

Abstract
RNA interference-mediated gene silencing has the potential to block gene expression. A synthetic double-stranded (ds) siRNA based on a sequence motif of 21 nts from human papillomavirus 16 (HPV16) E6E7 bicistronic RNA was found to be a potent small interfering RNA (siRNA) that suppresses expression of both the E6 and E7 oncogenes in HPV16+ CaSki and SiHa cells. When stably expressed as a short hairpin RNA (shRNA) in these cells, however, siRNA silencing of E6 and E7 expression was efficient only at early cell passages, but became inefficient with increased cell passages despite the continued expression of the siRNA at the same level. The loss of the siRNA function was duplicable in stable p53 siRNA cells, but not in stable lamin A/C siRNA cells, suggesting that it is gene-selective. The cells resistant to siRNA function retained normal siRNA processing, duplex unwinding and degradation of the unwound sense strand and RISC formation, suggesting that loss of the siRNA function occurred at a later step. Surprisingly, the siRNA-resistant cells were found to express notably a cytoplasmic protein of ~50 kDa that specifically and characteristically interacted with the unwound, antisense strand E7 siRNA. Altogether, our data indicate that a potent siRNA targeting to an essential or regulatory gene might induce a cell to develop siRNA-suppressive function.
Keywords: HPV16, E6, E7, oncogene, siRNA, cervical cancer
Introduction
RNA interference (RNAi) mediated through double-stranded (ds), small interfering RNA (siRNA) has been widely accepted as a gene-silencing tool in invertebrate and vertebrate cells and has potential clinical applications for therapeutic gene silencing (Dykxhoorn et al., 2003). One means of generating an siRNA is through the processing of a dsRNA by a ribonuclease-III-like enzyme, Dicer (Zamore et al., 2000; Elbashir et al., 2001; Zhang et al., 2002; Provost et al., 2002). In the resulting siRNA the antisense strand siRNA base-pairs with the targeted region of the mRNA in sequence-specific manner, leading to the degradation of the mRNAs (Martinez et al., 2002; Schwarz et al., 2003) or the inhibition of the mRNA translation (Doench et al., 2003; Zeng et al., 2003). Similarly, a synthetic 21-nt dsRNA containing 19 base pairs with 2-nt, 3′ overhanging ends can be introduced experimentally into cells, triggering siRNA-mediated gene silencing (Hammond et al., 2000; Elbashir et al., 2001; Yu et al., 2002).
Cervical infection with high-risk human papillomaviruses (HPVs), such as HPV16 and HPV18, is strongly associated with development of cervical cancer. Integration of the viral genomes into the cancer cell genome is characteristic of infection with these HPVs. Thus, the majority of cervical cancer cell lines established from cervical cancer permanently carry these virus genomes and express two viral oncoproteins, E6 and E7, which inactivate, respectively, p53 and pRb (Scheffner et al., 1990; Boyer et al., 1996; Gonzalez et al., 2001).
HPV16 E6 (16E6) and E7 (16E7) are expressed as a single bicistronic transcript (16E6E7) from the same promoter, p97 (Smotkin and Wettstein, 1986). The bicistronic transcript has three exons and two introns, which undergo alternative RNA splicing to produce both the E6 and E7 proteins (Zheng et al., 2004). Several approaches have been taken to block 16E6 and 16E7 expression experimentally in cervical cancer cell lines and some promising results have been obtained. These include viral E2-mediated suppression of the p97 promoter (Goodwin et al., 2000) and siRNA-mediated silencing of E6 or E7 expression (Jiang and Milner, 2002; Butz et al., 2003; Yoshinouchi et al., 2003). For this study, we chose an siRNA targeting a region downstream of all three alternative 3′ splice sites in the E7 coding region (Zheng et al., 2004). Theoretically, this siRNA should be potentially more 16E6E7-suppressive. In addition, we also attempted to obtain persistent suppression of HPV16 E6E7 expression by stably expressing the 16E7-specific siRNA in cervical cancer cell lines. To our surprise, we found that, although the stably transfected siRNA continued to be expressed in long-term culture, it retained its inhibitory function only for a short period.
Results
Transient transfection of synthetic HPV16 E7 siRNA silences expression of both the E6 and E7 oncogenes
The synthetic E7 dsRNA 198 used in this report has met six of eight criteria of siRNA design (Reynolds et al., 2004) and targets a region downstream of all three alternative 3′ splice sites in the 16E6E7 bicistronic RNA (Figure 1a). Unlike other HPV16 E6 or E7 siRNAs described (Jiang et al., 2002; Butz et al., 2003; Yoshinouchi et al., 2003), this siRNA duplex aims to degrade all unspliced and spliced isoforms of the E6E7 RNA and to suppress expression of both the E6 and E7 oncogenes, which are essential for HPV16 oncogenesis in the cervix (Howley and Lowy, 2001; zur Hausen H., 2002).
Figure 1.
