

Abstract
Geobacillus stearothermophilus T-6 utilizes an extensive and highly regulated hemicellulolytic system. The genes comprising the xylanolytic system are clustered in a 39.7-kb chromosomal segment. This segment contains a 6-kb transcriptional unit (xynDCEFG) coding for a potential two-component system (xynDC) and an ATP-binding cassette (ABC) transport system (xynEFG). The xynD promoter region contains a 16-bp inverted repeat resembling the operator site for the xylose repressor, XylR. XylR was found to bind specifically to this sequence, and binding was efficiently prevented in vitro in the presence of xylose. The ABC transport system was shown to comprise an operon of three genes (xynEFG) that is transcribed from its own promoter. The nonphosphorylated fused response regulator, His6-XynC, bound to a 220-bp fragment corresponding to the xynE operator. DNase I footprinting analysis showed four protected zones that cover the −53 and the +34 regions and revealed direct repeat sequences of a GAAA-like motif. In vitro transcriptional assays and quantitative reverse transcription-PCR demonstrated that xynE transcription is activated 140-fold in the presence of 1.5 μM XynC. The His6-tagged sugar-binding lipoprotein (XynE) of the ABC transporter interacted with different xylosaccharides, as demonstrated by isothermal titration calorimetry. The change in the heat capacity of binding (ΔCp) for XynE with xylotriose suggests a stacking interaction in the binding site that can be provided by a single Trp residue and a sugar moiety. Taken together, our data show that XynEFG constitutes an ABC transport system for xylo-oligosaccharides and that its transcription is negatively regulated by XylR and activated by the response regulator XynC, which is part of a two-component sensing system.
The natural degradation of the plant cell wall polysaccharides is a key step in the carbon cycle on Earth and is mediated mainly by microorganisms. The efficient breakdown of these polymers, cellulose and hemicellulose, is of great interest due to the need for renewable energy sources that contribute no net CO2 emission to the atmosphere (18, 53). Hemicelluloses, which can comprise up to 35% of the total cell wall dry mass, are a heterogeneous group of branched and linear polysaccharides bound to the cellulose microfibrils in the plant cell wall via hydrogen bonds, cross-linking them into a robust network (15, 25). Plant cell wall-degrading microorganisms utilize an impressive variety of modular enzyme structures and different physiological strategies for the degradation of the plant cell wall. Anaerobic bacteria, such as Clostridium spp., have evolved unique multienzyme complexes, called cellulosomes, that integrate many cellulolytic and hemicellulolytic enzymes that mediate the attachment of the cell to the crystalline polymer and its controlled hydrolysis (2, 24, 65, 71). Aerobic fungi, such as Trichoderma and Aspergillus, secrete numerous cellulases and hemicellulases that work synergistically to completely degrade the polymers into mono- or disaccharides which may be utilized by the surrounding microorganisms (14). Lastly, aerobic bacteria such as Bacillus and Cellvibrio (formerly Pseudomonas fluorescens subsp. cellulosa) secrete only a limited number of extracellular polysaccharide backbone-degrading enzymes that yield relatively large oligosaccharides. These oligosaccharides enter the cell via specific transporters, and their final breakdown is carried out by cell-associated or intracellular enzymes (5, 47, 73). This latter strategy has the advantage that the extracellular soluble products are not easily available to competing, nonhemicellulolytic microorganisms.
Although many hemicellulolytic enzymes have been studied extensively (70), very little is known about the sensing systems for the hemicellulolytic products and their uptake systems. Several microbial transport systems, including UhpT (81, 82), DcuB (88), UpgP (31, 48), and YxdLM (34), have been shown to be regulated by two-component systems (TCSs). This type of regulation enables the cell to respond to environmental or intracellular signals and to alter gene expression. TCSs comprise a sensor histidine protein kinase and a response regulator, usually found in separate proteins. The kinase, typically a membrane protein, becomes autophosphorylated at a conserved histidine residue using ATP as a substrate, in response to a specific signal. The phosphoryl group is then transferred to a conserved aspartate residue on the response regulator, altering its ability to bind target DNA sequences (75).
Geobacillus stearothermophilus T-6 is a soil bacterium that possesses a highly efficient and complete hemicellulolytic system. The 30 genes comprising the system appear to be organized in at least nine transcriptional units within a 39.7-kb chromosomal segment. When grown in the presence of xylan, strain T-6 secretes a single extracellular endo 1,4-β xylanase (XynA) that hydrolyzes the polymer's main backbone, producing short modified oligoxylose units of two or more sugars in length. These modified xylosaccharides enter the cell by specialized ATP-binding cassette (ABC) sugar transporters (73), and they are further degraded to monomers by intracellular hydrolases, including a glycoside hydrolase family 10 (GH10) xylanase (77), two GH51 α-l-arabinofuranosidases (29), an α-glucuronidase (GH67) (23, 68, 89), three β-xylosidases (GH39, GH43, and GH52) (6, 8-11), and two xylan acetyl esterases (CE4).
In this study, we demonstrate that in G. stearothermophilus strain T-6, a two-component sensing system, encoded by xynDC, positively regulates the expression of an adjacent ABC transporter system (xynEFG) designed for the uptake of xylo-oligosaccharides.
MATERIALS AND METHODS
Bacterial strains and plasmids.
G. stearothermophilus T-6 (NCIMB 40222) was isolated following an enrichment procedure for microbial strains capable of producing alkaline-tolerant, extracellular, thermostable xylanases (35, 72). Escherichia coli strains used were XL1-Blue (Stratagene) for general cloning and BL21(DE3) (Novagen, Madison, WI) for expression via the T7 RNA polymerase expression system with pET11d or pET9d (Novagen, Madison, WI).
Growth conditions.
Growth medium for G. stearothermophilus was basic salt medium (BSM) supplemented with 0.5% glucose or xylose. BSM contained the following per liter: KH2PO4, 0.4 g; MgSO4 · 7H2O, 0.1 g; (NH4)2SO4, 2 g; MOPS (N-morpholinepropanesulfonic acid) buffer, pH 7.0, 10.4 g; and trace elements solution, 4 ml. Trace elements solution contained the following per liter: CaCl2 · 7H2O, 0.92 g; FeSO4 · 7H2O, 1.51 g; MnSO4 · 4H2O, 0.148 g; ZnSO4 · 7H2O, 0.105 g; and CuSO4 · 5H2O, 0.156 g. The pH of the trace elements solution was adjusted to 2.0 with sulfuric acid.
DNA and RNA isolation and manipulation.
G. stearothermophilus T-6 genomic DNA was isolated by the procedure of Marmur (40) as outlined by Johnson (32). Plasmid DNA was purified using a QIAGEN plasmid kit (QIAGEN Inc., Chatsworth, CA). DNA was manipulated by standard procedures (58). The xynDCEFG coding sequence was revealed by chromosome walking, upstream of the previously reported xynB2 gene (73), using the inverse PCR method (49). Total RNA was isolated with an RNeasy kit (QIAGEN) according to the supplied protocol.
DNA sequencing and analysis.
DNA sequencing was performed on a Perkin-Elmer 377 automated sequencer (Weizmann Institute, Rehovot, Israel). Nucleotide and amino acid sequences were analyzed with either GeneRunner or the sequence analysis software package of the Genetics Computer Group, version 9 (University of Wisconsin, Madison, WI). Sequence homologies were searched with FASTA (50) or BLAST (3) algorithms. Protein sequences were analyzed with the Expert Protein Analysis System (ExPASy) proteomics server (http://www.expasy.org) of the Swiss Institute of Bioinformatics (SIB).
Cloning, production, and purification of G. stearothermophilus T-6 XylR.
The xylR gene was cloned via PCR using G. stearothermophilus T-6 chromosomal DNA as a template. The primers (Table 1) were designed based on the homologous gene from G. stearothermophilus 10 (http://www.genome.ou.edu/bstearo.html). The PCR product was cloned into the pET11d expression vector and thereby fused to a His6 tag at the N terminus of its coding sequence. Attempts to express the XylR protein in E. coli resulted in very low yields of soluble protein. To improve the solubility, growth was carried out at 18°C in the presence of 0.04 mM IPTG (isopropyl-β-d-thiogalactopyranoside) for 24 h. The XylR protein was purified from a crude cell extract by Ni+-nitrilotriacetic acid (NTA) chromatography, yielding 1.5 mg purified enzyme from a 1-liter culture. The purified XylR protein was dialyzed overnight against 2 liters of buffer containing 50 mM Tris-HCl, pH 7.0, and 100 mM KCl, followed by the addition of EDTA and glycerol to final concentrations of 1 mM and 10%, respectively.
TABLE 1.
