

Abstract
Mutations in the CSB gene cause Cockayne syndrome (CS), a DNA repair disorder characterized by UV sensitivity and severe physical and neurological impairment. CSB functions in the transcription-coupled repair subpathway of nucleotide excision repair. This function may explain the UV sensitivity but hardly clarifies the other CS symptoms. Many of these, including retinopathy, are associated with premature aging. We studied eye pathology in a mouse model for CS. Csbm/m mice were hypersensitive to UV light and developed epithelial hyperplasia and squamous cell carcinomas in the cornea, which underscores the importance of transcription-coupled repair of photolesions in the mouse. In addition, we observed a spontaneous loss of retinal photoreceptor cells with age in the Csbm/m retina, resulting in a 60% decrease in the number of rods by the age of 18 months. Importantly, when Csbm/m mice (as well as Csa−/− mice) were exposed to 10 Gy of ionizing radiation, we noticed an increase in apoptotic photoreceptor cells, which was not observed in wild-type animals. This finding, together with our observation that the expression of established oxidative stress marker genes is upregulated in the Csbm/m retina, suggests that (endogenous) oxidative DNA lesions play a role in this CS-specific premature-aging feature and supports the oxidative DNA damage theory of aging.
Cockayne syndrome (CS) (complementation groups A and B) is a rare autosomal recessive DNA repair disorder characterized by photosensitive skin and severely impaired physical and intellectual development (38). Patients generally show a postnatal growth defect (leading to cachectic dwarfism) and develop skeletal abnormalities such as a bird-like face (sunken eyes and a beaked nose), kyphosis, and, in older patients, osteoporosis. Characteristic neurological features are delayed psychomotor development, microcephaly, disturbed gait, ataxia, sensorineuronal hearing loss, and pigmentary retinopathy. Other CS features include dental caries, impaired sexual development, and cataract. The mean age of death in reported cases is 12.5 years (although patients as old as 55 years have been described), with the most common cause of death being pneumonia resulting from general atrophy and cachexia. Many of the CS features are progressive and resemble premature aging, for which reason CS is regarded a progeroid syndrome (34). In contrast to other photosensitive DNA repair disorders (i.e., xeroderma pigmentosum [XP]), CS is not associated with increased UV-induced skin cancer proneness.
CS originates from mutations in the CSA or CSB gene, which encode components of the nucleotide excision repair (NER) pathway. This DNA repair mechanism removes a wide variety of helix-distorting DNA lesions from the genome, including UV-induced cross-links between adjacent pyrimidines (24). The repair process involves the concerted action of more than 25 proteins that sequentially recognize the damaged nucleotide, locally unwind the helix, excise a 22- to 31-mer oligonucleotide containing the damage, and fill in the gap by DNA synthesis and ligation (13, 31). Two subpathways of NER are recognized: (i) global genome NER (GG-NER), which scans and repairs damage throughout the entire genome, which is thought to serve mainly to prevent mutations, and (ii) transcription-coupled NER (TC-NER), which selectively repairs lesions in the transcribed strand of active genes and thus may enable the cell to quickly resume transcription and prevent cell death (21). Cells from CS-A and CS-B patients are specifically defective in TC-NER, while GG-NER remains functional (49, 50). This may well provide an explanation for the cutaneous UV sensitivity and absence of skin cancer predisposition. However, the partial NER defect hardly clarifies the etiology of the other CS symptoms, since these features are absent in the related syndrome XP, even in those XP patients that completely lack both NER subpathways (XP-A patients). The CS-specific features may be attributed to other roles of CSA and CSB proteins, outside the context of NER. There are indications that these proteins are involved in transcription elongation (3, 18, 30, 42) and in the repair of other, non-NER types of DNA damage, e.g., oxidative DNA lesions (15-17, 39, 43, 45).
To facilitate experimental research on the etiology of CS, we have previously generated animal models for CS-A and CS-B by complete inactivation of the mouse Csa gene (Csa−/− mice) (47) and by mimicking a specific truncation (as found in CS-B patient CS1AN) in the mouse Csb gene (Csbm/m mice) (48). Both mouse models display a specific TC-NER defect and show increased photosensitivity of the skin (47, 48). In contrast to human CS patients, Csa−/− and Csbm/m mice display modest skin cancer susceptibility, which becomes apparent after chronic exposure of animals to daily doses of UV light (4, 48). This paradox is well explained by the fact that UV-induced cyclobutane pyrimidine dimers are the major causative lesion for UV skin cancer (27) and that rodents lack efficient GG-NER for the removal of this lesion from the DNA and thus rely primarily on TC-NER (5). Other CS features, like growth failure and neurological abnormalities, are present only in a mild form (47, 48). Interestingly, complete inactivation of NER (by concurrent inactivation of the Xpa gene) in the Csbm/m mouse dramatically aggravates the CS features (including premature aging). Csbm/m/Xpa−/− double mutant animals display dramatic postnatal growth retardation, kyphosis, ataxia, abnormal locomotor activity, and progressive weight loss and die before weaning (37). In view of the premature-aging features in DNA repair disorders and elaborating on Harman's “free-radical theory of aging” (22), we recently postulated that aging can result from (oxidative) DNA lesions that interfere with transcription and/or replication and cause cell death and/or cellular senescence, ultimately leading to the loss of tissue homeostasis and the onset of age-related disease (23, 35). According to this theory, the oxidation of DNA by endogenous free radicals, generated as a by-product of cellular metabolism, plays an important role in the etiology of the CS-specific features, notably premature aging. In line with this, Csbm/m mouse embryonic fibroblasts are susceptible to oxidative damage-producing agents like ionizing radiation (IR), which points to a defect in the repair of oxidative DNA lesions in addition to a TC-NER deficiency (16).
The present study addresses the eye pathology of CS, which includes photosensitivity and pigmentary retinopathy. The latter feature was first described by Cockayne in 1936 (9) and has since been considered a hallmark of the disease. Moreover, it has been reported in at least 55% of previously published cases (38). The CS retinopathy is one of the as-yet-unexplained CS-specific features that suggest that accelerated aging occurs in CS. Here, we show that the defect in the Csbm/m mouse not only causes corneal UV sensitivity and cancer susceptibility but also predisposes for spontaneous retinal degeneration. Importantly, we show that the Csbm/m mouse retina is hypersensitive to ionizing radiation, which suggests that oxidative lesions are at the basis of this premature-aging phenotype.
MATERIALS AND METHODS
Mice.
The generation and PCR-mediated genotyping of Csbm/m and Csa−/− mice has been described previously (4, 47, 48). Except for the chronic UV exposure study (performed with animals in an FVB and 129ola hybrid genetic background), experiments were performed with animals in a C57BL/6J background. Animals were housed in a controlled environment (food and water ad libitum, cycle of 12 h light-12 h dark, light intensity of 70 to 90 lx) at the Animal Resource Center (Erasmus University Medical Center), which operates in compliance with the Animal Welfare Act of the Dutch government. All animal studies were approved by an independent animal ethical committee (Dutch equivalent of the IACUC).
Chronic exposure to UV-B light.
