

Abstract
A bacterium isolated from patulin-contaminated apples was capable of degrading patulin to a less-toxic compound, ascladiol. The bacterium was identified as Gluconobacter oxydans by 16S rRNA gene sequencing, whereas ascladiol was identified by liquid chromatography-tandem mass spectrometry and proton and carbon nuclear magnetic resonance. Degradation of up to 96% of patulin was observed in apple juices containing up to 800 μg/ml of patulin and incubated with G. oxydans.
Patulin contamination of apple and other fruit-based foods and beverages is an important food safety issue, due to the high consumption of these commodities, especially by infants and children. This mycotoxin is mainly produced by species of filamentous fungi belonging to the genera Penicillium, Aspergillus, and Byssochlamis (5, 14). Penicillium expansum, in particular, is considered the main producer of patulin in fruits (7, 1).
At first, patulin was evaluated as a pharmacological agent due to its antimicrobial properties. However, the gastrointestinal and dermal irritation observed in human trials prevented its use as a pharmacological agent (6). Based on the experimental results available, it was concluded that patulin is genotoxic, but no adequate evidence of carcinogenicity in experimental animals exists (25). Within the food industry, patulin contamination is considered of greatest concern in apples and apple products, which are the main sources of human patulin consumption (11). Nevertheless, this mycotoxin has also been found at significant concentrations in other fruits, such as pears, peaches, strawberries, blueberries, cherries, apricots, and grapes, as well as in cheese (16, 10, 15).
High levels of patulin, up to 16,402 μg/kg and 44,572 μg/kg, have been found in conventional and organic rotten apple samples, respectively (15). The patulin concentration in apple juice can reach 2,500 μg/kg if the juice is obtained from rotten apples (13). Because of these data, there has been increasing interest in finding a safe and efficient detoxification process, since it is now evident that although a preventative strategy is the best approach, it is sometimes difficult to achieve.
The objectives of this work were (i) to isolate and identify microbial strains able to grow in the presence of high patulin concentrations, (ii) to select from those strains those capable of detoxifying patulin, and (iii) to isolate and chemically characterize the relevant degradation products.
MATERIALS AND METHODS
All the solvents used were of high-performance liquid chromatography (HPLC) grade from Baker (Deventer, The Netherlands). Water was obtained with a MilliQ system (Millipore, Bedford, MA). The patulin standard, Taq polymerase, deoxynucleotide triphosphates, and DNA molecular weight markers were purchased from Sigma (Milan, Italy). Salt-free, highly purified oligonucleotides were obtained from MGW Biotech S. R. L. (Florence, Italy). Cultural media were purchased from Difco (Detroit, MI).
Isolation and taxonomic identification of patulin-degrading bacteria.
Five apples with blue-spot symptoms were collected in January 2005 at local markets in Bari province (Italy). Each fruit was placed in a plastic bag and incubated at 24°C until it rotted (14 days). After the incubation period, the apples were pooled and homogenized in a blender (Lab Blender; Seaward, London, United Kingdom) for 1 min at room temperature. Aliquots (0.5 g) of apple purée were serially diluted with sterile distilled water and spread onto NYDA (10 g glucose, 8 g nutrient broth, 5 g yeast extract, and 18 g agar per liter), Rose Bengal agar containing 2 mg per liter of dichloran, and potato dextrose agar (PDA) and incubated at 25°C for 7 days. To increase the development of other microorganisms in addition to Aspergillia and Penicillia species, a nonselective enrichment was performed by mixing different 0.5-g aliquots of apple purée with 4.5 ml of Wickerham solution (24). The suspensions obtained were incubated for 12 h at 4°C, then serially diluted and spread onto PDA plates. After 6 days of incubation at 25°C, eight bacterial colonies were isolated according to their different morphologies and characterized by Gram staining and their catalase reactions.
The bacterial colonies were analyzed by using the two-step randomly amplified polymorphic DNA (RAPD)-PCR protocol, as described elsewhere (2), with three different primers: Lac1, 5′-AGCAGCGTGG-3′; M 13, 5′-GAGGGTGGCGGTTCT-3′; and Ope 7, 5′-AGATGCAGCC-3′.
When different isolates gave the same electrophoretic pattern, they were grouped together, and only one isolate from each group was chosen as the representative strain and used for further analyses. The selected bacterial isolates were identified by amplification and sequencing of the 16S rRNA gene, as previously described (8). The identified strains were stored at −80°C in the bacterial collection of the Institute of Sciences of Food Production. These bacteria were further characterized and tested for their abilities to grow in the presence of patulin and to degrade this mycotoxin. The remaining apple purée was analyzed for patulin as described elsewhere (9).
Determination of patulin in rotten fruit and bacterial cultures.
