

Abstract
Adult skeletal muscle retains the capability of transcriptional reprogramming. This attribute is readily observable in the non-weight-bearing (NWB) soleus muscle, which undergoes a slow-to-fast fiber type transition concurrent with decreased β-myosin heavy chain (βMyHC) gene expression. Our previous work showed that Sp3 contributes to decreased βMyHC gene expression under NWB conditions. In this study, we demonstrate that physical and functional interactions between Sp3, Purα, and Purβ proteins mediate repression of βMyHC expression under NWB conditions. Binding of Purα or Purβ to the single-stranded βMyHC distal negative regulatory element-sense strand (dβNRE-S) element is markedly increased under NWB conditions. Ectopic expression of Purα and Purβ decreased βMyHC reporter gene expression, while mutation of the dβNRE-S element increased expression in C2C12 myotubes. The dβNRE-S element conferred Pur-dependent decreased expression on a minimal thymidine kinase promoter. Short interfering RNA sequences specific for Sp3 or for Purα and Purβ decreased endogenous Sp3 and Pur protein levels and increased βMyHC reporter gene expression in C2C12 myotubes. Immunoprecipitation assays revealed an association between endogenous Purα, Purβ, and Sp3, while chromatin immunoprecipitation assays demonstrated Purα, Purβ, and Sp3 binding to the βMyHC proximal promoter region harboring the dβNRE-S and C-rich elements in vivo. These data demonstrate that Pur proteins collaborate with Sp3 to regulate a transcriptional program that enables muscle cells to remodel their phenotype.
A distinguishing feature of skeletal muscle is its intricate organization of contractile proteins into striated myofibrils composed of repeating units called sarcomeres, the smallest force-producing unit of a myofibril. Although each sarcomere displays the same precise structural organization, the amount and type of contractile protein comprising a given sarcomere can differ based on the differential use of fiber type-specific contractile protein isoforms. The functional implications of the differential use of contractile protein isoforms are exemplified by the sarcomeric myosin heavy chain (MyHC) gene family (26, 36). In the adult mouse, four MyHC isoforms (fast type IIb, IIx/d, IIa, and slow type I [or β]) are differentially expressed, and this expression pattern has been shown to contribute to the histochemical classification of four primary fiber types, termed IIb, IIx/d, IIa, and slow type I. Each of these fiber types displays unique functional properties with respect to size, metabolism, fatigability, and intrinsic contractile properties. Since MyHC is a major determinant of the maximum unloaded shortening velocity of skeletal muscle contraction, the type of MyHC comprising a sarcomere is of functional significance (2, 3, 24, 26, 29, 36). Consistent with the latter concept, a functional analysis of skeletal muscle from either MyHC IIb or MyHC IIx/d null mice revealed altered contractile properties that were unique to each null mutation (1, 25).
Although the sarcomeric structure of skeletal muscle must be maintained for efficient force production, the contractile protein composition can be remodeled in response to a broad range of physiological stimuli. In fact, variations in the amount and type of load bearing imposed on skeletal muscle are a potent external stimulus that induces a switch in fiber phenotype (3, 27, 29). Such plasticity is clearly demonstrated when skeletal muscle is subjected to increased loading (mechanical overload [MOV]) or extended periods of disuse (unloading) due to injury, disease, or exposure to the microgravity environment of space or in response to the ground-based experimental model of hind limb suspension (non-weight-bearing [NWB] model) (3, 13, 20-22, 27-34, 37, 38). Slow-twitch muscles such as the soleus, which are composed primarily of slow type I fibers, express high levels of the slow type I MyHC (βMyHC), and are used primarily in chronic activities such as postural maintenance, are most susceptible to the effects of muscle disuse as evidenced by a slow-to-fast fiber type conversion and decreased βMyHC gene expression (3, 20, 21, 28-30). Although this intriguing adaptation in phenotype has been well documented, the underlying transcriptional mechanisms are not well understood.
To gain insight into the transcriptional mechanisms that control NWB-induced genome reprogramming of the adult mouse soleus muscle, we have used the βMyHC gene as a model system (19-21, 23, 28). Our previous analyses of transgenes containing either the mouse or human βMyHC promoters led to the delineation of a 600-bp region that was sufficient to mimic endogenous βMyHC down-regulation in response to NWB (20). Further analysis of the 600-bp βMyHC promoter identified two muscle CAT (MCAT) sites (distal MCAT, [−290 to −284] and proximal MCAT [−210 to −203]), an E-box/nuclear factor of activated T-cells (−183 to −172) element, and three closely spaced GC-rich (GT/CACC) elements (C-rich A [−248 to −225], C-rich B [−160 to −140], and C-rich C [−61 to −41]) (30). The G/C-rich elements are functionally important for down-regulation of βMyHC in response to NWB, as electrophoretic mobility shift assay (EMSA) analyses displayed enriched binding of Sp3 isoproteins (115, 80, and 78 kDa) only with nuclear extract from soleus muscle exposed to NWB conditions (28). Overexpression of Sp3 resulted in decreased βMyHC reporter gene expression in both Drosophila SL-2 and mouse C2C12 myotubes (28).
In parallel work, we identified an additional element (βMyHC distal negative regulatory element-sense strand [dβNRE-S], −332 to −311) that displayed potent repressor activity in the context of a 350-base-pair βMyHC promoter/transgene in all transgenic lines examined (21). Further analysis of the dβNRE-S element revealed highly enriched binding of two distinct proteins, of approximately 50 and 52 kDa, when using nuclear extract prepared from NWB soleus muscle. A unique feature of these proteins was their marked preference for binding to the single-stranded dβNRE-S element. Although the identity of the dβNRE-S binding proteins is not known, our prior work has eliminated cellular nucleic acid binding protein (7) and the Y-box binding factor YB-1 (9) as candidates for the NWB-induced binding factors (21).
In this study, we have demonstrated that Purα and Purβ represent the functionally relevant NWB soleus dβNRE-S element binding proteins. Additionally, by using coimmunoprecipitation, immunoprecipitation, transient expression, and short interfering RNA (siRNA) assays, we demonstrate that Purα, Purβ, and Sp3 physically associate and collaborate to negatively regulate βMyHC reporter gene expression in C2C12 myotubes. Furthermore, chromatin immunoprecipitation (ChIP) assays revealed that the βMyHC proximal promoter region containing the dβNRE-S and C-rich elements is bound by Purα, Purβ, and Sp3 in C2C12 myotubes. These data provide the first evidence supporting the notion that the Pur proteins collaborate with Sp3 as important mediators of βMyHC gene transcription during skeletal muscle inactivity.
MATERIALS AND METHODS
Preparation of nuclear protein extract from adult skeletal muscle.
Nuclear extract was isolated from adult rat control soleus (CS), NWB soleus (NWB-S), control plantaris (CP), and MOV plantaris (MOV-P) muscles as described previously (21, 32). Protein concentrations were determined using the Bio-Rad protein reagent according to the method of Bradford (4).
Animal care and MOV and NWB procedures.
The MOV and NWB procedures used in this study were approved by the Animal Care Committee for the University of Missouri-Columbia, and the MOV mice were housed in an AAALAC-accredited animal facility. Rats were prepared for the NWB experiment by modification of the noninvasive tail traction procedure, as described previously (20). The imposition of a mechanical overload on the fast-twitch plantaris muscle was accomplished as described by us previously (31). All animals were provided with food and water ad libitum and were housed at room temperature (24°C) with a 12-h light-dark cycle in either standard filter top cages (control and MOV mice) or cages designed for head-down tilt suspension (hind limb suspension).
Plasmids and constructs.
βMyHC promoter constructs containing 1,285 bp of human βMyHC promoter sequence and 120 bp of 5′ untranslated region were cloned into the HindIII site of the pGL3 luciferase reporter gene vector. The βMyHC dβNRE-S element was mutated within the pGL3-β1285 plasmid by using the QuikChange site-directed mutagenesis kit (Stratagene) according to the manufacturer's recommendations. The sequence of the complementary oligonucleotide primers containing the desired mutations were as follows (mutated sites are in lowercase): primer 1, 5′-GCC AGG ACA TTG GCT GCC TGT Gtg Cgt caT aGT CGT GGT CAG TTC CC-3′; and primer 2, 5′-GGC TGC CTG TGT GCG TCA TAt gCc Tca TgA GTT CCC TCT CCT GCC AGC-3′. Novel transcription factor recognition sites were not created by these mutations, as determined by database analysis using the Eukaryotic Transcription Factor database (tfsites.dat) available from the Genetics Computer Group. The pCITE4-Sp3 expression vector used for in vitro transcription-translation (TNT) studies was constructed by inserting Sp3 cDNA into pCITE4 (Novagen) vectors in frame with the internal translation start site.
In vitro transcription-translation.
TNT reactions were performed using 1 μg of Sp3 expression plasmids in the T7 TNT rabbit reticulocyte lysate system or using 1 μg of Purα and Purβ expression plasmids (5, 15) in the T7 TNT coupled wheat germ extract system, according to the manufacturer's instructions (Promega). Efficient translation and expected molecular weights of the protein products were verified by resolving the radiolabeled reaction products on NuPage 4 to 12% bis-Tris gels (Invitrogen) and by Western blotting. Parallel reactions of unprogrammed lysate performed in the absence of plasmid DNA served as negative controls.
EMSAs.
All oligonucleotide probes used in this study are listed in Table 1, and the EMSAs were carried out as previously described (21). The single-stranded dβNRE-S (sense strand) oligonucleotide was end labeled with T4 polynucleotide kinase (New England Biolabs) and [γ-32P]ATP (Perkin-Elmer) and gel purified. Binding reactions were performed using 500 ng of CS or NWB-S, 20 ng of recombinant Purα or Purβ protein (18), and 20,000 cpm of labeled probe for 20 min at room temperature in a 25-μl total volume in binding buffer (50 mM Tris-HCl [pH 7.9], 50 mM KCl, 0.5 mM dithiothreitol, and 5% [wt/vol] glycerol). Supershift assays were performed with 2 μl preimmune serum or 0.5 μg of affinity-purified anti-Purα or Purβ antibody (15) in the binding reaction prior to the addition of probe. For competition EMSA experiments, double-stranded annealed probes were gel purified and used as binding competitors. Protein-DNA complexes were electrophoretically resolved from unbound oligonucleotide probe on a 5% (vol/vol) 0.5× TBE (25 mM Tris, 25 mM boric acid, 0.5 mM EDTA) nondenaturing polyacrylamide gel at 220 V for 2.5 h at 4°C.