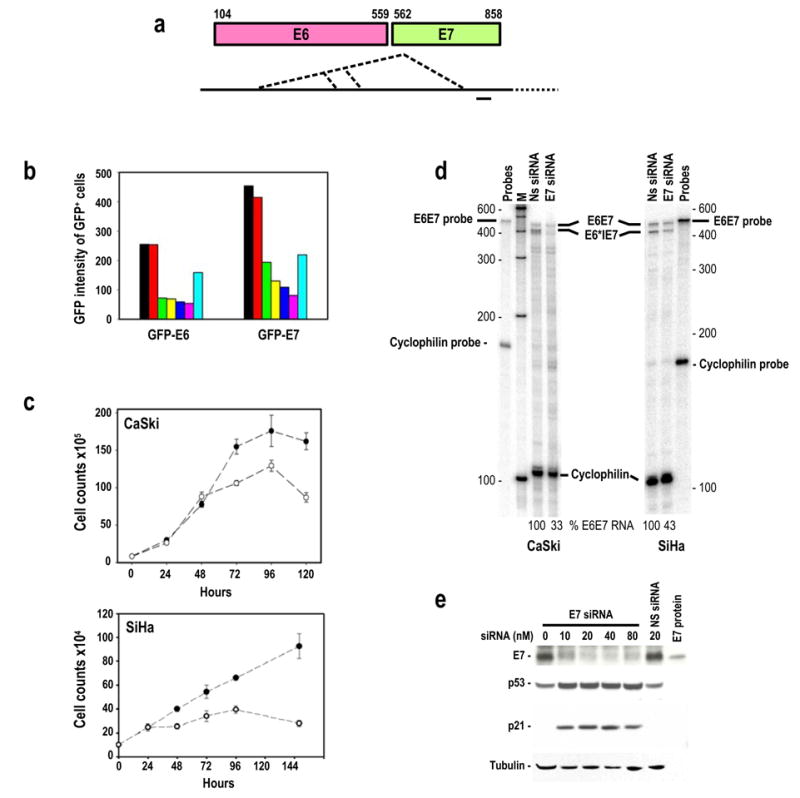
Suppression of HPV16 E6 and E7 expression by transient transfection of HPV16 E7 siRNA. (a) Diagram of HPV16 E6 and E7 gene structure (colored boxes) and their bicistronic transcripts (long heavy line) with alternative splice sites as depicted by the dotted lines. A small heavy dash immediately below the transcript indicates the region targeted by the E7 siRNA 198 duplex in this study. (b) Inhibition of GFP-E6 and GFP-E7 expression in 293 cells by co-transfection of GFP-E6 or GFP-E7 expression vector (1 μg/dish) with no siRNA (black), 20 nM non-specific (NS) siRNA (red), 10 (green), 20 (yellow), 40 (blue), or 80 nM (magenta) E7 siRNA, or 20 nM GFP siRNA (sky blue) as analyzed by flow cytometry. (c) Growth inhibition of cervical cancer–derived HPV16+ CaSki and SiHa cells in the presence of 40 nM E7 siRNA (open circles) or NS siRNA (closed circles). (d) HPV16 E7 siRNA-mediated degradation of HPV16 E6E7 RNAs in cervical cancer–derived CaSki and SiHa cells as analyzed by RPA. Total cell RNA was prepared after 24 hours of transfection with 20 nM E7 siRNA or NS siRNA. Cellular cyclophilin RNA was used for normalization of sampling as described (Tang and Zheng, 2002). (e) Inhibition of HPV16 E7 expression and accumulation of p53 and p21 proteins in CaSki cells 48 hours after transfection with HPV16 E7 siRNA or NS siRNA. HPV16 E7 protein is an E7 protein expressed from bacteria used as a control for the Western blot.
GFP expression was efficiently suppressed in a dose-dependent manner when 293 cells were co-transfected with the synthetic E7 siRNA 198 and a plasmid expressing GFP-E6 or GFP-E7 (Figure 1b). Transfection of HPV16+ CaSki and SiHa cells derived from cervical cancer with the synthetic E7 siRNA also showed efficient inhibition of E6E7 gene expression: cell growth was inhibited (Figure 1c), E6E7 RNA (Figure 1d) and E7 protein expression (Figure 1e) decreased, and cellular protein p53 and p21 expression (Figure 1e) increased. More than 94% of CaSki and SiHa cells could be efficiently transfected with siRNA 198 in our experimental conditions and the resulting decreases of both the E6 and E7 oncogene expression in these cervical cancer cells promoted cell cycle arrest at G1 for most transfected cells (77% for CaSki and 86% for SiHa) and consequently cell growth arrest. The majority of the transfected cells were viable (over 95%) by trypan blue exclusion assay and over 90% of the cells did not undergo apoptosis in our FACS analysis with annexin V staining. This observation is consistent with other reports demonstrating that silencing of HPV16 or 18 E6 and E7 oncogene expression by RNA interference confers no signs of significant apoptosis (Jiang et al., 2002; Hall and Alexander, 2003).
A set of multiprobes was also utilized to assess various effects of silencing HPV16 E6 and E7 oncogene expression in both CaSki and SiHa cells on expression of cell cycle-related genes by RNase protection assay and showed that, among all 11 genes examined, p21 was the only gene that had been up-regulated (Figure 2). Data suggest that the E7 siRNA-mediated E6 reduction in HPV16+ cervical cancer cells led to stabilize p53 and consequently an increased p53 transactivation of p21 transcription.
Figure 2.
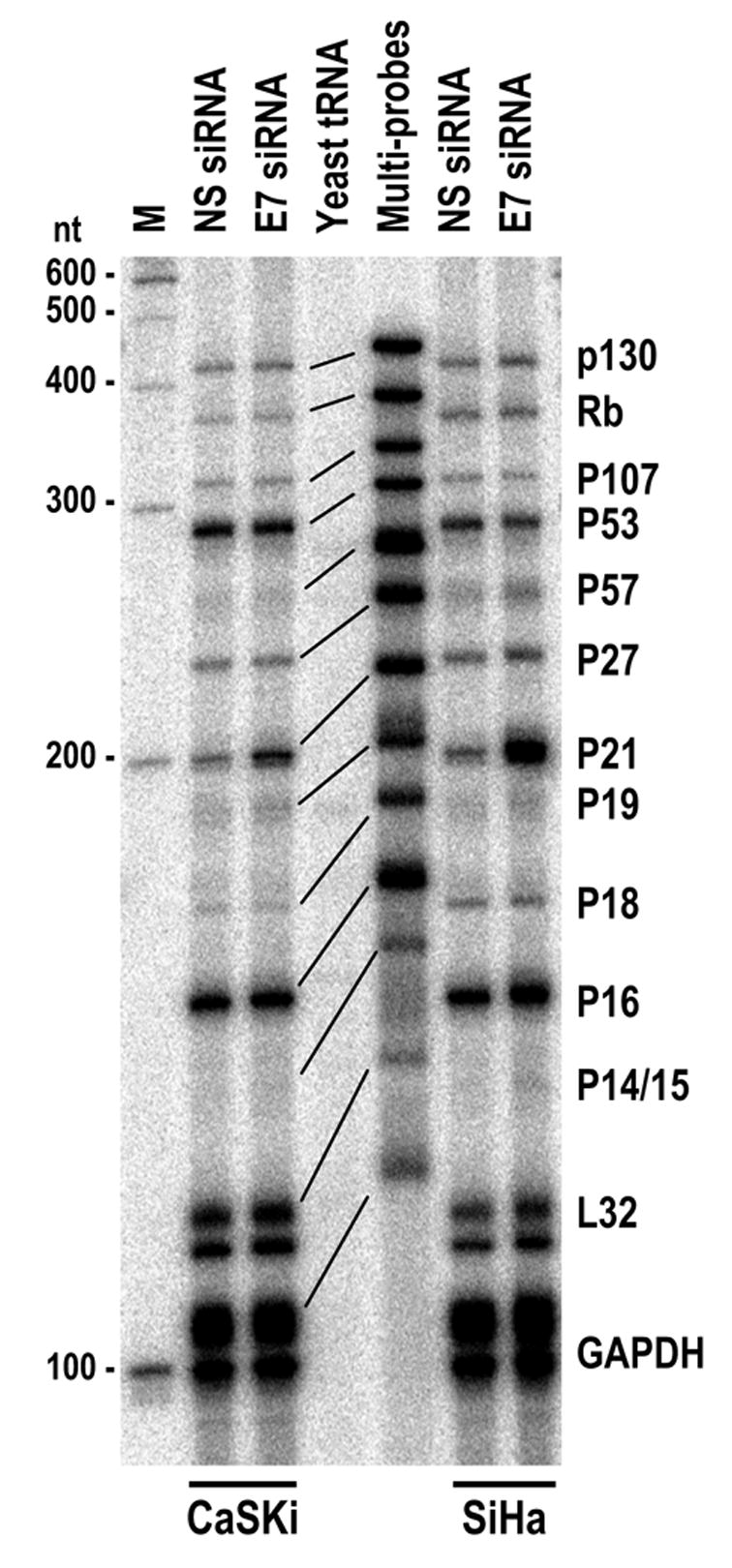
Effect of HPV16 E7 siRNA on cell cycle-related gene expression from cervical cancer cells by transient transfection. HPV16 E7 siRNA or NS siRNA at 40 nM was transfected into HPV16+ cervical cancer cell lines, CaSki or SiHa and total cell RNA was prepared from each cell line after 24 hours of transfection. Approximate 30 μg of total cell RNA were hybridized to hCC-2 multi-probes (~ 4 X 106 CPM) and then subjected to RNase A/T1 digestion. RNA fragments protected were resolved in a 8% denaturing PAGE gel. Multi-probes undigested were run in parallel as controls and their corresponding protected products from total cell RNA were indicated by lines.
