

Abstract
Key to understanding the structural biology of catalytic RNA is determining the underlying networks of interactions that stabilize RNA folding, substrate binding, and catalysis. Here we demonstrate the existence and functional importance of a Hoogsteen base triple (U300⋅A97-U277), which anchors the substrate helix recognition surface within the Tetrahymena group I ribozyme active site. Nucleotide analog interference suppression analysis of the interacting functional groups shows that the U300⋅A97-U277 triple forms part of a network of hydrogen bonds that connect the P3 helix, the J8/7 strand, and the P1 substrate helix. Product binding and substrate cleavage kinetics experiments performed on mutant ribozymes that lack this base triple (C A-U, U G-C) or replace it with the isomorphous C+⋅G-C triple show that the A97 Hoogsteen triple contributes to the stabilization of both substrate helix docking and the conformation of the ribozyme’s active site. The U300⋅A97-U277 base triple is not formed in the recently reported crystallographic model of a portion of the group I intron, despite the presence of J8/7 and P3 in the RNA construct [Golden, B. L., Gooding, A. R., Podell, E. R. & Cech, T. R. (1998) Science 282, 259–264]. This, along with other biochemical evidence, suggests that the active site in the crystallized form of the ribozyme is not fully preorganized and that substantial rearrangement may be required for substrate helix docking and catalysis.
To accomplish substrate helix docking and catalysis (1, 2), the Tetrahymena ribozyme (Fig. 1) must fold into a complex tertiary structure with a close-packed catalytic core (3) in which several structural elements come together to jointly recognize and interact with the P1 substrate helix. These elements include the J4/5 internal loop (4, 5), the J8/7 strand (6, 7), and the attacking guanosine nucleophile bound within the P7 helix (8). This complex interaction of multiple close-packed helical and single-stranded RNA elements (9) requires several layers of interactions to create the active site of the molecule. The molecular details of these interactions and the underlying principles of RNA tertiary structure stabilization and substrate recognition within the Tetrahymena ribozyme can be readily investigated from a functional perspective by using the techniques of nucleotide analog interference mapping (NAIM) and nucleotide analog interference suppression (NAIS) (7, 10).
Figure 1.

Tetrahymena group I intron secondary structure. This schematic diagram of the intron illustrates the secondary structure of the L-21 G414 version of the Tetrahymena group I self-splicing intron with a substrate oligonucleotide bound to the internal guide sequence (IGS). Heavy lines and letters indicate nucleic acid sequences while thin arrows trace the connections between sequence elements. Important paired (P) and joining (J) regions are labeled. Interacting nucleotides C(-5), G26, U300, A97, and U277 are marked with circles, and the mutations examined in this work are indicated. Individual nucleotides in regions P2-P2.1, P5abc, and P9-P9.2 have been omitted for clarity but are present in the RNAs characterized in this study.
Recently, the results of NAIM and NAIS experiments to identify the array of hydrogen bonding interactions that stabilize the interaction between the P1 substrate helix and the highly conserved J8/7 strand within the intron core were reported (7, 11). These data, along with other biochemical structural information (4–6, 12, 13), were used as restraints in building a model of a large section of the ribozyme active site. In conjunction with a G⋅U wobble receptor in the J4/5 region (4, 5), the J8/7 strand creates a complementary surface for recognition of the P1 substrate helix (6, 7). The data indicate that the J8/7 strand forms an extended minor groove triple helical interaction with four 2′-OH groups within the 5′-exon (P1) helix of the intron. This docking interface includes a tertiary interaction between the 2′-oxygen of U300 at the 5′-end of J8/7 and the 2′-OH of G26 within the P1 helix. However, the details of the interactions between J8/7 and the rest of the intron’s core remain largely unknown.
The single-stranded J8/7 segment is a particularly critical element of the group I active site. Its sequence contains some of the most phylogenetically conserved nucleotides within the intron (14), and it is covalently connected to the P8 and P7 helices (15). In addition to forming the extended triplex with the P1 duplex, it appears that the J8/7 strand may be tethered at both of its ends by forming base triples to two addition duplexes within the intron’s core. At the 3′ end of J8/7, nucleotide U305 forms a base triple with the G111-C209 Watson-Crick base pair in helix P4 (12, 13). At the 5′ end of J8/7, phylogenetic evidence suggests that U300 may form a triple with the A97-U277 base pair in the P3 helix (Figs. 1 and 2) (11, 16), although this has not been tested biochemically.
Figure 2.