TABLE 1.
Oligonucleotide probes
| Probe name | Sequencea | Position |
|---|---|---|
| dβNRE-S | 5′-GTGGTCTTGGTGGTCGTGGTCA-3′ | −322 to −311 |
| dβNRE-Sm1 | 5′-GTGtgCgTcaTatgCcTcaTgA-3′ | |
| dβNRE-Sm2 | 5′-aTatgCgTcGTGGTCGTGGTCA-3′ | |
| α-Actin | 5′-GGAGCAGAACAGAGGAATGCAGTGGAAGAG-3′ | −194 to −165 |
| αMyHC | 5′-ACCTAGAGGGAAAGTGTCTTCCCTGGAAGTGGGCT-3′ | +71 to +97 |
| HMG-CoA | 5′-GAAGCTTGTGCGGTGGAATTCTGCA-3′ | STEoctamer |
| C-richAb | 5′-TGAGCCACCCCGCCCCCTGGAACT-3′ | −248 to −255 |
| 3xdβNRE-S | 5′-C(GTGGTCTTGGTGGTCGTGGTCA)3-3′ | |
| 5′-(TGACCACGACCACCAAGACCAC)3GAGCT-3′ | ||
| 3xdβNRE-mut | 5′-C(GTGtgCgTcaTatgCcTcaTgA)3-3′ | |
| 5′-(TcAtgAgGcatAtgAcGcaCAC)3GAGCT-3′ |
Lowercase indicates mutations, and boldface indicates the consensus sterol regulatory element octamer.
Double-stranded DNA.
Western blots and antibodies.
Western blotting was carried out as previously described (13). TNT protein products (0.8 μl of Sp3, 2 μl of Purα, and 2 μl Purβ) or 50 μg C2C12, CS, NWB-S, CP, and MOV-P nuclear extracts were separated on NuPage 4 to 12% bis-Tris gels (Invitrogen) at 200 V and transferred to a polyvinylidene difluoride membrane (Bio-Rad Laboratories) at 30 V for 1 h. Following an overnight incubation at 4°C with 5% (wt/vol) nonfat dry milk in Tris-buffered saline with 0.1% Tween 20 (TBST), the blots were incubated with anti-Sp3 (1:1,000), anti-Purα (2 μg/ml), anti-Purβ (1 μg/ml), or anti-His (1:1,000) antibodies. The blots were washed with TBST and incubated with horseradish peroxidase-conjugated goat anti-rabbit immunoglobulin G (IgG) or horseradish peroxidase-conjugated goat anti-mouse IgG (1:2,000) (Cell Signal Technology). Following additional washes, the signal was detected using an enhanced chemiluminescence detection system (PicoWest SuperSignal substrate; Pierce) and subjected to autoradiography.
Cell culture, transfections, and reporter assays.
Mouse skeletal muscle C2C12 myoblasts (ATCC) were maintained in high-glucose Dulbecco's modified Eagle's medium (DMEM) (Gibco) supplemented with 10% (vol/vol) fetal bovine serum, 2 mM l-glutamine, sodium pyruvate, and antibiotics at 37°C in a humidified chamber containing 5% CO2 in air. Transfection experiments were carried out as previously described (13, 28). C2C12 cells (2 × 105) were plated onto 0.1% gelatin-coated 35-mm cell culture dishes. Transfections were carried out 24 h later using FuGENE 6 according to the manufacturer's manual (Roche), including cotransfection of 0.05 μg of pRL-TK expression vector (Promega) as the internal control. One microgram of a 1,285-bp wild-type βMyHC luciferase reporter gene (β1285 wt), 0.05 μg of pRL-TK Renilla luciferase reporter gene, and 0.5 μg expression plasmids were transfected, keeping the total amount of DNA at 2.5 μg with the addition of the promoterless plasmid pPac0 whenever necessary. Cells were washed 24 h following transfection with Hanks' balanced salt solution (Gibco), and differentiation medium (DMEM supplemented with 5% heat-inactivated horse serum) was added; 48 h later, the medium was replaced with fresh differentiation medium. Four days after transfection, extracts were prepared in passive lysis buffer according to the protocol supplied by Promega. Reporter gene assays were carried out using the dual-luciferase reporter assay system (Promega) and a Turner Designs model TD-20/20 luminometer.
siRNA transfections.
C2C12 myoblasts were plated at a density of 2 × 105 per well (6-well plates) or 8 × 104 per well (12-well plates). The following day, C2C12 cells were transfected at 60 to 80% confluence with plasmid DNA and siRNA by using Lipofectamine 2000 (Invitrogen) according to the manufacturer's protocol. The siRNA duplexes used in this study are from Dharmacon and Santa Cruz, and the sequences are as follows: Purα, ACA UGG AUC UCA AGG AGA AUU; Purβ, UGA AAG AGA UCC AGG AGC GUU; and siCONTROL Non-Targeting no. 1 and Sp3 (no sequence provided). Each siRNA was added to the cells at a final concentration of 50 nM. At 24 h following transfection, cells were washed with Hanks' balanced salt solution (Gibco) and changed into differentiation medium. Cells were harvested 48 h later and analyzed for β1285 wt reporter gene activity (luciferase) by using the DLR kit (Promega). In addition, parallel samples were tested by Western blot analysis for expression of endogenous Purα, Purβ, and Sp3 proteins by using anti-Purα (15), anti-Purβ (15), and anti-Sp3 (Santa Cruz) antibodies.
Coimmunoprecipitation analysis.
To monitor the interaction between Sp3 and Purα or Purβ protein, mouse C2C12 myoblasts were transiently transfected with various combinations of Sp3, Purα, and Purβ expression vectors. Nuclear lysates from C2C12 myotubes were precleared with protein G-agarose (Fast Flow; Upstate) for 30 min at 4°C. The precleared lysates were incubated overnight with 2 μg of anti-Sp3 antibody or 20 μl of anti-His probe conjugated to agarose (Santa Cruz). Twenty microliters of protein G-agarose was added to the lysate and incubated for 2 h at 4°C. Agarose beads were collected by centrifugation at 10,000 rpm for 30 s and washed three times. Samples were resuspended in 2× sample buffer for electrophoresis. To investigate interactions between endogenous Purα, Purβ, and Sp3, nuclear extract from C2C12 myotubes was used for immunoprecipitation assays as described above, using 2 μg of anti-Purα and anti-Purβ antibodies.
ChIP assay.
ChIP assays were performed using a CHIP-IT kit (Active Motif, Inc., Carlsbad, CA) as described by the manufacturer. All reagents and buffers are contained within the kit. Differentiated C2C12 cells were cross-linked with 1% formaldehyde for 10 min at room temperature, washed, and treated with glycine Stop-Fix solution. C2C12 myotubes were resuspended in cell lysis buffer and incubated on ice for 10 min. The pellets were resuspended in enzymatic shearing cocktail and incubated on ice for 10 min. The enzymatic shearing conditions were optimized to generate 250 to 400 base pairs of genomic DNA fragments. The conditions for enzymatic shearing were as follows. Chromatin was prewarmed at 37°C for 5 min and sheared with the enzymatic shearing cocktail for 10 min at 37°C. The chromatin was precleared using protein G beads. Precleared chromatin was incubated overnight at 4°C with anti-Purα (15), anti-Purβ (15), anti-Sp3 (Santa Cruz Biotechnology), or, for a negative control, IgG (Santa Cruz Biotechnology) or antihemagglutinin (anti-HA) (Covance). Protein G beads were then added to the antibody-chromatin complex and incubated for 3 h at 4°C. After extensive washings, the immunoprecipitated DNA complexes were eluted from the beads. Protein-DNA cross-linking was reversed by adding 5 M NaCl and RNase to the samples and incubating overnight at 65°C. DNA was purified by proteinase K digestion and phenol-chloroform extraction. The βMyHC promoter C-rich (−2 to −268) and dβNRE-S (−270 to −450) regions were amplified by PCR using the following primer sequences: C-rich, 5′-CTCGGTCTGGACCAGAGTC-3′ and 5′-CTCTATAAAAACGACGTGAAACTCGG-3′); and dβNRE-S, 5′-ACCTGACACGTCCCAGACTC-3′ and 5′-TCCCTCCTGTGACACCTTTT-3′. The products were resolved by electrophoresis in a 2% agarose gel.
Shift Southwestern analysis.
Shift Southwestern analysis was performed essentially as described by us previously (21). The specific protein-DNA complex formed when the distal portion of the dβNRE sense strand (dβNRE-S; −332 to −311) was incubated in a binding reaction with the 1.0 M KCl elution fraction, obtained following dβNRE-S element affinity binding using NWB soleus nuclear extracts was separated by EMSA. EMSA was performed essentially as described above except that the binding reaction was scaled up 10-fold, and 13 independent reaction mixtures were electrophoresed in a 0.75-mm-thick gel. Following EMSA, the section of the gel containing the protein-DNA complex was electrophoretically transferred to membranes (nitrocellulose and DEAE) placed in series using conditions described above for Western blot analysis. During transfer, the protein component of the protein-DNA complex bound to the nitrocellulose membrane and the dβNRE-S DNA probe bound to the DEAE membrane. Following localization of the bound protein by using the DEAE membrane, the protein was eluted from the nitrocellulose membrane by incubation in a 20% (vol/vol) acetonitrile solution for 3 h at 37°C. The eluate was centrifuged for 10 min to remove particulate material, lyophilized to remove solvent, and resuspended in 20 μl of 50 mM Tris-HCl, pH 7.5. The recovered protein was then solubilized in 6× sample buffer (350 mM Tris-HCl [pH 6.8], 30% [wt/vol] glycerol, 10% [wt/vol] sodium dodecyl sulfate [SDS], 0.93 mM dithiothreitol, 0.012% [wt/vol] bromophenol blue) and electrophoretically resolved by 12% (wt/vol) SDS-polyacrylamide gel electrophoresis at constant voltage (200 V) for 45 min at room temperature. The protein was electrophoretically transferred to a nitrocellulose membrane as described above for Western analysis. The membrane was incubated for 10 h at 4°C in a blocking solution composed of EMSA binding reaction buffer (minus glycerol) containing 5% (wt/vol) nonfat milk. Protein-DNA interaction occurred during incubation of the membrane in a blocking solution containing 0.25% nonfat milk and labeled dβNRE-S probe (2 × 106 cpm/ml) for 10 h at 4°C. Following hybridization, the membrane was washed three times for 5 min each at room temperature in a solution consisting of 50 mM Tris-HCl (pH 7.9), 30 mM KCl, 1 mM MgCl2, 0.5 mM EDTA, and 0.5 mM dithiothreitol; air dried; and exposed to film overnight.
