase site. J. Biol. Chem. 272:5128-5132. [DOI] [PubMed] [Google Scholar]
- 3.Aebersold, D. M., Y. D. Shaul, Y. Yung, N. Yarom, Z. Yao, T. Hanoch, and R. Seger. 2004. Extracellular signal-regulated kinase 1c (ERK1c), a novel 42-kilodalton ERK, demonstrates unique modes of regulation, localization, and function. Mol. Cell. Biol. 24:10000-10015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Akaike, M., W. Che, N. L. Marmarosh, S. Ohta, M. Osawa, B. Ding, B. C. Berk, C. Yan, and J. Abe. 2004. The hinge-helix 1 region of peroxisome proliferator-activated receptor γ1 (PPARγ1) mediates interaction with extracellular signal-regulated kinase 5 and PPARγ1 transcriptional activation: involvement in flow-induced PPARγ activation in endothelial cells. Mol. Cell. Biol. 24:8691-8704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Akiyama, T. E., C. T. Baumann, S. Sakai, G. L. Hager, and F. J. Gonzalez. 2002. Selective intranuclear redistribution of PPAR isoforms by RXR alpha. Mol. Endocrinol. 16:707-721. [DOI] [PubMed] [Google Scholar]
- 6.Berger, J., H. V. Patel, J. Woods, N. S. Hayes, S. A. Parent, J. Clemas, M. D. Leibowitz, A. Elbrecht, R. A. Rachubinski, J. P. Capone, and D. E. Moller. 2000. A PPARgamma mutant serves as a dominant negative inhibitor of PPAR signaling and is localized in the nucleus. Mol. Cell. Endocrinol. 162:57-67. [DOI] [PubMed] [Google Scholar]
- 7.Brummelkamp, T. R., R. Bernards, and R. Agami. 2002. A system for stable expression of short interfering RNAs in mammalian cells. Science 296:550-553. [DOI] [PubMed] [Google Scholar]
- 8.Bruna, A., M. Nicolas, A. Munoz, J. M. Kyriakis, and C. Caelles. 2003. Glucocorticoid receptor-JNK interaction mediates inhibition of the JNK pathway by glucocorticoids. EMBO J. 22:6035-6044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Burgermeister, E., M. Lanzendoerfer, and W. Scheuer. 2003. Comparative analysis of docking motifs in MAP-kinases and nuclear receptors. J. Biomol. Struct. Dyn. 20:623-634. [DOI] [PubMed] [Google Scholar]
- 10.Burgermeister, E., L. Tencer, and M. Liscovitch. 2003. Peroxisome proliferator-activated receptor-γ upregulates caveolin-1 and caveolin-2 expression in human carcinoma cells. Oncogene 22:3888-3900. [DOI] [PubMed] [Google Scholar]
- 11.Camp, H. S., S. R. Tafuri, and T. Leff. 1999. c-Jun N-terminal kinase phosphorylates peroxisome proliferator-activated receptor-gamma1 and negatively regulates its transcriptional activity. Endocrinology 140:392-397. [DOI] [PubMed] [Google Scholar]
- 12.Chen, D., T. Riedl, E. Washbrook, P. E. Pace, R. C. Coombes, J. M. Egly, and S. Ali. 2000. Activation of estrogen receptor alpha by S118 phosphorylation involves a ligand-dependent interaction with TFIIH and participation of CDK7. Mol. Cell 6:127-137. [PubMed] [Google Scholar]
- 13.Chen, F., M. Wang, J. P. O'Connor, M. He, T. Tripathi, and L. E. Harrison. 2003. Phosphorylation of PPARgamma via active ERK1/2 leads to its physical association with p65 and inhibition of NF-kappabeta. J. Cell. Biochem. 90:732-744. [DOI] [PubMed] [Google Scholar]
- 14.Diradourian, C., J. Girard, and J. P. Pegorier. 2005. Phosphorylation of PPARs: from molecular characterization to physiological relevance. Biochimie 87:33-38. [DOI] [PubMed] [Google Scholar]
- 15.Duesbery, N. S., C. P. Webb, S. H. Leppla, V. M. Gordon, K. R. Klimpel, T. D. Copeland, N. G. Ahn, M. K. Oskarsson, K. Fukasawa, K. D. Paull, and G. F. Vande Woude. 1998. Proteolytic inactivation of MAP-kinase-kinase by anthrax lethal factor. Science 280:734-737. [DOI] [PubMed] [Google Scholar]
- 16.Fields, S., and O. Song. 1989. A novel genetic system to detect protein-protein interactions. Nature 340:245-246. [DOI] [PubMed] [Google Scholar]
- 17.Floyd, Z. E., and J. M. Stephens. 2002. Interferon-gamma-mediated activation and ubiquitin-proteasome-dependent degradation of PPARgamma in adipocytes. J. Biol. Chem. 277:4062-4068. [DOI] [PubMed] [Google Scholar]