The quantification of patulin in bacterial cultures spiked with patulin and rotten apple purée was performed by HPLC-UV/diode array detector (DAD). The apple purée was analyzed according to the method described by MacDonald et al. (9), while bacterial cultures spiked with patulin and grown in potato dextrose broth (PDB), in YPM (5.0 g yeast extract, 3.0 g peptone, and 25.0 g mannitol per liter) or in commercial apple juice were centrifuged, filtered through a 0.22-μm cellulose acetate filter (Albet, Murcia, Spain), adequately diluted with water, and analyzed by HPLC-UV/DAD. The liquid chromatograph (Thermo Quest Inc., San Jose, CA) was equipped with a quaternary gradient pump (Spectra Series P4000), a vacuum membrane degasser (SCM 1000), an autosampler injection system with a 50-μl loop (AS 3000), a column oven set at 30°C, a DAD (UV 6000 LightPipe detector), and a chromatography data system for Windows 2000 (ChromQuest version 2.53). A Phenomenex C18 Synergi Hydro column (250 by 4.6 mm, 4-μm-particle size) was used, preceded by use of a 4-by-3-mm precolumn packed with the same stationary phase. The mobile phase was an isocratic mixture of water, acetonitrile, and perchloric acid (96:4:0.1) eluting at a flow rate of 0.8 ml/min for 20 min. At the end of each analysis, the column was washed for 7 min with an isocratic mixture of acetonitrile and water (65:35) and then equilibrated with water, acetonitrile, and perchloric acid (96:4:0.1) for 15 min before the next injection. Patulin was identified in sample extracts by comparing the retention time and UV spectrum of the peak recorded in the chromatogram with those of the authentic standard. UV spectra were recorded in the range of 200 to 400 nm. Quantification of the toxin levels was performed according to the external standard method, integrating peak areas, acquired at 275 nm, at the retention time of the corresponding patulin standard. A stock solution of patulin at a concentration of 5 mg/ml was prepared in distilled water acidified by glacial acetic acid at pH 4.0 and stored at 4°C. Four standard calibrated solutions of patulin at concentrations of 1, 2, 4, and 10 μg/ml were prepared by appropriately diluting the stock solution with acidified water. The volume injected ranged from 10 to 50 μl.
Patulin degradation assays.
The eight bacterial isolates were grown for 24 h in PDB at 30°C under shaking conditions (175 rpm) to provide an inoculum with an absorbance value ranging from 0.4 to 0.5 AU at 600 nm (Ultrospec 3100 Pro; Amersham Pharmacia Biotech). Sterile tubes containing 1.98 ml of PDB were spiked at 10 μg/ml of patulin with a sterile mycotoxin water solution, then inoculated with 20 μl of each bacterial suspension and incubated for 3 days at 30°C under shaking conditions. Controls without patulin or bacteria were prepared by adding sterile distilled water. The experiments were conducted in triplicate for a total of 51 samples. Three out of the 8 bacterial isolates were tested in PDB under the same conditions as above, with increasing patulin concentrations, i.e., 10, 20, 40, 70, and 100 μg/ml of toxin. The bacterial strain showing the best patulin degradation rate was cultured in 10 ml YPM spiked with 4 mg of patulin and incubated for 3 days at 30°C. This culture was used to produce and isolate adequate amounts of patulin degradation products to identify through liquid chromatography-tandem mass spectrometry (LC-MS-MS) and nuclear magnetic resonance (NMR).
The time course of patulin degradation by the selected strain was also checked in 30 ml of commercial apple juice spiked at 100 μg/ml of patulin and incubated for 72 h. The same bacterial strain was also tested in 10-ml aliquots of commercial apple juice and YPM with increasing patulin concentrations from 100 to 800 μg/ml. The number of viable cells for each aliquot in these experiments was calculated by plating the serially diluted culture on YPM agar.
The aliquots of each broth or apple juice culture were centrifuged for 4 min at 10,000 rpm (10,000 × g), and the supernatants were filtered through a 0.22-μm cellulose acetate filter (Albet, Murcia, Spain) and then analyzed by HPLC/UV-DAD to quantify the residual patulin content and the patulin degradation products.
Isolation of the patulin degradation products.
A 3-day-old, 10-ml bacterial culture incubated with 4 mg of patulin was centrifuged, and the supernatant was lyophilized and reconstituted with 2 ml of distilled water. This solution was split into two aliquots of 1 ml each that were purified through two Oasis-HLB columns containing 200 mg of stationary phase (Waters Co., Milford, MA) and placed onto a vacuum manifold (Supelco, Bellefonte PA). The columns were previously conditioned with methanol (4 ml) and water (4 ml), and then aliquots of 1 ml of the sample were applied to each column, and the eluates were discarded. The columns were washed with 4 ml of water, dried by passing air, and then eluted with 4 ml of methanol. The eluate was recovered, and 10 μl was analyzed by high-performance thin-layer chromatography (HPTLC) on plates precoated with silica gel 60 F 254 (10 by 10 cm, 0.1-mm thick plates; Merck, Darmstadt, Germany). The elution system was a mixture of toluene, ethyl acetate, and formic acid (TEF) (5:4:1). A spot with a Rf of 0.23 (the Rf of patulin was 0.55), detected under UV light at 254 nm, was scraped off, and the compound was eluted from the silica gel, using 1 ml of acetone, dried, and then dissolved in 1 ml of HPLC mobile phase and analyzed by HPLC-UV/DAD as described above. This compound showed two resolved peaks in HPLC with retention times of 9.8 min and 10.6 min (the retention time of patulin was 16.0 min), both having the same UV spectrum with an absorbance maximum at 268 nm, similar to that of ascladiol, which is not commercially available. To confirm this hypothesis, adequate amounts of this compound were obtained by purifying the methanol fractions collected from the Oasis columns. The compound was purified on semipreparative thin-layer chromatographic (TLC) plates precoated with silica gel 60 F-254 (20-by-20-cm, 0.5-mm-thick plates; Merck, Darmstadt, Germany), using TEF (5:4:1) as the elution system. The band of ascladiol detected under UV light at 254 nm at Rf 0.27 was scraped off from each semiprepared TLC plate, and the compound was eluted from the silica gel with 9 ml of acetone. The purified compound was dried under a nitrogen stream to yield about 3 mg of white powder that was submitted to HPLC-UV/DAD, LC-MS-MS, and proton (1H) and carbon (13C) NMR analyses for chemical characterization. An aliquot of this compound was used to prepare a stock solution and four standard calibrated solutions in water acidified by glacial acetic acid at pH 4. These solutions were used to identify and quantify ascladiol in bacterial cultures analyzed by HPLC-UV/DAD.