Wild-type and mutant ribozyme base triples involving nucleotides 300, 97, and 277. Functional groups assayed by NAIM are indicated by rectangles (interference) and ovals (enhancement). (A) Wild-type Tetrahymena intron base triple, U300⋅A97-U277, in which the H3 of U300 hydrogen bonds with the N7 of A97, and the O4 of U300 hydrogen bonds with the N6 amine of A97. Both the N7 and N6 positions show interference in the wild-type ribozyme (11). (B) U300C (1X) mutant. By replacing U300 with a C, the interaction with the Hoogsteen face of A97 is perturbed. (C) A97G + U277C (2X) mutant. The base triple is disrupted by replacing the A97-U277 base pair with a Watson-Crick G-C pair. (D) U300C + A97G − U277C (3X) mutant. This set of mutations restores the predicted base triple with an isomorphous triple that is the next most phylogenetically conserved sequence in the IC1 and IC2 intron subgroups (11). In every case that U300 is changed to a C within these subgroups, the A97-U277 base pair co-varies as a G-C pair.
Confirmation of the J8/7-P3 triple would provide a valuable experimental test of structural models of the intron active site. Recently, a model of the Tetrahymena group I intron was created based on 5-Å resolution crystallographic data collected on a version of the intron lacking the substrate helix (P1) and several peripheral elements (P2, P2.1, P9.1, P9.2) (17). Based on the docking of an RNA duplex representing the P1 helix into the active site of their model, Golden et al. (17) concluded that the active site was largely preorganized to bind P1. However, the observation that the phylogenetically predicted U300⋅A97-U277 base triple is not formed within this model illustrates why it is important to examine the putative triple biochemically.
In this paper, we report the results from substrate binding, kinetics, and NAIS experiments to demonstrate the formation and functional significance of the U300⋅A97-U277 base triple. These experiments identify the specific functional groups involved in hydrogen bonds between the bases and demonstrate that the U300⋅A97-U277 triple forms part of an interconnected series of hydrogen bonds that includes nucleotides in helix P3, helix P1, and the J8/7 strand. This network contributes to the stabilization of the substrate helix binding interaction and to the conformation of the ribozyme’s active site. The lack of the U300⋅A97-U277 triple in the 5-Å structural model of Golden et al. (17) argues that the crystallographic model may not reflect a fully preorganized active site.
MATERIALS AND METHODS
Ribozymes and Substrates.
Plasmids for run-off T7 RNA polymerase transcription of mutant ribozymes U300C (1X), A97G + U277C (2X), and U300C + A97G + U277C (3X) were created from the parent pUCL-21G414 plasmid (10) containing the Tetrahymena L-21 G414 sequence by using the GeneEditor multiple oligo-directed mutagenesis kit (Promega). The sequences of these plasmids were confirmed by dideoxy sequencing (18).
Wild-type and mutant L-21 G414 and L-21 ScaI ribozyme RNAs were synthesized by T7 RNA polymerase transcription of EarI- and ScaI-digested plasmid DNA, respectively, and were purified on denaturing polyacrylamide gels (11). For NAIM and NAIS experiments, AαS, 7dAαS, m6AαS, GαS, 7dGαS, CαS, and dCαS were randomly incorporated at low levels (≤5%) into wild-type and mutant ribozymes as described (11). 5′-O-(1-thio)zebularine monophosphate (ZαS) was randomly incorporated (at ≤5%) into the ribozymes by using the Y639F mutant T7 RNA polymerase (19) in transcription buffer containing 40 mM Tris⋅HCl (pH 7.5), 4 mM spermidine, 10 mM DTT, 15 mM MgCl2, 1 mM Mn(OAc)2, 0.05% Triton X-100, 0.05 μg/μl DNA template, and 1 mM ZTPαS in the presence of 1 mM ATP, UTP, and GTP and 0.5 mM CTP. Substrate oligonucleotides 5′-CCCUCdTAAAAA-3′ (d1S), 5′-CdCdCUCdTAAAAA-3′ (d145S), pro-RP 5′-CCCUCdTSA-3′ (d1RpS), and the product oligonucleotide 5′-CCCUCU-3′ (rP) were synthesized by using 2′-ACE chemistry and were deprotected as recommended by the manufacturer (Dharmacon Research, Boulder, CO) (20).
Gel Shift Oligonucleotide Binding Experiments.
Gel shift experiments were performed following the general procedures of Pyle and Cech (21). Ribozymes were diluted to concentrations ranging from 20 pM to 4 μM in buffer containing 50 mM Tris⋅HCl (pH 7.5), 0.1 mM EDTA, 100 mM NaCl, and 4 mM MgCl2 in a total volume of 5 μl and were heated for 10 min at 50°C to ensure proper folding. 5′-32P radiolabeled CCCUCU (≤10 pM) was dissolved in 5 μl of the same buffer plus 0.1% xylene cyanol and 20% glycerol. Both oligonucleotides and ribozymes were equilibrated at 40°C for 2 min, were mixed, and were incubated for another 10 min at 40°C before being loaded on a 10% native polyacrylamide gel to separate bound and free oligonucleotide. Gels contained 34 mM Tris⋅HCl, 66 mM Hepes (pH 7.0), 0.1 mM EDTA, and 4 mM MgCl2. Band intensities of the bound and free forms were measured by using a Molecular Dynamics Storm 820 PhosphorImager, and the data were fit to a two-state binding curve.