Stable expression of an HPV16 E7 siRNA in cervical cancer cell lines silences both E6 and E7 expression at early cell passages
When inserted downstream of the pol III promoter in a mammalian expression vector, pSUPER (Brummelkamp et al., 2002), the siRNA 198 was expressed as a short hairpin RNA (shRNA) structure (Figure 3a) and was processed into siRNAs (Figure 3b) in the cells with stable E7 siRNA transfection derived from the HPV16+ cervical cancer cell lines, CaSki and SiHa. The antisense (As) strand of the siRNA with the correct size (21 nts) was detected at variable levels by Northern blot (Figure 3b) using a 32P-labeled sense oligo, while the sense (S) strand was barely detectable with a 32P-labeled antisense oligo, suggesting efficient unwinding of the duplex RNA and degradation of the released sense strand (Schwarz et al., 2003). Consistent with this, E7 expression was suppressed to various degrees among the stably transfected cells, with CaSki C-2 cells conferring the best E7 silencing (Figure 3c). CaSki C-3 and SiHa S-3 cells had little E7 siRNA expression (Figure 3b) and did not show decreased HPV16 E7 expression (Figure 3c). However, it is notable that both CaSki C-1 and SiHa S-2 cells expressed abundant E7 siRNA (Figure 3b), but provided only limited inhibition of the E7 expression (Figure 3c), indicating that siRNA function was restrained in these cells.
Figure 3.
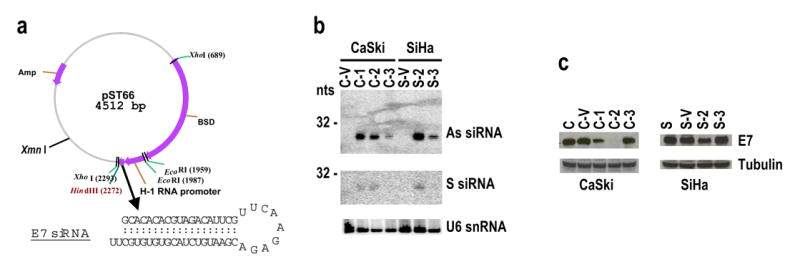
Stable expression of HPV16 E7 siRNA in HPV16+ cervical cancer cells. (a) Diagram of the HPV16 E7 siRNA expression vector, a pSUPER-derived pST66, and the expressed shRNA 198 sequence. BSD is a blasticidin S deaminase gene used as a mammalian selection marker. HPV16+ cervical cancer cells, CaSki and SiHa, transfected with the plasmid pST66 or pSUPER vector control, were grown in the presence of blasticidin S in the culture medium and the blasticidin S-resistant cells were selected to grow into a confluent monolayer for further analysis. Total cell RNA extracted from those cells stably transfected with the plasmid pST66 (CaSki for C-1 to C-3 and SiHa for S-2 to S-3) or with the pSUPER vector only (CaSki for C-V and SiHa for S-V) was tested for the presence of HPV16 E7 siRNA by Northern blot (b). Protein samples extracted from the stable siRNA cells were examined for HPV16 E7 expression by Western blot (c). Sample loading was normalized with U6 snRNA for Northern blot (b) and β-tubulin for Western blot (c).
Suppression of siRNA function in cells with stable expression of E7 siRNA
Due to existence of restrained siRNA function in both CaSki C-1 and SiHa S-2 cells even at the initial selection, CaSki-derived C-2 cells were chosen for further study of how silencing both E6 and E7 expression affects expression of other cellular genes. However, loss of the siRNA-mediated E7 silencing became evident over time, as E7 expression reappeared in the C-2 cells (Figure 4a) despite the continued expression of the E7 siRNA at the same level as at earlier times (Figure 4b). Seven single-cell subclones were subsequently isolated from the C-2 cells. Among the single-cell subclones, three (B12SC, D4SC, and H7SC) retained strong siRNA activity for silencing E6E7 expression with altered expression of p53 and p21 proteins, whereas four other subclones, A3SC, B3SC, C2SC, and H8SC, had no or only weak siRNA activity, even though all of which expressed a high level of the E7 siRNA (Figure 4c and 4d). Subclone D4SC was chosen to determine whether the loss of siRNA function in the C-2 cells could be replicated with increasing numbers of D4SC cell passages. The data shown in Figure 4e confirmed that the efficiency of the siRNA-mediated silencing of E7 expression in the D4SC subclone at passage 4 was reduced substantially by passage 8. This was further supported by changes in the cellular cyclin A level, which correlated with E7 expression upon increased cell passage. Cyclin A is a downstream target of E2F and its expression can be activated through reactivation of E2F by E7 (Schulze et al., 1998).
Figure 4.