Statistical analysis.
Statistical analyses were performed using the SPSS Graduate Pack 10.0 program (SPSS, Chicago, IL) A Levene test for equality of variances was performed, followed by a two-tailed independent-sample t test to assess differences between group means. Where the Levene test was rejected (significance of ≤0.05), the separate variance t test for means was used, where equal variances were not assumed. The lowest significance level accepted was a P value of <0.05. All data are reported as the mean ± standard error.
RESULTS
EMSA analysis reveals comparable binding patterns of dβNRE-S affinity-enriched and whole NWB soleus nuclear extract.
Our previous transgenic analyses have provided in vivo evidence that the single-stranded dβNRE-S element (−332 to −311) functions as a negative regulator of βMyHC gene expression (21) (Fig. 1). In addition, our studies on the binding properties of the dβNRE-S element using EMSA, UV cross-linking, and shift Southwestern analyses revealed the formation of a highly enriched binding complex comprised of two distinct proteins only when nuclear extract prepared from adult soleus muscle following 2 weeks of NWB was used (21). Because NWB leads to decreased βMyHC gene expression, the latter observation is supportive of the notion that the dβNRE-S may play a regulatory role in response to NWB-imposed muscle inactivity.
FIG. 1.

The βMyHC dβNRE-S element is highly conserved in sequence and position across species. The nucleotide sequence comparison of the βMyHC proximal promoters of various species reveals high conservation of the dβNRE-S and adjacently located CG-rich, A/T-rich, muscle CAT, and E-box/nuclear factor of activated T-cell elements (shaded).
To determine the identity of the nuclear protein(s) that associated with the dβNRE-S element, we performed a DNA sequence-specific protein affinity binding assay using nuclear extract prepared from adult NWB soleus muscle and three tandem copies of the single-stranded biotinylated wild-type dβNRE-S sequence as target DNA (Table 1). As a control for nonspecific protein binding, parallel binding reactions using three tandem copies of a single-stranded biotinylated mutant βMyHC dβNRE-S sequence were performed (data not shown). Following incubation of the biotinylated βMyHC dβNRE-S sequence with nuclear extract isolated from adult NWB soleus muscle, the reaction mixture was centrifuged and the supernatant was collected and designated the flowthrough fraction. The pellet containing the concatenated single-stranded biotinylated βMyHC dβNRE-S element was thoroughly washed, and the interacting proteins were eluted with buffer with increasing salt concentrations (0.1, 1.0, or 2.0 M KCl). Eluted protein fractions corresponding to each salt concentration were pooled, concentrated, and analyzed by SDS-polyacrylamide gel electrophoresis (Fig. 2A).
FIG. 2.

Affinity enrichment of dβNRE-S binding protein. (A) SDS-polyacrylamide gel electrophoresis of approximately 1% of the total protein (silver-stained gel) eluted from the concatenated biotinylated dβNRE-S element following incubation with adult NWB soleus nuclear extract. F.T., flowthrough. (B) Electrophoretic mobility shift assay showing binding complex similarity when using either the 1.0 M KCl elution fraction (SC1 and SC2) or three distinct nonfractionated adult NWB soleus nuclear extracts (NE1, NE2, and NE3). Eluates 1A and 1B and eluates 3A and 3B represent distinct 1.0 M KCl elution fractions obtained following incubation with either NE1 or NE3, respectively. (C) Shift Southwestern (SW) analysis of dβNRE-S/1.0 M elution fraction binding complex. Two protein bands of approximately 45 and 50 kDa were detected.
Electrophoretic fractionation using a 12% polyacrylamide separating gel matrix revealed that the flowthrough and the 0.1 M KCl elution fraction contained a majority of the nuclear proteins (Fig. 2A, lanes 1 and 2). The 1.0 M KCl elution fractions contained several proteins that were not observed in the 2 M KCl elution fraction (Fig. 2A, lane 3 versus lane 4). We next performed an EMSA analysis to determine if the 1.0 M KCl elution fractions would form an enriched dβNRE-S binding complex comparable to that obtained when using nonfractionated nuclear extract prepared from adult NWB soleus muscle (Fig. 2B). Incubation of 32P-labeled human βMyHC single-stranded dβNRE-S oligonucleotide with the 1.0 M KCl elution fractions from three independent nuclear extract preparations revealed the formation of two protein-DNA complexes (SC1 and SC2). The migration pattern of SC1 and SC2 closely resembled that of the protein-DNA complexes observed when the dβNRE-S sequence was reacted with aliquots from the nonfractionated nuclear extracts (Fig. 2B, lanes 1 to 5 versus lanes 6, 8, and 10). A protein-DNA complex was not observed when control soleus nuclear extract was used (Fig. 2B, lane 12).
To further characterize proteins that bound to the dβNRE-S motif, we performed a shift Southwestern blot analysis (21). In this assay, a 32P-labeled dβNRE-S oligonucleotide was incubated with the 1.0 M KCl elution fraction, and the binding reaction was then fractionated by native gel electrophoresis. Proteins that bound to the oligonucleotide were electroeluted from the native gel and subjected to Southwestern analysis using a 32P-labeled dβNRE-S probe (Fig. 2C). This analysis revealed that two proteins of approximately 45 and 50 kDa comprised the enriched dβNRE-S binding complex (Fig. 2C).
A number of recent observations raise the intriguing possibility that Purα and Purβ may bind to the dβNRE-S motif. For example, Purα and Purβ have estimated molecular masses of 46 and 44 kDa, respectively (10, 16). In addition, both of these proteins have been shown to bind single-stranded purine-rich repeats (consensus, GGN) similar to the sequence of the βMyHC dβNRE-S element (5′-GTGGTCTTGGTGGTCGTGGTCA-3′; boldface indicates consensus GGN repeats) (5, 10, 11, 14). Furthermore, the binding of Purα and Purβ to purine-rich elements resembling the βMyHC dβNRE-S element has been shown to negatively regulate the expression of both the cardiac-specific αMyHC and vascular smooth muscle α-actin reporter genes (5, 10).
As an initial step towards determining whether Purα and Purβ represent the enriched βMyHC dβNRE-S binding proteins, we performed a competition EMSA experiment using nuclear extract prepared from adult NWB soleus muscle and single-stranded DNA oligonucleotides previously shown to bind Purα and Purβ. Incubation of the 32P-labeled single-stranded βMyHC dβNRE-S oligonucleotide with nuclear extract prepared from nonfractionated adult NWB soleus muscle resulted in a protein-DNA complex that was effectively competed away by the addition of a 100-fold molar excess of cold wild-type single-stranded dβNRE-S oligonucleotide to the binding reaction mixture (Fig. 3A, lane 1 versus lane 2). In contrast, complex formation was not abolished by addition of a 100-fold molar excess of cold single-stranded βMyHC dβNRE-S mutant oligonucleotide (Fig. 3A, lane 1 versus lanes 3 and 4; Table 1). Addition of a 100-fold molar excess of cold single-stranded oligonucleotide containing the Purα and Purβ binding site from either the smooth muscle α-actin or cardiac αMyHC to the binding reaction mixture completely abolished complex formation (Fig. 3A, lane 1 versus lanes 5 and 6).
FIG. 3.