Identification of ascladiol.
LC-MS-MS analyses were performed by an LC system with a 1,100-capillary pump, a microautosampler, and a UV detector set at 268 nm (Agilent Technologies, Waldbrom, Germany) and interfaced with a QTrap mass spectrometer MS-MS system equipped with an electrospray interface. A Harvard 22 syringe pump was used for infusion experiments (Applied Biosystems, Foster City, CA). The mass spectrometer was used in negative- and positive-ion mode, employing electrospray ionization. A 150-by-2.1-mm, 4-μm Synergi Hydro-RP column (Phenomenex) was used, preceded by use of a guard column with the same packing material. The mobile phase, eluting at a flow rate of 200 μl/min, consisted of an isocratic mixture of water and acetonitrile (98:2). The mass spectrometric conditions (negative ionization) were as follows: nebulizer gas and heater gas (air), 20 and 40 lb/in2, respectively; curtain gas (nitrogen), 20 lb/in2; temperature, 380°C; mass range, 50.0 to 200.0 AMU; scan time, 2 s; needle voltage, −4,500 V; declustering potential, −19 V; entrance potential voltage, −9 V. The mass spectrometer was used in triple quadrupole and ion trap modes.
NMR analysis.
1H NMR and 13C NMR results were recorded on a Bruker DRX500 Advance instrument equipped with probes for inverse detection and with a z gradient for gradient-accelerated spectroscopy. Standard Bruker automation programs were used for two-dimensional NMR experiments. Two-dimensional correlation spectroscopy (COSY) experiments were performed using COSY double-quantum-filtered, phase-sensitive and COSY gradient-accelerated sequences. Spectra from nuclear Overhauser effect spectroscopy (NOESY) were acquired using mixing times of 0.6 to 0.9 s. Inverse-detected normal and long-range 1H and 13C heterocorrelated two-dimensional NMR spectra were obtained by using the gradient sensitivity-enhanced pulse sequences INVIEAGSSI and INV4GPLRND, respectively. CDCl3 was used as the solvent in all of the NMR experiments. Residual 1H and 13C peaks of the solvent (δH 7.26, δC 77.0) were used as internal standards to calculate chemical shifts with reference to those of tetramethylsilane.
RESULTS
The purée obtained from rotten apples proved to be contaminated with patulin at a level of 2,130 μg/kg. Preliminary attempts to isolate microorganisms other than fungi from this purée by using NYDA, Rose Bengal agar, and PDA as substrates were unsuccessful. By contrast, nonselective enrichment by Wickerham solution before apple purée plating allowed the growth of several colonies with bacterial morphology. Eight colonies in particular, which were constituted of rod-shaped, catalase-positive, gram-negative cells, were isolated from the apple purée.
The eight bacterial isolates significantly reduced the concentration of patulin when tested in PDB, as shown in Table 1. The analysis of RAPD-PCR fingerprints, obtained with a Lac1 primer, showed that four isolates (M3, M4, M5, and M6) belonged to the same strain, whereas the remaining four fingerprints (M1, M2, M8, and M13) were different from each other, leading to the isolation of five different biotypes within the eight isolates (Fig. 1).
TABLE 1.
Residual patulin concentrations and relevant percent reductions of patulin in PDB spiked at 10 μg/ml of patulin, inoculated with the isolated bacteria, and incubated for 3 days at 30°C and 175 rpma
| Bacterial isolate | Residual patulin (μg/ml) | % Patulin reduction |
|---|---|---|
| M 1 | 1.01 ± 0.05 | 89.9 |
| M 2 | 1.60 ± 0.13 | 84.0 |
| M 3 | 0.44 ± 0.03 | 95.6 |
| M 4 | 0.52 ± 0.04 | 94.8 |
| M 5 | 0.92 ± 0.05 | 90.8 |
| M 6 | 0.41 ± 0.03 | 95.9 |
| M 8 | 0.39 ± 0.03 | 96.1 |
| M 13 | 0.95 ± 0.07 | 90.5 |
Each result is the mean ± the SE of three replicates. Negative controls containing patulin at 10 μg/ml without bacteria and incubated at the same experimental conditions used for the samples showed a mean patulin recovery of 90%.
FIG. 1.