- 18.Fukuda, M., I. Gotoh, M. Adachi, Y. Gotoh, and E. Nishida. 1997. A novel regulatory mechanism in the mitogen-activated protein (MAP) kinase cascade. Role of nuclear export signal of MAP kinase kinase. J. Biol. Chem. 272:32642-32648. [DOI] [PubMed] [Google Scholar]
- 19.Gardner, O. S., B. J. Dewar, H. S. Earp, J. M. Samet, and L. M. Graves. 2003. Dependence of peroxisome proliferator-activated receptor ligand-induced mitogen-activated protein kinase signaling on epidermal growth factor receptor transactivation. J. Biol. Chem. 278:46261-46269. [DOI] [PubMed] [Google Scholar]
- 20.Gurnell, M., J. M. Wentworth, M. Agostini, M. Adams, T. N. Collingwood, C. Provenzano, P. O. Browne, O. Rajanayagam, T. P. Burris, J. W. Schwabe, M. A. Lazar, and V. K. Chatterjee. 2000. A dominant-negative peroxisome proliferator-activated receptor gamma (PPARgamma) mutant is a constitutive repressor and inhibits PPARgamma-mediated adipogenesis. J. Biol. Chem. 275:5754-5759. [DOI] [PubMed] [Google Scholar]
- 21.Ho, A., S. R. Schwarze, S. J. Mermelstein, G. Waksman, and S. F. Dowdy. 2001. Synthetic protein transduction domains: enhanced transduction potential in vitro and in vivo. Cancer Res. 61:474-477. [PubMed] [Google Scholar]
- 22.Hu, E., J. B. Kim, P. Sarraf, and B. M. Spiegelman. 1996. Inhibition of adipogenesis through MAP kinase-mediated phosphorylation of PPARgamma. Science 274:2100-2103. [DOI] [PubMed] [Google Scholar]
- 23.Jaaro, H., H. Rubinfeld, T. Hanoch, and R. Seger. 1997. Nuclear translocation of mitogen-activated protein kinase kinase (MEK1) in response to mitogenic stimulation. Proc. Natl. Acad. Sci. USA 94:3742-3747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kyriakis, J. M. 2000. MAP kinases and the regulation of nuclear receptors. Sci. STKE 2000:PE1. [DOI] [PubMed] [Google Scholar]
- 25.Lenormand, P., C. Sardet, G. Pages, G. L'Allemain, A. Brunet, and J. Pouyssegur. 1993. Growth factors induce nuclear translocation of MAP kinases (p42mapk and p44mapk) but not of their activator MAP kinase kinase (p45mapkk) in fibroblasts. J. Cell Biol. 122:1079-1088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lu, M. L., M. C. Schneider, Y. Zheng, X. Zhang, and J. P. Richie. 2001. Caveolin-1 interacts with androgen receptor. A positive modulator of androgen receptor mediated transactivation. J. Biol. Chem. 276:13442-13451. [DOI] [PubMed] [Google Scholar]
- 27.Marenda, D. R., A. D. Vrailas, A. B. Rodrigues, S. Cook, M. A. Powers, J. A. Lorenzen, L. A. Perkins, and K. Moses. 2006. MAP kinase subcellular localization controls both pattern and proliferation in the developing Drosophila wing. Development 133:43-51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Perry, R. L., M. H. Parker, and M. A. Rudnicki. 2001. Activated MEK1 binds the nuclear MyoD transcriptional complex to repress transactivation. Mol. Cell 8:291-301. [DOI] [PubMed] [Google Scholar]
- 29.Raviv, Z., E. Kalie, and R. Seger. 2004. MEK5 and ERK5 are localized in the nuclei of resting as well as stimulated cells, while MEKK2 translocates from the cytosol to the nucleus upon stimulation. J. Cell Sci. 117:1773-1784. [DOI] [PubMed] [Google Scholar]
- 30.Reginato, M. J., S. L. Krakow, S. T. Bailey, and M. A. Lazar. 1998. Prostaglandins promote and block adipogenesis through opposing effects on peroxisome proliferator-activated receptor gamma. J. Biol. Chem. 273:1855-1858. [DOI] [PubMed] [Google Scholar]
- 31.Rosen, E. D., and B. M. Spiegelman. 2001. PPARgamma: a nuclear regulator of metabolism, differentiation, and cell growth. J. Biol. Chem. 276:37731-37734. [DOI] [PubMed] [Google Scholar]
- 32.Rubinfeld, H., T. Hanoch, and R. Seger. 1999. Identification of a cytoplasmic-retention sequence in ERK2. J. Biol. Chem. 274:30349-30352. [DOI] [PubMed] [Google Scholar]