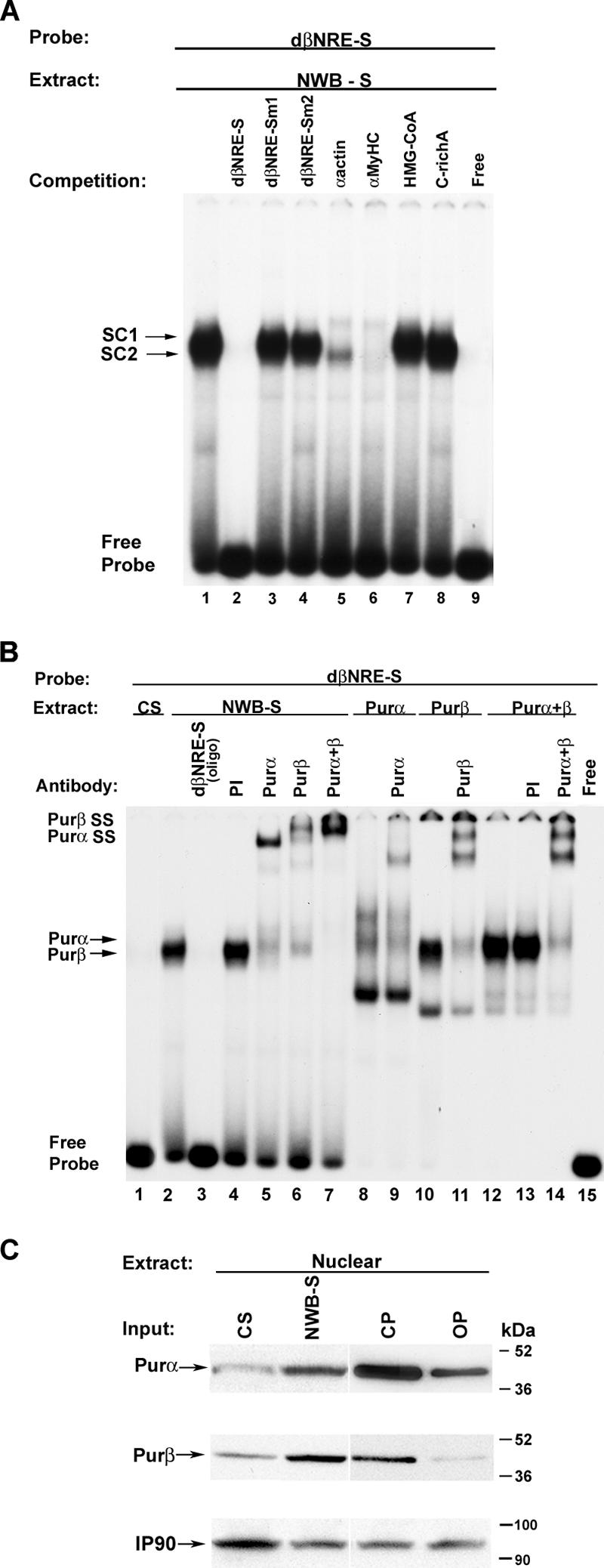
Competition and antibody EMSA analysis of sequence-specific protein-DNA interactions at the dβNRE-S element. (A) Five hundred nanograms of NWB-S nuclear extract was incubated in the presence of 20,000 cpm of the 32P-labeled dβNRE-S element (lanes 1 to 9). For competition assays, the following nonradioactive competitor oligonucleotides were added to the reaction mixture at a 100-fold molar excess prior to the addition of the 32P-labeled dβNRE-S probe: dβNRE-S wt (lane 2), dβNRE-Sm1 (lane 3), dβNRE-Sm2 (lane 4), α-actin (lane 5), αMyHC (lane 6), HMG-CoA (lane 7), and C-rich A (lane 8). Free probe (lane 9) represents the dβNRE-S probe resolved in the absence of nuclear extract. SC, specific complex. (B) Antibody EMSA analysis of the specific dβNRE-S binding complex formed when using NWB-S nuclear extract. The 32P-labeled βMyHC dβNRE-S element was incubated with 500 ng of either CS (lane 1) or NWB-S (lanes 2 to 7) nuclear extract or with 20 ng of purified Purα or Purβ protein (lanes 8 to 14). Addition of anti-Purα or anti-Purβ antibody to the binding reaction mixture containing NWB-S nuclear extract resulted in a supershift (lanes 5 to 7). The addition of anti-Purα or anti-Purβ antibody to the binding reaction mixture containing purified Purα and Purβ protein similarly resulted in a supershifted binding complex (lanes 9, 11, and 14), supporting the notion that the enriched dβNRE-S binding complex obtained when using NWB-S nuclear extract is comprised of Purα and Purβ. The lack of a detectable binding complex when using CS nuclear extract is consistent with our previous findings. (C) Pur protein expression pattern in CS, NWB-S, CP, and MOV-P. Western blot results of rat CS and NWB-S nuclear extract (50 μg; lanes 1 and 2) and CP and MOV-P nuclear extract (50 μg; lanes 3 and 4) using rabbit polyclonal Purα or Purβ antibody are shown. IP90 was used as a loading control. Note that the qualitative levels of Purα and Purβ increase in response to NWB and decrease with MOV.
In contrast, addition of a 100-fold molar excess of the single-stranded 3-hydroxy-3-methylglutanyl coenzyme A (HMG-CoA) oligonucleotide did not interfere with complex formation, indicating that cellular nucleic acid binding protein is not a component of the dβNRE-S binding activity within NWB soleus nuclear extract (Fig. 3A, lane 1 versus lane 7). Complex formation was also not altered by the addition of a 100-fold molar excess of cold double-stranded βMyHC Sp1/Sp3 oligonucleotide (−255 to −248) (31) (Fig. 3A, lane 1 versus lane 8). Taken together, these data suggest that Purα and Purβ are components of the dβNRE-S binding activity in NWB soleus nuclear extract.
The single-stranded DNA binding proteins Purα and Purβ represent the enriched βMyHC dβNRE-S binding activity in NWB adult soleus nuclear extract.
To directly establish whether Purα and Purβ comprise the enriched βMyHC dβNRE-S binding activity identified within adult NWB soleus nuclear extract, we performed supershift EMSA analysis using polyclonal antibodies that specifically recognize either Purα or Purβ (Fig. 3B). Incubation of 32P-labeled human βMyHC dβNRE-S oligonucleotide with adult NWB soleus nuclear extract revealed the formation of a protein-DNA binding complex that was competed away by the addition of a 100-fold molar excess of cold dβNRE-S oligonucleotide (Fig. 3B, lane 2 versus lane 3). Formation of this protein-DNA complex was not altered by addition of preimmune serum (Fig. 3B, lane 2 versus lane 4), whereas either anti-Purα or anti-Purβ antibody supershifted the protein-DNA complex (Fig. 3B, lane 4 versus lanes 5 and 6). The simultaneous addition of both the anti-Purα and anti-Purβ antibodies to binding reaction mixtures resulted in a complete supershift of the protein-DNA complex (Fig. 3B, lane 2 versus lane 7). When the βMyHC dβNRE-S sequence was reacted with purified Purα protein, a protein-DNA complex was observed which had a migration pattern resembling that of the protein-DNA complex that formed when adult NWB soleus nuclear extract was used (Fig. 3B, lane 2 versus lane 8). The addition of anti-Purα antibody to binding reaction mixtures containing purified Purα protein led to a supershifted band (Fig. 3B, lanes 8 versus lane 9). When the dβNRE-S sequence was reacted with either purified Purβ protein or a combination of purified Purα and Purβ proteins, a protein-DNA complex formed that resembled the binding complex formed when adult NWB soleus nuclear extract was used (Fig. 3B, lane 2 versus lanes 10 and 12). As expected, the addition of antibodies against either Purβ or both Purα and Purβ to binding reaction mixtures containing purified Purβ protein or purified Purα and Purβ proteins completely supershifted the protein-DNA complexes (Fig. 3B, lane 10 versus lane 11 and lane 12 versus lane 14). Collectively, these experiments indicate that Purα and Purβ represent the single-stranded βMyHC dβNRE-S-specific binding activity found within adult NWB soleus nuclear extract.
Western blot analysis suggests that Purα and Purβ protein levels are regulated in response to NWB.
To determine if there were qualitative differences in Purα and Purβ nuclear protein levels between adult control and NWB soleus nuclear extracts, we performed a Western blot analysis (Fig. 3C). When either anti-Purα or anti-Purβ specific antibodies were used, a band in the predicted size range for Purα and Purβ (≈46 and 44 kDa) was detected in CS nuclear extract. The intensity of these bands was considerably higher when NWB soleus nuclear extract was used (Fig. 3C, lane 1 versus lane 2). Western blot analysis using nuclear extract prepared from the fast-twitch CP muscle revealed a band whose intensity decreased when nuclear extract from MOV-P muscle was used (Fig. 3C, lane 3 versus lane 4). Collectively, these data demonstrate that the levels of nuclear Purα and Purβ proteins differ between slow-twitch and fast-twitch skeletal muscles and that the nuclear abundance of these proteins is regulated in response to altered mechanical loads.
Mutation of selected nucleotides comprising the dβNRE-S sequence significantly increases βMyHC promoter activity in C2C12 myotubes.
To characterize the functional role of the βMyHC dβNRE-S element in C2C12 muscle cells, we generated a 1,285-bp wild-type βMyHC luciferase reporter gene (β1285 wt) and a mutant version that carried site-directed mutations of selected nucleotides that comprised the dβNRE-S element (β1285dβNRE-Sm1) (Fig. 4A; Table 1). In transient-transfection assays using C2C12 myotubes, the basal activity of the β1285 wt promoter was markedly higher than that of the promoterless pGL3 basic luciferase plasmid (Fig. 4B). Mutation of the dβNRE-S element significantly increased β1285dβNRE-Sm1 activity (Fig. 4B). These data demonstrate that the βMyHC dβNRE-S element functions as a negative regulator of the 1,285-bp βMyHC reporter gene in mouse C2C12 myotubes.
FIG. 4.

Functional role of the dβNRE-S element. (A) Schematic of β1285 wt and β1285dβNRE-Sm1 forms of a 1,285-bp human βMyHC proximal promoter. Site-directed mutations involved nucleotides previously shown to comprise consensus Pur protein binding sites (GGN)n. (B) Promoter activities of wild-type and dβNRE-S mutant reporter genes (1 μg) determined in C2C12 myotubes. Mutation of the dβNRE-S element increased expression of the 1,285-bp βMyHC Luc reporter gene expression in C2C12 muscle cells. Data are reported as luciferase-normalized relative light units (RLU) (firefly/Renilla) and are expressed as the mean ± standard error (n = 8). *, P < 0.0001; all comparisons are to β1285 wt. (C) siRNA targeting either Purα or Purβ led to decreased levels of its target. C2C12 myoblasts were transfected with control siRNA (nontargeting [NT]) or siRNA targeting Purα, Purβ, or Purα and Purβ as described in Methods and Materials. IP90 was used as a loading control. Western blot analysis revealed that Purα and Purβ siRNAs effectively decreased endogenous levels of both Purα and Purβ. (D) Knockdown of endogenous Purα or Purβ protein results in increased expression of the β1285 wt reporter gene in C2C12 myotubes. Data are reported as luciferase-normalized RLU (firefly/Renilla) and are expressed as the mean ± standard error (n = 3). *, P < 0.0001; all comparisons are against β1285 wt activity in the presence of NT siRNA.
Pur siRNA decreases endogenous Pur protein levels and increases βMyHC reporter gene expression in C2C12 muscle cells.
Since C2C12 myotubes express endogenous Purα and Purβ proteins, we wished to determine whether transient knockdown of endogenous Pur protein levels by using siRNA would increase βMyHC reporter gene expression in C2C12 muscle cells. In these experiments, C2C12 myoblasts were transfected with siRNA against either Purα or Purβ, with siRNA against Purα and Purβ (Purα + Purβ), or with an equivalent amount of nontargeting siRNA as a control. Western blot analysis of C2C12 myotube cytoplasmic extracts using anti-Purα and anti-Purβ specific antibodies revealed that siRNA directed against Purα, Purβ, or Purα + Purβ markedly reduced the endogenous levels of Purα and Purβ (Fig. 4C, lanes 1 and 3 versus lanes 2 and 4). In parallel experiments, treatment of C2C12 muscle cells with either Purα- or Purβ-specific siRNA led to 3.8- and 5.8-fold increases in β1285 wt luciferase reporter gene expression, while the simultaneous treatment with Purα and Purβ (Purα + Purβ)-specific siRNAs resulted in a 7.2-fold increase in β1258 wt luciferase reporter gene activity (Fig. 4D). These experiments provide clear evidence that Purα and Purβ act as negative regulators of βMyHC reporter gene expression in C2C12 myotubes and are consistent with the notion that increased expression of these proteins may repress βMyHC gene expression in the adult soleus muscle under NWB conditions.