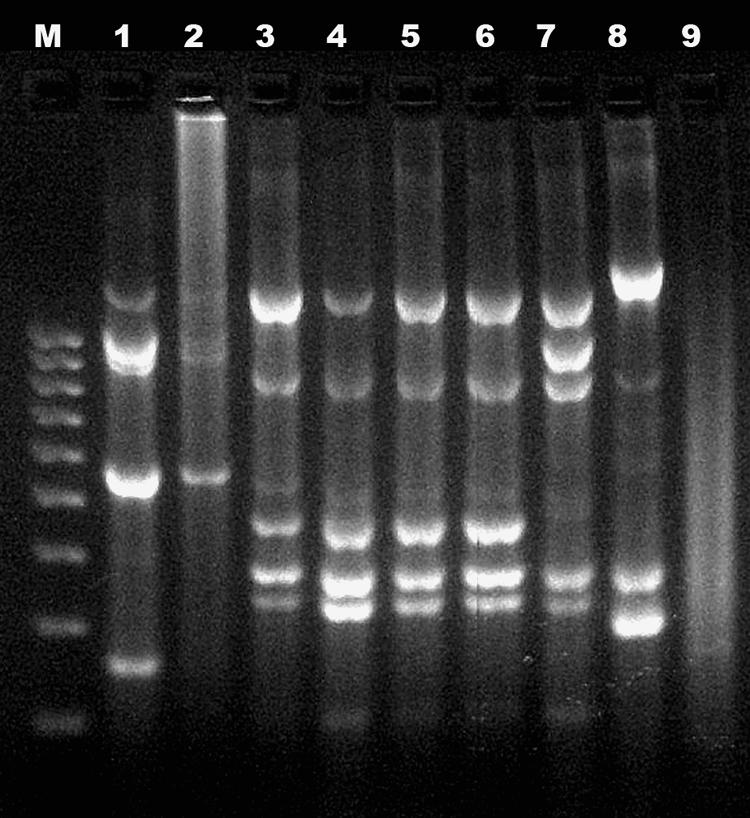
RAPD-PCR patterns of patulin-degrading bacterial isolates from PDA plates. Lanes: M, DNA molecular weight marker (Sigma); 1 to 8, isolates M1, M2, M3, M4, M5, M6, M8, M13; 9, PCR negative control.
The taxonomic positions of the M3, M8, and M13 biotypes, determined by partial rRNA gene sequence analysis (Fig. 2), made it possible to classify them as strains of Gluconobacter oxydans, a gram-negative aerobic bacterium.
FIG. 2.

Dendrogram of Gluconobacter strains based on 16S rRNA gene sequence analysis, using the neighbor-joining algorithm and 0.75 as the maximum sequence difference with the “distance tree of results,” which is freely available at http://www.ncbi.nlm.nih.gov/BLAST/BLAST.cgi.
Figure 3 shows the HPLC chromatograms of a liquid medium (YPM) spiked with patulin and incubated at 30°C for 3 days at 175 rpm with and without the bacterial strain M3. In the presence of viable bacterial cells, the reduction of the patulin peak in the chromatogram corresponded with the appearance of a new peak that eluted before patulin, with a retention time of 9.8 min. A minor peak with a retention time of 10.6 min was also observed. These two peaks had the same UV spectrum, with a UV absorbance maximum at 268 nm, as measured by the UV diode array detector. A suitable amount of these two compounds was produced by incubating 4 mg of patulin for 3 days in 10 ml of YPM broth inoculated with the strain M3.
FIG. 3.

HPLC-UV/DAD chromatograms of YPM liquid medium spiked at 200 μg/ml patulin (3) (upper trace) and YPM liquid medium spiked at 200 μg/ml patulin and inoculated with G. oxydans (lower trace), containing E ascladiol (1), Z ascladiol (2) and residual patulin.
The MS-MS spectrum of patulin ([M − H]−, m/z 153.0), acquired in negative-ion mode, showed main fragments at m/z 109.0 (loss of CO2) and m/z 125.0 and 97.0 (loss of two CO molecules) and fragments at m/z 135.0 (loss of H2O). Mass spectra recorded at the retention time of the two peaks monitored by the UV detector (retention time, 8.1 min and 9.3 min) showed the same main peak at m/z 155.0, presumably corresponding to the deprotonated ([M − H]−) ion of ascladiol. LC-MS-MS analysis showed the same fragmentation pattern for the two compounds, suggesting the presence of two isomers, as proposed in Fig. 4. A common fragment with patulin was observed at m/z 125.0, whereas fragments at m/z 137.1 (loss of H2O) and 111.1 (loss of CO2) indicated the 2-AMU difference with respect to patulin, due to the diol instead of a pyran ring.
FIG. 4.

Structures of patulin, E ascladiol, and Z ascladiol.
The E,Z mixture of 5-(2-hydroxy-ethylidene)-4-hydroxymethyl-5H-furan-2-one, ascladiol, was characterized by multinuclear multidimensional NMR experiments. 1H and 13C data for the two isomers depicted in Fig. 4 are reported in Table 2. Integration data (1H spectrum) for the collected sample dissolved in CDCl3 show the presence of the two isomers E and Z in a 3.5:1 ratio (Fig. 4). 1H NMR spectra for the E,Z mixture of ascladiol dissolved in more-polar solvents such as d6 acetone have already been reported (13, 19). The high-field NMR instrument used in the present work also allowed complete 1H and 13C characterizations in CDCl3, where the ascladiol isomers are sparingly soluble. Identification of the two isomers has previously been carried out on the basis of the long-range J5 1H coupling between CH(6) and CH(3) reported to be present (E ascladiol) or absent (Z ascladiol) (19). In our case, we found the presence of J5 coupling between CH(6) and CH(3) for both E and Z ascladiol. As expected, due to the fixed-saw conformation of the carbon chain linking CH(6) and CH(3), such a J5 coupling is higher in the case of the E (1.52 Hz) with respect to the Z (0.65 Hz) isomer. By contrast, we found a very similar J4 coupling between CH(3) and CH2(8) for both isomers (1.52 and 1.53 Hz for E and Z, respectively). The complete coupling network for the two isomers can easily be assessed from the COSY spectrum (Fig. 5). An even more convincing isomer assignment can be made in the present work on the basis of the dipolar coupling network observed in the NOESY spectrum, where the spatial proximity between CH2(7) and CH2(8) and between CH(6) and CH2(8) originates cross peaks selectively in E and Z, respectively (Fig. 5).