L-21 ScaI Ribozyme Kinetics.
Ribozyme cleavage kinetics were measured in a single turnover assay under conditions of saturating ribozyme such that the exponential decay rate constant is equivalent to kcat (22). A limiting amount of oligonucleotide substrate, d1S labeled at the 5′ end with γ-32P-ATP, was dissolved in 20 μl of reaction buffer containing 50 mM Hepes (pH 7.0) and 10 mM MgCl2. GMP (4 mM) was included in the oligo mix. Ribozymes were dissolved in 20 μl of the same buffer without GMP and were folded at 50°C for 10 min. Both ribozyme and substrate oligo mixes then were preequilibrated at 30°C for 2 min before being mixed to initiate the reaction. Final ribozyme concentrations were 100 nM wild-type, 100 nM 3X mutant, 500 nM 1X mutant, and 500 nM 2X mutant. To confirm that the reactions were saturated, both the ribozyme and GMP concentrations were doubled, and no change in the measured rates were observed. Reaction progress was monitored by removing 3-μl aliquots at various times and quenching the reaction with 6 μl of 95% formamide containing EDTA and loading dyes. Cleavage products were separated by 20% denaturing PAGE, and band intensities were quantitated as described above. The fraction of unreacted substrate was plotted vs. time, and the data were fit to an exponential decay with a non-zero endpoint to obtain kcat for each enzyme.
Nucleotide Analog Interference Mapping and Interference Suppression.
NAIM experiments were performed as described (10, 11) by using the L-21 G414 form of the Tetrahymena group I intron and substrate oligonucleotides in a 3′-ligation assay that is analogous to the reverse of the second step of splicing (23). Ribozymes were synthesized with low level incorporation of each of the nucleotide analogs listed above. Oligonucleotides d1S and d145S were 3′-end labeled by using T4 RNA ligase with α-32P-pCp generated from Cp and γ-32P-ATP (24). Analog interference was assayed for wild-type and 3X mutants by reacting the ribozyme with the appropriate substrate for 10 min at 50°C in buffer containing 50 mM Hepes (pH 7.0), 3 mM MgCl2, and 1 mM Mn(OAc)2. The 1X mutant ribozyme was much less reactive and was therefore incubated for 90 min at 30°C in the same buffer. To eliminate miscleavage observed because of the slow reaction rate of d1S and the 1X ribozyme, the d145S substrate was used for 1X mutant NAIM ligation reactions. The presence of 2′-deoxyribose sugars at positions −4 and −5 in this substrate prevents miscleavage but does not destabilize helix docking (21). To facilitate a direct comparison, wild-type ribozyme reactions also were carried out with d145S. To ensure that the substrate was completely bound, ribozyme concentrations were severalfold higher than the dissociation constants measured for each mutant. Wild-type and 1X reactions used 50 nM and 400 nM ribozyme, respectively. Experiments with the 3X mutant used d1S and 100 nM ribozyme. To control for the extent of analog incorporation at each nucleotide position, the L-21 G414 RNAs were 5′-end 32P labeled and cleaved with iodine (10). Iodine cleaved RNA fragments were analyzed by 5% and 6% denaturing PAGE. Individual band intensities were measured on a PhosphorImager and interference (κ) values for each nucleotide position were calculated as described (10, 11) using the formula
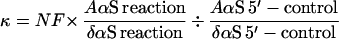 |
1 |
where NF represents the cumulative normalization factor necessary to correct for loading differences and variability in the extent of the ligation reaction (NF is typically between 0.8 and 1.2).
RESULTS
To study the phylogenetically predicted U300⋅A97-U277 triple interaction between J8/7 and P3, three variants of the L-21 G414 ribozyme were created by site-directed mutagenesis (Figs. 1 and 2). Two of these mutations are expected to perturb the base triple whereas the third mutation should restore it. The U300C (1X) mutation creates a C A-U juxtaposition whereas the A97G + U277C (2X) mutation creates a U G-C (Fig. 2). Both mutations destabilize potential hydrogen bonding interactions between the pyrimidine at position 300 and the purine at position 97. The third mutation, U300C + A97G + U277C (3X), changes the identity of all three potentially interacting nucleotides to create a base triple that is isomorphous with the original U⋅A-U (25). The C+⋅G-C is expected to restore the hydrogen bonding array if the triple is protonated, as has been observed in DNA triplexes (25) and in RNA triples (26). To determine whether the predicted U300⋅A97-U277 triple is in fact formed within the intron’s core and, if so, what functional significance this interaction might have, we compared the wild-type and mutant ribozymes in terms of product oligonucleotide binding affinity, the rate of substrate oligonucleotide cleavage, and the nucleotide analog interference pattern of the base triple mutations.