Oligonucleotides used in this study
| Primer | Sequence (5′-3′)a | Application |
|---|---|---|
| xylR N-ter | CCAATTGCATATGCCATGGCTCACCATCATCACCATCAC AATAAACAGCTCGTCTTAAAATTG | Cloning into T7 expression vectors |
| xylR C-ter | ACGTGCTAAGCTGGATCCGCCTCTTTTCGCATGCGCT | |
| xynC N-ter | GTTAACGGAGGTGAAAAACCATGGATCATCACCATCACCATCACGAGAAGACCATTTTAGTTG | |
| xynC C-ter | GAAAAGTACCAGTATTGGATCC TTACTTTTCTTTTTGAC | |
| xynE N-ter | GTTCCTCAAATCCATGGCTCATCATCATCATCATTCTTCTTCGTCGAGCGATTC | |
| xynE C-ter | GGCTGCAGGGATCCTTATTTGTTGTTGGCTTCCTCCGCC | |
| ABC C-ter1 | CATAAACTTTACCCCCTTCTCTATTCTTTG | Determination of transcriptional start points |
| HKCW2 | CAGCTACTCCATGATCCATTCTACCG | |
| 1IR_TCS | CTAATCAACCTTTGTTTATATTCTATACAAATCATTTCAGAG | DNA-binding and footprinting assays |
| 2IR_TCS | CTCTGAAATGATTTGTATAGAATATAAACAAAGGTTGATTAG | |
| ABC N-ter | CAGAGATTGCGGAAGAAGCTTGTTACAAAAC | |
| ABC C-ter | CATAAACTTTACCCCCATGGCTATTCTTTG | |
| Primer1 | TTAAAAAGAAAAGTAATCTAAAAATACTGGTACTTTTCCTAAT | |
| Primer2 | TTATTAGGAAAAGTACCAGTATTTTTAGATTACTTTTCTTTTT | |
| INV2 | CGTGATGTTCATCTCCATTGG | In vitro transcription assays |
| IVT1 xynA | GTGCTGTCCGTTTTCACTG | |
| IVT2 xynA | GAAGCGAATATCCATGCC | |
| RT-PCR 1xynE | GCAAGCAAAGAATAGAGAAGG | Real-time RT-PCR |
| RT-PCR 2 xynE | GAAGAACAACCAGACAGCACAATC | |
| RT-PCR 1 xynA | GCGGCAGTAGAACCTTA | |
| RT-PCR 2 xynA | AGGTTGAATGCTGATCG |
Boldface bases indicate engineered restriction sites.
Cloning, production, and purification of His6-tagged XynE and XynC.
The xynE and xynC genes were cloned into pET11d and pET9d vectors (Novagen), respectively, yielding plasmids pET11d-xynE and pET9d-xynC. All the primers were designed to allow in-frame cloning of the genes into the T7 polymerase expression vector by use of an NcoI restriction site at the 5′ terminus and a BamHI restriction site at the 3′ terminus (Table 1). The N-terminal primers contained six histidine codons to provide His6-fused products. DNA sequencing verified the sequences of the cloned genes. E. coli BL21(DE3)(pET9d-xynC) cultures were grown overnight in Terrific broth (58) with kanamycin (25 μg ml−1) and E. coli BL21(DE3)(pET11d-xynE) with ampicillin (100 μg ml−1) without induction (1× 500 ml in 2-liter shake flasks) at 37°C. The cultures were harvested, resuspended in 30 ml of buffer (10 mM imidazole, 50 mM Tris-HCl, 100 mM NaCl, pH 7.0), disrupted by two passages through a French press (Spectronic Instruments, Inc., Rochester, NY), and centrifuged (14,000 × g for 15 min) for the production of clear crude protein extracts.
The His-tagged proteins were isolated using Ni+-NTA resin (QIAGEN) at a ratio of 2 ml of resin per 50 ml of supernatant fluid. The resin was loaded onto a 0.8- by 4-cm plastic column (Bio-Rad) and was then washed with 2 column volumes of binding buffer (300 mM Tris-HCl buffer, pH 7.5, 300 mM KCl, and 20 mM imidazole). Proteins were eluted using elution buffer supplemented with 350 mM imidazole. The purified XynC protein was dialyzed and stored as described for XylR above. Samples (100 μl) of the purified proteins were stored at −80°C until further use.
Transcriptional analyses.
Transcriptional analyses were performed on total RNA extracted from exponentially growing cells. Primer extension reactions were conducted as described previously (44) with avian myeloblastosis virus reverse transcriptase (Promega), 40 μg of total RNA, and the primers listed in Table 1. Northern blot analysis was conducted following the procedure described by Moran (44).
Mobility shift DNA-binding assays.
Five unique radioactive DNA probes were used for gel retardation assays. For His6-XylR, a 42-bp double-stranded DNA fragment containing the putative xyl operator (from position −25 to position −5 relative to the transcriptional start site of the xynDC operon) was prepared. The double-stranded probe was composed of two synthetic complementary oligonucleotides (Table 1) designed to have two noncomplementary T nucleotides at the 5′ end for end labeling with Klenow fragment (Fermentas) in the presence of [α-32P]dATP.
For His6-XynC, three DNA fragments of 220, 140, and 121 bp corresponding, respectively, to positions −120 to +100, −120 to +20, and −21 to +100 with respect to the transcriptional start site of xynE were generated via PCR with primers listed in Table 1. These PCR products were purified with a Wizard SV gel PCR clean-up system (Promega) and end labeled using [γ-32P]ATP and T4 polynucleotide kinase (New England Biolabs) as described by the manufacturer. A fourth probe was a 41-bp double-stranded DNA fragment containing a putative inverted repeat (from position −21 to position +20 relative to the transcriptional start site of the xynE operon). The double-stranded probe was composed of two synthetic complementary oligonucleotides (Table 1) designed to have two noncomplementary T nucleotides at the 5′ end for end labeling with Klenow fragment (Fermentas) in the presence of [α-32P]dATP.
The binding reaction mixture (30-μl total volume) contained 20 μl of solution comprised of 50 mM Tris-HCl (pH 7.5), 100 mM KCl, 10% glycerol, 1 mM EDTA, 2 μg of salmon sperm DNA, 0.66 mM dithiothreitol, 33 μg of bovine serum albumin, 0.08 ng of labeled probe (about 50,000 cpm), and the indicated amount of protein. The binding mixture was incubated for 30 min at 45°C and then separated on a 6.6% nondenaturing polyacrylamide gel prepared in Tris-borate-EDTA buffer (58). Gels were dried under vacuum and exposed to a phosphorimager screen before analysis with a Fuji BAS2000 phosphorimager.
DNase I footprinting assay.
The probes for DNase I footprinting assays were PCR products corresponding to positions −100 to +120 relative to the xynE transcriptional start site. Labeled DNA fragments were prepared via PCR using two primers, ABC N-ter and ABC C-ter, which were designed to contain HindIII and NcoI restriction sites, respectively (Table 1). The PCR product was purified from 4% MetaPhor agarose gel (Bio Science Rockland, Rockland, ME). Digestion with NcoI or HindIII permitted end labeling of the coding or noncoding strands, respectively. Labeled DNA fragments (400,000 cpm) were incubated for 30 min at 45°C in 30 μl of the binding reaction mixture described above with various concentrations of XynC. After adjusting the MgCl2 and CaCl2 concentrations to 16 mM and 5 mM, respectively, DNA-protein complexes were treated with 1.2 units of RQ1 DNase I (Promega) for 1 min at room temperature and the digest was stopped by addition of EDTA to 30 mM. The samples were purified using a QIAquick PCR purification kit (QIAGEN) and eluted with 30 μl of 10 mM Tris-HCl (pH 8.5) followed by 10 μl of formamide-loading buffer (95% [vol/vol] formamide, 20 mM EDTA, 0.05% [wt/vol] bromophenol blue, 0.05% [wt/vol] xylene cyanol FF). After being heated for 5 minutes at 90°C, the samples were applied to a denaturing 6% polyacrylamide gel containing 7 M urea. Sanger sequencing reaction products of the same fragments with the primers indicated above and α-35S-labeled dATP were loaded on the same gel (Sequenase version 2.0 DNA sequencing kit; Amersham Biosciences). After electrophoresis, the gel was dried and visualized using a Fuji BAS2000 PhosphorImager.
Preparation of G. stearothermophilus T-6 cell extract.
Cell extract from strain T-6 was prepared for the in vitro transcription reaction, which was followed by quantitative real-time PCR. Strain T-6 cell culture was grown at 55°C in Terrific broth medium (58) (0.5 liters medium in 2.5-liter baffled shake flask) to an optical density at 600 nm of 3.5. After growth, the culture was left on ice for 25 min and then harvested by centrifugation (10,000 × g, 10 min). The cell pellet was washed once with 500 ml of cold, diethyl pyrocarbonate-treated S-30 buffer (10 mM Tris-acetate, pH 7.4, 1 mM dithiothreitol, 14 mM magnesium acetate, 60 mM potassium acetate, and 7 mM 2-mercaptoethanol per liter of freshly added buffer). The washed pellet was resuspended in 10 ml of cold S-30 buffer, and the cells were broken by one passage through a French press (Spectronic Instruments, Inc., Rochester, NY). Following centrifugation (14,000 × g for 30 min), dithiothreitol was added to a final concentration of 1 mM to the clear supernatant. Aliquots (100 μl) were frozen immediately in liquid nitrogen and stored at −80°C for the in vitro transcription experiments.
In vitro transcription reaction.
Template DNAs (800 bp) of the target gene (xynE) and of the reference (ref) gene (xynA, encoding T-6 extracellular xylanase) were amplified by PCR using specific primers (Table 1) and T-6 chromosomal DNA as the template. The in vitro transcription reaction mixture (33 μM of each nucleoside triphosphate, 4 μl of RNasin [Promega], 12 μl of commercial 5× RNA polymerase buffer [Fermentas] to a final volume of 60 μl) was kept on ice for 10 min. Fifteen microliters of fresh strain T-6 cell extract and the indicated amounts of the response regulator, His6-XynC, were added. The reaction was initiated by the addition of 5 μl of template DNAs (1 μg/μl) of the target gene and the reference gene, and the mixture was incubated at 45°C for 50 min. Following incubation, the template DNA was digested with 25 units of RQ1 DNase I (Promega) for 25 min at 37°C. mRNA transcripts were purified with an RNeasy Mini kit (QIAGEN) as outlined by the manufacturer. The mRNA level transcribed in the in vitro transcription reactions was determined following reverse transcription and quantitative real-time PCR as described below.