TABLE 2.
1H and 13C NMR data for E and Z ascladiola
| Atomic numbering | NMR data for:
|
||||||
|---|---|---|---|---|---|---|---|
|
E ascladiol
|
Z ascladiol
|
||||||
|
1H
|
13C
|
1H
|
13C, this workb | ||||
| This workb | Moss and Longc | This workb | Moss and Longc | This workb | Sekiguchi et al.d | ||
| CO(2) | 167.9 | 168.9 | —e | ||||
| CH(3) | 6.34 td (1.52, 1.52) | 6.30 d (1.5) | 119.7 | 118.3 | 6.258 td (1.53, 0.61) | 6.16 s | 116.2 |
| C(4) | 156.0 | 160.9 | 157.8 | ||||
| C(5) | 150.0 | 149.4 | 147.8 | ||||
| CH(6) | 6.020 td (7.24, 1.52) | 5.87 td (7.67, 1.6) | 113.0 | 115.9 | 5.454 td (6.72, 0.61) | 5.52 t (7) | 110.7 |
| CH2(7) | 4.380 d (7.24) | 4.32 d (7.71) | 56.3 | 56.74 | 4.517 d (6.72) | 4.36 d (7) | 57.1 |
| CH2(8) | 4.767 d (1.52) | 4.76 s | 58.6 | 59.04 | 4.667 d (1.53) | 4.62 d (7) | 57.0 |
Assignment was also based on COSY, NOESY, heteronuclear single quantum coherence (HSQC), and heteronuclear multiple bond correlation (HMBC) spectra. Atomic numbers are according to Fig. 4. td, triplet of doublet.
Data from this work (solvent, CDCl3).
Data from Moss and Long (13) (solvent, d6 acetone).
Data from Sekiguchi et al. (19) (solvent, d6 acetone). A broad 1H signal for both CH2OH groups in the molecule was also observed at 4.30 δ.
Not observed due to low concentration and small long-range 1H 13C couplings.
FIG. 5.

(A) 1H NMR COSY spectrum showing the scalar coupling network for the E- and Z-ascladiol isomers. (B) 1H NMR NOESY spectrum showing the dipolar coupling network for the E- and Z-ascladiol isomers. Cross-peaks due to the spatial proximity between CH2(7) and CH2(8) (a) and between CH(6) and CH2(8) (b) are selectively observed for the E and Z isomers, respectively.
The results of the capabilities of three out of the eight isolates (M3, M8, and M13) to transform increasing concentrations of patulin (10, 20, 40, 70, and 100 μg/ml) into ascladiol are reported in Table 3. All of the tested strains quantitatively transformed patulin at concentrations of up to 70 μg/ml, whereas at 100 μg/ml only the M3 and M8 strains were capable of degrading patulin at a similar level.
TABLE 3.
Reduction of patulin and formation of ascladiol caused by three isolates of G. oxydans (M3, M8, and M13) incubated with increasing concentrations of patulin in PDBa
| Amt of patulin added (μg/ml) | Residual patulin (μg/ml) for indicated isolate
|
Ascladiol (μg/ml) for indicated isolate
|
OD600 (A-U) for indicated isolate
|
||||||
|---|---|---|---|---|---|---|---|---|---|
| M3 | M8 | M13 | M3 | M8 | M13 | M3 | M8 | M13 | |
| None | 0 | 0 | 0 | 0 | 0 | 0 | 0.179 ± 0.05 | 0.203 ± 0.03 | 0.169 ± 0.03 |
| 10 | 0.19 ± 0.01 | 0.69 ± 0.04 | 2.07 ± 0.14 | 9.97 ± 0.61 | 8.65 ± 0.61 | 8.21 ± 0.49 | 0.130 ± 0.03 | 0.188 ± 0.02 | 0.110 ± 0.02 |
| 20 | 0.22 ± 0.02 | 1.39 ± 0.11 | 0.61 ± 0.05 | 21.87 ± 1.75 | 21.45 ± 1.77 | 20.35 ± 1.71 | 0.095 ± 0.08 | 0.167 ± 0.04 | 0.137 ± 0.01 |
| 40 | 1.64 ± 0.11 | 2.62 ± 0.18 | 2.15 ± 0.17 | 37.35 ± 1.87 | 40.38 ± 1.98 | 42.80 ± 2.79 | 0.153 ± 0.03 | 0.166 ± 0.03 | 0.107 ± 0.05 |
| 70 | 4.71 ± 0.36 | 6.31 ± 0.50 | 1.31 ± 0.09 | 67.06 ± 4.71 | 65.41 ± 4.84 | 63.36 ± 5.07 | 0.139 ± 0.04 | 0.176 ± 0.03 | 0.101 ± 0.07 |
| 100 | 9.26 ± 0.78 | 12.87 ± 1.03 | 102.1 ± 8.17 | 102.87 ± 8.23 | 109.34 ± 7.65 | 4.90 ± 0.35 | 0.127 ± 0.05 | 0.154 ± 0.05 | 0.053 ± 0.02 |
The mixture was incubated for 3 days at 30°C and 175 rpm. The cell growth of G. oxydans is reported as optical density at 600 nm (OD600). Each result is the mean ± the SE of three replicates. Negative controls containing patulin from 10 to 100 μg/ml without bacteria and incubated at the same experimental conditions used for the samples showed mean patulin recoveries ranging from 90 to 104%.