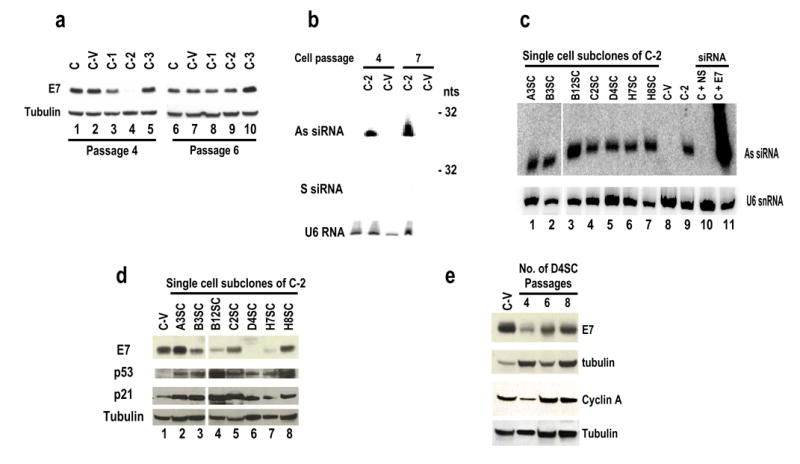
Development of suppression to siRNA function in CaSki-derived cells with stable expression of E7 siRNA. (a) Reappearance of 16E7 in CaSki-derived C-2 cells stably expressing HPV16 E7 siRNA as an indication of loss of E7 siRNA function with additional cell passages. E7 expression in protein samples prepared at passage 4 and at passage 6 was compared by Western blot. (b) Loss of E7 siRNA function in the C-2 cells at later cell passages was not due to decreased expression of E7 siRNA. Total cell RNA was probed for sense (S) or antisense (As) strand siRNA expression by Northern blot with a 32P-labeled As or S oligo or a U6-specific oligo as a normalization of sampling. (c) Expression of E7 siRNA in single-cell subclones. Total cell RNA extracted from each single-cell subclone or their parent C-2 cells (lane 9) or vector-control C-V cells (lane 8) was examined for antisense (As) strand siRNA expression by Northern blot using a 32P-labeled sense oligo or a U6-specific oligo for sampling control. Total cell RNA isolated from CaSki cells (C, lanes 10–11) transiently transfected with NS (lane 10) or E7 siRNA (lane 11) at 10 nM was included as a cell control and a siRNA size control, respectively. (d) Expression of E7, p53, and p21 in each single-cell subclone was analyzed by Western blot with antibodies against HPV16 E7, p53, or p21. (e) Cell passage affects E7 siRNA function in single-cell subclone D4SC cells. Protein samples prepared from cell passage 4, 6, and 8 were examined by Western blot using antibodies against HPV16 E7 or cyclin A and compared with the protein levels in CaSki-derived vector control C-V cells. The membranes in panels a, d, and e were also blotted for β-tubulin as a sample loading control.
Suppression of siRNA function in cells with stable expression of siRNAs is gene-selective
To explore whether the observed suppression of E7 siRNA function in our stable cells could be generalized to other gene-specific siRNAs, we chose CaSki cells carrying wild type p53 (Yaginuma and Westphal, 1991; Kessis et al., 1993) for stably expressing shRNA-derived p53 siRNA and examined the correlation of the p53 protein levels with the expression of the p53 siRNA in the cells. Surprisingly, a loss of p53 siRNA function also became evident with increased cell passages (Figure 5a and b).
Figure 5.
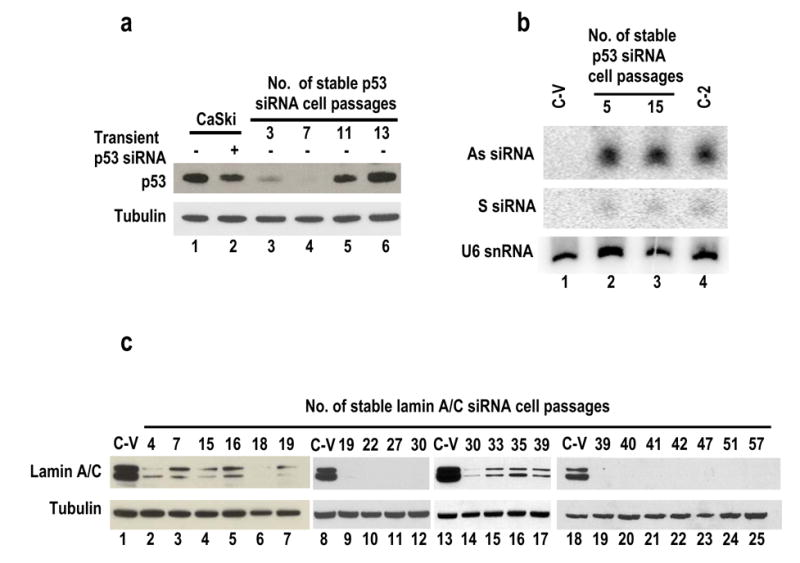
Development of suppression to siRNA function in CaSki-derived cells stably expressing p53 siRNA, but not in the cells stably expressing lamin A/C siRNA. (a) CaSki cells were transfected with the modified pSUPER-p53 siRNA expression vector and selected in the presence of BSD. Protein samples (lanes 3–6) were prepared from the selected stable p53 siRNA cells at each passage and analyzed by Western blot for p53 and β-tubulin. The CaSki cells with or without transient transfection of p53 siRNA duplex (5 nM) were used as transient p53 siRNA controls (lanes 1 and 2). (b) Total cell RNA prepared from vector control C-V cells (lane 1) or stable p53 siRNA cells at passages 5 (lane 2) and 15 (lane 3) was examined by Northern blot for expression of antisense (As) or sense (S) strand p53 siRNA and U6 snRNA. RNA isolated from stable E7 siRNA cells (C-2, lane 4) was examined as a control by Northern blot using E7 siRNA-specific probes as described in Figure 4b. (c) CaSki cells were stably transfected with modified pSUPER-lamin A/C siRNA expression vector (pST83) and were selected in the presence of BSD. Protein samples were prepared at each cell passage as indicated and examined by Western blot using anti-lamin A/C antibody. The same membrane was reprobed with anti-β tubulin. C-V, protein samples from CaSki cells stably transfected with an empty vector only.
When the same strategy was used to stably express lamin A/C siRNA in CaSki cells, we were unable to observe any development of suppression to lamin A/C siRNA in cells with stable expression of lamin A/C siRNA till the 57th cell passage (9 months) (Figure 5c).