Whole-body UV exposure studies were performed according to an incremental-dose protocol as described previously (48). In brief, nine Csbm/m, seven Csb+/m, and seven Csb+/+ mice (age, 20 weeks) received an initial daily dose of 100 J/m2 UV-B (250 to 400 nm; American Philips F40 sunlamps; dose rate, 8.3 J/m2/min), gradually increasing up to 250 J/m2. Animals were thoroughly screened twice a week for the occurrence of skin and eye abnormalities. Mice that developed tumors were sacrificed by CO2 inhalation and subsequent cervical dislocation. Eyes were isolated and processed for histopathological examination as described below. After 38 weeks (cumulative dose received, ≈50 kJ/m2), the remaining animals were sacrificed, and eyes were processed for histopathological analysis.
Exposure to ionizing radiation.
Animals (5 to 8 weeks old; n = 4 to 6 mice/genotype) were anesthetized by intraperitoneal injection of pentobarbital (50 mg/kg), positioned with the left eye in the focus of a 50-kV X-ray source (diameter, 1.5 cm; 1-mm aluminum filter; 40-mm final optic assembly; dose rate, 830 R/min, 7.47 Gy/min), and exposed at doses as indicated in the text. Animals were sacrificed 7 days after exposure, and the exposed eye was further processed for histopathological analysis. In a second experiment, mice (8 to 10 weeks old; n = 6 mice/genotype) received a brief (13-min) total body irradiation of 10 Gy using a 137Cs source. After 20 h, animals were sacrificed, and eyes were processed for further analysis.
Isolation of eyes.
Animals were anesthetized by CO2 inhalation, followed by cervical dislocation. Eyes were marked on the nasal side with Alcian blue (5% Alcian blue in 96% ethanol) and subsequently enucleated and fixed in 2.5% glutaraldehyde in 0.1 M cacodylate buffer. After rinsing in buffer, the anterior segment was removed, and the eye cup was postfixed in 1% OsO4 and embedded in epon. Horizontal sections with a thickness of 1 μm were cut, stained with toluidine blue, and examined with a light microscope. Alternatively, for the study of chronic UV exposure, enucleated eyes were fixed in 2% glutaraldehyde and 2% paraformaldehyde in 0.1 M cacodylate buffer and either embedded in paraffin or postfixed in 1% OsO4 in Na-cacodylate buffer and embedded in epon. For terminal deoxynucleotidyltransferase-mediated dUTP nick-end labeling (TUNEL) staining (see below), eyes were fixed in 4% paraformaldehyde in 0.1 M phosphate buffer and embedded in paraffin.
Quantification of retinal cell loss and visualization of apoptosis.
Digital images of the retina (horizontal, central sections, passing through the optic nerve head) were taken at 200 μm nasally and temporally of the optic nerve head using a microscope equipped with a high-resolution camera (Prog/Res/3012; Kontron, Germany). The numbers of nuclei in the outer nuclear layer (ONL) and inner nuclear layer (INL) within a rectangular field of 500 μm2 were counted. For each mouse, two sections (separated by 15 μm) were analyzed, and counts were averaged. In addition, an estimate of the number of cones was obtained by specifically counting ONL profiles with the characteristic heterochromatin distribution (7). For an estimate of the number of ganglion cells, we counted the nuclei in the ganglion cell layer in the whole horizontal section.
Apoptotic cells were visualized in horizontal paraffin sections (5 μm thick) of paraformaldehyde-fixed eyes using the TUNEL method according to the specifications of the manufacturer of the kit (fluorescein apoptosis detection system [Promega] and Apoptag Plus Peroxidase in situ apoptosis detection kit [Chemicon]).
The observed differences in cell densities and the number of apoptotic cells were tested for significance using a Student's t test and one-way analysis of variance (ANOVA). In case of statistical significance, ANOVA was followed by the post hoc test of Student-Newman-Keuls. Two-way ANOVA was used to determine the independent effects of ionizing radiation and genotype on apoptosis in the various retinal layers as well as the interaction between these two variables. Significance was accepted at a P value of <0.05.
The decrease in photoreceptor number in Csbm/m mice with increasing age (see Fig. 2C, dotted line) was fitted to a mathematical model with constant risk of cell death [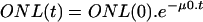 ] as well as to a model with an age-related increasing risk of cell death [
] as well as to a model with an age-related increasing risk of cell death [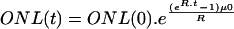 ] using nonlinear regression (SPSS version 11.5.1). A model was considered appropriate when all parameter estimates differed significantly from zero (8).
] using nonlinear regression (SPSS version 11.5.1). A model was considered appropriate when all parameter estimates differed significantly from zero (8).
FIG. 2.

Progressive loss of photoreceptors with age in the Csbm/m mouse. (A) Representative micrographs of the central region of the retina of young (3 months) and old (18 months) wt and Csbm/m mice. Note the specific loss of ONL nuclei and distortion of the outer segment layer in the 18-month-old Csbm/m mouse. Bar, 25 μm. (B) Quantification of the number of nuclei in the various layers of the retina of 18-month-old wt and Csbm/m mice demonstrating a specific loss of rods in the aged Csbm/m retina (ANOVA, followed by t test). (C) Kinetics of photoreceptor cell loss in Csbm/m mice. The relative number of rod nuclei in wt and Csbm/m mice is plotted as the percentage of nuclei relative to that observed in 3-month-old wt mice. Open squares, wt; closed diamonds, Csbm/m. Error bars indicate standard deviations. (D) Micrographs of the retinas of 3-month-old wt and Csbm/m mice stained for apoptosis using the TUNEL method. Arrows indicate fluorescein isothiocyanate (FITC) staining for TUNEL-positive cells in the ONL (right panels). Nuclei were visualized using the DAPI (4′,6′-diamidino-2-phenylindole) staining method (left panels).
Q-PCR evaluation.
Total RNA was isolated from the retina of ∼5-month-old wild-type (wt) (n = 7) and Csbm/m mice (n = 8) using a Total RNA isolation kit (QIAGEN) according to the manufacturer's instructions. Quantitative PCR (Q-PCR) was performed with a DNA Engine Opticon device according to the instructions of the manufacturer (MJ Research). Primer pairs designed to generate intron-spanning products of 180 to 210 bp were as follows: 5′-GGG ACA ATA CAC AAG GCT GT-3′ and 5′-GCC AAT GAT GGA ATG CTC TC-3′ for superoxide dismutase (Sod1), 5′-CCG CCT GAA CAC CAT CTA T-3′ and 5′-TTC CCA TTG ACT TCC ACC G-3′ for glutathione S-reductase (Gsr1), 5′-GCC CAA GTC CAC GAA TAC CT-3′ and 5′-CTC TGT TCC GTT CCA CCT TC-3′ for glutathione theta transferase 2 (Gstt2), 5′-AAC ACT CTG GAG ATG ACA CCT-3′ and 5′-TGT GAG GGA CTC TGG TCT TTG-3′ for heme oxygenase (Hmox1), 5′-GCT GGA AAC CCT ACA CAA GT-3′ and 5′-GAG TCT CTG CTT CTG GAC CA-3′ for 8-oxoguanine DNA glycosylase (Ogg1), and 5′-CTG GAG CAG TTC ATT GGC TTT-3′ and 5′-TCA GAA GAA GGG CAG TGT CA-3′ for uracil-N-glycosylase (Ung). The generation of specific PCR products was confirmed by melting curve analysis (which measures product specificity by the decrease in fluorescence signal when the PCR product is denatured) and gel electrophoresis (using Roche Agarose MS for analyzing small PCR products). Each primer pair was tested with a logarithmic dilution of a cDNA mix to generate a linear standard curve (crossing point [CP] plotted versus log of template concentration), which was used to calculate the primer pair efficiency [E = 10(−1/slope)]. Glyceraldehyde-3-phosphate dehydrogenase (Gapdh) and hypoxanthine guanine phosphoribosyltransferase 1 (Hprt-1) mRNAs were used as external standards. For data analysis, the second-derivative maximum method was applied: [E1 gene of interestΔCP (cDNA of wt mice − cDNA of Csbm/m) gene of interest]/[Ehprt-1ΔCP (cDNA wt mice − cDNA of Csbm/m) hprt-1]. All Q-PCR experiments were repeated at least four times.