The U300⋅A97-U277 Triple Stabilizes Substrate Helix Binding.
The J8/7 single stranded region contributes significantly to P1 helix docking by interacting with 2′-OHs along the length of the helix (7, 21). One of the J8/7 nucleotides that recognizes a 2′-OH in P1 is U300, which is also implicated as the third nucleotide in the U300⋅A97-U277 triple. Therefore, disrupting the base triple interaction with A97 might reduce P1 binding affinity by destabilizing U300 and the rest of J8/7. To test this hypothesis, the affinity of each of the ribozyme variants for a product oligonucleotide was measured. The binding affinities of CCCUCU to the L-21 ScaI form of the ribozymes were measured by gel shift experiments, which reflect the affinity of the ribozyme for the P1 substrate helix (21). In 100 mM NaCl and 4 mM MgCl2 at 40°C, both the 1X and 2X mutant ribozymes bind CCCUCU ≈1 kcal⋅mol−1 less tightly than the wild-type ribozyme (Table 1). In contrast, the 3X mutant binds as well as the wild-type ribozyme. The simplest interpretation of this result is that disrupting the U300⋅A97-U277 base triple destabilizes the tertiary structure of the ribozyme and therefore interferes with 5′-exon binding. The base triple interaction is restored within the 3X mutant, and, as a consequence, wild-type substrate affinity is recovered. The magnitude of the loss in 5′-exon affinity is consistent with an indirect effect, as might be expected for the loss of this triple, which does not directly contact the P1 helix.
Table 1.
Cleavage rate and dissociation constants for L-21 ScaI mutant ribozymes
| Ribozyme | Product binding*
|
Substrate cleavage†
|
|||
|---|---|---|---|---|---|
| Kd, nM | Rel. | ΔΔG, kcal/mol | kcat, min−1 | Rel. | |
| Wild-type | 11 ± 2 | 1.0 | 0.00 | 0.0450 ± 0.0008 | 1.0 |
| 1X | 92 ± 3 | 8.4 | 1.32 | 0.00240 ± 0.00014 | 18 |
| 2X | 50 ± 9 | 4.5 | 0.94 | 0.0195 ± 0.0007 | 2.3 |
| 3X | 12 ± 3 | 1.1 | 0.05 | 0.0425 ± 0.0006 | 1.1 |
Average of at least three independent CCCUCU-binding experiments.
Average of at least two independent CCCUCdTAAAAA-cleavage experiments. Reported errors are the standard deviation of the sample.
The U300⋅A97-U277 Triple Also Helps Stabilize the Active Site.
To determine whether removing the A97 triple interaction affects more than just 5′-exon binding, we examined the rate of substrate cleavage by the L-21 ScaI form of the three mutant ribozymes (Fig. 3A). At saturating ribozyme concentrations (100 nM for the wild-type and 3X mutants, and 500 nM for the 1X and 2X ribozymes) the single turnover rate for cleavage of the d1S substrate was measured. In this assay, cleavage of bound substrate oligonucleotides proceeds relatively slowly, and the chemical step is limiting, such that kcat = kchem (27, 28). The 1X mutant ribozyme reacts 18-fold slower than wild-type whereas kcat for the 2X mutant is only reduced by a factor of 2.3 (Table 1). As was observed for substrate binding, the 3X mutant reacts with the same rate as the wild type. No increases in rates were detected when the ribozyme concentrations were doubled, indicating that ribozyme concentration is saturating and the rate is equal to kchem. The unreactive fractions of all mutant ribozymes were equally small (<15%), eliminating misfolding as a potential cause for the difference in reaction rates. To further confirm that kcat is equal to kchem, experiments were performed with a substrate oligonucleotide, d1RpS, containing a pro-RP phosphorothioate at the cleavage site. The presence of a phosphorothioate should reduce the rate of chemistry by ≈2- to 10-fold because of the inherent reactivity of the sulfur-substituted substrate (27). A phosphorothioate effect of this magnitude was observed for all four ribozymes (data not shown), indicating that all of the rates measured are equal to kchem. These results show that disrupting the A97 triple slows the rate of catalysis, and, thus, this interaction must help stabilize the active form of the group I intron’s catalytic core.
Figure 3.

Group I ribozyme reactions used in this work. (A) L-21 ScaI cleavage reaction (31) used for kinetics and binding studies. In the presence of guanosine, the bound substrate oligonucleotide is cleaved by the ribozyme into two fragments in a reaction that can be monitored kinetically (22). Product binding affinity can be studied by gel shift analysis (21). (B) L-21 G414 ligation reaction used for NAIM and NAIS experiments, in which G414, bound in the G-binding site, attacks a 3′-labeled substrate oligonucleotide. The reaction is analogous to the reverse of the second step of splicing (23), in which the AAAAAA* is covalently transferred onto the 3′ ends of the active ribozyme variants.