RT and quantitative real-time PCR.
Reverse transcription (RT) was performed following the manufacturer's protocol (ABgene, United Kingdom) by use of a first-strand synthesis kit and 2 μl of purified mRNA obtained from the in vitro transcription assays. In order to check for DNA contamination, control reactions were carried out in the absence of reverse transcriptase. Real-time RT-PCR primers were designed with the aid of the Primers3 program (http://www-genome.wi.mit.edu/cgi-bin/primer/primer3_www.cgi) to generate amplicons ranging in size from 100 to 150 bp (Table 1). Gene quantification was performed with a Rotor-Gene 3000 (Corbett Research, Sydney, Australia) instrument. Each 25-μl reaction mixture included template cDNA, 180 nM of each primer, and ABsolute 2× SYBR green mix (ABgene) containing SYBR green, deoxynucleoside triphosphates, MgCl2, and Thermo-Start DNA polymerase. The activation (n-fold) of the target gene (xynE) was determined based on its real-time PCR efficiencies (E) and the crossing point difference (change in cycle threshold [ΔCT]) for a treated sample versus a control sample (51) and calculated using the following equation: activation (n-fold) = E(target)ΔCTtarget(control − treated)/E(ref)ΔCTref(control − treated). For each gene, cDNA dilution curves were generated and used to calculate the individual real-time PCR efficiencies: E = 10−1/slope. Typical efficiencies for xynE and xynA were 1.90 and 1.88, respectively. cDNA and RT negative controls were included in each run. Melt curves were analyzed to ensure specificity of primer annealing and lack of primer secondary structure.
Phosphorylation of His6-XynC.
Phosphorylation of His6-XynC was accomplished by following the protocol of Lukat et al. (39). The XynC protein (6 mg) was incubated in 10 ml of a buffer containing 100 mM Tris-HCl (pH 7.0), 5 mM MgCl2, 1 mM dithiothreitol, and 10 mM acetyl phosphate (Fluka).
Microcalorimetry titration studies.
Titration calorimetry measurements were performed with a VP-ITC calorimeter (Microcal; Northampton, MA) as described by Wiseman et al. (85). Protein solutions for isothermal titration calorimetry (ITC) were dialyzed extensively overnight against buffer A (50 mM Tris-HCl, pH 7.0, 100 mM NaCl, 0.02% NaN3). Ligand solutions of xylobiose (X2), xylotriose (X3), xylotetraose (X4), xylopentaose (X5), and xylohexaose (X6) (Megazyme; Wicklow, Ireland) were prepared by dilution with the protein dialysis buffer. Aliquots (10 μl) of the ligand solution at 8.5 to 20 times the binding-site concentration were added to the reaction cell containing 1.41 ml of the 0.02 to 0.1 mM protein solution by the controlled action of a 250-μl rotating stirrer-syringe. The heat of dilution was determined to be negligible in separate titrations of the ligand into the buffer solution. Calorimetric data analysis was carried out with ORIGIN 5.0 software (MicroCal). Binding parameters, including binding stoichiometry (n), the binding constant (KB [M−1]), and the binding enthalpy (ΔHB [kcal/mol of bound ligand]), were determined by fitting the experimental binding isotherms. KB was determined primarily by the slope of the isotherm at the equivalence point (17).
Nucleotide sequence accession number.
The 39,692-bp sequence of the xylan utilization region and the sequence of the xylR gene from G. stearothermophilus T-6 have been deposited in the GenBank under accession numbers DQ868502 and DQ868501, respectively.
RESULTS
For the cloning of the xylan utilization-related genes from G. stearothermophilus T-6, we combined two main strategies: screening of genomic libraries for hemicellulolytic activities (73) and PCR-based chromosome walking. By using those methods, the entire xylan utilization cluster has been cloned and sequenced. The cluster is 39.7 kb long and contains 30 open reading frames; many of the products have been characterized structurally and biochemically (6-8, 10, 11, 23, 29, 66-69, 76, 77, 89). In this cluster, a putative TCS, xynDC, lies just upstream of a putative ABC transport system (xynEFG). Similar combinations of a TCS and an adjacent transport system that is regulated by the TCS have been described for several systems (31, 34, 88), suggesting that the putative TCS encoded by xynDC regulates the ABC transport system (xynEFG).
Sequence analysis of the two-component system (xynDC) and the ABC transport system (xynEFG).
The putative xynD gene product exhibits characteristic features of bacterial histidine kinase proteins, including two transmembrane (TM) helices (TM1 residues 15 to 35 and TM2 residues 255 to 306) flanking an extracellular domain (residues 36 to 254) and a conserved C-terminal cytoplasmic region containing the ATP-binding kinase domain. Downstream to xynD lies xynC, which encodes a protein with strong similarity to response regulators. As in the case for many response regulators, the xynC gene product has a predicted two-domain architecture, with an N-terminal signal receiver domain linked to a C-terminal effector domain (55). The N-terminal signal receiver domain (positioned at residues 5 to 122) shares homology with the CheY superfamily (83), whereas the C-terminal domain from position 157 to position 225 contains a putative helix-turn-helix motif that resembles the AraC-type DNA-binding domain (20). The neighboring genes, xynEFG, are likely to code for an ABC transport system. XynE appears to correspond to an extracellular sugar-binding protein, with characteristic features of signal peptides of bacterial lipoproteins. Its N-terminal region contains a putative cleavage site with an almost perfect match to the consensus cleavage site sequence for the lipoprotein signal peptidase II (Leu-Ser-Gly/Ala↓Cys) (84). Hydropathy analyses for XynF and XynG show patterns of hydrophobic and hydrophilic regions that suggest that these proteins are likely to have membrane-spanning regions. Hydrophobic-moment analysis of XynF and XynG predict six transmembrane helices. The two proteins show homology to several integral cytoplasmic membrane proteins involved in sugar transport and contain a conserved hydrophilic segment with the consensus sequence, EAAX3GX9IXLP; this sequence is typical of integral membrane proteins from binding protein-dependent transport systems (27). On the basis of this signature, XynF and XynG can be assigned to the disaccharide subcluster of bacterial binding protein-dependent permeases. This subcluster contains proteins involved in the transport of short sugars and oligosaccharides. Thus, it is likely that the xynEFG cluster encodes three of the components of an ABC transport system for xylo-oligosaccharides. The gene for the ATP-binding protein is presumably located elsewhere on the chromosome.
The substrate-binding protein XynE binds xylo-oligosaccharides.
Based on sequence homology, xynE encodes a substrate-binding protein. In gram-negative bacteria, substrate-binding proteins are located in the periplasmic space, whereas in gram-positive bacteria, they were found to be linked to the cytoplasmic membrane by a lipid anchor (22, 80). These proteins interact with various substrates, such as sugars, amino acids, oligopeptides, and vitamins. The ability of the XynE protein to bind xylosaccharides was demonstrated using ITC (Fig. 1A). ITC provides a direct measure of binding enthalpy, ΔHB, and allows the simultaneous determination of the binding parameters, which include the binding constant (KB), entropy (ΔSB), the free energy of binding (ΔGB), and the binding stoichiometry (n). Thermodynamic binding parameters of xylo-oligosaccharides (X2 to X6) to XynE are summarized in Table 2, showing that all of the binding interactions were exothermic and enthalpy driven. The highest KB values were gained with xylotriose, suggesting that the binding protein XynE prefers trisaccharides. For other xylo-oligosaccharides, the order of binding affinities was as follows: X2<X3>X4>X5>X6. Large negative heat capacity (ΔCp) values are thought to be associated with hydrophobic stacking interactions and result from the dehydration of the aromatic side chains in the protein and the rings of the carbohydrate (38, 74, 78). From the thermodynamic parameters obtained at different temperatures, the heat capacity change (ΔCp = ΔHB/ΔT, where HB is binding enthalpy and T is temperature) for the binding reaction could be extracted. Plotting ΔHB versus temperature for xylotriose gave a linear correlation, indicating that ΔCp for binding is constant in the temperature range used (Fig. 1B). The yielded slope was ΔCp = −158 cal mole−1 K−1, suggesting one stacking interaction in the binding site that can be provided by a single Trp residue interacting with a sugar ring (90).
FIG. 1.

(A) Representative isothermal calorimetric titration curve of XynE with xylotriose. Titration calorimetry measurements were performed with a Microcal VP-ITC titration calorimeter (Microcal Inc., Northampton, MA). The top half shows the calorimetric titration of binding protein with ligand, and the lower half displays the integrated injection heats from the upper half, corrected for control dilution heats. The solid line is the curve of best fit that was used to derive binding parameters. (B) Changes in the binding enthalpies (ΔHB) at different temperatures for interaction of xylotriose with XynE. The value of ΔCp, −158 cal mole−1 K−1, was derived from the slope of the linear correlation.
TABLE 2.