RESULTS
Csb deficiency predisposes to UV-induced cornea pathology.
Chronically UV-B-exposed Csbm/m mice have been shown to develop skin cancer, notably squamous cell carcinomas (4, 48). To establish the effect of chronic UV exposure on the eyes of wild-type (Csb+/+), heterozygous (Csb+/m), and homozygous mutant (Csbm/m) mice, animals (n = 7 to 9 mice per genotype) were exposed to low daily doses of UV-B light for up to 38 weeks. After 5 weeks, Csbm/m mice started to develop photophobia, while eyes appeared irritated with red eye lids. Ten weeks after the first exposure (cumulative UV dose of ±9 kJ/m2), the cornea of Csbm/m mice started to become opaque (Fig. 1A), while some eyes became enlarged and bulging. After 29 weeks (cumulative UV dose of ±35 kJ/m2), the corneal surface of the eyes of Csbm/m mice became distorted, and bleedings occurred (Fig. 1A). These animals were euthanized, and their eyes were processed for histology. After 38 weeks of UV exposure (cumulative dose of ±50 kJ/m2), the experiment was terminated, and all remaining animals were euthanized. By that time, all irradiated Csbm/m mice had developed macroscopic eye abnormalities (Table 1). In marked contrast, UV-exposed Csb+/+ and Csb+/m animals as well as non-UV-treated Csbm/m mice did not show any eye abnormalities visible to the naked eye.
FIG. 1.

Increased UV sensitivity of the eyes of Csbm/m mice. (A) Representative example of a UV-B-exposed Csbm/m mouse with macroscopic ocular abnormalities. The photograph was taken 29 weeks after the start of the experiment (cumulative dose, ±35 kJ/m2). The right eye is enlarged and opaque; the left eye is red with an irregular corneal surface. (B to D) Micrographs of toluidine-blue-stained plastic sections of eyes of UV-exposed Csbm/m mice. (B) Micrograph of the anterior part (cornea, iris, and part of the lens) of an eye with an opaque macroscopic appearance revealing extensive epithelial hyperplasia (arrowheads) of the corneal epithelium. The hyperplasia is largely confined to the central part of the cornea. (C) Micrographs of the cornea of a wild-type mouse (left) and a Csbm/m mouse (right), both after UV exposure. The thickness of corneal epithelium and stroma is increased in the Csbm/m mouse (as indicated by black lines). Note the extensive vascularization of the corneal stroma (normally not containing blood vessels) of the Csbm/m mouse. (D) Example of a corneal squamous cell carcinoma infiltrating into other parts of the eye (arrows point at remnants of the retina).
TABLE 1.
UV-induced pathology of the corneas of wt and Csbm/m mice
| Genotype (no. of mice) | UV exposure (wk) | Macroscopic appearance (no. of eyes/total no. of eyes) | Histopathology |
|---|---|---|---|
| Csb+/+ (7) | 38 | Normal (14/14) | No pathology (6 out of 7 eyes investigated), neovascularization (1 out of 7 eyes investigated) |
| Csb+/m (7) | 38 | Normal (14/14) | No pathology (7 out of 7 eyes investigated) |
| Csbm/m (9) | 28-38 | Opaque (8/18) | Stroma vascularization and epithelial hyperplasia (7 out of 8 eyes investigated), bulla (1 out of 8 eyes investigated) |
| Enlarged, bulging eye, often with bleedings (10/18) | Severe hyperplasia of epithelium (5 out of 10 eyes investigated), squamous cell carcinoma (4 out of 10), severe neovascularization in the stroma (1 out of 10) | ||
| Csbm/m | Unirradiated | Normal (10/10) | No pathology (4 out of 4 eyes investigated) |
Subsequent histopathological analysis of eyes collected from UV-exposed Csb+/+ and Csb+/m animals did not show any abnormalities, except for the presence of a few capillaries in the corneal stroma in one out of seven Csb+/+ eyes investigated (Table 1). In marked contrast, UV-exposed Csbm/m eyes disclosed many abnormalities, localizing in the cornea. Opaque Csbm/m eyes revealed epithelial hyperplasia in the central part of the cornea, along with a reactive stroma and neovascularization. In addition, Bowman's membrane was increased in thickness. Enlarged, bulging eyes generally displayed severe hyperplasia of the corneal epithelium (Fig. 1B), accompanied by local disruptions and hemorrhages at the corneal surface. At least four of the bulging eyes carried a corneal squamous cell carcinoma with infiltrations into other regions of the eye, including the retina (Fig. 1D). One of the enlarged bulging eyes showed a different pathology. In this eye, there was only a modest hyperplasia of the epithelium. Most striking in this eye was the extensive vascularization of the corneal stroma (Fig. 1C). In contrast to the cornea, the lens of UV-exposed Csbm/m animals appeared to be normal.
These findings demonstrate that the TC-NER defect renders the eyes of Csbm/m mice sensitive to semiacute (hyperplasia) and long-term (carcinogenesis) effects of UV exposure.
Csb deficiency predisposes to spontaneous retinal degeneration.
While analyzing the UV-induced eye pathology in the CS mouse model, we noticed a severe reduction in the number of photoreceptor cells (hereafter referred to as photoreceptors) in the eyes of UV-exposed as well as in nonexposed (58-week-old) Csbm/m mice (data not shown). This finding, together with the notion that retinal degeneration is a characteristic clinical feature of Cockayne syndrome patients (38), prompted us to study the spontaneous changes in the morphology of the retina of Csbm/m mice. To this end, we isolated the eyes of wt and Csbm/m mutant mice of defined ages (all in a C57BL/6J genetic background and housed under identical light conditions) and quantified the number of photoreceptors and other cell types in defined regions of stained sections of the eye. At 3 months of age, we could not detect any difference between the wt and Csbm/m retinas, which excludes developmental abnormalities as the primary cause of the reduced number of photoreceptors in the retina of old Csbm/m animals. However, in 18-month-old Csbm/m mice, the ONL (containing the nuclei of the photoreceptor cells) and the outer segment layer (containing the rod and cone moiety of the photoreceptor cells) were clearly reduced in thickness compared to those of wt mice (Fig. 2A). Quantification of the number of nuclei in the various layers of the retina did not yield significant differences between wt and Csbm/m mice with respect to cell number in the ganglion cell layer and the INL (containing the cell bodies of bipolar, horizontal, amacrine, and Müller cells) (Fig. 2B). In marked contrast, the number of rod nuclei in the ONL of Csbm/m mice was significantly reduced (P < 0.01) to less than 40% of the wt level. The number of cones, identified on the basis of their characteristic distribution of heterochromatin in the nucleus, appeared unchanged. We next determined the kinetics of photoreceptor loss in the retina of Csbm/m animals. Quantification of the ONL nuclei showed that the number of photoreceptors gradually decreased with age in Csbm/m mice (Fig. 2C). We did not observe an obvious photoreceptor loss in wt mice, although there may be a tendency (but not statistically significant) toward an aging-related decline in the number of ONL nuclei (Fig. 2C). Other retinal layers appeared to be unaffected (data not shown). Thus, Csbm/m mice specifically lose rods during aging.