NAIM Confirms the Predicted Base Triple Structure.
To examine in atomic detail the hydrogen bonding interactions between U300 and A97, we performed NAIM experiments using the L-21 G414 versions of each ribozyme with nucleotide analogs randomly incorporated at a low level throughout the RNA. The L-21 G414 form of the intron performs a 3′-exon ligation reaction that is analogous to the reverse of the second step of splicing. At substoichiometric concentrations of substrate, only the most reactive molecules in the population become radiolabeled (Fig. 3B). We used this reaction in a NAIM assay to identify the functional groups important for stabilizing the active conformation of the ribozyme. By assaying interference in the context of the different mutants, NAIS makes it possible to identify the hydrogen bonding partners within the base triple interaction.
In the wild-type form of the group I ribozyme, interference is observed at A97 with 7-deazaadenosine (7dAαS), N-methyladenosine (m6AαS), and purine riboside (PurαS) (Figs. 4 and 5) (11). Such a pattern of interference is consistent with a hydrogen bonding interaction to the Hoogsteen face of A97, as all of these analogs modify or delete functional groups in the major groove of the P3 helix. Interference also is observed at U300 with dUαS in the context of the wild-type ribozyme (Fig. 5) because of disruption of a hydrogen bond between the 2′-OHs of U300 and G26. This is one of the interactions that is required for efficient substrate helix docking of P1 into J8/7 (7).
Figure 4.

Nucleotide analog interference mapping experiments. Shown here is a sample set of denaturing polyacrylamide gels that resolve the active ribozyme I2-cleaved RNA fragments resulting from NAIM experiments. Each gel lane is labeled to indicate the specific mutation and the incorporated nucleotide analog in each ribozyme. Nucleotide numbers are indicated to the left of the autoradiographs. Asterisks mark bands of interest. (A) Interference at position 300 in the 3X and 1X mutant ribozymes. Lanes: 1 and 4, CαS phosphorothioate interference controls; 2 and 5, 3X and 1X ribozyme dCαS interference patterns; 3 and 6, 3X and 1X ribozyme ZαS interference patterns. (B) Interference at position 97 in the wild-type and 1X mutant ribozymes. Lanes: 1 and 5, AαS controls; 2, 3, and 4, wild-type m6AαS, PurαS, and 7dAαS interference; 6, 7, and 8, 1X mutant ribozyme m6AαS, PurαS, and 7dAαS interference suppression. The 5′-end labeled control reactions are not shown [similar data have been previously published (11)].
Figure 5.

Interference effects for wild-type and mutant ribozymes. Each ribozyme’s nucleotide triple is listed on the left (see Fig. 2 for the structure of each triple). The height of each bar represents the interference value (κ) at each position for a given analog, calculated as described in Materials and Methods. Bars extending below the baseline correspond to enhancement and have a magnitude of 1/κ. The label “na” indicates that no corresponding analog was available for that nucleotide.
Interference experiments were performed with the 3X and 1X ribozymes to confirm the formation of a C+⋅G-C triple and to identify sites of interference suppression, respectively. The analog 7-deazaguanosine (7dGαS), like 7dAαS, modifies the Hoogsteen face of the purine base and should show interference when incorporated at G97 if a base triple is formed in the 3X mutant. In contrast, if the J8/7-P3 triple does not form in the 1X mutant, as predicted by the 5′-exon binding experiments, the major groove modifying analogs 7dAαS, PurαS, and m6AαS should not show interference at A97. The U300C mutation should therefore suppress the Hoogsteen interference because the N7 and the N6 functional groups no longer contribute to activity if the triple cannot form.
As predicted by the wild-type levels of binding and cleavage observed for the 3X mutant, 7dGαS interference is observed at G97 (Fig. 5), consistent with the formation of the C+⋅G-C triple diagrammed in Fig. 2. In contrast, there was no interference at A97 with 7dAαS, m6AαS, or PurαS in the context of the 1X mutant. In fact, 7dAαS and PurαS actually show enhancement at A97, suggesting that a 1X ribozyme with these groups modified is more reactive than the unmodified form. 7dAαS and PurαS each would eliminate a potentially disruptive interaction between C300 and A97. PurαS eliminates a clash of two amino groups whereas 7dAαS eliminates any possible negative interaction attributable to juxtaposition of two lone electron pairs. Together, this provides high resolution biochemical data to suggest that both the N7 and the N6 position are interacting with U300 in the wild-type ribozyme and that C300 does not form hydrogen bonds to A97 in the 1X ribozyme.