Desensitization of mammalian cells by chronic siRNA treatment
One possible shortcoming of these experiments is that suppression of siRNA function observed in stable E7 siRNA and p53 siRNA cells developed under strong selection pressure that would not exist in a physiological environment. To address this issue, we chronically treated two different cell types, CaSki and 293 cells, with a low dose of siRNA by transient transfection every three days for 45 days. This approximates a constant siRNA level in the cells because transient siRNA functions most efficiently within 72 hours after transfection of mammalian cells (our unpublished data). Subsequently, CaSki cells with or without chronic siRNA treatment were examined for expression levels of endogenous E7 and p53 as an indication of developing desensitization to siRNA function. The 293 cells with or without chronic siRNA treatment were co-transfected with a GFP-E6E7 expression vector along with a gene-specific siRNA and examined for efficiency of the siRNA in silencing the GFP-fusion protein. Our experiments further demonstrated that desensitization to a siRNA was inducible by chronic siRNA treatment in both types of cells. The CaSki cells receiving chronic E7 siRNA treatment responded poorly to cognate siRNA, with little change in E7 and p53 protein expression (compare lane 3 to lane 4 in Figure 6a and their correspondent bars in Figure 6b). In contrast, the cells that had not previously received E7 siRNA treatment responded efficiently to transient transfection of this siRNA, with decreased E7 and increased p53 expression (compare lanes 1 to 2 and 5 to 6 in Figure 6a and their correspondent bars in Figure 6b).
Figure 6.
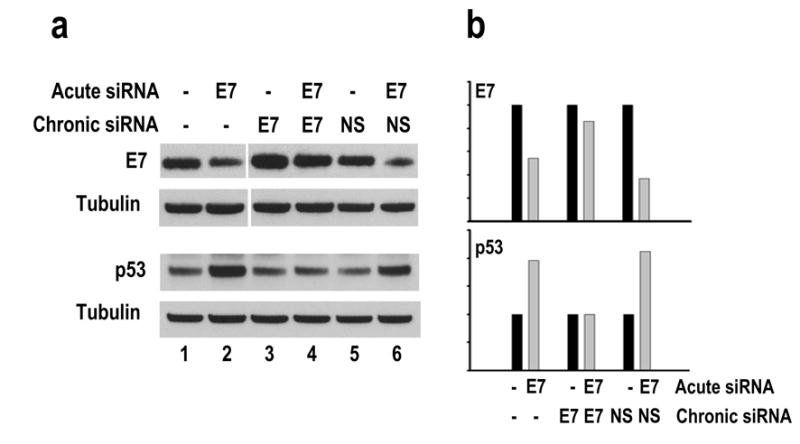
Desensitization of CaSki cells by chronic treatment with a low dose of siRNA duplex. CaSki cells treated with 10 nM E7 siRNA (lanes 3–4) or NS siRNA (lanes 5–6) every three days for 45 days (lanes 3–4 and 5–6) were compared on the last day of the chronic treatment to CaSki cells without chronic treatment (lanes 1–2) for their responses to 5 nM E7 siRNA. The protein samples were collected 24 hours after siRNA transfection and analyzed by Western blot for expression of 16E7, p53, and β-tubulin as shown in (a). The relative levels of 16E7 and p53 in each sample were normalized to β-tubulin and compared to their corresponding controls without acute siRNA treatment (b).
The same was true for the 293 cells treated chronically with the siRNA. The 293 cells receiving chronic E7 siRNA treatment responded poorly to E7 siRNA, and did not effectively silence GFP-E6 expression (data not shown).
Cells suppressing an endogenous siRNA are sensitive to exogenously provided synthetic siRNAs by transient transfection
To determine whether a cell suppressing one particular shRNA-derived siRNA is also sensitive to another synthetic siRNA duplex, E7 siRNA–suppressive C-2 cells were compared with E7 siRNA-sensitive C-V cells for their sensitivity to several transiently transfected synthetic siRNAs. We first compared siRNA levels in CaSki cells by transient transfection and stable transfection. Although only 1 nM siRNA was used for transient transfection, amount of the synthetic siRNA associated with the cells was much greater than the shRNA-derived endogenous siRNA expressed in C-2 or D4SC cells (Figure 7). Despite this difference, C-2 cells required a little higher dose of synthetic E7 siRNA to mediate a similar level of E7 silencing conferred by a relatively low dose of synthetic E7 siRNA in C-V cells by transient transfection (Figure 8a). Furthermore, C-2 cells were found to be sensitive to other synthetic siRNA 210 (data not shown), which targets another region of 16E6E7 (Yoshinouchi et al., 2003), and to a low dose (5 nM) of synthetic p53 siRNA duplex (Figure 8b), resulting in silencing of these individual genes. The same is true for D4SC cells in responding to a low dose (5 nM) of synthetic p53 siRNA (Figure 8b). This indicates that the cells insensitive to a stably expressed siRNA are still sensitive to other synthetic siRNAs by transient transfection.
Figure 7.
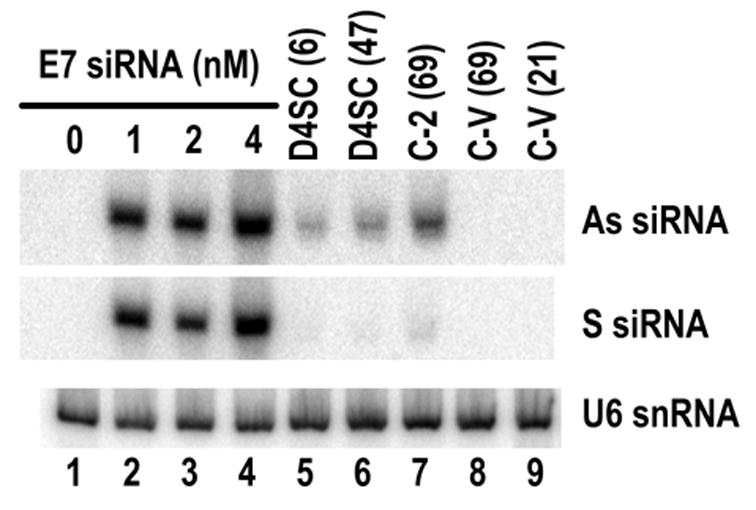
Comparison of siRNA levels in CaSki cells by transient (lanes 2–4) or stable (lane 5–7) E7 siRNA transfection. Transiently transfected cells were washed 3 times with PBS, and total cell RNA was prepared 24 hours after transient siRNA transfection, or prepared from the individual CaSki-derived cells with stable expression of E7 siRNA (D4SC and C-2), or from cells transfected with pSUPER vector only (C-V, lanes 8 and 9). The RNA was analyzed by Northern blot. Numbers in the parentheses of lanes 5–9 indicate the number of cell passages at the time used for the assays. The same membrane used for antisense and sense siRNA strand detection was also blotted for U6 snRNA to control for sample loading.
Figure 8.