To investigate whether the process of spontaneous photoreceptor loss involves cell death via apoptosis, we next performed TUNEL staining on horizontal sections of the retinas of 3- and 11.5-month-old wt and Csbm/m mice. In line with the nearly constant number of nuclei in the ONL and other layers of the retinas of aging wt animals, we could hardly detect any TUNEL-positive cells (0.3 ± 0.2 and 0.6 ± 0.1 stained nuclei per section at 3 and 11.5 months, respectively [mean ± standard error of the mean]). In marked contrast, the retinas of 3-month-old Csbm/m mice contain 14.1 ± 1.6 TUNEL-positive cells per section, which localized almost exclusively to the ONL (Fig. 2D) without overt regional specificity for the central or peripheral retina. In the retinas of 11.5-month-old Csbm/m mice, we observed 10.7 ± 2.3 positive cells, which translates into approximately 17 positive cells when corrected for the loss of photoreceptors at this age (≈40%) (Fig. 2C) and does not significantly differ from the number of apoptotic photoreceptor cells in 3-month-old mutant animals (t test, P = 0.48). Similar results were obtained when cells were stained for caspase 3 (data not shown).
Taken together, these findings indicate that a CSB deficiency makes retinal photoreceptors (notably the rods) more sensitive to apoptosis, resulting in progressive spontaneous photoreceptor loss with age.
Enhanced expression of genes associated with antioxidant defense and oxidative damage repair in the Csbm/m retina.
To investigate the relevance of oxidative DNA damage in Csb-related retinal degeneration, we quantified the expression levels of genes that represent well-characterized markers for oxidative stress (i.e., superoxide dismutase [Sod1], heme oxygenase [Hmox1], glutathione theta transferase 2 [Gstt2], and glutathione S-reductase [Gsr]) (19, 20, 41) as well as genes that are implicated in the repair of (oxidative) DNA lesions (i.e., uracil-N-glycosylase [Ung] and 8-oxoguanine DNA glycosylase [Ogg1]) (2, 11) in the isolated retinas of 5-month-old wt (n = 7) and Csbm/m (n = 8) mice. As shown in Fig. 3, quantitative PCR evaluation revealed a moderate to substantial increase in the expression of these established oxidative stress markers in the Csbm/m mouse retina compared to the wt retina (P = 0.07 for Hmox1 and Gsst2, P < 0.05 for Ung, and P < 0.01 for Gsr, Gstt2, and Ogg1 [one-sided t test]). This finding strongly suggests that Csbm/m photoreceptor cells are subject to increased oxidative stress and point to elevated levels of unrepaired oxidative DNA lesions as the underlying cause of the spontaneous photoreceptor loss.
FIG. 3.
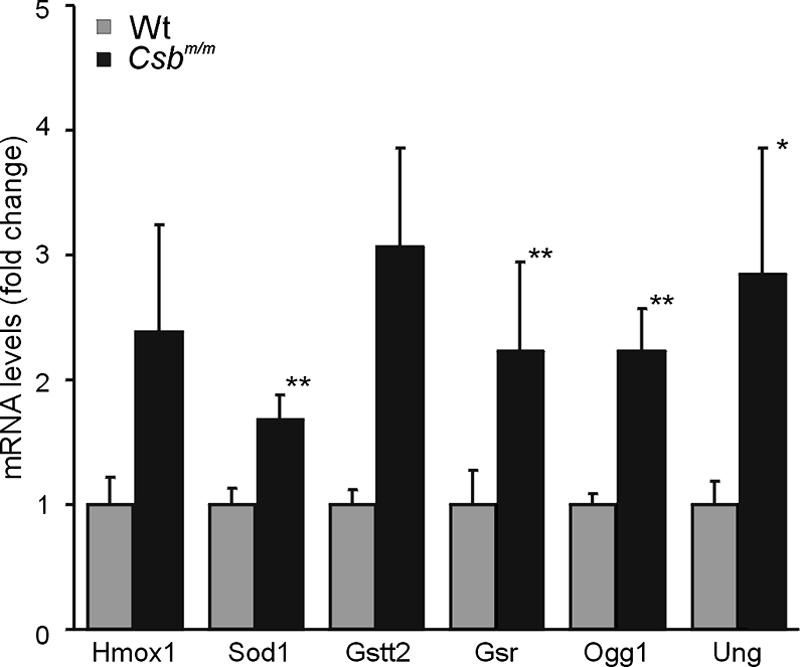
Upregulation of oxidative stress marker gene expression in the retinas of 5-month-old Csbm/m mice. mRNA levels were determined by Q-PCR; values were normalized (wild-type value is set at 1). Error bars indicate standard errors of the means. The primer set consisted of heme oxygenase (Hmox1), superoxide dismutase (Sod1), glutathione theta transferase 2 (Gstt2), and glutathione S-reductase (Gsr), and genes specific for repair of oxidative DNA lesions included 8-oxoguanine DNA glycosylase (Ogg1) and uracil-N-glycosylase (Ung). For all these genes, we measured higher expression values in Csbm/m than in wt retinas. (one-sided t test; * indicates significance at P < 0.05; ** indicates significance at P < 0.01, and the other P values are <0.07).
The retinas of Csbm/m and Csa−/− mice are hypersensitive to ionizing radiation.
Since oxidative DNA damage may play a role in CS as well as in aging, we next explored the IR sensitivities of the retinas of wt and Csbm/m mice. In addition to DNA strand breaks (which are repaired normally by CS cells) (43), IR is known to induce oxidative DNA damage via reactive free radicals that originate from the radiolysis of water (10, 51). Animals were locally exposed to various doses of X rays and sacrificed 7 days after treatment. Next, eye pathology was examined in horizontal sections of the retina. For wt mice, no overt pathology was noticed in the retina at any time point after irradiation at 10 or 15 Gy. In marked contrast, damage to photoreceptors became apparent 7 days after exposure to 18.5 Gy, culminating in extensive photoreceptor loss at doses of 22 and 25 Gy (Fig. 4A). The cell loss was most prominent in the periphery, where hardly any photoreceptor cells were left. This observation is well explained by the fact that the periphery was closest to the irradiation source and accordingly received the highest dose of X rays. At the highest doses (22 and 25 Gy), we also observed damage to the retinal pigment epithelium (RPE), as evident from the swollen appearance of RPE cells, containing large vacuoles (Fig. 4A). These findings fit well with a previous study of the acute effects of ionizing radiation on the rat retina, which identified photoreceptors as the most sensitive cell type, followed by the RPE cell (1). Surprisingly, Csbm/m mice showed essentially the same pathology at each given dose. Quantification of the cell loss (by counting the surviving cells in the periphery) did not reveal a statistically significant difference in photoreceptor loss between Csbm/m mice and wild-type mice (Fig. 4B).