Thermodynamic parameters of binding of xylo-oligosaccharides to XynE
| Ligand | T (°C) | KB × 106 (M−1) | KD (1/KB) (μM) | ΔHB (kcal/mol) | TΔSB (kcal/mol) | ΔGB (kcal/mol) |
|---|---|---|---|---|---|---|
| X2 | 30 | 2.3 ± 0.5 | 0.44 | −18.6 ± 0.2 | −9.8 | −8.8 |
| X3 | 10 | 21 ± 4.3 | 0.05 | −15.5 ± 0.2 | −6.1 | −9.4 |
| 20 | 9 ± 1.2 | 0.11 | −17.6 ± 0.1 | −8.2 | −9.4 | |
| 30 | 12 ± 2 | 0.08 | −20.0 ± 0.2 | −10.2 | −9.8 | |
| 40 | 10 ± 1.3 | 0.1 | −20.7 ± 0.2 | −10.7 | −10.0 | |
| 50 | 7.3 ± 0.8 | 0.14 | −21.8 ± 0.1 | −11.7 | −10.1 | |
| X4 | 30 | 8.3 ± 1.2 | 0.12 | −13.0 ± 0.1 | −3.4 | −9.6 |
| X5 | 30 | 1.8 ± 0.2 | 0.56 | −8.1 ± 0.1 | 0.6 | −8.7 |
| X6 | 30 | 0.7 ± 0.1 | 1.4 | −8.8 ± 0.4 | −0.7 | −8.1 |
Transcriptional analysis of the xynDCEFG cluster.
The xynDCEFG genes are all transcribed in the same direction and potentially constitute a polycistronic operon. The intergenic spacer region between xynD and xynC is only 3 bp long and lacks any obvious rho-independent hairpin-like secondary structure, suggesting that the two genes are cotranscribed. The space between xynC and xynE is 110 bp long and also lacks any obvious transcriptional terminator. However, this region does contain a potential promoter. Downstream from the last gene, xynG, there is a potential transcription terminator with a calculated ΔG of −4.2 kcal/mol followed by T nucleotides typical of a rho-independent terminator. To determine whether the xynDCEFG genes are cotranscribed, Northern blot analysis was performed. Total RNA was isolated from T-6 cultures grown in the presence or absence of xylose (a potential inducer) and annealed to a DNA probe for xynD. As shown in Fig. 2A, the probe hybridized to RNAs of many different sizes, with a maximum length of about 6 kb. This mRNA size is close to the calculated size (5.95 kb) of the presumed xynDCEFG transcript, suggesting that the five genes are cotranscribed from the xynD promoter. Hybridization to smaller RNA species is presumed to reflect, at least in part, the degradation of the 6-kb transcript. No hybridization was detected with mRNA from cultures grown without xylose, suggesting that xylose can function as a molecular inducer. Using primer extension analysis, the apparent transcriptional start point of the xynD promoter was identified and assigned to a T nucleotide 205 bases upstream from the ATG initiation codon of xynD (Fig. 2B). As expected, no extension product was detected with mRNA from glucose-grown cultures. The apparent −35 sequence, TTCTAA, differs from the Bacillus subtilis consensus sequence, TTGACA, at three positions and is separated by 17 bp from the potential −10 region, TCTATA, which differs by 3 nucleotides from the σA consensus sequence, TATAAT (26, 30, 45). An inverted repeat sequence (5′-TTTGTTTATATTCTATACAAA-3′) resembling the xylR operator sites from Bacillus and other gram-positive bacteria (56) overlaps the −10 region, suggesting that the xynD promoter is negatively regulated by XylR (Fig. 2C). Two potential operator sites for catabolite-responsive regulation (catabolite-responsive elements [CREs]) are located between the promoter and the ATG initiation codon (41, 42) (Fig. 2C).
FIG. 2.

Transcriptional analyses of the xynDC. (A) Determination of the xynDC transcript length using Northern blot analysis. Total RNA was extracted from mid-exponential-phase cultures of G. stearothermophilus T-6 grown in BSM supplemented with 0.5% glucose (lane 1) as the sole carbon source or with 0.5% xylose (lane 2). The RNA was subjected to electrophoresis, transferred to nitrocellulose (BA85; Schleicher & Schuell), and annealed to [32P]DNA for xynD. (B) Mapping the 5′ termini of the xynDC cluster by primer extension analysis. Total RNA was extracted as mentioned above. Extension products resulting from RNA obtained from cultures grown on 0.5% xylose are shown in lane 1 or with 0.5% glucose as the sole carbon source (lane 2). Dideoxynucleotide sequence reactions were carried out with the same primer used for the reverse transcriptase reactions. The position of the transcriptional start point is indicated with an asterisk on the inferred nontemplate strand sequence. (C) Sequence data for the regulatory region. The transcriptional start point (+1) is indicated by a vertical arrowhead. The −35 and −10 regions, the proposed ribosome-binding site (RBS), the initiating methionine codon, and the potential CREs are in boldface. The CRE sequence is TGT/AAANC GNTNA/TCA, where underlined letters represent the most critical bases, N is any base, and the vertical line denotes an axis of symmetry (41). The xylR operator is indicated by horizontal arrowheads above the inverted repeat.
XylR binds the xynD promoter.
We have recently cloned and sequenced the xylose repressor gene, xylR, from G. stearothermophilus T-6 via PCR based on the homologous gene from strain G. stearothermophilus 10 (37). The His6-tagged gene product was expressed in E. coli, and the purified protein was used for gel mobility shift assays. The electrophoretic mobility of a 42-bp DNA fragment containing the putative xynD operator sequence was retarded when the fragment was incubated with 1 μM His6-XylR (Fig. 3A). To test whether xylose can act as the inducer of the xynDC operon, the binding of XylR to the xyl operator was assayed in the presence of d-xylose or d-arabinose. Binding was prevented in the presence of 10 mM xylose, while l-arabinose showed no effect (Fig. 3B). These results are consistent with the Northern blot analysis indicating that xylose can act as the molecular inducer of the xynD promoter.
FIG. 3.

Gel retardation of a 32P-labeled xynD operator fragment by His6-XylR. (A) All lanes contained about 0.08 ng of radioactively labeled DNA fragment containing the synthetic xynD operator. Lane 1 contained no protein; lanes 2 to 4 contained different concentrations of XylR. (B) Binding of XylR to the xynD operator in the presence of xylose (Xyl) or arabinose (Ara). All lanes contained 0.08 ng of radioactively labeled DNA and 1 μM of the purified XylR. Lanes 1, 2, and 3 contained 5 mM, 10 mM, and 15 mM xylose, respectively; lane 4 contained 15 mM arabinose.
Mapping the 5′ end of xynEFG.
As shown above, the xynEFG genes can be cotranscribed from the xynD promoter. However, the 110-bp spacer between xynC and xynE contains sequences that resemble potential promoter and regulatory elements as well. To test whether the xynEFG genes can also be transcribed from an additional promoter, primer extension analysis was performed. The apparent transcriptional start point corresponded to an A residue, which is 96 bp upstream of the translational initiation codon of xynE (Fig. 4A) and was detected only in xylose-grown cultures. The −35 sequence (TATCGA) differs from the B. subtilis consensus, TTGACA, by four nucleotides and is separated by 17 bp from the potential −10 region (AGTAAT), which differs from the σA consensus, TATAAT, by two nucleotides (45). The poor adherence of these putative promoter sequences to consensus sequences is typical of promoters that depend on a positive regulator. In addition, the promoter region contains an inverted repeat positioned at −17 to +15 with respect to the transcriptional start point and several direct repeats of the sequence GAAA (Fig. 4B). Interestingly, although the xynE promoter is induced by xylose (based on the primer extension analysis), the promoter region does not contain the typical XylR-binding motif, suggesting that transcription from this promoter is not controlled by XylR. However, the inverted and direct repeats identified in the promoter region suggest that an additional transcriptional regulator may function on the xynE promoter.
FIG. 4.

Mapping the 5′ termini of the xynEFG cluster by primer extension analysis. (A) Total RNA was extracted from mid-exponential-phase cultures of G. stearothermophilus T-6 grown in BSM supplemented with 0.5% xylose or with 0.5% glucose as the sole carbon source. Extension products resulted from RNA extracted from cultures grown on 0.5% xylose are shown in lane 1, and those from growth on 0.5% glucose are shown in lane 2. Dideoxynucleotide sequence reactions were carried out with the same primer used for the reverse transcriptase reactions. The position of the transcriptional start point is indicated with an asterisk on the inferred nontemplate strand sequence. (B) Sequence data for the regulatory region. The transcriptional start point (+1) is indicated by a vertical arrowhead. The −35 and −10 regions, the direct repeat sequences of GAAA, the proposed ribosome-binding site (RBS), and the initiating methionine codon are in boldface. An inverted repeat sequence is indicated by horizontal arrowheads.
XynC binds to the xynE promoter region.
To test whether the putative response regulator, XynC, can bind to the xynE promoter region, the xynC gene was fused to an His6 sequence and the gene product was purified from E. coli by Ni+-NTA chromatography. Based on sodium dodecyl sulfate-polyacrylamide gel electrophoresis analysis, the purity of the final protein solution was over 97%. A 220-bp DNA fragment corresponding to positions −120 to +100 with respect to the xynE transcriptional start site was used as a probe for testing XynC-DNA interaction. Gel mobility shift assays showed that the purified His6-XynC protein can bind to the xynE promoter region; a nearly complete shift was seen in the presence of 0.7 μM XynC (Fig. 5). This relatively high concentration of XynC may reflect the fact that the protein was not phosphorylated, as was observed for other response regulators (1). The binding seemed to be specific, since XynC did not shift an unrelated 200-bp fragment from the xynA promoter region (data not shown). To map more precisely the boundaries of the binding region, two DNA probes representing regions upstream and downstream of the apparent transcriptional start site were used. With these two probes, retardation was visible only at relatively high protein concentrations (4 μM). In addition, XynC failed to bind to a 42-bp DNA probe that contained only the inverted repeat positioned at −17 to +15 with respect to the transcription start point. To delineate the sequence within the xynE promoter region that is bound by XynC, DNase I footprinting experiments were performed on the 220-bp fragment that was used in the gel shift experiments (Fig. 6A). Analysis of the coding strand footprint showed four protected regions at positions −53 to −46, −33 to −22, −12 to +12, and +34 to +38 (Fig. 6B and C). Inspection of these regions revealed that they all contain direct repeats of the sequence GAAA (Fig. 6B). XynC also caused certain sites to become hypersensitive to DNase I (Fig. 6A). Such hypersensitivity may indicate that binding of XynC induces DNA bending. To test whether phosphorylation of XynC promotes binding to DNA, we attempted to phosphorylate XynC in vitro with acetyl phosphate as a phosphate donor. Unfortunately, incubation of XynC with acetyl phosphate resulted in protein precipitation, presumably due to oligomerization. Indeed, native polyacrylamide gel electrophoresis analysis indicated that XynC treated with acetyl phosphate appears as a high-molecular-weight protein (data not shown). This phenomenon was also observed for other response regulators (1).