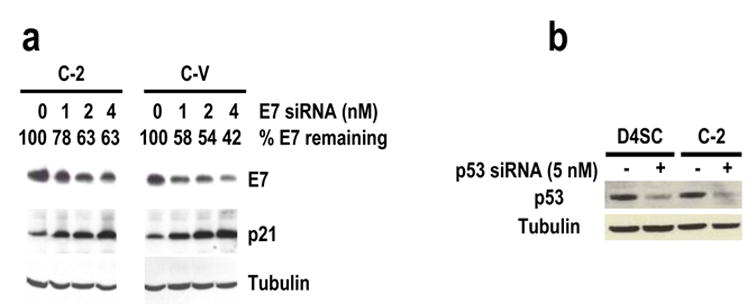
Cells insensitive to endogenous siRNA are sensitive to exogenous siRNA duplexes. Transient transfection was used to compare E7 siRNA-insensitive cells, C-2 and D4SC subclone cells, with E7 siRNA–sensitive C-V cells, for their sensitivities to E7 siRNA 198 (a) at 1, 2, and 4 nM and p53 siRNA at 5 nM (b). The protein samples were prepared 24 hours after transfection and analyzed by Western blot for E7, p21, or p53. The same membrane was also examined by Western blot for β-tubulin as a control for sample loading.
Loss of endogenous siRNA function occurs after RISC formation and siRNA duplex unwinding
Various approaches were taken to explore the mechanism that leads to loss of endogenous siRNA function in these stable E7 siRNA cells. Sequence analysis of the targeted E6 and E7 genes as well as the vector insert in these siRNA stable cells showed no mutation of the nucleotide sequences (data not shown), suggesting that the loss of siRNA function in these cells was not due to any change in the targeted sequences or the sequences of the shRNA insert. We also cloned and sequenced the siRNA expressed from these stable E7 siRNA cells and found no RNA sequence change by RNA editing (Scadden and Smith, 2001; Knight and Bass, 2002; Yang et al., 2005). E7 siRNA expressed from these stable siRNA cells was processed correctly, with a size of 19 nts plus two uridines as a 3′ overhang (Figure 9a).
Figure 9.
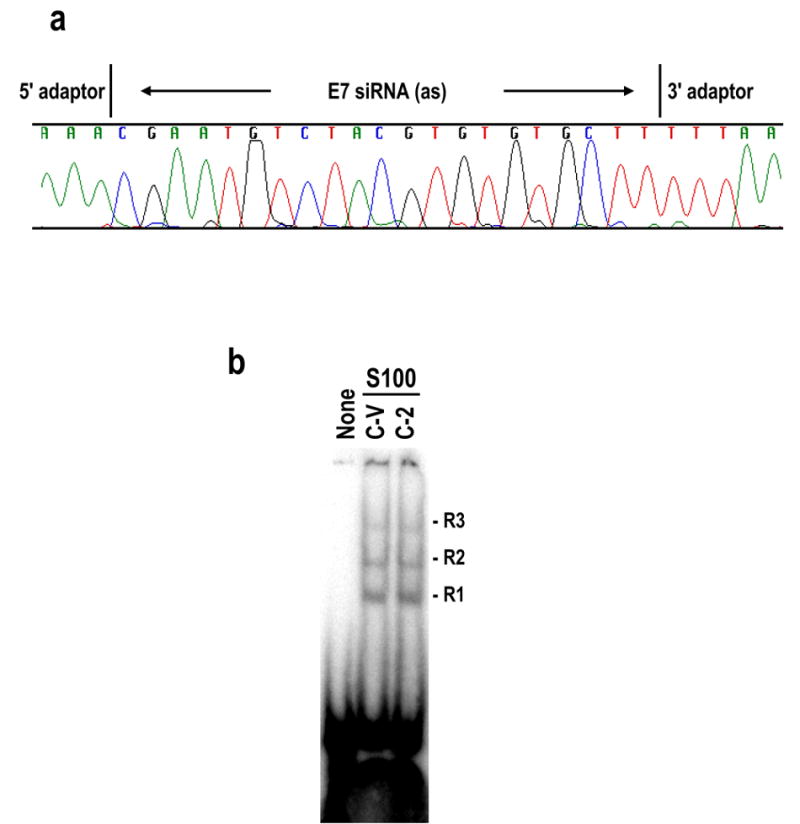
Loss of siRNA function in cells stably expressing E7 siRNA is not due to changes of siRNA sequence and occurs after RISC formation. (a) The E7 siRNA sequence was verified by cloning of small RNA molecules from C-2 cells. An antisense strand E7 siRNA cloned and sequenced by this approach was shown in the chromatogram. (b) Cytoplasmic S100 extracts prepared from stable E7 siRNA-suppressive C-2 cells and -sensitive control C-V cells were compared for their capacities for RISC formation in the presence of 32P-labeled E7 siRNA. None, E7 siRNA with no S100. Both C-V and C-2 S100 extracts conferred three RISC-like complexes (R1, R2, and R3) with E7 siRNA.
Suppression to E7 siRNA expressed in these stable cells did not appear due to possible activation of an interferon response by stably expressed siRNA (Bridge et al., 2003; Sledz et al., 2003) since the expression of 2′5′-oligoadenylate synthetase mRNA in the stable 16E7 siRNA cells remained the same level as detected in the vector control cells (data not shown).
We also wondered whether the cells lacking E7 siRNA function were capable of forming RISC (RNA-induced silencing complex). RISC is a multiprotein complex (Haley and Zamore, 2004; Pham et al., 2004) into which 21- to 23-nt siRNAs are incorporated after Dicer cleavage (Zhang et al., 2002; Provost et al., 2002). Before the incorporation, the siRNA needs to be 5′ phosphorylated (Nykanen et al., 2001; Martinez et al., 2002) and unwound by an ATP-dependent RNA helicase (Tomari et al., 2004). This process leads the antisense strand into RISC to guide RISC to its target complementary mRNA for endonucleolytic cleavage at the center of the duplexed siRNA:mRNA region, 10 nt from the siRNA 5′ end (Elbashir et al., 2001; Schwarz et al., 2003). Since all of our stable cells expressed duplex 21-nt siRNAs, we assumed that Dicer functions well in these cells, but suppression of siRNA function in these cells might be attributable to a deficiency of RISC formation. We examined in vitro RISC formation using the synthetic E7 siRNA 198 labeled with 32P and cytoplasmic S100 extracts prepared from C-2 (E7 siRNA–suppressive) or C-V (E7 siRNA–sensitive) cells. We found that both C-2 and C-V S100 extracts conferred a likelihood of three complexes (R1, R2, and R3) (Pham et al., 2004) with the E7 siRNA (Figure 9b), suggesting that suppression of E7 siRNA function in C-2 cells occurred at steps after RISC formation. Moreover, Northern blots, probed separately with antisense and sense probes, demonstrated the presence of much more (>10 fold) of the siRNA antisense strand than the sense strand in the cells with siRNA suppression (Figs. 4b and 5b), implying that unwinding of the E7 siRNA duplex and degradation of the unwound sense strand had occurred normally in these cells. Thus, the observed endogenous siRNA suppression in these cells must occur at yet a later step.