FIG. 4.

Hypersensitivity of the Csbm/m and Csa−/− retinas to ionizing radiation. (A) Micrographs of the retinas of wt and Csbm/m mice before and 7 days after focused exposure of the eye to a high dose of ionizing radiation (25 Gy; X rays). Note the nearly complete loss of photoreceptors in the exposed wt and Csbm/m retinas. The arrow indicates swollen RPE cells with large vacuoles. (B) Quantification of photoreceptor cell loss in wt and Csbm/m mice exposed to various doses (15 to 25 Gy) of X rays. ONL nuclei were counted in the peripheral retina and expressed as a percentage of the number of ONL nuclei present in the unexposed wt retina. Open squares, wt; closed diamonds, Csbm/m. (C and D) Effect of lower-dose (10 Gy; gamma rays) whole-body IR exposure to the retina of wt and Csbm/m mice (six animals/genotype, 8 to 10 weeks of age). (C) Example of apoptotic cells (recognized by a brown nucleus), as visualized by TUNEL staining. (D) Quantification of apoptotic (TUNEL-positive) cells in the retinal layers. Two-way ANOVA was used to test the independent effects of gamma rays and genotype on the number of TUNEL-positive cells as well as the interaction between these variables. (E) Comparison of ionizing radiation sensitivities of wt, Csbm/m, and Csa−/− retinas expressed as a change (n-fold) in the number of TUNEL-positive photoreceptor cells in the ONL of irradiated (10 Gy) versus unirradiated animals. Note the comparable hypersensitivities of Csa−/− and Csbm/m mice (two-way ANOVA; P value of interaction between genotype and irradiation is <0.001 for Csbm/m versus wt and 0.03 for Csa−/− versus wt).
We reasoned that the failure to detect a difference in ionizing radiation sensitivity between the wt and Csbm/m retinas might have been caused by the design of the experiment. At a high IR dose, the heavy damage load could have masked a possible subtle effect of the Csb deficiency, whereas at lower IR doses, the method of counting surviving cells may not be sensitive enough. To overcome this problem, we performed a whole-body exposure experiment in which wt and Csbm/m animals were exposed to gamma rays at a dose of 10 Gy and quantified the number of apoptotic cells in sections stained with the TUNEL assay 20 h after exposure. As shown in Fig. 4C and D, the frequency of apoptotic cells in the ONL (photoreceptor cells), INL, and ganglion cell layer of the wt retina did not significantly increase after exposure (P > 0.5 for each layer), indicating that the wt mouse eye is not sensitive to a single dose of ionizing radiation at doses up to 10 Gy. In marked contrast, the already higher frequency of apoptotic photoreceptors in the ONL of the Csbm/m retina (compared to the wt retina; P = 0.0003 [two-way ANOVA, followed by t test]) increased over twofold (P = 0.002) after exposure. Similarly, IR exposure of the Csbm/m retina resulted in a higher frequency of apoptotic INL cells (P = 0.024). These findings clearly indicate that retinal cells in Csbm/m mice are hypersensitive to ionizing radiation (two-way ANOVA, P < 0.05) and point to unrepaired DNA damage (originating from oxidative stress) as the underlying trigger.
In humans and mice, both complementation groups of CS are associated with retinal degeneration (38, 47). As we recently reported the presence of cell type- and genotype-specific differences in IR sensitivity in Csbm/m and Csa−/− mice (with Csbm/m cells being more sensitive than Csa−/− cells) (15, 16), we next compared the IR sensitivities of the retinas of Csa−/− and Csbm/m mice (n = 6 mice per genotype per treatment) using the TUNEL assay described above. In line with the above-described experiment (Fig. 4D), exposure to 10 Gy of gamma rays caused a more-than-twofold increase in the number of apoptotic cells in the ONL of the Csbm/m mouse, while the number of TUNEL-positive cells in the wt retina did not significantly increase (Fig. 4E). Importantly, IR exposure also caused a twofold increase in apoptotic ONL cells in the Csa−/− retina, which indicates that both Csbm/m and Csa−/− photoreceptor cells are hypersensitive to IR radiation (two-way ANOVA, P < 0.05 relative to wt).
DISCUSSION
The present study shows that the absence of a functional CSB protein in the Csbm/m mouse model for the progeroid DNA repair disorder Cockayne syndrome increases the UV sensitivity of the cornea. Effects are noticed in both the stroma and epithelium, with epithelial hyperplasia and carcinoma formation as the most prominent pathological consequence. This finding fits well with our previous observation that Csbm/m (and Csa−/−) mice are photosensitive and predisposed to develop skin cancer upon UV irradiation (4, 48) and underscores the potential carcinogenicity of photolesions in sun/UV-exposed areas of the body. However, neither skin nor corneal neoplasms have been reported to occur in human CS patients. This human-rodent paradox is believed to originate at least in part from the inability of rodent cells to upregulate the p48 subunit of the XP-E p48-p125 UV-DNA damage binding heterodimer required to enhance CPD recognition by the GG-NER machinery (25, 44). TC-NER-deficient rodents, like Csbm/m mice, thus lack the slower GG-NER backup mechanism to remove CPDs from transcribed strands of active genes, allowing this lesion to contribute to mutagenesis and carcinogenesis at doses that do not yet result in apoptosis.
Retinal degeneration is regarded as a hallmark of CS. Based on the fundus appearance, the retinopathy of CS patients is generally referred to as a pigmentary retinopathy. However, case reports with detailed morphological data on CS eye pathology are scarce (38). To our knowledge, the literature contains only one report on a 44-month-old boy documenting loss of photoreceptors, in addition to other pathological changes such as ganglion cells and nerve fiber loss, and irregular RPE pigmentation (29). We have shown here that, similar to this patient, Csbm/m mice undergo spontaneous retinal degeneration, which consists of a gradual loss of rods with age. Other cell types, like cones and ganglion cells, are spared, at least up to an age of 18 months. As such, the Csbm/m mouse model does not exactly mimic the retinopathy of the human syndrome, although it currently cannot be excluded that additional aging-related pathologies (e.g., in the RPE) will develop at a later age.
CS is considered to be a progeroid syndrome since many of the CS symptoms resemble premature aging. In this respect, it is interesting that a gradual and selective loss of photoreceptors with age (with rods degenerating earlier than cones) is also observed in the aging human central retina and in patients with age-related macular degeneration (12, 26). In a primate study involving 6- to 34-year-old rhesus monkeys, the thickness of the ONL was shown to decrease with age as a result of the apoptosis of photoreceptors, which was distributed equally over all ages, except for an increase in the number of TUNEL-positive cells in the oldest animals (28). This, together with the notion that wild-type mice may also lose photoreceptors due to old age, indicates that the selective loss of rods in the Csbm/m mouse and in CS patients reflects accelerated aging.