- 33.Sato, H., S. Ishihara, K. Kawashima, N. Moriyama, H. Suetsugu, H. Kazumori, T. Okuyama, M. A. Rumi, R. Fukuda, N. Nagasue, and Y. Kinoshita. 2000. Expression of peroxisome proliferator-activated receptor (PPAR) gamma in gastric cancer and inhibitory effects of PPARgamma agonists. Br. J. Cancer 83:1394-1400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Schlegel, A., C. Wang, B. S. Katzenellenbogen, R. G. Pestell, and M. P. Lisanti. 1999. Caveolin-1 potentiates estrogen receptor alpha (ERalpha) signaling. caveolin-1 drives ligand-independent nuclear translocation and activation of ERalpha. J. Biol. Chem. 274:33551-33556. [DOI] [PubMed] [Google Scholar]
- 35.Shibuya, A., K. Wada, A. Nakajima, M. Saeki, K. Katayama, T. Mayumi, T. Kadowaki, H. Niwa, and Y. Kamisaki. 2002. Nitration of PPARgamma inhibits ligand-dependent translocation into the nucleus in a macrophage-like cell line, RAW 264. FEBS Lett. 525:43-47. [DOI] [PubMed] [Google Scholar]
- 36.Sumanasekera, W. K., E. S. Tien, J. W. Davis II, R. Turpey, G. H. Perdew, and J. P. Vanden Heuvel. 2003. Heat shock protein-90 (Hsp90) acts as a repressor of peroxisome proliferator-activated receptor-alpha (PPARalpha) and PPARbeta activity. Biochemistry 42:10726-10735. [DOI] [PubMed] [Google Scholar]
- 37.Takeda, K., T. Ichiki, T. Tokunou, N. Iino, and A. Takeshita. 2001. 15-Deoxy-delta 12,14-prostaglandin J2 and thiazolidinediones activate the MEK/ERK pathway through phosphatidylinositol 3-kinase in vascular smooth muscle cells. J. Biol. Chem. 276:48950-48955. [DOI] [PubMed] [Google Scholar]
- 38.Tanoue, T., M. Adachi, T. Moriguchi, and E. Nishida. 2000. A conserved docking motif in MAP kinases common to substrates, activators and regulators. Nat. Cell Biol. 2:110-116. [DOI] [PubMed] [Google Scholar]
- 39.Thuillier, P., R. Baillie, X. Sha, and S. D. Clarke. 1998. Cytosolic and nuclear distribution of PPARgamma2 in differentiating 3T3-L1 preadipocytes. J. Lipid Res. 39:2329-2338. [PubMed] [Google Scholar]
- 40.Tolwinski, N. S., P. S. Shapiro, S. Goueli, and N. G. Ahn. 1999. Nuclear localization of mitogen-activated protein kinase kinase 1 (MKK1) is promoted by serum stimulation and G2-M progression. Requirement for phosphorylation at the activation lip and signaling downstream of MKK. J. Biol. Chem. 274:6168-6174. [DOI] [PubMed] [Google Scholar]
- 41.Tugwood, J. D., I. Issemann, R. G. Anderson, K. R. Bundell, W. L. McPheat, and S. Green. 1992. The mouse peroxisome proliferator activated receptor recognizes a response element in the 5′ flanking sequence of the rat acyl CoA oxidase gene. EMBO J. 11:433-439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Varley, C. L., J. Stahlschmidt, W. C. Lee, J. Holder, C. Diggle, P. J. Selby, L. K. Trejdosiewicz, and J. Southgate. 2004. Role of PPARgamma and EGFR signalling in the urothelial terminal differentiation programme. J. Cell Sci. 117:2029-2036. [DOI] [PubMed] [Google Scholar]
- 43.Wolf, I., H. Rubinfeld, S. Yoon, G. Marmor, T. Hanoch, and R. Seger. 2001. Involvement of the activation loop of ERK in the detachment from cytosolic anchoring. J. Biol. Chem. 276:24490-24497. [DOI] [PubMed] [Google Scholar]
- 44.Xu, B. E., S. Stippec, F. Robinson, and M. H. Cobb. 2001. Hydrophobic as well as charged residues in both MEK1 and ERK2 are important for their proper docking. J. Biol. Chem. 276:26509-26515. [DOI] [PubMed] [Google Scholar]
- 45.Yao, Z., I. Flash, Z. Raviv, Y. Yung, Y. Asscher, S. Pleban, and R. Seger. 2001. Non-regulated and stimulated mechanisms cooperate in the nuclear accumulation of MEK1. Oncogene 20:7588-7596. [DOI] [PubMed] [Google Scholar]
- 46.Yoon, S., and R. Seger. 2006. The extracellular signal-regulated kinase: multiple substrates regulate diverse cellular functions. Growth Factors 24:21-44. [DOI] [PubMed] [Google Scholar]
- 47.Yung, Y., Z. Yao, T. Hanoch, and R. Seger. 2000. ERK1b, a 46-kDa ERK isoform that is differentially regulated by MEK. J. Biol. Chem. 275:15799-15808. [DOI] [PubMed] [Google Scholar]