Presence of a 50-kDa protein in 16E7 siRNA–resistant cells and its interaction with 16E7 siRNA
We therefore proposed that there might be one or more proteins in the siRNA–resistant cells interacting with the E7 siRNA and counteracting the siRNA function after RISC formation and duplex unwinding. A UV cross-linking assay was conducted to explore whether the cytoplasmic S100 extracts contain specific E7 siRNA-binding protein(s). The E7 siRNA 198 and NS siRNA duplexes, end-labeled with 32P, were examined in the presence of C-2 (16E7 siRNA–resistant) or C-V (16E7 siRNA–susceptible) cytoplasmic S100 or total cell extracts. Notably, a protein with a molecular weight of approximately 50 kDa was found specifically cross-linked to the E7 siRNA in the C-2 S100 extract, but not much so in the C-V S100 (Figure 10a, compare lane 1 to lane 2). This S100 cross-linking profile between E7-resistant and E7-susceptible cell lines also existed in SiHa-derived cells (data not shown). In contrast, the C-2 S100 and C-V S100 were not different in their cross-linking to the NS siRNA which did bind a smaller protein (s) with an equal affinity for both extracts (Figure 10a, compare lane 5 to lane 6). The total cell extracts from either C-2 or C-V cells showed the same cross-linking profile for both siRNAs (Figure 10a, compare lane 3 to lane 4 and lane 7 to lane 8). The binding of the ~50-kDa protein in the C-2 S100 to the 32P-labeled E7 siRNA was ATP-dependent (Figure 10b, compare lane 1 to lane 2) and could be competed out only with cold E7 siRNA (left panel of Figure 10c, compare lane 4 to lane 5), implying a specific interaction with the E7 siRNA. In contrast, the interaction of the smaller protein(s) with the NS siRNA in the C-2 S100 was not ATP-dependent (Figure 10b, compare lane 3 to lane 4) and appeared to be non-specific, since the interaction was inhibited by tRNA or the NS siRNA, but not by the E7 siRNA (left panel of Figure 10d, compare lanes 2–4 and 6 to lane 5). Moreover, the NS siRNA-binding protein(s) retained their RNA binding affinity in high salt (600 mM) (right panel of Figure 10d), whereas the E7 siRNA-binding protein(s) had an optimal RNA binding affinity only in salt conditions of less than 200 mM (right panel of Figure 10c).
Figure 10.
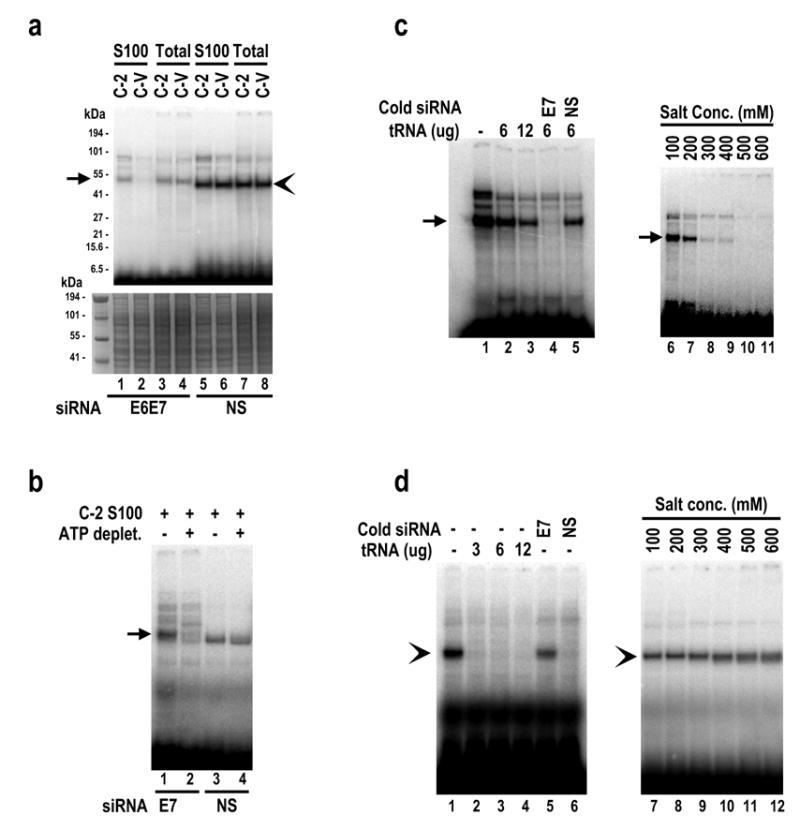
Identification of a cellular protein that interacts with HPV16 E7 siRNA 198 in E7 siRNA-resistant cells. Total cell extracts and cytoplasmic S100 extracts were prepared from E7 siRNA-resistant C-2 cells and vector control C-V (siRNA susceptible) cells and were compared for their capacities for siRNA binding in the presence of 32P-labeled siRNA duplexes. Arrows and arrowheads indicate, respectively, specific and nonspecific protein binding. (a) A ~50 kDa protein in the cytoplasmic S100 extract of E7 siRNA-resistant C-2 cells binds specifically to the E7 siRNA. Cytoplasmic S100 and total cell extracts were compared in siRNA binding and cross-linking assays using 32P-labeled E7 siRNA or NS siRNA (top panel). The same gel was stained by Coomassie blue for protein sampling in each binding assay (bottom panel). (b) Binding of the ~50 kDa protein in C-2 S100 extract to the E7 siRNA is ATP-dependent. ATP in the C-2 S100 was depleted by treatment of the extract with 0.05 U/ul hexokinase in the presence of 20 mM glucose for 20 min at RT (Nykanen et al., 2001). The C-2 S100 with or without depletion of ATP was used for RNA-protein interactions and UV cross-linking to 32P-labeled E7 or NS siRNA. (c) Cross-linking of the ~50 kDa protein in C-2 S100 to 32P-E7 siRNA can be competed out with cold HPV16 E7 siRNA, but not with tRNA or 100-fold excess of NS siRNA (left panel). Cross-linking was affected by the presence of high concentrations of NaCl (right panel). (d) Cross-linking of a protein smaller than 50 kDa in C-2 S100 to NS siRNA is nonspecific and can be competed out with tRNA or 100-fold excess of NS siRNA, but not with 100-fold excess of E7 siRNA (left panel). This nonspecific protein-siRNA interaction was not affected by the presence of high concentrations of NaCl (right panel).