What is the trigger for the photoreceptor loss in the Csbm/m mouse? Preliminary data from a cohort study with the GG-NER/TC-NER-deficient Xpa mouse model (14) revealed that 7-month-old Xpa−/− mice contain the normal number of photoreceptors, while the thickness of the ONL in Csbm/m animals of the same age was already reduced by 30% (T. G. Gorgels, H. van Steeg, and G. T. van der Horst, unpublished data). This finding suggests that a NER defect is not the major driving force behind the CS mouse retinopathy. Rather, it appears that the severe photoreceptor loss in Csbm/m animals represents a CS-specific trait related to a function of the CSB protein outside the context of NER. The nature of this non-NER function is not yet clear, but there are indications that the CSB protein has an auxiliary function in transcription elongation, notably the bypass of pause sites and RNA secondary structures (3, 18, 30, 42), as well as in the repair of other (non-NER-type) oxidative DNA lesions (16, 17, 39, 43). Csbm/m mouse embryonic fibroblasts, embryonic stem cells, and keratinocytes are IR sensitive (16). While IR induces a variety of DNA lesions, it is likely that oxidative DNA modifications determine the observed hypersensitivity. Indeed, gamma-ray-irradiated human fibroblasts of CS-B patients accumulate more 8-oxo-guanine (8-oxoG) damage than control cells (45). In addition, Spivak and Hanawalt (43) recently showed that plasmids containing 8-oxoG are less well repaired by CS patient cells, whereas strand breaks (as also produced by ionizing radiation) are repaired at a normal rate. Furthermore, Csbm/m mice are sensitive to the pro-oxidant di-(2-ethylhexyl)phthalate, and exposed animals contain higher levels of 8-oxoG in the liver than wt mice (15, 16). In view of this, and noting that the photoreceptor layer is oxygen rich and has a high metabolic activity, it is tempting to speculate that endogenous, oxidative DNA modifications contribute to the CS retinopathy.
Experimental support for this hypothesis was obtained from two independent experiments. First, we measured the expression of a set of genes known to be involved in the defense against oxidative stress and, specifically, in the repair of oxidative DNA damage and observed an upregulation of these genes in the Csbm/m retina. Second, we determined the IR sensitivity of the mouse retina in vivo and found that exposure of animals to 10 Gy of gamma rays caused increased cell death in the Csbm/m retina. Together, these findings support the hypothesis that oxidative DNA damage is at the basis of photoreceptor cell loss in the aging Csbm/m retina. Moreover, as shown for Csbm/m cells in vitro (16), these data provide in vivo evidence for ionizing radiation sensitivity in CS for the first time.
Since retinal degeneration is a general feature of CS-A and CS-B patients, and Csbm/m and Csa−/− mice both show spontaneous photoreceptor loss, the above-described scenario predicts that the Csa−/− mouse retina should also be IR hypersensitive. Indeed, we noticed enhanced apoptosis of photoreceptor cells in the retina of gamma-ray-exposed Csa−/− animals. Previous in vitro experiments with Csa−/− mouse embryonic fibroblasts failed to demonstrate IR sensitivity (15), but since cultured cells are already under high oxygen stress (6, 40), small differences may have been masked.
How would oxidative DNA lesions contribute to cell death in the Csbm/m retina? As shown for UV lesions, stalled RNA polymerase II forms a major trigger for apoptosis (33), and it has been hypothesized that transcription might serve as a DNA damage dosimeter where the severity of blockage acts as a go/no-go decision point for the induction of cell death (32). One can envision a scenario in which some oxidative lesions may remain unrepaired in the absence of the CSB protein and, accordingly, will accumulate over time until the damage load reaches a threshold that sets off apoptosis. Such a mechanism would imply that the probability of cell death (and thus the frequency of apoptotic photoreceptors) would increase with age. In view of the wild-type level of TUNEL-positive cells in the ONL of 3-week-old Csbm/m animals (I. van der Pluijm et al., unpublished data) and the significantly increased frequency of apoptosis in 8- to 10-week-old Csbm/m animals, such a scenario might apply to young adult mutant animals. However, our observation that the frequency of apoptotic photoreceptors does not significantly increase between the ages of 3 and 11.5 months suggests that gradually accumulating DNA damage is not the major driving force in adult animals (at least until the age of 1 year). Alternatively, it could be that backup mechanisms such as GG-NER and GG-BER still enable the CSB-deficient cell to repair oxidative DNA lesions, albeit at a slower pace. In this case, the CSB-deficient mouse would have higher steady-state levels of oxidative DNA damage, which would result in slower transcription rates with a higher (stochastic) risk of stalling RNA polymerase II and subsequent induction of apoptosis. Since retinal photoreceptors are postmitotic cells and are not replaced by dormant stem/progenitor cells (36), analysis of the kinetics of cell loss might discriminate between the two models. Curve fitting on the data set provided in Fig. 2C showed that the relationship between the number of photoreceptors and age is well described by a mathematical model in which the chance of cell death is constant in time (R2 = 0.945). In contrast, fitting to a model in which the risk of cell death increases with age did not yield a significant regression result, as the parameter reflecting the increasing risk did not significantly differ from zero (P = 0.505). This analysis suggests that, at least within the age range investigated, the retinal photoreceptor loss in the Csbm/m mouse is related to higher steady-state levels of oxidative DNA damage rather than to the age-related accumulation of this type of lesions.
In conclusion, we found spontaneous retinal degeneration in the Csb mouse model for Cockayne syndrome along with an increased sensitivity of photoreceptor cells for ionizing radiation. This finding underscores the importance of DNA repair and control of oxidative stress levels for long-term survival of photoreceptor cells in the retina and supports Harman's “free-radical theory of aging” (22). In addition, it warrants further investigation of these mechanisms in the etiology of (age-related) retinal disease and other brain disorders. The recent finding that a polymorphism in the promoter region of CSB is associated with age-related macular degeneration susceptibility strongly supports this view (46). Finally, the spontaneous and induced loss of terminally differentiated, postmitotic photoreceptor cells in the Csbm/m retina may serve as a sensitive readout for the screening of compounds that may retard or prevent the onset of pathology in the (aging) retina and brain.
Acknowledgments
We thank Rob Berg for technical assistance with the UV experiment.
This research was supported by the Rotterdamse Vereniging Blindenbelangen and the Algemene Nederlandse Vereniging ter Voorkoming van Blindheid, The Netherlands Organization for Scientific Research (NWO), through the foundation of the Research Institute Diseases of the Elderly as well as the National Institutes of Health (NIH 1PO1 AG17242-02).
Footnotes
Published ahead of print on 4 December 2006.