Pur proteins are negative mediators of βMyHC reporter gene expression in C2C12 myotubes.
To further examine the functional significance of Pur protein binding to the βMyHC dβNRE-S element, we conducted transient-expression assays in which expression vectors for His-tagged Purα and Purβ were cotransfected with the wild-type β1285 wt luciferase reporter gene into C2C12 myoblasts (Fig. 5). Western blot analysis confirmed nuclear expression of the His-tagged Purα and Purβ proteins (Fig. 5A). The promoterless pGL3 basic luciferase plasmid was not highly expressed in C2C12 myotubes and did not exhibit regulated expression in response to increased Purα or Purβ expression (Fig. 5B). The basal expression level of the β1285 wt luciferase reporter gene in C2C12 myotubes was significantly higher than that of pGL3 basic luciferase (Fig. 5B). Expression levels of the β1285 wt luciferase reporter gene decreased significantly when the cells were cotransfected with Purα, Purβ, or Purα and Purβ expression vectors (Fig. 5B). These data show that Purα and Purβ are negative regulators of the 1,285-bp βMyHC promoter in C2C12 myotubes.
FIG. 5.
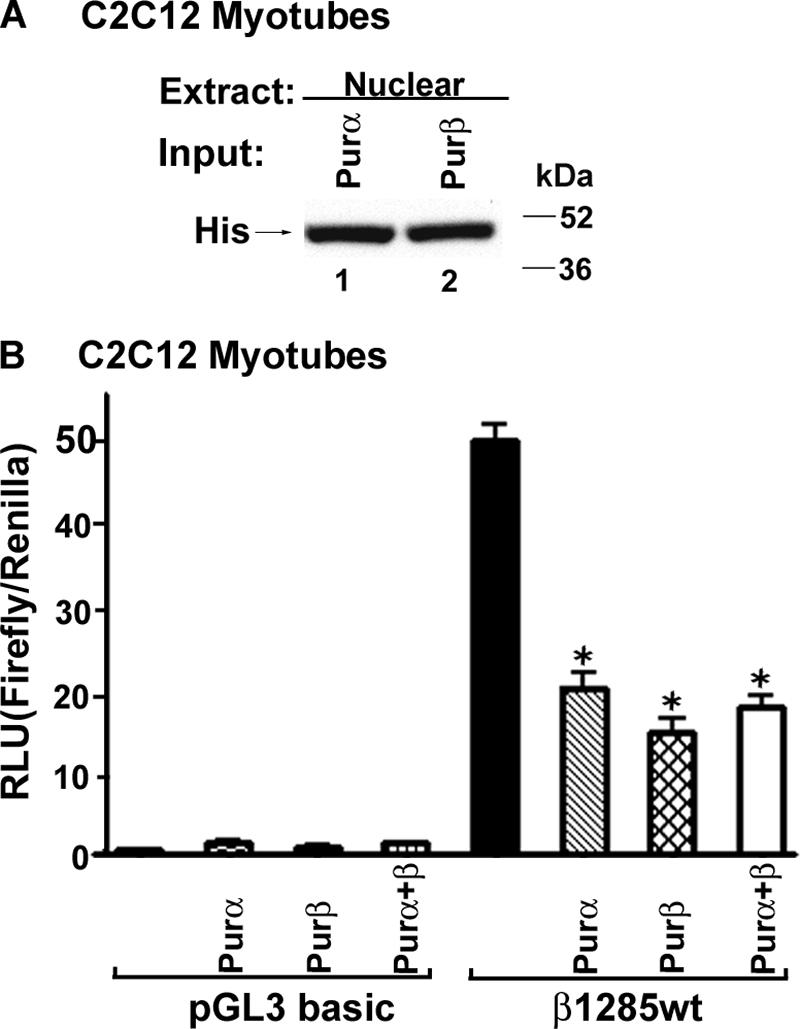
βMyHC promoter activity is decreased by ectopic expression of Purα or Purβ. (A) C2C12 myoblasts were transfected with the β1285 wt reporter gene (1 μg) and Purα (0.5 μg), Purβ (0.5 μg), or Purα and Purβ (0.5 μg total) expression plasmids. C2C12 myotube nuclear extracts were collected 48 h after transfection and assessed for His-tagged Purα or His-tagged Purβ expression by Western blotting. (B) Ectopic expression of Purα and Purβ significantly decreased βMyHC reporter gene activity in C2C12 myotubes. Forced expression of Purα and Purβ did not regulate the pGL3 basic plasmid. Data are reported as luciferase-normalized relative light units (RLU) (firefly/Renilla) and are expressed as the mean ± standard error (n = 8). *, P < 0.0001; all comparisons are against β1285 wt activity.
The βMyHC dβNRE-S element confers Pur-dependent expression on a heterologous promoter.
To determine whether the βMyHC dβNRE-S element could confer Pur protein-dependent expression on a heterologous promoter, three tandem copies were fused upstream of a minimal thymidine kinase (TK) promoter (Fig. 6A). Transient-expression assays using C2C12 muscle cells revealed that the pGL3-TK plasmid was not regulated by the concurrent cotransfection of Purα and Purβ expression vectors (Fig. 6B). However, the addition of three concatenated dβNRE-S elements to the minimal wild-type TK-luciferase reporter gene (TK-3x dβNRE-S wt) resulted in decreased expression, whereas mutation of the dβNRE-S elements (TK-3x dβNRE-Sm1) significantly up-regulated expression of the reporter gene in C2C12 myotubes (Fig. 6B). As expected, the concurrent cotransfection of Purα and Purβ expression vectors did not significantly decrease expression of the mutant TK-3x dβNRE-Sm1 reporter gene, confirming the specific actions of the dβNRE-S element (Fig. 6B). These experiments demonstrate that the βMyHC dβNRE-S element functions as a negative element that can confer Pur-dependent regulation on a heterologous promoter.
FIG. 6.

βMyHC dβNRE-S element confers Pur-dependent expression on a minimal TK promoter. (A) Schematic representation of wild-type (TK-3x dβNRE-S wt) and mutant (TK-3x dβNRE-Sm1) heterologous reporter genes. (B) C2C12 myoblasts were cotransfected with various combinations of either TK-3x dβNRE-S wt or TK-3x dβNRE-Sm1 reporter genes and Purα, Purβ, or Purα and Purβ expression plasmids and allowed to differentiate. Forty-eight hours later, C2C12 myotube cellular extracts were collected and assayed for luciferase activity. Data are reported as luciferase-normalized relative light units (RLU) (firefly/Renilla) and are expressed as the mean ± standard error (n = 8). *, P < 0.0001; all comparisons are against TK-3x-dβNRE-S wt activity. ns, not significant.
Ectopic and endogenously expressed Purα, Purβ, and Sp3 physically associate within C2C12 myotubes.
Our previous work has implicated the Sp3 proteins (115, 80, and 78 kDa) as negative regulators of βMyHC gene expression (28). To determine if Sp3 physically interacts with Purα and/or Purβ, we performed coimmunoprecipitation assays. In these experiments C2C12 myoblasts were cotransfected with His-tagged Purα and Sp3 (Purα + Sp3) or His-tagged Purβ and Sp3 (Purβ + Sp3), or with HA-tagged-Nrf2 as a negative control, and then allowed to differentiate. Western blot analysis using C2C12 myotube nuclear extract and anti-His antibody revealed that both anti-Purα antibody and anti-Sp3 antibody immunoprecipitated His-tagged Purα protein (Fig. 7A, lanes 1 and 2). Likewise, both anti-Purβ antibody and anti-Sp3 antibody precipitated His-tagged Purβ protein (Fig. 7B, lanes 1 and 2). In contrast, anti-HA antibody and IgG did not precipitate His-tagged Purα or Purβ protein (Fig. 7A and B, lanes 3 and 4). In parallel coimmunoprecipitation experiments, Western blot analysis using C2C12 myotube nuclear extracts and anti-Sp3 antibody revealed that anti-His antibody precipitated three proteins which displayed the same migration pattern as in vitro-synthesized Sp3 and endogenous Sp3 from nontransfected C2C12 muscle cells (Fig. 7C, lanes 1 and 2 versus lanes 3 and 4). In contrast, anti-His and IgG did not coprecipitate endogenous nuclear Sp3 from nontransfected C2C12 muscle cells (Fig. 7C, lanes 5 and 6).
FIG. 7.

Purα and Purβ associate with Sp3 in C2C12 myotubes. (A and B) C2C12 myoblasts were cotransfected simultaneously with His-tagged Purα and Sp3 (Purα + Sp3), His-tagged Purβ and Sp3 (Purβ + Sp3), His-tagged Purα and HA-Nrf2 (Purα + HA-Nrf2), or His-tagged Purβ and HA-Nrf2 (Purβ + HA-Nrf2) or with Purα and Purβ alone and then allowed to differentiate. Western blot analysis using an anti-His antibody (Ab) revealed that the anti-Sp3 antibody coprecipitated His-Purα and His-Purα but not HA-Nrf2. Neither IgG nor the anti->HA antibody could coimmunoprecipitated His-tagged Purα or Purβ. (C)> C2C12 myoblasts were cotransfect with His-tagged Purα and Sp3 (Purα + Sp3) or His-tagged Purβ and Sp3 (Purβ + Sp3) and allowed to differentiate. Western blot analysis using C2C12 myotube nuclear extract and an anti-Sp3 antibody revealed that the anti-His antibody coimmunoprecipitated three proteins which displayed the same migration pattern as in vitro-synthesized Sp3 and endogenous nuclear Sp3 from nontransfected C2C12 muscle cells. In contrast, anti-His antibody and IgG did not coimmunoprecipitate endogenous nuclear Sp3 from nontransfected C2C12 muscle cells. (D) Nuclear extract was obtained from C2C12 myotubes and used for immunoprecipitation (IP) of endogenous Sp3 with anti-Purα and anti-Purβ antibodies. Western blot analysis revealed that both anti-Purα and anti-Purβ antibodies immunoprecipitated a protein which displayed the same migration pattern as in vitro-synthesized Sp3 (lane 1 versus lanes 2 and 3). In contrast, IgG did not immunoprecipitate endogenous nuclear Sp3 from C2C12 myotube nuclear extract (lane 4).