The time course of patulin degradation by G. oxydans M3 in commercial apple juice containing 100 μg/ml of patulin is shown in Fig. 6. It is evident that the reduction in patulin concentration corresponded to the appearance of ascladiol. The maximum rate of ascladiol formation was observed at 36 to 48 h, whereas quantitative transformation (91% degradation) occurred after 72 h. At higher concentrations (200 and 400 μg/ml), patulin was also quantitatively degraded (90% degradation), whereas at 800 μg/ml the transformation occurred in only one of the three replicates (85% degradation). The absence of patulin degradation corresponded to the absence of bacterial growth and vice versa. In particular, after 3 days of incubation, where transformation occurred the viable cell count was 107 CFU/ml in juice samples containing patulin at 100, 200, and 400 μg/ml and 105 CFU/ml in juice samples containing 800 μg/ml of patulin. Where no transformation occurred no viable cells were detected at the end of the incubation period (3 days) in the two replicates containing 800 μg/ml of patulin. When tested in YPM broth, G. oxydans M3 was able to quantitatively (90%) degrade 800 μg/ml of patulin in all three replicates.
FIG. 6.

Time course of patulin reduction and ascladiol formation on commercial apple juice spiked with patulin at 100 μg/ml, inoculated with G. oxydans, and incubated for 72 h at 30°C at 175 rpm. Each result is the mean ± the standard error (SE) of three replicates.
The effect of patulin (100 μg/ml) on the cell viability of G. oxydans incubated in commercial apple juice is more clearly shown in Fig. 7. It is evident that bacterial growth is affected by patulin. In particular, patulin-treated cells have a 6-h lag phase. In addition, while the control cells reached their maximum number after 24 h of incubation, the patulin-treated cells reached their maximum number after 48 h, with a significantly lower total cell number than that of the control. Interestingly, the last 12-h log phase of the treated cells (36 to 48 h) corresponded to the maximum rate of ascladiol formation (Fig. 6).
FIG. 7.

Comparison between G. oxydans growth with and without patulin at 100 μg/ml for 72 h at 30°C on apple juice. The cell count was performed by plating diluted aliquots of the samples on YPM agar plates. Each result is the mean ± the SE of three replicates.
The body of data reported above demonstrates that patulin transformation by the assayed G. oxydans M3 strain leads to the formation of ascladiol.
DISCUSSION
The transformation of patulin into E ascladiol was reported by Moss and Long (13), who used the commercial yeast Saccharomyces cerevisiae, in 2002. The transformation occurred under fermentative conditions, while only a limited transformation was observed when the culture was grown aerobically in shake flasks. The present work is the first report on bacterial biotransformation of patulin. This finding is noteworthy, considering that patulin has a broad antibacterial spectrum and is active against both gram-positive and gram-negative microorganisms at concentrations of 1 to 5 μg/ml (18). All the bacterial isolates tested in this study were able to grow and degrade patulin at a concentration of 10 μg/ml. The origin of these bacteria (i.e., from rotted apples containing patulin) could explain this characteristic, since the presence of patulin could be a selective factor on the apple surface for these strains. This could be the reason why other bacteria were not isolated from this sample. The M3 strain was able to grow and degrade much-higher patulin concentrations (i.e., 400 to 800 μg/ml), even though we cannot exclude the possibility that the M8 strain may also degrade patulin at the same concentration. The M3 strain was chosen because it belongs to the most-representative group of patulin-degrading isolates. Moreover, the M3 strain showed the same degradation capability as M8 with lower cell proliferation.
The M13 isolate, probably being more susceptible to the bacteriostatic activity of patulin than the M3 or M8 isolates (Table 3), was not able to grow and did not significantly degrade the mycotoxin at 100 μg/ml. Moreover, no significant formation of ascladiols was observed, which indicates that ascladiol is a result of patulin degradation produced by growing G. oxydans.