REFERENCES
- 1.Amoaku, W. M., L. Frew, G. J. Mahon, T. A. Gardiner, and D. B. Archer. 1989. Early ultrastructural changes after low-dose X-irradiation in the retina of the rat. Eye 3:638-646. [DOI] [PubMed] [Google Scholar]
- 2.An, Q., P. Robins, T. Lindahl, and D. E. Barnes. 2005. C→T mutagenesis and gamma-radiation sensitivity due to deficiency in the Smug1 and Ung DNA glycosylases. EMBO J. 24:2205-2213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Balajee, A. S., A. May, G. L. Dianov, E. C. Friedberg, and V. A. Bohr. 1997. Reduced RNA polymerase II transcription in intact and permeabilized Cockayne syndrome group B cells. Proc. Natl. Acad. Sci. USA 94:4306-4311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Berg, R. J., H. Rebel, G. T. van der Horst, H. J. van Kranen, L. H. Mullenders, W. A. van Vloten, and F. R. de Gruijl. 2000. Impact of global genome repair versus transcription-coupled repair on ultraviolet carcinogenesis in hairless mice. Cancer Res. 60:2858-2863. [PubMed] [Google Scholar]
- 5.Bohr, V. A., C. A. Smith, D. S. Okumoto, and P. C. Hanawalt. 1985. DNA repair in an active gene: removal of pyrimidine dimers from the DHFR gene of CHO cells is much more efficient than in the genome overall. Cell 40:359-369. [DOI] [PubMed] [Google Scholar]
- 6.Busuttil, R. A., M. Rubio, M. E. Dolle, J. Campisi, and J. Vijg. 2003. Oxygen accelerates the accumulation of mutations during the senescence and immortalization of murine cells in culture. Aging Cell 2:287-294. [DOI] [PubMed] [Google Scholar]
- 7.Carter-Dawson, L. D., and M. M. LaVail. 1979. Rods and cones in the mouse retina. II. Autoradiographic analysis of cell generation using tritiated thymidine. J. Comp. Neurol. 188:263-272. [DOI] [PubMed] [Google Scholar]
- 8.Clarke, G., R. A. Collins, B. R. Leavitt, D. F. Andrews, M. R. Hayden, C. J. Lumsden, and R. R. McInnes. 2000. A one-hit model of cell death in inherited neuronal degenerations. Nature 406:195-199. [DOI] [PubMed] [Google Scholar]
- 9.Cockayne, E. A. 1936. Dwarfism with retinal atrophy and deafness. Arch. Dis. Child. 11:1-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cooke, M. S., M. D. Evans, M. Dizdaroglu, and J. Lunec. 2003. Oxidative DNA damage: mechanisms, mutation, and disease. FASEB J. 17:1195-1214. [DOI] [PubMed] [Google Scholar]
- 11.Cortina, M. S., W. C. Gordon, W. J. Lukiw, and N. G. Bazan. 2005. Oxidative stress-induced retinal damage up-regulates DNA polymerase gamma and 8-oxoguanine-DNA-glycosylase in photoreceptor synaptic mitochondria. Exp. Eye Res. 81:742-750. [DOI] [PubMed] [Google Scholar]
- 12.Curcio, C. A., C. L. Millican, K. A. Allen, and R. E. Kalina. 1993. Aging of the human photoreceptor mosaic: evidence for selective vulnerability of rods in central retina. Investig. Ophthalmol. Vis. Sci. 34:3278-3296. [PubMed] [Google Scholar]
- 13.de Laat, W. L., N. G. Jaspers, and J. H. Hoeijmakers. 1999. Molecular mechanism of nucleotide excision repair. Genes Dev. 13:768-785. [DOI] [PubMed] [Google Scholar]
- 14.de Vries, A., C. T. van Oostrom, F. M. Hofhuis, P. M. Dortant, R. J. Berg, F. R. de Gruijl, P. W. Wester, C. F. van Kreijl, P. J. Capel, H. van Steeg, et al. 1995. Increased susceptibility to ultraviolet-B and carcinogens of mice lacking the DNA excision repair gene XPA. Nature 377:169-173. [DOI] [PubMed] [Google Scholar]
- 15.de Waard, H., J. de Wit, J. O. Andressoo, C. T. van Oostrom, B. Riis, A. Weimann, H. E. Poulsen, H. van Steeg, J. H. Hoeijmakers, and G. T. van der Horst. 2004. Different effects of CSA and CSB deficiency on sensitivity to oxidative DNA damage. Mol. Cell. Biol. 24:7941-7948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.de Waard, H., J. de Wit, T. G. Gorgels, G. van den Aardweg, J. O. Andressoo, M. Vermeij, H. van Steeg, J. H. Hoeijmakers, and G. T. van der Horst. 2003. Cell type-specific hypersensitivity to oxidative damage in CSB and XPA mice. DNA Repair (Amsterdam) 2:13-25. [DOI] [PubMed] [Google Scholar]
- 17.Dianov, G., C. Bischoff, M. Sunesen, and V. A. Bohr. 1999. Repair of 8-oxoguanine in DNA is deficient in Cockayne syndrome group B cells. Nucleic Acids Res. 27:1365-1368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dianov, G. L., J. F. Houle, N. Iyer, V. A. Bohr, and E. C. Friedberg. 1997. Reduced RNA polymerase II transcription in extracts of Cockayne syndrome and xeroderma pigmentosum/Cockayne syndrome cells. Nucleic Acids Res. 25:3636-3642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dong, A., J. Shen, M. Krause, H. Akiyama, S. F. Hackett, H. Lai, and P. A. Campochiaro. 2006. Superoxide dismutase 1 protects retinal cells from oxidative damage. J. Cell. Physiol. 208:516-526. [DOI] [PubMed] [Google Scholar]
- 20.Ganea, E., and J. J. Harding. 2006. Glutathione-related enzymes and the eye. Curr. Eye Res. 31:1-11. [DOI] [PubMed] [Google Scholar]
- 21.Hanawalt, P. C. 2002. Subpathways of nucleotide excision repair and their regulation. Oncogene 21:8949-8956. [DOI] [PubMed] [Google Scholar]
- 22.Harman, D. 1956. Aging: a theory based on free radical and radiation chemistry. J. Gerontol. 11:298-300. [DOI] [PubMed] [Google Scholar]
- 23.Hasty, P., J. Campisi, J. Hoeijmakers, H. van Steeg, and J. Vijg. 2003. Aging and genome maintenance: lessons from the mouse? Science 299:1355-1359. [DOI] [PubMed] [Google Scholar]
- 24.Hoeijmakers, J. H. 2001. Genome maintenance mechanisms for preventing cancer. Nature 411:366-374. [DOI] [PubMed] [Google Scholar]
- 25.Hwang, B. J., J. M. Ford, P. C. Hanawalt, and G. Chu. 1999. Expression of the p48 xeroderma pigmentosum gene is p53-dependent and is involved in global genomic repair. Proc. Natl. Acad. Sci. USA 96:424-428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jackson, G. R., C. Owsley, and C. A. Curcio. 2002. Photoreceptor degeneration and dysfunction in aging and age-related maculopathy. Ageing Res. Rev. 1:381-396. [DOI] [PubMed] [Google Scholar]