We also attempted to probe the pyrimidine side of the base triple interaction by incorporating zebularine (ZαS, a C analog that lacks the N4 amine) into the 1X and 3X mutant ribozymes. If the base triple is restored in the 3X mutant, incorporation of ZαS at C300 should result in interference. In contrast, ZαS interference should be suppressed in the 1X mutant that lacks a base triple. As expected, ZαS interference is seen at C300 in the context of the 3X mutant, consistent with an interaction between the N4 C300 amino group and the O6 of G97. In the context of the 1X mutant, ZαS incorporation at C300 results in substantially enhanced ribozyme activity (Figs. 4 and 5). Thus, eliminating the clash of amino groups by deleting an amine from either side of the triple interaction (C300 or A97) enhances reactivity.
Finally, to analyze the nature of the J8/7 interaction with P1, 2′-deoxy interference at position 300 was assayed. Both the 3X and 1X mutant ribozymes showed interference at C300 with 2′-deoxycytidine (dCαS), indicating that the important 2′-hydroxyl interaction between the nucleotide at position 300 and the substrate helix is maintained, whether or not the base triple is formed. This is a valuable control because it demonstrates that binding affinity is not reduced simply because of the loss of the hydrogen bond between the G26 and U300 2′-OHs. Instead, it argues that the P1-J8/7 complex is not properly aligned against P3 to achieve a stable complex in the absence of a base triple interaction between nucleotides 300 and 97.
DISCUSSION
Implications for Intron Structure and Function.
The U300⋅A97-U277 base triple diagrammed in Fig. 2A was proposed by Michel and Westhof in their original group I structural model, based on phylogenetic data (16). These experiments provide biochemical evidence that the U⋅A-U base triple is functionally important. Disruption of the base triple results in decreased ribozyme affinity for the 5′-exon helix and a reduced rate of chemistry for the L-21 ScaI cleavage reaction. Replacing the U300⋅A97-U277 triple with the isomorphous C300+⋅G97-C277 triple completely restores binding and catalysis to wild-type levels. Thus, the triple interaction with U300 is important for stabilizing both substrate binding and the conformation of the ribozyme active site. This is consistent with the predicted role of U300 in substrate helix binding and the importance of the J8/7 strand in forming the intron core (7).
The pattern of analog interference in the mutant and wild-type ribozymes further supports the existence of the A97 triple and helps to explain the differences in substrate affinity and reactivity of the three mutants. The greatest reduction in binding affinity and reactivity is seen with the 1X mutant, in which the amino groups of C300 and A97 are opposed (Fig. 2B). Lack of interference in the 1X mutant when the N7 of A97 is deleted argues against a hydrogen bond between C300 and the N7 of A97. In fact, several analog enhancements are observed (Fig. 5), which suggest that removal of one of the two opposed lone pairs of electrons, or elimination of steric clash between the two amines, alleviates a negative interaction within the 1X mutant. Another explanation for the observed enhancements could be that analog incorporation disrupts an alternative non-native base triple in the 1X mutant.
The interference data also support the formation of a C+⋅G-C triple in the 3X mutant. Interference caused by deleting the N7 of G97 in the 3X mutant suggests that the G accepts a hydrogen bond. The most likely hydrogen bonding partner is the protonated N3 of C300, which has been observed in a similar RNA base triple by NMR spectroscopy (26). Strong interference at C300 has been observed from nucleotide analogs that reduce the pKa of the N3 of C (A.K.O. and S.A.S., unpublished results). The effect can be rescued by reducing the pH of the reaction solution, which provides further evidence that the N3 position is protonated in the C+-G-C triple. Retention of 2′-deoxy interference at position 300 in both the 1X and 3X mutants is consistent with preservation of the hydrogen bond between the 2′-oxygen of U300 and the 2′-OH of G26, even in the absence of a base triple interaction. However, the fact that substrate affinity is reduced in the 1X and 2X mutants implies that substrate binding depends not only on the hydrogen bonds between J8/7 and P1 but also on the stability of the entire tertiary structure at the center of the intron.
These and other biochemical data indicate that, within its 7-nucleotide length, J8/7 mediates the close approach of five helices (P1, P3, P4, P7, and P8) within the core of the intron’s active site (4–7, 12, 13). The U300⋅A97-U277 base triple is part of a hydrogen bonding network that juxtaposes the A97-U277 base pair in helix P3 against nucleotide U300 in the J8/7 strand, which in turn interacts with the C(-5)-G26 base pair in helix P1 (Fig. 6). These hydrogen bonds provide a set of stabilizing interactions that help to define not only the substrate binding surface but also the conformation of the catalytic site. In this way, a single nucleotide, U300, organizes two different helical elements (P1 and P3). Such a hydrogen bond network may be a common mechanism for mediating the close approach of helices in other RNAs.