We next wished to determine whether endogenously expressed Purα, Purβ, and Sp3 physically associate within C2C12 myotubes. For these experiments, we isolated nuclear extract from differentiated C2C12 muscle cells and performed an immunoprecipitation assay using either anti-Purα or anti-Purβ antibody or IgG. Western blot evaluation of the precipitated material using anti-Sp3 antibody revealed that anti-Purα and anti-Purβ antibodies precipitated a protein (≈115 kDa) which displayed the same migration pattern as in vitro-synthesized Sp3 (Fig. 7D, lane 1 versus lanes 2 and 3). In contrast, IgG did not precipitate endogenous nuclear Sp3 from nontransfected C2C12 muscle cells (Fig. 7D, lane 4). Taken together, these experiments provide evidence that endogenous Purα, Purβ, and Sp3 physically interact within the nuclear compartment of C2C12 myotubes.
Purα, Purβ, and Sp3 collaborate to down-regulate βMyHC reporter gene expression in C2C12 myotubes.
To determine if Purα, Purβ, and Sp3 collaborate to repress βMyHC gene expression, we performed transient-coexpression studies using C2C12 muscle cells and various combinations of Sp3 with Purα, Purβ, or Purα and Purβ. In transient-expression studies, ectopic expression of Sp3 decreased β1285 wt luciferase reporter gene activity compared to basal β1285 wt expression levels (Fig. 8A). Importantly, a further decrease in β1285 wt reporter gene expression was measured when Sp3 was coexpressed with either Purα or Purβ (Fig. 8A), and these decreases were greater than those measured when either Purα, Purβ, or Sp3 was expressed independently (Fig. 8A).
FIG. 8.
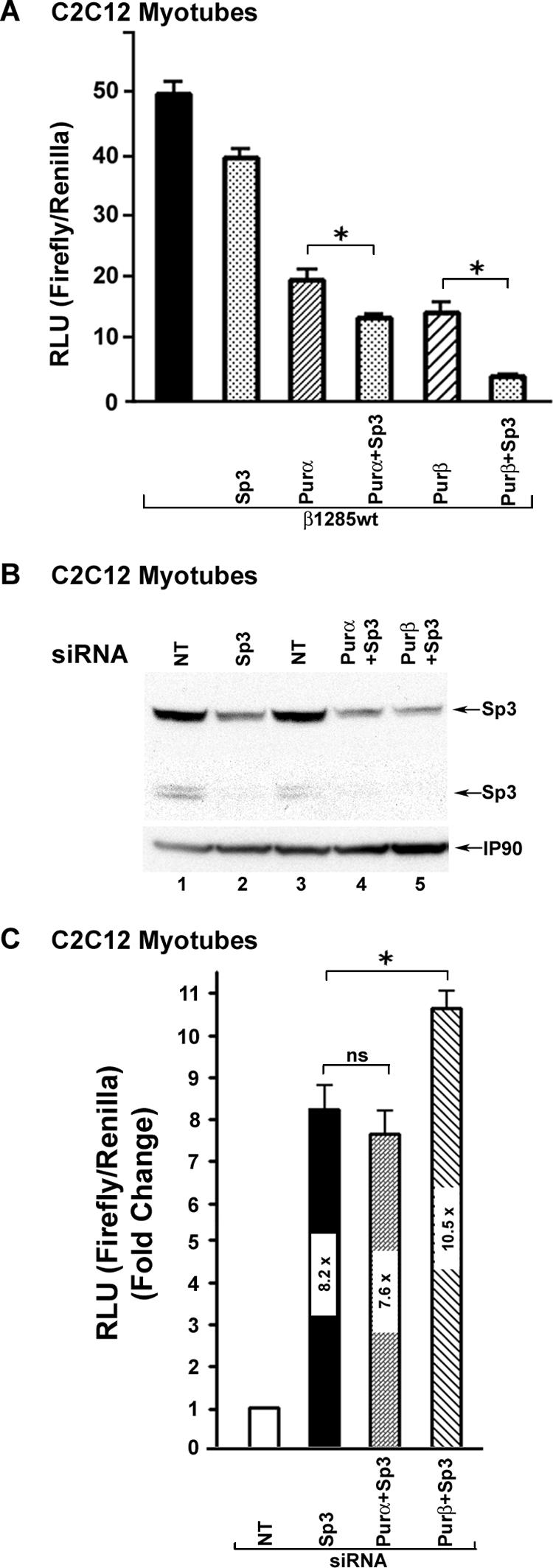
Sp3 collaborates with Purα and Purβ to repress βMyHC reporter gene expression. (A) C2C12 myoblasts were transfected with the 1,285-bp βMyHC Luc reporter gene (β1285 wt; 1 μg) and Purα (0.5 μg), Purβ (0.5 μg), Purα and Sp3 (0.5 μg total) or Purβ and Sp3 (0.5 μg total) expression plasmids. C2C12 myotube nuclear extract was collected 48 h after transfection and assessed for luciferase activity. Data are reported as luciferase-normalized relative light units (RLU) (firefly/Renilla) and are expressed as the mean ± standard error (n = 8). *, P < 0.05 (Purα versus Purα + Sp3 and Purβ versus Purβ + Sp3). All comparisons against β1285 wt activity P, < 0.0001 (asterisk not shown). (B) C2C12 myoblasts were transfected with control siRNA (nontargeting [NT]), or siRNA targeting Sp3, Sp3 and Purα, or Sp3 and Purβ and were allowed to differentiate. Cell lysates were harvested 48 h after transfection and assessed for endogenous Sp3 levels by Western blotting. (C) Knockdown of endogenous Sp3 protein resulted in increased expression of the β1285 wt reporter gene in C2C12 myotubes, and further increases were seen when Sp3 and Purβ siRNAs were cotransfected. IP90 represents a loading control. Data are reported as luciferase-normalized RLU (firefly/Renilla) and are expressed as the mean ± standard error (n = 3). *, P < 0.05 (β1285 wt activity in the presence of Sp3 siRNA versus Sp3 + Purβ siRNA); P < 0.0001 for all comparisons against β1285 wt activity in the presence of NT siRNA (asterisk not shown).
Next, we examined whether transient knockdown of endogenous Sp3 protein levels by using siRNA would result in increased βMyHC reporter gene expression. In these experiments, C2C12 myoblasts were transfected with siRNAs specific for Sp3, Sp3 + Purα, or Sp3 + Purβ or with an equivalent amount of nontargeting siRNA as a control. Western blot analysis of C2C12 myotube nuclear extract using anti-Sp3 specific antibody revealed that siRNA against Sp3, Sp3 + Purα, or Sp3 + Purβ markedly reduced the endogenous levels of the Sp3 proteins (115, 80, and 78 kDa) compared to those in nuclear extracts isolated from C2C12 muscle cells treated with control nontargeting siRNA (Fig. 8B, lanes 2, 4, and 5 versus lanes 1 and 3). Transient-expression assays revealed that treatment of C2C12 muscle cells with Sp3-specific siRNA led to an 8.2-fold increase in β1285 wt luciferase reporter gene expression, while the simultaneous treatment with siRNAs specific for Sp3 and Purα (Sp3 + Purα) or Sp3 and Purβ (Sp3 + Purβ) resulted in 7.6- and 10.5-fold increases in β1258 wt luciferase reporter gene activity (Fig. 8C). These data show that Sp3, Purα, and Purβ can collaborate to mediate repression of the 1,285-bp βMyHC promoter in C2C12 myotubes.
ChIP assays reveal βMyHC proximal promoter cis element-specific in vivo binding of Purα, Purβ, and Sp3.
We have demonstrated that both ectopic and endogenously expressed Purα, Purβ, and Sp3 can physically associate (Fig. 7D). In addition, we have shown that ectopically expressed Purα, Purβ, and Sp3 collaborate to negatively regulate βMyHC reporter gene expression (8A to C). To directly determine whether Purα and/or Purβ binds to the βMyHC dβNRE-S element and whether Sp3 binds to the βMyHC C-rich (A to C) elements in vivo, we performed ChIP assays on chromatin prepared from C2C12 myotubes (Fig. 9). Oligonucleotide primers designed to amplify the mouseβMyHC proximal promoter region harboring either the dβNRE-S or C-rich elements were used for PCR on DNA purified after chromatin immunoprecipitation. In PCRs, sheared genomic DNA template served as a positive control, while water without template DNA served as a negative control (Fig. 9, lanes 1 and 2). Immunoprecipitation reactions with no antibody, mouse anti-HA antibody, or mouse IgG were used as controls for nonspecific immunoprecipitations (Fig. 9, lanes 3 to 5). The βMyHC proximal promoter region was amplified from immunoprecipitation reactions when anti-Purα, anti-Purβ, or anti-Sp3 antibodies were used but was not amplified in the control reactions (Fig. 9, lanes 6 to 8 versus lanes 3 to 5). This result demonstrates that both Pur proteins and Sp3 bind to the βMyHC proximal promoter region. Collectively, our experiments provide evidence consistent with the notion that Purα, Purβ, and Sp3 occupy their cognate binding elements in vivo and that these transcription factors cooperate to negatively regulate chromosomally located βMyHC gene expression.
FIG. 9.

Purα, Purβ, and Sp3 bind to the βMyHC proximal promoter region in vivo. (A) ChIP assay demonstrating that Purα, Purβ, and Sp3 associate with the βMyHC proximal promoter region in vivo. ChIP assays were performed on chromatin prepared from C2C12 myotubes. Two distinct sets of oligonucleotide primers to amplify the mouse βMyHC proximal promoter region harboring either the dβNRE-S or C-rich elements were used for PCR on DNA purified after chromatin immunoprecipitation (IP). Immunoprecipitation reactions using no antibody (no Ab), mouse anti-HA, or mouse IgG were used as controls for nonspecific immunoprecipitations. PCRs with sheared genomic DNA template (input) served as positive controls, while water without template DNA served as a negative control (lane 2). The ChIP patterns obtained on C2C12 myotube chromatin when using anti-Purα or anti-Purβ antibody demonstrate that Purα and Purβ were associated with the endogenous βMyHC dβNRE-S element, while the use of anti-Sp3 antibody revealed that Sp3 was associated with the endogenous βMyHC C-rich sites). (B) Schematic of the βMyHC proximal promoter region. Arrows represent the specific PCR primer sets used to amplify the βMyHC proximal promoter dβNRE-S and C-rich sites on DNA purified after chromatin immunoprecipitation. Because the dβNRE-S and C-rich elements are separated by only 84 nucleotides, sheared genomic DNA fragments (ranging from 250 to 400 nucleotides) containing both the dβNRE-S and C-rich elements were amplified from IP reactions using either anti-Purα, anti-Purβ, or anti-Sp3 antibodies.