The ability of G. oxydans to transform patulin was maintained even when the strains were serially subcultured on patulin-free media, suggesting that this is a stable characteristic of these strains. Since the patulin concentration did not significantly decrease in either the spiked synthetic liquid medium or the apple juice incubated without G. oxydans, it is reasonable to conclude that the formation of ascladiol can be ascribed to the biotransformation of patulin operated by G. oxydans. The products of patulin degradation isolated in this study were a mixture of E ascladiol and Z ascladiol in a 3.5:1 ratio, as determined by NMR. E ascladiol has been reported to be a direct precursor of patulin, whereas Z ascladiol has been reported to be the product of a nonenzymatic transformation of E ascladiol catalyzed by the sulfhydryl compounds homocysteine, cysteine, and glutathione or dithiothreitol (19). The results of this study seem to confirm these findings. It seems that G. oxydans produces only E ascladiol as the transformation product of patulin, whereas the Z ascladiol observed in this study is the product of a nonenzymatic transformation of E ascladiol. In fact, only E ascladiol, not Z ascladiol, was observed in the HPLC chromatograms recorded during the time course experiments, when the transformation of patulin started. The transformation of patulin to ascladiol observed in this study was quantitative, and no additional metabolites were noted in the HPLC chromatograms of either synthetic liquid medium or apple juice spiked with patulin and incubated with G. oxydans. Ascladiol maintains the chromophore characteristics of patulin (i.e., the furan ring and the double-bond position), which explain the similarities in their UV spectra, whereas the ipsochromic shift from 275 nm to 268 nm could be explained by the opening of the pyran ring.
Although patulin reduced the growth of G. oxydans at 100 μg/ml, as shown in Fig. 7, its ability to transform patulin into ascladiol was not affected. In the presence of patulin, the maximum growth of G. oxydans occurred between 24 and 48 h, which corresponds to the quantitative transformation of patulin into ascladiol, as shown in Fig. 6.
At high patulin concentrations (800 μg/ml), G. oxydans M3 performed better in YPM than in commercial apple juice, suggesting that YPM is a better synthetic medium for G. oxydans, even though it also grows actively and degrades patulin at concentrations of up to 400 μg/ml in apple juice.
E ascladiol has been reported to be a precursor of patulin in the biosynthetic pathway of this mycotoxin and to be produced by Aspergillus clavatus isolated from wheat flour (19, 21). The toxicity of ascladiol has not been fully investigated, since only acute toxicity has been reported, amounting to only one-fourth of the strength of patulin (21, 12). G. oxydans M3 could be used to produce adequate quantities of ascladiols so that exhaustive studies of its toxicity could be carried out.
Within the Acetobacteriaceae, there are three genera: Gluconobacter, Gluconacetobacter, and Acetobacter (23, 17). The genus Gluconobacter, whose taxonomy is at present under worldwide study, is made up of five different species (20, 22) that do not show health risks and are commonly used in food manufacturing. Gluconobacter species are usually unable to oxidize acetate and lactate to carbon dioxide, unlike Acetobacter and many Gluconacetobacter strains (3). Gluconobacter species, mainly G. oxydans strains, incompletely oxidize many substrates, such as sugars, alcohols, and polyols (4). In this study, patulin was reduced to E ascladiol, and this represents a new kind of transformation by G. oxydans, leading to opening of the pyran ring.
This is the first report of patulin degradation produced by G. oxydans. The data in this study extend our knowledge of the sources of patulin degradation and confirm that ascladiol plays a role in the degradation pathway of patulin, in addition to the biosynthetic one. Work is in progress to ascertain whether the degradation of patulin into ascladiol produced by G. oxydans is an enzymatically driven process. In terms of practical applications, this bacterium seems to have the potential to detoxify patulin-contaminated fruit juice. Apple juice inoculated with this bacterium and incubated for 3 days still tasted like juice and was drinkable. However, use of this bacterium at the industrial level must follow a full examination of the potential toxicity of ascladiol in order to exclude any risk to human health.
Acknowledgments
We thank Veronica Lattanzio for LC-MS-MS analyses of ascladiol and patulin, Leonardo Caputo for advice on the isolation of G. oxydans from apple purée, and Stanislao Mastrorosa for providing apples with rot symptoms.
Footnotes
Published ahead of print on 17 November 2006.
REFERENCES
- 1.Andersen, B., J. Smedsgaaed, and J. C. Frisvad. 2004. Penicillium expansum: consistent production of patulin, chaetoglobusin, and other secondary metabolites in culture and their natural occurrence in fruit products. J. Agric. Food Chem. 52:2421-2428. [DOI] [PubMed] [Google Scholar]
- 2.Baruzzi, F., M. Morea, A. Matarante, and P. S. Cocconcelli. 2000. Changes in the Lactobacillus community during ricotta forte cheese natural fermentation. J. Appl. Microbiol. 89:807-814. [DOI] [PubMed] [Google Scholar]
- 3.De Ley, J., M. Gillis, and J. Swings. 1984. The genus Gluconobacter, p. 267-278. In N. R. Krieg and J. G. Holt (ed.), Bergey's manual of systematic bacteriology, vol. 1. Williams and Wilkins, Baltimore, MD. [Google Scholar]
- 4.Deppenmeier, U., M. Hoffmeister, and C. Prust. 2002. Biochemistry and biotechnological applications of Gluconobacter strains. Appl. Microbiol. Biotechnol. 60:233-242. [DOI] [PubMed] [Google Scholar]
- 5.Draughon, F. A., and J. C. Ayres. 1980. Insecticide inhibition of growth and patulin production in Penicillium expansum, Penicillium urticae, Aspergillus clavatus, Aspergillus terreus and Byssochlamis nivea. J. Agric. Food Chem. 28:115-1117. [Google Scholar]
- 6.Friedman, L. 1990. Patulin: mycotoxin or fungal metabolite? Biodeterioration Res. 3:21-54. [Google Scholar]
- 7.Frisvad, J. C., and U. Thrane. 1996. Mycotoxin production by food-borne fungi, p. 251-260. In R. A. Samson, E. S. Hoekstra, J. C. Frisvad, and O. Filtenborg (ed.), Introduction to food-borne fungi, 5th ed. Centraalbureau voor Schimmelcultures, Baarn, The Netherlands.