Figure 6.

Three-dimensional model of the group I ribozyme core in the region near the U300⋅A97-U277 base triple (7). Dotted lines indicate hydrogen bonds. The interference and phylogenetic data suggest that the base triple is part of a hydrogen bonding network that connects the A97-U277 base pair in helix P3 with U300 in the J8/7 strand and with the C(-5)-G26 base pair in the P1 substrate helix. In this way, U300 within the J8/7 strand mediates the close approach of two helices in the ribozyme active site.
Comparison to the Crystallographic Model of the Intron Active Site.
Golden et al. have reported a model for the Tetrahymena group I intron based on 5-Å resolution x-ray diffraction data of a truncated ribozyme that includes the J8/7 and P3 elements but lacks the P1 substrate helix and the P2, P2.1, P9.1, and P9.2 peripheral elements (17). Based primarily on the similarity of the two molecules in the asymmetric unit and the observation that, when a P1 helix is modeled into J4/5, the splice site ends up in proximity to the guanosine binding site in P7, they concluded that the ribozyme’s active site is largely preorganized to bind its substrate.
The U300⋅A97-U277 base triple is not formed within the model of Golden et al. (17), although all three nucleotides are present in the crystallized construct. This triple significantly constrains the relative location of the P3 and P1 helices. The location of the P1 helix has been defined relative to the P4-P6 domain [these constraints were used by Golden et al. (17) to build the docked P1 complex] (4, 5). Thus, this network of tertiary interactions suggests that the P3 helix must move as much as 20–25 Å from its current position in the 5-Å model toward the P4-P6 domain to reach the docked conformation.
This raises the question of how well the structure fits with other previously published biochemical data regarding the active site of the ribozyme. Golden et al. (17) argue that their model places the 5′-splice site into proximity with the terminal guanosine (ωG) binding site (8). Inspection of the structural coordinates suggests that ωG is several angstroms out of its binding site in P7 and still 10 Å away from the 5′-splice site in P1. For the ribozyme to achieve catalysis, it appears that P7 must be 15–25 Å closer to J4/5 than it is in the crystallographic model. Additionally, the bases of several nucleotides within J8/7 (A301, A302, G303) that have been shown to interact with the docked P1 helix (6, 7) are pointed in the opposite direction toward the P3 helix, and the J8/7 strand is compressed along its ribose backbone. For these bases to make ground state interactions with P1, it seems that the phosphate backbone of J8/7 must be extended and rotated 180°.
Although the global fold of the intron appears to be roughly preorganized in the structure, helical and single stranded rearrangements of this magnitude seem inconsistent with an active site that is preorganized before substrate binding. If the structure of Golden et al. (17) reflects the open conformation of the ribozyme (the active site lacking the substrate helix), then these data argue that significant structural changes must occur between the open and closed ribozyme complexes. This would be consistent with the observation that substrate helix docking within the group I intron is entropically driven (29) and with other results that suggest that the group I intron experiences large scale internal motions (4, 30). Two alternative explanations are that the 5-Å electron density map provides insufficient detail to accurately position the RNA or that a non-native conformation of the active site in the truncated intron may have been crystallized because of the lack of the substrate helix and other peripheral elements. In either case, the absence of the U300⋅A97-U277 triple in the crystallographic model suggests that additional structural and biochemical data are needed to reach conclusions regarding the extent of preorganization of the group I intron active site.
Acknowledgments
We thank Sean Ryder for helpful discussions and Lara Weinstein for a critical reading of the manuscript. A.K.O. is the recipient of a postdoctoral fellowship from the Jane Coffin Childs Memorial Fund for Medical Research. This work was supported by National Institutes of Health Grant GM 54839, a Beckman Young Investigator award, and a Searle Scholar award to S.A.S.