DISCUSSION
Adult skeletal muscle retains the ability to adapt its phenotype in accordance to altered load-bearing conditions. This adaptive response is readily measured in the non-weight-bearing slow-twitch soleus muscle, which undergoes a slow-to-fast fiber type transition that is underscored by a marked decrease in βMyHC gene expression (3, 20, 21, 28-30). Nevertheless, little is known about the identities, abundances, or activities of specific transcription factors that mediate skeletal muscle fiber type shifts in response to altered states of muscle activity. This study reports the novel finding that Purα and Purβ mediate repression of βMyHC gene expression by directly binding to the single-stranded dβNRE-S as demonstrated by EMSA and ChIP experiments. Moreover, our experiments show that endogenous Purα and Purβ physically associate with Sp3 and that Purα, Purβ, and Sp3 cooperatively mediate a decrease in βMyHC gene expression (Fig. 1 to 9). These data support the notion that interactions between Purα, Purβ, and Sp3 are important determinants for skeletal muscle fiber type switches in response to NWB conditions.
A new biological role for the multifunctional Pur proteins.
In both humans and mice, Purα and Purβ function as sequence-specific single-stranded DNA and RNA binding proteins that are encoded by distinct genes (PURA and PURB) and are broadly expressed in adult tissues (8, 11, 17). Although there are differences in amino acid composition between these two proteins, they both contain a highly conserved, centrally located DNA binding domain (8, 11). Both Purα and Purβ specifically recognize single-stranded, purine-rich cis-acting elements with the consensus (GGN)n, where N is not a G. An interesting feature of Purα and Purβ is their ability to unwind DNA in an ATP-independent manner (6, 11, 39), which is likely to contribute to their participation in diverse cellular processes (8, 11, 12, 17, 35). Despite the observation that both Purα and Purβ are expressed in adult skeletal muscle, their regulatory role in skeletal muscle function has never been investigated. Our experiments indicate that Purα and Purβ serve a gene regulatory role in transcriptional reprogramming of skeletal muscle under NWB conditions.
Purα and Purβ are cognate βMyHC dβNRE-S element binding factors that repress βMyHC gene expression.
Our previous transgenic and protein-DNA interaction studies have provided evidence that the βMyHC dβNRE-S element (−332 to −311) acts as a repressor of βMyHC gene expression during NWB (21). In this report, we have provided multiple lines of evidence that Purα and Purβ represent the cognate dβNRE-S element binding factors and that Purα and Purβ contribute to decreased βMyHC gene transcription in response to NWB. First, shift Southwestern analysis detected two proteins that ranged from 45 to 50 kDa, which is consistent with the apparent sizes (46 and 44 kDa) of Purα and Purβ, respectively (10, 16). Second, competition and antibody supershift EMSA analyses convincingly demonstrated that the enriched nuclear protein-dβNRE-S binding complex that formed when using non-weight-bearing soleus nuclear extract was comprised exclusively of Purα and Purβ proteins. Third, chromatin immunoprecipitation assays demonstrated that the Pur proteins interact directly with the βMyHC proximal promoter dβNRE-S element within the chromatin context. Fourth, the forced expression of both Purα and Purβ in C2C12 muscle cells resulted in decreased expression of a 1,285-bp βMyHC reporter gene and a minimal TK promoter fused to multiple copies of the dβNRE-S element (TK-3x dβNRE-S wt). Importantly, mutation of the dβNRE-S element within either the 1,285-bp βMyHC reporter gene or the heterologous promoter construct (TK-3x dβNRE-Sm1) resulted in increased expression, and negative regulation of the heterologous reporter gene was not restored by the forced expression of Purα and Purβ. Finally, in C2C12 muscle cells, Purα- and Purβ-specific siRNAs resulted in a qualitative decrease in endogenous Purα and Purβ protein levels and a concurrent 3.8- to 7.2-fold increase in βMyHC reporter gene expression. Our findings that Purα and Purβ act as repressors of gene transcription are consistent with those of Knapp et al. (18), who demonstrated by RNA interference that Purα and Purβ act as negative regulators of the smooth muscle α-actin promoter in cultured fibroblasts. Moreover, several other studies have provided experimental evidence consistent with a negative transcriptional role for the Pur proteins (8, 10, 11, 14, 18).
Sp3 collaborates with Purα and Purβ to negatively regulate βMyHC reporter gene activity in C2C12 myotubes.
Our previous work has provided evidence that increased binding of Sp3 to three highly conserved and closely spaced βMyHC proximal promoter GC-rich elements is a critical event for down-regulation of βMyHC gene expression under NWB conditions (Fig. 1))28). When those results are considered with our current data showing that Purα and Purβ mediate decreased βMyHC gene transcription by directly binding to the βMyHC dβNRE-S element, it is reasonable to consider that Purα, Purβ, and Sp3 form a nucleoprotein complex that favors decreased βMyHC gene expression under NWB conditions. Consistent with this notion, our coimmunoprecipitation assays demonstrated physical interactions between Purα, Purβ, and Sp3 in nuclear extracts isolated from C2C12 myotubes, while our chromatin immunoprecipitation assays revealed that Purα, Purβ, and Sp3 bind to the βMyHC proximal promoter region.
The concept of a collaborative functionality between Sp3, Purα, and Purβ is further supported by our transient-cotransfection experiments, in which coexpression of Sp3 with either Purα or Purβ resulted in greater reduction in βMyHC reporter gene expression than was achieved with expression of the individual proteins. Furthermore, a simultaneous decrease in expression of Sp3 and the Pur proteins by siRNA resulted in elevated expression of the βMyHC reporter gene, compared to siRNA-mediated decreases in expression of the individual proteins.
Collectively, our data support three possible mechanisms that could account for a collaborative interaction between the Pur proteins and Sp3 to negatively regulate expression of the βMyHC promoter. First, Purα, Purβ, and Sp3 bind in a linear manner to their cognate elements and exert a negative influence on the transcription initiation complex. Second, Purα, Purβ, and Sp3 can indirectly associate with DNA by interaction with another DNA binding protein via protein-protein interactions. For example, in our study Purα and Purβ would interact indirectly with the C-rich elements by association with bound Sp3, and Sp3 could interact with the dβNRE-S element by association with bound Purα and Purβ. Third, Purα, Purβ and Sp3 bind to their cognate elements and interact with each other due to strand separation and looping of the Pur binding site (dβNRE-S). The last mechanism is feasible since the Pur proteins have been shown to unwind duplex DNA in an ATP-independent manner (6, 11, 39). It should be noted that while our data do not specify one of these molecular mechanisms, they do not need to operate in a mutually exclusive manner.
Purα and Purβ may serve a functional role in skeletal muscle fiber type gene expression.
We propose that the Sp and Pur family of proteins function as part of a complex gene regulatory network that responds to various levels of skeletal muscle activity. In terms of cellular remodeling, our current study and previous studies have shown that nuclear Sp3 and Pur protein levels and DNA binding activity increase in response to non-weight-bearing conditions and decrease in response to mechanical overload. Although our experiments are limited to the analysis of βMyHC gene expression in skeletal muscle, the combined effects of Pur-Sp3-mediated repression of gene expression may be more broadly relevant, since the Pur and Sp3 proteins are widely expressed across tissues. For example, Purα and Purβ have recently been shown to regulate smooth muscle α-actin in fibroblast and vascular smooth muscle cells by a mechanism involving both sequence-specific single-stranded DNA binding and cell type-dependent protein-protein interactions (see reference 18 and references therein). Moreover, the levels of both Purα and Purβ were shown to increase in the failing heart and to participate in regulation of αMyHC gene transcription and translation in cardiac myocytes (10). Additional work will be necessary to determine the scope of collaborative Pur-Sp3 interaction for regulation of gene expression in response to various physiological and/or pathological stimuli. A better understanding of the transcriptional mechanisms that are activated during skeletal muscle inactivity is critical to the development of novel drug targets aimed at halting the deleterious effects of various disease states on skeletal muscle mass and function.
Acknowledgments
This work was supported by Public Health Service grants AR41464 and AR47197 (to R.T.) from the National Institute of Arthritis and Musculoskeletal and Skin Disease and by grant HL54281 (to R.J.K.).
We thank Mark Hannink for critical review of the manuscript.
This paper is in loving memory of Gretchen L. Tsika.
Footnotes
Published ahead of print on 4 December 2006.
REFERENCES
- 1.Acakpo-Satchivi, I., W. Edelmann, C. A. Sartorius, B. D. Lu, P. A. Wahr, S. C. Watkins, J. M. Metzger, L. Leinwand, and R. Kucherlapati. 1997. Growth and muscle defects in mice lacking adult myosin heavy chain genes. J. Cell Biol. 139:1219-1229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Barany, M. 1967. ATPase activity of myosin correlated with speed of muscle shortening. J. Gen. Physiol. 5:197-218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Booth, F. W., and K. M. Baldwin. 1996. Muscle plasticity: energy demand and supply processes, p. 1075-1123. In L. B. Rowell and J. T. Shepard (ed.), Handbook of physiology. Exercise: regulation and integration of multiple systems. American Physiological Society, Bethesda. MD.