FIG. 5.

Gel mobility shift assay for binding of XynC to the xynE promoter region. XynC and a radioactively labeled 220-bp DNA fragment containing the xynE promoter were incubated as described in Materials and Methods. Lane 1 contains no protein; lanes 2 to 6 contain increasing concentrations of purified XynC. The shifted band is indicated by arrow a, and free DNA by arrow b.
FIG. 6.

DNase I footprint assay of XynC interaction with the xynE regulatory region. (A) A 220-bp PCR product, corresponding to positions −120 to +100 with respect to the xynE transcriptional start site and labeled at the 5′ end of the template (noncoding) strand or the nontemplate (coding) strand, was incubated with increasing amounts of purified XynC-His6. Following treatment with DNase I, the DNA was denatured and subjected to electrophoresis. Sanger sequencing reactions were primed with the same oligonucleotide used for synthesis of the PCR product and used to establish the positions of the protected bands. Open boxes indicate regions protected by XynC. The apparent transcription start site is shown by the bent arrow on the coding strand. Black arrows indicate hypersensitive sites in the DNA induced by binding XynC to the DNA. Lanes 1 contained no XynC; other lanes contained increasing concentrations of purified XynC: 0.7 μM (lanes 2), 1 μM (lanes 3), and 1.5 μM (lanes 4). (B) Sequence of the xynE regulatory region. The promoter −35 and −10 regions are underlined. The transcriptional start point (+1) is indicated by a vertical arrowhead. The direct repeated GAAA motif is shown in bold, and the protected regions are in open dashed boxes. Arrows indicate hypersensitive sites at positions −16 and +42 on the noncoding strand and −16 on the coding strand. (C) Schematic map summarizing the footprinting results and gel shift experiments. The positions of the xynC and xynE genes are shown. The protected regions are shown by gray boxes, and black horizontal bars indicate the DNA fragments (a, b, and c) tested by gel shift analysis. The asterisk indicates the fragment that was retarded by XynC.
XynC activates xynE transcription in vitro.
The direct interaction of XynC with the xynE promoter suggests that XynC acts as a transcriptional regulatory protein. The effect of XynC on xynE transcription was tested in vitro and quantified by real-time RT-PCR. An in vitro transcriptional assay was preformed by mixing nucleoside triphosphates, fresh cell extract of strain T-6 (as a source of RNA polymerase), purified XynC, and a DNA template corresponding to the promoter region and the coding sequence of the xynE gene. An unrelated gene (xynA) was used as a reference gene to take account of any experimental conditions that affect transcription efficiency. Real-time RT-PCR was carried out on the transcription products by use of primers that amplified internal regions within the xynE and xynA genes and gave efficiencies of about 90% for both genes. The transcript level of xynE increased 3- to 140-fold in the presence of XynC at 0.5 to 1.5 μM, respectively (Table 3) . As expected, the transcription level of the reference gene (xynA) was not influenced by the presence of XynC. These results indicate that XynC activates the transcription of xynEFG.
TABLE 3.
Real-time RT-PCR analysis demonstrating in vitro transcriptional activation of xynE by XynCa
| XynC concn (μM) | Activation, fold
|
CT for target gene
|
CT for ref gene
|
||||
|---|---|---|---|---|---|---|---|
| n-fold | Ref gene | Target gene | −XynC | +XynC | −XynC | +XynC | |
| 0 | 1.0 ± 0.4 | 1.0 ± 0.2 | 1.0 ± 0.08 | 19.6 ± 0.6 | 19.6 ± 0.6 | 20.5 ± 0.4 | 20.5 ± 0.4 |
| 0.5 | 2.8 ± 1.7 | 1.6 ± 0.3 | 4.6 ± 1.6 | 19.6 ± 0.6 | 17.2 ± 1.1 | 20.5 ± 0.4 | 19.7 ± 0.3 |
| 1 | 26.5 ± 7.1 | 1.8 ± 0.3 | 47.8 ± 1.4 | 19.2 ± 0.5 | 13.2 ± 1.3 | 20.6 ± 0.2 | 19.6 ± 0.09 |
| 1.5 | 140.8 ± 42.0 | 1.2 ± 0.4 | 169.0 ± 3.0 | 19.6 ± 0.8 | 11.6 ± 2.6 | 20.5 ± 0.4 | 20.1 ± 0.2 |
CT is the cycle number at which the fluorescence crosses an arbitrary threshold line. To calculate the efficiency of the real-time RT-PCR, a cDNA dilution curve was generated and calculated by the equation E = 10−1/slope. The efficiencies for the target gene and the reference gene were determined to be 1.90 and 1.88, respectively. Activation (n-fold) of the target gene (xynE) was calculated using the method of Pfaffl (51) and was based on the amplification efficiency (E) and the crossing point differences (ΔCT) for the control (without XynC) and for the treated sample (with XynC) by the following equation: activation (n-fold) = 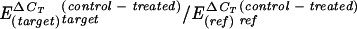 . Results are average of three independent experiments ± standard deviations and were conducted using specific primers for the target gene (xynE) and for the reference gene (xynA).
. Results are average of three independent experiments ± standard deviations and were conducted using specific primers for the target gene (xynE) and for the reference gene (xynA).
DISCUSSION
G. stearothermophilus T-6 possesses a complete system for degrading and utilizing hemicellulose. This system is highly regulated and is mediated via several mechanisms, including induction-repression, catabolite repression, transition phase regulation, and quorum sensing at low cell densities (21, 73). In this work we reveal that a regulatory TCS is involved in regulating an ABC transport system for xylo-oligosaccharides.
XynEFG constitutes an ABC transporter for xylo-oligosaccharides.
The xynEFG genes appear to encode an ABC transport system that belongs to the carbohydrate uptake transporter-1 (CUT1) family. This family mainly contains transporters for di- and oligosaccharides, in addition to glycerol phosphate and polyols (http://www.tcdb.org) (12, 64) and consists of two integral membrane domains/proteins, an ATPase subunit, and an extracytoplasmic solute-binding protein. The extracellular binding protein determines the specificity of the transporter and in gram-positive bacteria is present either as a lipoprotein, tethered to the external surface of the cytoplasmic membrane, or as a cell surface-associated protein, bound to the external membrane surface via electrostatic interactions (80). The ability of XynE to bind xylosaccharides was demonstrated using ITC. The binding constants, KB, of XynE for different xylo-oligosaccharides were all within the range of 106 M−1, corresponding to equilibrium dissociation constant, KD (KD = 1/KB), values of 0.08 μM for X3 and 1.4 μM for X6. Similar KD values have been reported for other bacterial sugar-binding proteins by use of different methodologies, such as equilibrium dialysis (63), sugar uptake by whole cells (86), and surface plasmon resonance analysis (79). The xylobiose-binding lipoprotein (BxlE) from Streptomyces thermoviolaceus has a KD of 0.008 μM (79), whereas the cellodextrin lipid-anchored binding protein from Streptomyces reticuli showed a KD of 1.5 μM (63). A KD value of 1.6 μM was obtained for trehalose/maltose with the binding protein from Thermococcus litoralis (28), and recently a KD of 0.03 μM was measured for chitobiose with NgcE from Streptomyces olivaceoviridis (87). The ITC measurements of XynE and xylotriose allowed us to extract the heat capacity change of the interaction. Changes in the binding enthalpies (ΔHB) at different temperatures yielded a slope of ΔCp = −158 cal mole−1 K−1, suggesting that stacking interactions are involved in the binding of the sugar to the protein. The negative ΔCp value can be provided by a single Trp residue (90), and indeed a conserved Trp residue (Trp285) is found in XynE. This residue corresponds to Trp230 in the maltodextrin-binding protein from Pyrococcus furiosus, and the crystal structure of this protein complexed with trehalose reveals that this aromatic residue is located in the binding site and interacts with the sugar ring (16). This conserved tryptophan residue is also found in the crystal structures of two other maltose-maltodextrin-binding proteins from E. coli and Alicyclobacillus acidocaldarius (60). In the N-acetyl-d-glucosamine-binding protein (NgcE), the corresponding tryptophan residue (Trp280) was shown to be essential for binding (57).
Like all membrane-spanning subunits of the ABC importers, the XynF and XynG proteins contain the EAA motif (EAAX3GX9IXLP) that is located ∼100 residues from the C terminus (13, 59). In gram-negative bacteria, the ATP-binding protein gene is usually part of the ABC transporter operon, whereas in gram-positive bacteria the gene is often located elsewhere on the chromosome (52, 62, 73). It is likely that in this case a single ATP-binding protein can serve several ABC transport systems, as was demonstrated for S. reticuli, in which the ATP-binding protein (MsiK) is involved in the transport of both cellobiose and maltose (61, 62). Recently, we have cloned and sequenced the putative ATP-binding protein (XynK) from G. stearothermophilus T-6. This protein has a significant homology to MsiK, but it remains to be determined whether it is involved in the transport of xylo-oligosaccharides.