ABBREVIATIONS
- NAIM
nucleotide analog interference mapping
- NAIS
nucleotide analog interference suppression
References
- 1.Cech T R, Herschlag D, Piccirilli J A, Pyle A M. J Biol Chem. 1992;267:17479–17482. [PubMed] [Google Scholar]
- 2.Bevilacqua P C, Kierzek R, Johnson K A, Turner D H. Science. 1992;258:1355–1358. doi: 10.1126/science.1455230. [DOI] [PubMed] [Google Scholar]
- 3.Latham J A, Cech T R. Science. 1989;245:276–282. doi: 10.1126/science.2501870. [DOI] [PubMed] [Google Scholar]
- 4.Wang J F, Downs W D, Cech T R. Science. 1993;260:504–508. doi: 10.1126/science.7682726. [DOI] [PubMed] [Google Scholar]
- 5.Strobel S A, Ortoleva-Donnelly L, Ryder S P, Cate J H, Moncoeur E. Nat Struct Biol. 1998;5:60–66. doi: 10.1038/nsb0198-60. [DOI] [PubMed] [Google Scholar]
- 6.Pyle A M, Murphy F L, Cech T R. Nature (London) 1992;358:123–128. doi: 10.1038/358123a0. [DOI] [PubMed] [Google Scholar]
- 7.Szewczak A A, Ortoleva-Donnelly L, Ryder S, Strobel S A. Nat Struct Biol. 1998;5:1037–1042. doi: 10.1038/4146. [DOI] [PubMed] [Google Scholar]
- 8.Michel F, Hanna M, Green R, Bartel D P, Szostak J W. Nature (London) 1989;342:391–395. doi: 10.1038/342391a0. [DOI] [PubMed] [Google Scholar]
- 9.Cech T R, Herschlag D. In: Nucleic Acids and Molecular Biology. Eckstein F, Lilley D M J, editors. Vol. 10. New York: Springer; 1996. pp. 1–17. [Google Scholar]
- 10.Strobel S A, Shetty K. Proc Natl Acad Sci USA. 1997;94:2903–2908. doi: 10.1073/pnas.94.7.2903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ortoleva-Donnelly L, Szewczak A A, Gutell R R, Strobel S A. RNA. 1998;4:498–519. doi: 10.1017/s1355838298980086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tanner M A, Anderson E M, Gutell R R, Cech T R. RNA. 1997;3:1037–1051. [PMC free article] [PubMed] [Google Scholar]
- 13.Tanner M A, Cech T R. Science. 1997;275:847–849. doi: 10.1126/science.275.5301.847. [DOI] [PubMed] [Google Scholar]
- 14.Damberger S H, Gutell R R. Nucleic Acids Res. 1994;22:3508–3510. doi: 10.1093/nar/22.17.3508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Waring R B, Davies R W, Brown T A, Scazzocchio C. Gene. 1984;28:277–291. [Google Scholar]
- 16.Michel F, Westhof E. J Mol Biol. 1990;216:585–610. doi: 10.1016/0022-2836(90)90386-Z. [DOI] [PubMed] [Google Scholar]
- 17.Golden B L, Gooding A R, Podell E R, Cech T R. Science. 1998;282:259–264. doi: 10.1126/science.282.5387.259. [DOI] [PubMed] [Google Scholar]
- 18.Saenger F, Nicklen S, Coulson A R. Proc Natl Acad Sci USA. 1977;74:5463–5467. doi: 10.1073/pnas.74.12.5463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sousa R, Padilla R. EMBO J. 1995;14:4609–4621. doi: 10.1002/j.1460-2075.1995.tb00140.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Scaringe S A, Wincott F E, Caruthers M H. J Am Chem Soc. 1998;120:11820–11821. [Google Scholar]
- 21.Pyle A M, Cech T R. Nature (London) 1991;350:628–631. doi: 10.1038/350628a0. [DOI] [PubMed] [Google Scholar]
- 22.Herschlag D, Cech T R. Biochemistry. 1990;29:10159–10171. doi: 10.1021/bi00496a003. [DOI] [PubMed] [Google Scholar]
- 23.Beaudry A A, Joyce G F. Science. 1992;257:635–641. doi: 10.1126/science.1496376. [DOI] [PubMed] [Google Scholar]
- 24.England T E, Bruce A G, Uhlenbeck O C. Methods Enzymol. 1980;65:65–74. doi: 10.1016/s0076-6879(80)65011-3. [DOI] [PubMed] [Google Scholar]
- 25.Moser H E, Dervan P B. Science. 1987;238:645–650. doi: 10.1126/science.3118463. [DOI] [PubMed] [Google Scholar]
- 26.Brodsky A S, Erlacher H A, Williamson J R. Nucleic Acids Res. 1998;26:1991–1995. doi: 10.1093/nar/26.8.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Herschlag D, Piccirilli J A, Cech T R. Biochemistry. 1991;30:4844–4854. doi: 10.1021/bi00234a003. [DOI] [PubMed] [Google Scholar]
- 28.Herschlag D, Eckstein F, Cech T R. Biochemistry. 1993;32:8312–8321. doi: 10.1021/bi00083a035. [DOI] [PubMed] [Google Scholar]
- 29.Li Y, Bevilacqua P C, Mathews D, Turner D H. Biochemistry. 1995;34:14394–14399. doi: 10.1021/bi00044a016. [DOI] [PubMed] [Google Scholar]
- 30.Cohen S B, Cech T R. J Am Chem Soc. 1997;119:6259–6268. [Google Scholar]
- 31.Zaug A J, Grosshans C A, Cech T R. Biochemistry. 1988;27:8924–8931. doi: 10.1021/bi00425a008. [DOI] [PubMed] [Google Scholar]