- 27.Jans, J., W. Schul, Y. G. Sert, Y. Rijksen, H. Rebel, A. P. Eker, S. Nakajima, H. van Steeg, F. R. de Gruijl, A. Yasui, J. H. Hoeijmakers, and G. T. van der Horst. 2005. Powerful skin cancer protection by a CPD-photolyase transgene. Curr. Biol. 15:105-115. [DOI] [PubMed] [Google Scholar]
- 28.Lambooij, A. C., M. Kliffen, R. W. Kuijpers, A. B. Houtsmuller, J. J. Broerse, and C. M. Mooy. 2000. Apoptosis is present in the primate macula at all ages. Graefes Arch. Clin. Exp. Ophthalmol. 238:508-514. [DOI] [PubMed] [Google Scholar]
- 29.Levin, P. S., W. R. Green, D. I. Victor, and A. L. MacLean. 1983. Histopathology of the eye in Cockayne's syndrome. Arch. Ophthalmol. 101:1093-1097. [DOI] [PubMed] [Google Scholar]
- 30.Licht, C. L., T. Stevnsner, and V. A. Bohr. 2003. Cockayne syndrome group B cellular and biochemical functions. Am. J. Hum. Genet. 73:1217-1239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lindahl, T., and R. D. Wood. 1999. Quality control by DNA repair. Science 286:1897-1905. [DOI] [PubMed] [Google Scholar]
- 32.Ljungman, M., and D. P. Lane. 2004. Transcription—guarding the genome by sensing DNA damage. Nat. Rev. Cancer 4:727-737. [DOI] [PubMed] [Google Scholar]
- 33.Ljungman, M., F. Zhang, F. Chen, A. J. Rainbow, and B. C. McKay. 1999. Inhibition of RNA polymerase II as a trigger for the p53 response. Oncogene 18:583-592. [DOI] [PubMed] [Google Scholar]
- 34.Martin, G. M. 2005. Genetic modulation of senescent phenotypes in Homo sapiens. Cell 120:523-532. [DOI] [PubMed] [Google Scholar]
- 35.Mitchell, J. R., J. H. Hoeijmakers, and L. J. Niedernhofer. 2003. Divide and conquer: nucleotide excision repair battles cancer and ageing. Curr. Opin. Cell Biol. 15:232-240. [DOI] [PubMed] [Google Scholar]
- 36.Moshiri, A., J. Close, and T. A. Reh. 2004. Retinal stem cells and regeneration. Int. J. Dev. Biol. 48:1003-1014. [DOI] [PubMed] [Google Scholar]
- 37.Murai, M., Y. Enokido, N. Inamura, M. Yoshino, Y. Nakatsu, G. T. van der Horst, J. H. Hoeijmakers, K. Tanaka, and H. Hatanaka. 2001. Early postnatal ataxia and abnormal cerebellar development in mice lacking seroderma pigmentosum group A and Cockayne syndrome group B DNA repair genes. Proc. Natl. Acad. Sci. USA 98:13379-13384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nance, M. A., and S. A. Berry. 1992. Cockayne syndrome: review of 140 cases. Am. J. Med. Genet. 42:68-84. [DOI] [PubMed] [Google Scholar]
- 39.Osterod, M., E. Larsen, F. Le Page, J. G. Hengstler, G. T. Van Der Horst, S. Boiteux, A. Klungland, and B. Epe. 2002. A global DNA repair mechanism involving the Cockayne syndrome B (CSB) gene product can prevent the in vivo accumulation of endogenous oxidative DNA base damage. Oncogene 21:8232-8239. [DOI] [PubMed] [Google Scholar]
- 40.Parrinello, S., E. Samper, A. Krtolica, J. Goldstein, S. Melov, and J. Campisi. 2003. Oxygen sensitivity severely limits the replicative lifespan of murine fibroblasts. Nat. Cell Biol. 5:741-747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Risom, L., P. Moller, U. Vogel, P. E. Kristjansen, and S. Loft. 2003. X-ray-induced oxidative stress: DNA damage and gene expression of HO-1, ERCC1 and OGG1 in mouse lung. Free Radic. Res. 37:957-966. [DOI] [PubMed] [Google Scholar]
- 42.Selby, C. P., and A. Sancar. 1997. Cockayne syndrome group B protein enhances elongation by RNA polymerase II. Proc. Natl. Acad. Sci. USA 94:11205-11209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Spivak, G., and P. C. Hanawalt. 2006. Host cell reactivation of plasmids containing oxidative DNA lesions is defective in Cockayne syndrome but normal in UV-sensitive syndrome fibroblasts. DNA Repair (Amsterdam) 5:13-22. [DOI] [PubMed] [Google Scholar]
- 44.Tang, J. Y., B. J. Hwang, J. M. Ford, P. C. Hanawalt, and G. Chu. 2000. Xeroderma pigmentosum p48 gene enhances global genomic repair and suppresses UV-induced mutagenesis. Mol. Cell 5:737-744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tuo, J., P. Jaruga, H. Rodriguez, V. A. Bohr, and M. Dizdaroglu. 2003. Primary fibroblasts of Cockayne syndrome patients are defective in cellular repair of 8-hydroxyguanine and 8-hydroxyadenine resulting from oxidative stress. FASEB J. 17:668-674. [DOI] [PubMed] [Google Scholar]
- 46.Tuo, J., B. Ning, C. M. Bojanowski, Z. N. Lin, R. J. Ross, G. F. Reed, D. Shen, X. Jiao, M. Zhou, E. Y. Chew, F. F. Kadlubar, and C. C. Chan. 2006. Synergic effect of polymorphisms in ERCC6 5′ flanking region and complement factor H on age-related macular degeneration predisposition. Proc. Natl. Acad. Sci. USA 103:9256-9261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.van der Horst, G. T., L. Meira, T. G. Gorgels, J. de Wit, S. Velasco-Miguel, J. A. Richardson, Y. Kamp, M. P. Vreeswijk, B. Smit, D. Bootsma, J. H. Hoeijmakers, and E. C. Friedberg. 2002. UVB radiation-induced cancer predisposition in Cockayne syndrome group A (Csa) mutant mice. DNA Repair (Amsterdam) 1:143-157. [DOI] [PubMed] [Google Scholar]
- 48.van der Horst, G. T., H. van Steeg, R. J. Berg, A. J. van Gool, J. de Wit, G. Weeda, H. Morreau, R. B. Beems, C. F. van Kreijl, F. R. de Gruijl, D. Bootsma, and J. H. Hoeijmakers. 1997. Defective transcription-coupled repair in Cockayne syndrome B mice is associated with skin cancer predisposition. Cell 89:425-435. [DOI] [PubMed] [Google Scholar]
- 49.van Hoffen, A., A. T. Natarajan, L. V. Mayne, A. A. van Zeeland, L. H. Mullenders, and J. Venema. 1993. Deficient repair of the transcribed strand of active genes in Cockayne's syndrome cells. Nucleic Acids Res. 21:5890-5895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Venema, J., L. H. Mullenders, A. T. Natarajan, A. A. van Zeeland, and L. V. Mayne. 1990. The genetic defect in Cockayne syndrome is associated with a defect in repair of UV-induced DNA damage in transcriptionally active DNA. Proc. Natl. Acad. Sci. USA 87:4707-4711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ward, J. 1975. Molecular mechanisms of radiation-induced damage to nucleic acids. Adv. Rad. Biol. 5:181-239. [Google Scholar]