Regulation of the xynDCEFG transcript.
The xynDCEFG transcriptional unit codes for a TCS and an ABC transport system. The xynD promoter contains a 16-bp inverted repeat that also appears in promoters of four other xylan utilization genes in strain T-6, including xylM, xynA, xynX, and xylA. DNA fragments containing this motif were shown recently to bind XylR by gel retardation assays (37). Similar XylR inverted repeat motifs were identified in other gram-positive bacteria (56). The regulatory region of xynD contains two potential CREs, suggesting that the transcriptional unit is also negatively regulated by the CcpA protein. The ccpA gene from strain T-6 was previously cloned and sequenced, and it showed 68% identity to B. subtilis CcpA (36). In B. subtilis, several TCSs are located near ABC transport systems on the chromosome but do not appear to be part of the same transcriptional unit (33). In strain T-6, the l-arabinan utilization cluster also has a TCS located near an ABC transport system.
Positive regulation of xynE by XynC.
As shown here, XynC binds to the regulatory region of xynEFG and activates transcription from the xynE promoter. DNase I footprint analysis with XynC identified four protected regions located between positions −53 and +34 with respect to the transcriptional start point (Fig. 6). Interestingly, the positions of the binding sites do not follow the classic pattern for transcriptional activators. In the usual case, the binding site is located upstream of the −35 region, allowing direct interaction of the activator with the carboxy-terminal domain of the RNA polymerase α subunit (4, 54). There are only a few examples of bacterial regulators that activate when bound to both upstream and downstream regions with respect to the transcriptional start point (46). However, it is also possible that the nonphosphorylated protein binds somewhat differently than the phosphorylated form.
A sensor/regulator mechanism controls xylo-oligosaccharide uptake.
Based on their primary sequences, the xynD and xynC products appear to constitute a TCS. XynD would be classified as a class I histidine kinase (19). XynD has two putative transmembrane segments and a conserved C-terminal cytoplasmic region containing the ATP-binding kinase domain. As expected, the N-terminal sensing domain has no significant homology to any other sensor domains, reflecting the many different signals to which histidine kinases are responsive. Attempts to express the XynD sensor domain as a soluble protein were unsuccessful, preventing the determination of the chemical nature of the substrates that bind to XynD. However, it is likely that the XynD sensor domain binds to xylo-oligosaccharides, thus allowing the cell to respond to xylosaccharides in the local environment.
The genes for the ABC transport system (xynEFG) are regulated by both XylR and XynC. This type of regulation allows the cells to rapidly amplify the expression of the transport system and thus be able to utilize xylo-oligosaccharides efficiently when they are available. Such a sensing mechanism, used by a xylan-degrading microorganism, has been described only recently (43). The XynR protein from Prevotella bryantii B14 contains both a histidine kinase and response regulator domains. This protein was found to activate the expression of genes coding for a xylanase, β-xylosidase, and a putative symporter.
Acknowledgments
This research was supported by the Israel Science Foundation (grant 1006/05 to Y.S.) and the United States-Israel Binational Science Foundation, Jerusalem, Israel (grants 93-171 and 96-178 to A.L.S. and Y.S.). Additional support was provided by the Otto Meyerhof Minerva Center for Biotechnology, established by the Minerva Foundation (Munich, Germany). Y.S. holds the Erwin and Rosl Pollak Chair in Biotechnology.
Footnotes
Published ahead of print on 1 December 2006.
REFERENCES
- 1.Abo-Amer, A. E., J. Munn, K. Jackson, M. Aktas, P. Golby, D. J. Kelly, and S. C. Andrews. 2004. DNA interaction and phosphotransfer of the C4-dicarboxylate-responsive DcuS-DcuR two-component regulatory system from Escherichia coli. J. Bacteriol. 186:1879-1889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Adelsberger, H., C. Hertel, E. Glawischnig, V. V. Zverlov, and W. H. Schwarz. 2004. Enzyme system of Clostridium stercorarium for hydrolysis of arabinoxylan: reconstitution of the in vivo system from recombinant enzymes. Microbiology 150:2257-2266. [DOI] [PubMed] [Google Scholar]
- 3.Altschul, S. F., W. Gish, W. Miller, E. W. Myers, and D. J. Lipman. 1990. Basic local alignment search tool. J. Mol. Biol. 215:403-410. [DOI] [PubMed] [Google Scholar]
- 4.Barnard, A., A. Wolfe, and S. Busby. 2004. Regulation at complex bacterial promoters: how bacteria use different promoter organizations to produce different regulatory outcomes. Curr. Opin. Microbiol. 7:102-108. [DOI] [PubMed] [Google Scholar]
- 5.Beylot, M. H., K. Emami, V. A. McKie, H. J. Gilbert, and G. Pell. 2001. Pseudomonas cellulosa expresses a single membrane-bound glycoside hydrolase family 51 arabinofuranosidase. Biochem. J. 358:599-605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bravman, T., V. Belakhov, D. Solomon, G. Shoham, B. Henrissat, T. Baasov, and Y. Shoham. 2003. Identification of the catalytic residues in family 52 glycoside hydrolase, a beta-xylosidase from Geobacillus stearothermophilus T-6. J. Biol. Chem. 278:26742-26749. [DOI] [PubMed] [Google Scholar]
- 7.Bravman, T., A. Mechaly, S. Shulami, V. Belakhov, T. Baasov, G. Shoham, and Y. Shoham. 2001. Glutamic acid 160 is the acid-base catalyst of beta-xylosidase from Bacillus stearothermophilus T-6: a family 39 glycoside hydrolase. FEBS Lett. 495:115-119. [DOI] [PubMed] [Google Scholar]
- 8.Bravman, T., G. Zolotnitsky, V. Belakhov, G. Shoham, B. Henrissat, T. Baasov, and Y. Shoham. 2003. Detailed kinetic analysis of a family 52 glycoside hydrolase: a beta-xylosidase from Geobacillus stearothermophilus. Biochemistry 42:10528-10536. [DOI] [PubMed] [Google Scholar]
- 9.Bravman, T., G. Zolotnitsky, S. Shulami, V. Belakhov, D. Solomon, T. Baasov, G. Shoham, and Y. Shoham. 2001. Stereochemistry of family 52 glycosyl hydrolases: a beta-xylosidase from Bacillus stearothermophilus T-6 is a retaining enzyme. FEBS Lett. 495:39-43. [DOI] [PubMed] [Google Scholar]
- 10.Brux, C., A. Ben-David, D. Shallom-Shezifi, M. Leon, K. Niefind, G. Shoham, Y. Shoham, and D. Schomburg. 2006. The structure of an inverting GH43 beta-xylosidase from Geobacillus stearothermophilus with its substrate reveals the role of the three catalytic residues. J. Mol. Biol. 359:97-109. [DOI] [PubMed] [Google Scholar]
- 11.Czjzek, M., A. Ben David, T. Bravman, G. Shoham, B. Henrissat, and Y. Shoham. 2005. Enzyme-substrate complex structures of a GH39 beta-xylosidase from Geobacillus stearothermophilus. J. Mol. Biol. 353:838-846. [DOI] [PubMed] [Google Scholar]
- 12.Dassa, E., and P. Bouige. 2001. The ABC of ABCS: a phylogenetic and functional classification of ABC systems in living organisms. Res. Microbiol. 152:211-229. [DOI] [PubMed] [Google Scholar]
- 13.Davidson, A. L., and J. Chen. 2004. ATP-binding cassette transporters in bacteria. Annu. Rev. Biochem. 73:241-268. [DOI] [PubMed] [Google Scholar]
- 14.de Vries, R. P., and J. Visser. 2001. Aspergillus enzymes involved in degradation of plant cell wall polysaccharides. Microbiol. Mol. Biol. Rev. 65:497-522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Eriksson, K. E. L., R. A. Blanchette, and P. Ander. 1990. Microbial and enzymatic degradation of wood and wood components. Springer-Verlag, Berlin, Germany.
- 16.Evdokimov, A. G., D. E. Anderson, K. M. Routzahn, and D. S. Waugh. 2001. Structural basis for oligosaccharide recognition by Pyrococcus furiosus maltodextrin-binding protein. J. Mol. Biol. 305:891-904. [DOI] [PubMed] [Google Scholar]
- 17.Faergeman, N. J., B. W. Sigurskjold, B. B. Kragelund, K. V. Andersen, and J. Knudsen. 1996. Thermodynamics of ligand binding to acyl-coenzyme A binding protein studied by titration calorimetry. Biochemistry 35:14118-14126. [DOI] [PubMed] [Google Scholar]
- 18.Farrell, A. E., R. J. Plevin, B. T. Turner, A. D. Jones, M. O'Hare, and D. M. Kammen. 2006. Ethanol can contribute to energy and environmental goals. Science 311:506-508. [DOI] [PubMed] [Google Scholar]
- 19.Foussard, M., S. Cabantous, J. Pedelacq, V. Guillet, S. Tranier, L. Mourey, C. Birck, and J. Samama. 2001. The molecular puzzle of two-component signaling cascades. Microbes Infect. 3:417-424. [DOI] [PubMed] [Google Scholar]
- 20.Gallegos, M. T., R. Schleif, A. Bairoch, K. Hofmann, and J. L. Ramos. 1997. Arac/XylS family of transcriptional regulators. Microbiol. Mol. Biol. Rev. 61:393-410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gat, O., A. Lapidot, I. Alchanati, C. Regueros, and Y. Shoham. 1994. Cloning and DNA sequence of the gene coding for Bacillus stearothermophilus T-6 xylanase. Appl. Environ. Microbiol. 60:1889-1896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gilson, E., G. Alloing, T. Schmidt, J. P. Claverys, R. Dudler, and M. Hofnung. 1988. Evidence for high affinity binding-protein dependent transport systems in gram-positive bacteria and in Mycoplasma. EMBO J. 7:3971-3974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Golan, G., D. Shallom, A. Teplitsky, G. Zaide, S. Shulami, T. Baasov, V. Stojanoff, A. Thompson, Y. Shoham, and G. Shoham. 2004. Crystal structures of Geobacillus stearothermophilus alpha-glucuronidase complexed with its substrate and products: mechanistic implications. J. Biol. Chem. 279:3014-3024. [DOI] [PubMed] [Google Scholar]
- 24.Han, S. O., H. Y. Cho, H. Yukawa, M. Inui, and R. H. Doi. 2004. Regulation of expression of cellulosomes and noncellulosomal (hemi)cellulolytic enzymes in Clostridium cellulovorans during growth on different carbon sources. J. Bacteriol. 186:4218-4227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hazlewood, G. P., and H. J. Gilbert. 1993. Hemicellulose and hemicellulases. In M. P. Coughlan and G. P. Hazlewood (ed.), Molecular biology of hemicellulases. Portland Press, London, United Kingdom.