- 8.Klijn, N., A. H. Weerkamp, and W. M. de Vos. 1995. Detection and characterization of lactose-utilizing Lactococcus spp. in natural ecosystems. Appl. Environ. Microbiol. 61:788-792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.MacDonald, S., M. Long, J. Gilbert, and I. Felgueiras. 2000. Liquid chromatography method for determination of patulin in clear and cloudy apple juices and apple puree: collaborative study. J. AOAC Int. 83:1387-1394. [PubMed] [Google Scholar]
- 10.Majerus, P., and K. Kapp. 2002. Assessment of dietary intake of patulin by the population of EU member states. Reports on tasks for scientific cooperation, task 3.2.8. SCOOP report, Brussels, Belgium. http://ec.europa.eu/food/chemicalsafety/contaminants/3.2.8_en.pdf.
- 11.Moake, M. M., O. I. Padilla-Zakour, and R. W. Worobo. 2005. Comprehensive review of patulin control methods in foods. Comp. Rev. Food Sci. Food Safety 1:8-21. [DOI] [PubMed]
- 12.Moss, M. O. 1998. Recent studies of mycotoxins. J. Appl. Microbiol. Sympos. Suppl. 84:62S-76S. [DOI] [PubMed] [Google Scholar]
- 13.Moss, M. O., and M. T. Long. 2002. Fate of patulin in the presence of the yeast Saccharomyces cerevisiae. Food Addit. Contam. 19:387-399. [DOI] [PubMed] [Google Scholar]
- 14.Northolt, M. D., H. P. Van Egmond, and W. E. Paulsch. 1979. Patulin production by some fungal species in relation to water activity and temperature. J. Food Prot. 41:885-890. [DOI] [PubMed] [Google Scholar]
- 15.Piemontese, L., M. Solfrizzo, and A. Visconti. 2005. Occurrence of patulin in conventional and organic fruit products in Italy and subsequent exposure assessment. Food Addit. Contam. 22:437-442. [DOI] [PubMed] [Google Scholar]
- 16.Pittet, A. 1998. Natural occurrence of mycotoxins in food and feeds: an updated review, p. 479-492. In J. L. Bars and P. Galtier (ed.), Mycotoxins in the food chain: processing and toxicological aspects. Revue de Medecine Veterinaire, Toulouse, France.
- 17.Ruiz, A., M. Poblet, A. Mas, and J. M. Guillamon. 2000. Identification of acetic acid bacteria by RFLP of PCR-amplified 16S-23S rDNA intergenic spacer. Int. J. Syst. Evol. Microbiol. 50:1981-1987. [DOI] [PubMed] [Google Scholar]
- 18.Scott, P. M. 1974. Patulin, p. 383-403. In Mycotoxins. Health Protection Branch, Department of Health and Welfare, Ottawa, Canada.
- 19.Sekiguchi, J., T. Shimamoto, Y. Yamada, and G. M. Gaucher. 1983. Patulin biosynthesis: enzymatic and nonenzymatic transformations of the mycotoxin (E)-Ascladiol. Appl. and Environ. Microbiol. 45:1939-1942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sievers, M., I. C. Gaberth, C. Boesch, W. Ludwig, and M. Teuber. 1995. Phylogenetic position of Gluconobacter species as a coherent cluster separated from all Acetobacter species on the basis of 16S ribosomal RNA sequences. FEMS Microbiol. Lett. 126:123-126. [DOI] [PubMed] [Google Scholar]
- 21.Suzuki, T., T. Takeda, and H. Tanabe. 1971. A new mycotoxin produced by Aspergillus clavatus. Chem. Pharm. Bull. 19:1780-1788. [DOI] [PubMed] [Google Scholar]
- 22.Tanasupawat, S., C. Thawai, P. Yukphan, D. Moonmangmee, T. Itoh, O. Adachi, and Y. Yamada. 2004. Gluconobacter thailandicus sp. nov., an acetic acid bacterium in the a-Proteobacteria. J. Gen. Appl. Microbiol. 50:159-167. [DOI] [PubMed] [Google Scholar]
- 23.Yamada, Y., K. Hoshino, and T. Ishikawa. 1997. The phylogeny of acetic acid bacteria based on the partial sequences of 16S ribosomal RNA: the elevation of the subgenus Gluconoacetobacter to the generic level. Biosci. Biotechnol. Biochem. 61:1244-1251. [DOI] [PubMed] [Google Scholar]
- 24.Wickerham, L. J. 1951. Taxonomy of yeast. Technical bulletin 1029. U.S. Department of Agriculture, Washington, DC.
- 25.Wouters, M. F. A., and G. J. A. Speijers. 1996. Patulin food additives, p. 337-402. In Toxicological evaluation of certain food additives and contaminants. WHO food additives series 35. World Health Organization, Geneva, Switzerland. http://www.inchem.org/documents/jecfa/jecmono/v35je16.htm.