- 4.Bradford, M. M. 1976. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 72:248-254. [DOI] [PubMed] [Google Scholar]
- 5.Carlini, L. E., M. J. Getz, A. R. Strauch, and R. J. Kelm. 2002. Cryptic MCAT enhancer regulation in fibroblast and smooth muscle cells. J. Biol. Chem. 277:8682-8692. [DOI] [PubMed] [Google Scholar]
- 6.Darbinian, N., G. L. Gallia, and K. Khalili. 2001. Helix-destabilizing properties of the human single-stranded DNA- and RNA-binding protein Purα. J. Cell Biochem. 80:589-595. [PubMed] [Google Scholar]
- 7.Flink, I., and E. Morkin. 1995. Alternative processed isoforms of cellular nucleic acid-binding protein interact with a suppressor region of the human βMyHC. J. Biol. Chem. 270:6959-6965. [DOI] [PubMed] [Google Scholar]
- 8.Gallia, G. L., E. M. Johnson, and K. Khalili. 2000. Purα, a multifunctional single-stranded DNA- and RNA-binding protein. Nucleic Acids Res. 28:3197-3205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gore, M. T., T. Sumbandam, and P. K. Umeda. 1993. A Y-box DNA binding protein associates with the thyroid hormone binding region of the rabbit βMyHC promoter. Circ. Suppl. 88:1261. [Google Scholar]
- 10.Gupta, M., V. Sueblinvong, J. Raman, V. Jeevanandam, M. P. Gupta. 2003. Single-stranded DNA-binding proteins Purα and Purβ bind to a purine-rich negative regulatory element of the α-myosin heavy chain gene and control transcriptional and translational regulation of the gene expression. J. Biol. Chem. 278:44935-44948. [DOI] [PubMed] [Google Scholar]
- 11.Johnson, E. M. 2003. The Pur protein family: clues to function from recent studies on cancer and AIDS. Anticancer Res. 23:2093-2100. [PubMed] [Google Scholar]
- 12.Kanai, Y., N. Dohmae, and N. Hirokawa. 2004. Kinesin transports RNA: isolation and characterization of an RNA-transporting granule. Neuron 43:513-525. [DOI] [PubMed] [Google Scholar]
- 13.Karasseva, N., G. L. Tsika, J. Ji, A. Zhang, X. Mao, and R. W. Tsika. 2003. Transcription enhancer factor 1 binds multiple MEF2 and A/T-rich elements during fast-to-slow skeletal muscle fiber type transitions. Mol. Cell Biol. 23:5143-5164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kelm, R. J., Jr., S. X. Wang, J. A. Polikandriotis, and A. R. Strauch. 2003. Structure/function analysis of mouse purβ, a single-stranded DNA-binding repressor of vascular smooth muscle α-actin gene transcription. J. Biol. Chem. 278:38749-38757. [DOI] [PubMed] [Google Scholar]
- 15.Kelm, R. J., Jr., J. G. Cogan, P. K. Elder, A. R. Strauch, and M. J. Getz. 1999. Molecular interactions between single-stranded DNA-binding proteins associated with an essential MCAT element in the mouse smooth muscle α-actin. J. Biol. Chem. 274:14238-14245. [DOI] [PubMed] [Google Scholar]
- 16.Kelm, R. J., Jr., P. K. Elder, A. R. Strauch, and M. J. Getz. 1997. Sequence of cDNAs encoding components of vascular actin single-stranded DNA-binding factor 2 establish identity to Purα and Purβ. J. Biol. Chem. 272:26727-26733. [DOI] [PubMed] [Google Scholar]
- 17.Khalili, K., L. D. Valle, V. Muralidharan, W. J. Gault, N. Darbinian, J. Otte, E. Meier, E. M. Johnson, D. C. Daniel, Y. Kinosshita, S. Amini, and J. Gordon. 2003. Purα is essential for postnatal brain development and developmentally coupled cellular proliferation as revealed by genetic inactivation in the mouse. Mol. Cell. Biol. 23:6857-6875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Knapp, A. M., J. E. Ramsey, S.-X. Wang, K. E. Godburn, A. R. Strauch, and R. J. Kelm, Jr. 2006. Nucleoprotein interactions governing cell cycle type-dependent repression of the mouse smooth muscle α-actin promoter by single-stranded DNA-binding Purα and Purβ. 281:7907-7918. [DOI] [PubMed]
- 19.Knotts, S. H. Rindt, J. Neumann, and J. Robbins. 1994. In vivo regulation of the mouse β myosin heavy chain gene. 269:31275-31282. [PubMed]
- 20.McCarthy, J. J., A. M. Fox, G. L. Tsika, L. Gao, and R. W. Tsika. 1997. β-MHC transgene expression in suspended and mechanically overloaded/suspended soleus muscle of transgenic mice. Am. J. Physiol. 272:R1552-R1561. [DOI] [PubMed] [Google Scholar]
- 21.McCarthy, J. J., D. R. Vyas, G. L. Tsika, and R. W. Tsika. 1999. Segregated regulatory elements direct β-myosin heavy chain expression in response to altered muscle activity. J. Biol. Chem. 274:14270-14279. [DOI] [PubMed] [Google Scholar]
- 22.Parsons, S. A., S. A. D. P. Millary, B. J. Wilkins, O. F. Bueno, G. L. Tsika, J. R. Neilson, C. M. Liberatore, K. E. Yutzey, G. R. Crabtree, R. W. Tsika, and J. D. Molkentin. 2004. Genetic loss of calcineurin blocks mechanical overload-induced skeletal muscle fiber-type switching but not hypertrophy. J. Biol. Chem. 279:26192-26200. [DOI] [PubMed] [Google Scholar]
- 23.Rindt, H., J. Gulick, and J. Robbins. 1995. Segregation of cardiac and skeletal muscle-specific regulatory elements of the β-myosin heavy chain gene. Proc. Natl. Acad. Sci. USA 92:1540-1544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Roy, R. R., K. M. Baldwin, and V. R. Edgerton. 1991. The plasticity of skeletal muscle: effects of neuromuscular activity. Exercise Sport Sci. Rev. 19:269-313. [PubMed] [Google Scholar]
- 25.Sartorius, C. A., B. D. Lu, I. Acakpo-Satchivi, R. P. Jacobsen, W. C. Bynes, and L. Leinwand. 1998. Myosin heavy chains IIa and IId are functionally distinct in the mouse. J. Cell Biol. 141:943-953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schiaffino, S., and A. L. Serrano. 1996. Molecular diversity of myofibrillar proteins: gene regulation and functional significance. Physiol. Rev. 76:371-432. [DOI] [PubMed] [Google Scholar]
- 27.Tsika, G. L., J. L. Wiedenman, L. Gao, J. J. McCarthy, K. Sheriff-Carter, I. D. Rivera-Rivera, and R. W. Tsika. 1996. Induction of β-MHC transgene in overloaded skeletal muscle is not eliminated by mutation of conserved elements. Am. J. Physiol. 271:C690-C699. [DOI] [PubMed] [Google Scholar]
- 28.Tsika, G., J. Ji, and R. Tsika. 2004. Sp3 proteins negatively regulate βMyHC gene expression during skeletal muscle inactivity. Mol. Cell. Biol. 24:10777-10791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tsika, R. 2006. The muscular system: the control of muscle mass, p. 161-177. In C. M. Tipton (ed.), Graduate text in exercise physiology. Williams & Wilkins, Baltimore, MD.
- 30.Tsika, R. W., J. J. McCarthy, N. Karasseva, Y. Ou, and G. L. Tsika. 2002. Divergence in species and regulatory role of β-myosin heavy chain proximal promoter muscle-CAT elements. Am. J. Physiol. Cell Physiol. 283:C1761-C1775. [DOI] [PubMed] [Google Scholar]
- 31.Tsika, R. W., S. D. Hauschka, and L. Gao. 1995. M-creatine kinase gene expressionin mechanically overloaded skeletal muscle of transgenic mice. Am. J. Physiol. 269:C665-C674. [DOI] [PubMed] [Google Scholar]
- 32.Vyas, D. R., J. J. McCarthy, and R. W. Tsika. 1999. Nuclear protein binding at the βMyHC A/T-rich element is enriched following increased skeletal muscle activity. J. Biol. Chem. 274:30832-30842. [DOI] [PubMed] [Google Scholar]
- 33.Vyas, D. R., J. J. McCarthy, G. L. Tsika, and R. W. Tsika. 2001. Multiprotein complex formation at the β-myosin heavy chain distal muscle CAT element correlates with slow muscle expression but not mechanical overload responsiveness. J. Biol. Chem. 276:1173-1184. [DOI] [PubMed] [Google Scholar]
- 34.Vyas, D. R., J. J. McCarthy, G. L. Tsika, and R. W. Tsika. 2000. Dissimilar nuclear protein binding at human β-myosin heavy chain proximal and distal MCAT elements in response to increased skeletal muscle activity. Basic Appl. Myol. 10:5-16. [Google Scholar]
- 35.Wang, S. X., P. K. Elder, Y. Zhang, A. R. Strauch, and R. J. Kelm, Jr. 2005. Cell cycle-mediated regulation of smooth muscle α-actin gene transcription in fibroblasts and vascular smooth muscle cells involves multiple adenovirus E1A-interacting cofactors. J. Biol. Chem. 280:6204-6214. [DOI] [PubMed] [Google Scholar]
- 36.Weiss, A., L. A. Leinwand. 1996. The mammalian myosin heavy chain gene family. Annu. Rev. Cell Dev. Biol. 12:417-439. [DOI] [PubMed] [Google Scholar]
- 37.Wiedenman, J. L., G. L. Tsika, L. Gao, J. J. McCarthy, I. D. Rivera-Rivera, D. Vyas, K. Sheriff-Carter, and R. W. Tsika. 1996. Muscle-specific and inducible expression of 293-base pair β-myosin heavy chain promoter in transgenic mice. Am. J. Physiol. 271:R688-R695. [DOI] [PubMed] [Google Scholar]
- 38.Wiedenman, J. L., I. Rivera-Rivera, D. Vyas, G. Tsika, L. Gao, K. Sheriff-Carter, X. Wang, L. Y. Kwan, and R. W. Tsika. 1996. βMHC and SMLC1 transgene induction in overloaded skeletal muscle of transgenic mice. Am. J. Physiol. 270:C1111-C1121. [DOI] [PubMed] [Google Scholar]
- 39.Wortman, J., E. M. Johnson, and A. D. Bergemann. 2005. Mechanism of DNA binding and localized strand separation by Purα and comparison with Pur family member, Purβ. Biochim. Biophys. Acta 1743:64-78. [DOI] [PubMed] [Google Scholar]