- 26.Helmann, J. D. 1995. Compilation and analysis of Bacillus subtilis sigma A-dependent promoter sequences: evidence for extended contact between RNA polymerase and upstream promoter DNA. Nucleic Acids Res. 23:2351-2360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Higgins, C. F., S. C. Hyde, M. M. Mimmack, U. Gileadi, D. R. Gill, and M. P. Gallagher. 1990. Binding protein-dependent transport systems. J. Bioenerg. Biomembr. 22:571-592. [DOI] [PubMed] [Google Scholar]
- 28.Horlacher, R., K. B. Xavier, H. Santos, J. DiRuggiero, M. Kossmann, and W. Boos. 1998. Archaeal binding protein-dependent ABC transporter: molecular and biochemical analysis of the trehalose/maltose transport system of the hyperthermophilic archaeon Thermococcus litoralis. J. Bacteriol. 180:680-689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hovel, K., D. Shallom, K. Niefind, V. Belakhov, G. Shoham, T. Baasov, Y. Shoham, and D. Schomburg. 2003. Crystal structure and snapshots along the reaction pathway of a family 51 alpha-l-arabinofuranosidase. EMBO J. 22:4922-4932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jarmer, H., T. S. Larsen, A. Krogh, H. H. Saxild, S. Brunak, and S. Knudsen. 2001. Sigma A recognition sites in the Bacillus subtilis genome. Microbiology 147:2417-2424. [DOI] [PubMed] [Google Scholar]
- 31.Jiang, S. Q., G. Q. Yu, Z. G. Li, and J. S. Hong. 1988. Genetic evidence for modulation of the activator by two regulatory proteins involved in the exogenous induction of phosphoglycerate transport in Salmonella typhimurium. J. Bacteriol. 170:4304-4308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Johnson, J. L. 1981. Genetic characterization, p. 450-472. In P. Gerhardt, R. G. E. Murray, E. W. Costilow, W. A. Nester, N. R. Wood, R. Krieg, and G. B. Philips (ed.), Manual of methods for general bacteriology. American Society for Microbiology, Washington, DC.
- 33.Joseph, P., G. Fichant, Y. Quentin, and F. Denizot. 2002. Regulatory relationship of two-component and ABC transport systems and clustering of their genes in the Bacillus/Clostridium group, suggest a functional link between them. J. Mol. Microbiol. Biotechnol. 4:503-513. [PubMed] [Google Scholar]
- 34.Joseph, P., A. Guiseppi, A. Sorokin, and F. Denizot. 2004. Characterization of the Bacillus subtilis YxdJ response regulator as the inducer of expression for the cognate ABC transporter YxdLM. Microbiology 150:2609-2617. [DOI] [PubMed] [Google Scholar]
- 35.Khasin, A., I. Alchanati, and Y. Shoham. 1993. Purification and characterization of a thermostable xylanase from Bacillus stearothermophilus T-6. Appl. Environ. Microbiol. 59:1725-1730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kraus, A., E. Kuster, A. Wagner, K. Hoffmann, and W. Hillen. 1998. Identification of a co-repressor binding site in catabolite control protein CcpA. Mol. Microbiol. 30:955-963. [DOI] [PubMed] [Google Scholar]
- 37.Langut, Y. 2006. Characterization of XylR, the repressor of the xylanolytic system in Geobacillus stearothermophilus. M. Sc. thesis, Technion-Israel Institute of Technology, Haifa, Israel.
- 38.Livingstone, J. R., R. S. Spolar, and M. T. Record, Jr. 1991. Contribution to the thermodynamics of protein folding from the reduction in water-accessible nonpolar surface area. Biochemistry 30:4237-4244. [DOI] [PubMed] [Google Scholar]
- 39.Lukat, G. S., W. R. McCleary, A. M. Stock, and J. B. Stock. 1992. Phosphorylation of bacterial response regulator proteins by low molecular weight phospho-donors. Proc. Natl. Acad. Sci. USA 89:718-722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Marmur, J. 1961. A procedure for the isolation of deoxyribonucleic acid from micro-organisms. J. Mol. Biol. 3:208-218. [Google Scholar]
- 41.Miwa, Y., K. Nagura, S. Eguchi, H. Fukuda, J. Deutscher, and Y. Fujita. 1997. Catabolite repression of the Bacillus subtilis gnt operon exerted by two catabolite-responsive elements. Mol. Microbiol. 23:1203-1213. [DOI] [PubMed] [Google Scholar]
- 42.Miwa, Y., A. Nakata, A. Ogiwara, M. Yamamoto, and Y. Fujita. 2000. Evaluation and characterization of catabolite-responsive elements (cre) of Bacillus subtilis. Nucleic Acids Res. 28:1206-1210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Miyazaki, K., H. Miyamoto, D. K. Mercer, T. Hirase, J. C. Martin, Y. Kojima, and H. J. Flint. 2003. Involvement of the multidomain regulatory protein XynR in positive control of xylanase gene expression in the ruminal anaerobe Prevotella bryantii B(1)4. J. Bacteriol. 185:2219-2226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Moran, C. P. 1990. Measuring gene expression in Bacillus, p. 267-293. In C. R. Harwood and S. M. Cutting (ed.), Molecular biological methods for bacillus. John Wiley & Sons, Chichester, United Kingdom.
- 45.Moran, C. P., Jr., N. Lang, S. F. LeGrice, G. Lee, M. Stephens, A. L. Sonenshein, J. Pero, and R. Losick. 1982. Nucleotide sequences that signal the initiation of transcription and translation in Bacillus subtilis. Mol. Gen. Genet. 186:339-346. [DOI] [PubMed] [Google Scholar]
- 46.Munson, G. P., and J. R. Scott. 2000. Rns, a virulence regulator within the AraC family, requires binding sites upstream and downstream of its own promoter to function as an activator. Mol. Microbiol. 36:1391-1402. [DOI] [PubMed] [Google Scholar]
- 47.Nagy, T., K. Emami, C. M. Fontes, L. M. Ferreira, D. R. Humphry, and H. J. Gilbert. 2002. The membrane-bound α-glucuronidase from Pseudomonas cellulosa hydrolyzes 4-O-methyl-d-glucuronoxylooligosaccharides but not 4-O-methyl-d-glucuronoxylan. J. Bacteriol. 184:4925-4929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Niu, S., S. Q. Jiang, and J. Hong. 1995. Salmonella typhimurium pgtB mutants conferring constitutive expression of phosphoglycerate transporter pgtP independent of pgtC. J. Bacteriol. 177:4297-4302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ochman, H., A. S. Gerber, and D. L. Hartl. 1988. Genetic applications of an inverse polymerase chain reaction. Genetics 120:621-623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Pearson, W. R., and D. J. Lipman. 1988. Improved tools for biological sequence comparison. Proc. Natl. Acad. Sci. USA 85:2444-2448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Pfaffl, M. W. 2001. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 29:e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Quentin, Y., G. Fichant, and F. Denizot. 1999. Inventory, assembly and analysis of Bacillus subtilis ABC transport systems. J. Mol. Biol. 287:467-484. [DOI] [PubMed] [Google Scholar]
- 53.Ragauskas, A. J., C. K. Williams, B. H. Davison, G. Britovsek, J. Cairney, C. A. Eckert, W. J. Frederick, Jr., J. P. Hallett, D. J. Leak, C. L. Liotta, J. R. Mielenz, R. Murphy, R. Templer, and T. Tschaplinski. 2006. The path forward for biofuels and biomaterials. Science 311:484-489. [DOI] [PubMed] [Google Scholar]
- 54.Rhodius, V. A., and S. J. Busby. 1998. Positive activation of gene expression. Curr. Opin. Microbiol. 1:152-159. [DOI] [PubMed] [Google Scholar]
- 55.Robinson, V. L., D. R. Buckler, and A. M. Stock. 2000. A tale of two components: a novel kinase and a regulatory switch. Nat. Struct. Biol. 7:626-633. [DOI] [PubMed] [Google Scholar]
- 56.Rodionov, D. A., A. A. Mironov, and M. S. Gelfand. 2001. Transcriptional regulation of pentose utilisation systems in the Bacillus/Clostridium group of bacteria. FEMS Microbiol. Lett. 205:305-314. [DOI] [PubMed] [Google Scholar]
- 57.Saito, A., and H. Schrempf. 2004. Mutational analysis of the binding affinity and transport activity for N-acetylglucosamine of the novel ABC transporter Ngc in the chitin-degrader Streptomyces olivaceoviridis. Mol. Genet. Genomics. 271:545-553. [DOI] [PubMed] [Google Scholar]
- 58.Sambrook, J., and D. W. Russell. 2001. Molecular cloning: a laboratory manual, 3rd ed. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- 59.Saurin, W., W. Koster, and E. Dassa. 1994. Bacterial binding protein-dependent permeases: characterization of distinctive signatures for functionally related integral cytoplasmic membrane proteins. Mol. Microbiol. 12:993-1004. [DOI] [PubMed] [Google Scholar]
- 60.Schafer, K., U. Magnusson, F. Scheffel, A. Schiefner, M. O. Sandgren, K. Diederichs, W. Welte, A. Hulsmann, E. Schneider, and S. L. Mowbray. 2004. X-ray structures of the maltose-maltodextrin-binding protein of the thermoacidophilic bacterium Alicyclobacillus acidocaldarius provide insight into acid stability of proteins. J. Mol. Biol. 335:261-274. [DOI] [PubMed] [Google Scholar]
- 61.Schlosser, A. 2000. MsiK-dependent trehalose uptake in Streptomyces reticuli. FEMS Microbiol. Lett. 184:187-192. [DOI] [PubMed] [Google Scholar]
- 62.Schlosser, A., T. Kampers, and H. Schrempf. 1997. The Streptomyces ATP-binding component MsiK assists in cellobiose and maltose transport. J. Bacteriol. 179:2092-2095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Schlosser, A., and H. Schrempf. 1996. A lipid-anchored binding protein is a component of an ATP-dependent cellobiose/cellotriose-transport system from the cellulose degrader Streptomyces reticuli. Eur. J. Biochem. 242:332-338. [DOI] [PubMed] [Google Scholar]
- 64.Schneider, E. 2001. ABC transporters catalyzing carbohydrate uptake. Res. Microbiol. 152:303-310. [DOI] [PubMed] [Google Scholar]
- 65.Schwarz, W. H. 2001. The cellulosome and cellulose degradation by anaerobic bacteria. Appl. Microbiol. Biotechnol. 56:634-649. [DOI] [PubMed] [Google Scholar]
- 66.Shallom, D., V. Belakhov, D. Solomon, S. Gilead-Gropper, T. Baasov, G. Shoham, and Y. Shoham. 2002. The identification of the acid-base catalyst of alpha-arabinofuranosidase from Geobacillus stearothermophilus T-6, a family 51 glycoside hydrolase. FEBS Lett. 514:163-167. [DOI] [PubMed] [Google Scholar]
- 67.Shallom, D., V. Belakhov, D. Solomon, G. Shoham, T. Baasov, and Y. Shoham. 2002. Detailed kinetic analysis and identification of the nucleophile in alpha-l-arabinofuranosidase from Geobacillus stearothermophilus T-6, a family 51 glycoside hydrolase. J. Biol. Chem. 277:43667-43673. [DOI] [PubMed] [Google Scholar]
- 68.Shallom, D., G. Golan, G. Shoham, and Y. Shoham. 2004. Effect of dimer dissociation on activity and thermostability of the alpha-glucuronidase from Geobacillus stearothermophilus: dissecting the different oligomeric forms of family 67 glycoside hydrolases. J. Bacteriol. 186:6928-6937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Shallom, D., M. Leon, T. Bravman, A. Ben-David, G. Zaide, V. Belakhov, G. Shoham, D. Schomburg, T. Baasov, and Y. Shoham. 2005. Biochemical characterization and identification of the catalytic residues of a family 43 beta-d-xylosidase from Geobacillus stearothermophilus T-6. Biochemistry 44:387-397. [DOI] [PubMed] [Google Scholar]
- 70.Shallom, D., and Y. Shoham. 2003. Microbial hemicellulases. Curr. Opin. Microbiol. 6:219-228. [DOI] [PubMed] [Google Scholar]
- 71.Shoham, Y., R. Lamed, and E. A. Bayer. 1999. The cellulosome concept as an efficient microbial strategy for the degradation of insoluble polysaccharides. Trends Microbiol. 7:275-281. [DOI] [PubMed] [Google Scholar]
- 72.Shoham, Y., Z. Schwartz, A. Khasin, O. Gat, Z. Zosim, and E. Rosenberg. 1992. Delignification of wood pulp by a thermostable xylanase. Biodegradation 3:161-170. [Google Scholar]
- 73.Shulami, S., O. Gat, A. L. Sonenshein, and Y. Shoham. 1999. The glucuronic acid utilization gene cluster from Bacillus stearothermophilus T-6. J. Bacteriol. 181:3695-3704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Spolar, R. S., and M. T. Record, Jr. 1994. Coupling of local folding to site-specific binding of proteins to DNA. Science 263:777-784. [DOI] [PubMed] [Google Scholar]
- 75.Stock, A. M., V. L. Robinson, and P. N. Goudreau. 2000. Two-component signal transduction. Annu. Rev. Biochem. 69:183-215. [DOI] [PubMed] [Google Scholar]
- 76.Teplitsky, A., A. Mechaly, V. Stojanoff, G. Sainz, G. Golan, H. Feinberg, R. Gilboa, V. Reiland, G. Zolotnitsky, D. Shallom, A. Thompson, Y. Shoham, and G. Shoham. 2004. Structure determination of the extracellular xylanase from Geobacillus stearothermophilus by selenomethionyl MAD phasing. Acta Crystallogr. Sect. D 60:836-848. [DOI] [PubMed] [Google Scholar]
- 77.Teplitsky, A., S. Shulami, S. Moryles, Y. Shoham, and G. Shoham. 2000. Crystallization and preliminary X-ray analysis of an intracellular xylanase from Bacillus stearothermophilus T-6. Acta Crystallogr. Sect. D 56:181-184. [DOI] [PubMed] [Google Scholar]
- 78.Tomme, P., A. L. Creagh, D. G. Kilburn, and C. A. Haynes. 1996. Interaction of polysaccharides with the N-terminal cellulose-binding domain of Cellulomonas fimi CenC. 1. Binding specificity and calorimetric analysis. Biochemistry 35:13885-13894. [DOI] [PubMed] [Google Scholar]
- 79.Tsujibo, H., M. Kosaka, S. Ikenishi, T. Sato, K. Miyamoto, and Y. Inamori. 2004. Molecular characterization of a high-affinity xylobiose transporter of Streptomyces thermoviolaceus OPC-520 and its transcriptional regulation. J. Bacteriol. 186:1029-1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.van der Heide, T., and B. Poolman. 2002. ABC transporters: one, two or four extracytoplasmic substrate-binding sites? EMBO Rep. 3:938-943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Verhamme, D. T., J. C. Arents, P. W. Postma, W. Crielaard, and K. J. Hellingwerf. 2001. Glucose-6-phosphate-dependent phosphoryl flow through the Uhp two-component regulatory system. Microbiology 147:3345-3352. [DOI] [PubMed] [Google Scholar]
- 82.Verhamme, D. T., J. C. Arents, P. W. Postma, W. Crielaard, and K. J. Hellingwerf. 2002. Investigation of in vivo cross-talk between key two-component systems of Escherichia coli. Microbiology 148:69-78. [DOI] [PubMed] [Google Scholar]
- 83.Volz, K. 1993. Structural conservation in the CheY superfamily. Biochemistry 32:11741-11753. [DOI] [PubMed] [Google Scholar]
- 84.von Heijne, G. 1989. The structure of signal peptides from bacterial lipoproteins. Protein Eng. 2:531-534. [DOI] [PubMed] [Google Scholar]
- 85.Wiseman, T., S. Williston, J. F. Brandts, and L. N. Lin. 1989. Rapid measurement of binding constants and heats of binding using a new titration calorimeter. Anal. Biochem. 179:131-137. [DOI] [PubMed] [Google Scholar]
- 86.Xavier, K. B., L. O. Martins, R. Peist, M. Kossmann, W. Boos, and H. Santos. 1996. High-affinity maltose/trehalose transport system in the hyperthermophilic archaeon Thermococcus litoralis. J. Bacteriol. 178:4773-4777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Xiao, X., F. Wang, A. Saito, J. Majka, A. Schlosser, and H. Schrempf. 2002. The novel Streptomyces olivaceoviridis ABC transporter Ngc mediates uptake of N-acetylglucosamine and N,N′-diacetylchitobiose. Mol. Genet. Genomics 267:429-439. [DOI] [PubMed] [Google Scholar]
- 88.Yurgel, S. N., and M. L. Kahn. 2004. Dicarboxylate transport by rhizobia. FEMS Microbiol. Rev. 28:489-501. [DOI] [PubMed] [Google Scholar]
- 89.Zaide, G., D. Shallom, S. Shulami, G. Zolotnitsky, G. Golan, T. Baasov, G. Shoham, and Y. Shoham. 2001. Biochemical characterization and identification of catalytic residues in alpha-glucuronidase from Bacillus stearothermophilus T-6. Eur. J. Biochem. 268:3006-3016. [DOI] [PubMed] [Google Scholar]
- 90.Zolotnitsky, G., U. Cogan, N. Adir, V. Solomon, G. Shoham, and Y. Shoham. 2004. Mapping glycoside hydrolase substrate subsites by isothermal titration calorimetry. Proc. Natl. Acad. Sci. USA 101:11275-11280. [DOI] [PMC free article] [PubMed] [Google Scholar]