Finding of ATP requirement in E7 siRNA duplex and the ~50 kDa protein interaction (Figure 10b) implies that the siRNA-protein interaction in the C-2 S100 might exert on a unwound single strand siRNA since unwinding of siRNA duplex is an ATP-dependent process (Nykanen et al., 2001). To confirmed this presumption, both sense and antisense single strand E7 siRNA were synthesized, labeled with 32P at their 5′ ends, and then compared to double-stranded E7 siRNA duplex for protein binding and cross-linking assays. As shown in Figure 11a, the ~50 kDa protein in the C-2 S100 was able to bind strongly to the antisense strand E7 siRNA (lane 2), but only weakly to the double-stranded E7 siRNA duplex (lanes 3 and 4). The similarity of the binding profiles between the antisense strand E7 siRNA (lane 2) and double-stranded E7 siRNA duplex (lanes 3 and 4), along with no migration retardation of the cross-linked products in the binding of double stranded E7 duplex, indicates that the ~50 kDa protein cross-linked siRNA was indeed the unwound, single strand antisense E7 siRNA. Consistently with this conclusion, the ~50 kDa protein in the C-2 S100 had no binding activity for the sense-strand (lane 1) E7 siRNA. Instead, a protein larger than 50 kDa in the C-2 S100 was found to cross-link with sense-strand E7 siRNA (lane 1). All double-stranded siRNAs used for the assays were very stable (Figure 11b), with only a trace amount of the single strand E7 siRNAs leftover in our annealing reactions (Figure 11b, lane 3), further confirming the existence of active unwinding activity in the reactions. Together, these data indicate that the ~50 kDa protein in the C-2 S100 is E7 siRNA-specific and interacts preferentially with the antisense E7 siRNA unwound in an ATP-dependent manner.
Figure 11.
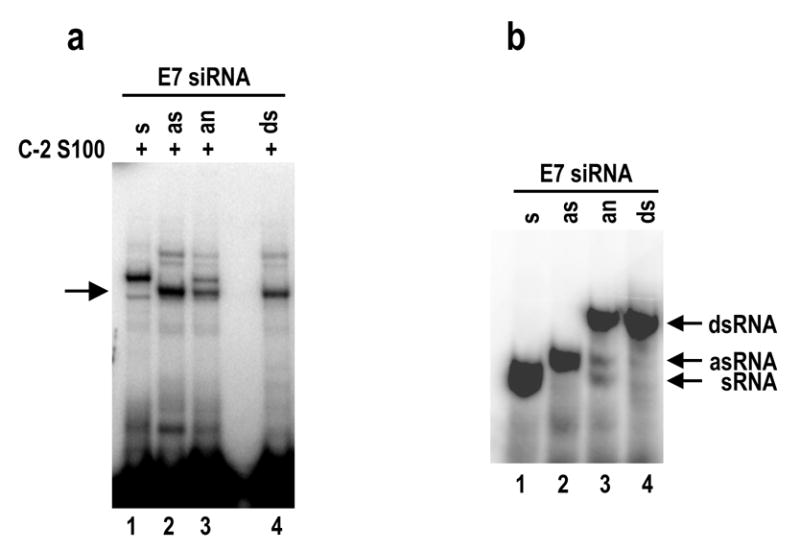
The ~50 kDa protein in C-2 S100 preferentially interacts with an antisense strand E7 siRNA. (a) Interaction of C-2 S100 proteins with single or double strand siRNAs. All of 32P-labeled, synthetic single strand E7 siRNA (sense [s] and antisense [as] strand) and their annealed (an) products (s + as) as well as synthetic, double strand (ds) E7 siRNA 198 were compared in an equal amount for their capabilities to interact with protein(s) in C-2 S100 using UV cross-linking assays. Arrow indicates a specific siRNA binding of the ~50 kDa protein. (b) Electrophoretic profiles of the 32P-labeled, corresponding siRNAs used in (a) on a 12% native PAGE gel.
Discussion
Here we have shown that a specifically targeted siRNA can effectively silence both E6 and E7 expression when transiently or stably transfected in cells derived from cervical cancer. The siRNA inhibited cell growth, decreased E6E7 RNA and E7 protein expression, and increased cellular protein p53 and p21 expression, but induced no signs of significant cell apoptosis as reported from other laboratories (Jiang et al., 2002; Hall et al., 2003). The E7 siRNA-mediated enhancement of p53 and p21 expression in these cervical cancer cells indicates that a reduced E6 protein expression leads to stabilization of p53 followed by p53-stimulated p21 expression (Howley et al., 2001). However, the function of this siRNA was selectively suppressed with increasing time in culture.
Suppression of siRNA function by viral suppressors has been described in plant cells (Li et al., 2002; Mallory et al., 2002; Kasschau et al., 2003), Drosophila (Li et al., 2002) and mosquito cells (Li et al., 2004). It has been assumed that suppression of siRNA silencing could also exist in mammalian cells, which may function as a natural defense against siRNA silencing in mammals. Our report supports this presumption and provides a direct evidence that human cells can develop a function to counteract siRNA-mediated long-term silencing of critical genes. This suppressive function appears unrelated to a viral suppressor or possible activation of an interferon response by stable expression of the siRNA (Bridge et al., 2003; Sledz et al., 2003), nor to RNA editing (Scadden et al., 2001; Knight et al., 2002) Yang & Nishikura, JBC 280: 3946–3953, 2005). While we are preparing our manuscript for publication, other groups also reported suppression of siRNA or miRNA function by viral suppressors in mammalian cells with medically important RNA or DNA virus infections (Lu and Cullen, 2004; Lecellier et al., 2005; Soldan et al., 2005).
Expression of both 16E6 and E7 oncogenes is critical for survival of cervical cancer cells. Silencing of HPV16 E6 and E7 expression by the shRNA-derived E7 siRNA might provide an additional selection pressure for the cells to overcome siRNA function, because all stable E7 siRNA cells were already undergoing blasticidin selection. Further experimental data support this presumption by demonstrating that a cell with stable expression of shRNA-derived p53 siRNA could become suppressive, but a cell with stable expression of lamin A/C siRNA did not. Although p53 is not an essential gene and cells without p53 have a growth advantage, p53 is a cell cycle regulator and involves many signal pathways (Vousden, 2000). Lamin A/C is an intermediate filament protein that is not essential for cell survival and plays no role in regulation of cell function (Aebi et al., 1986). Together, these data indicate that suppression of siRNA function in these stable cells is a selective process and depends upon the commission of a targeted gene. Perhaps, suppression to a specific siRNA in a cell may be an acquired cell immunity for cell survival. Data from desensitization of CaSki cells by chronic siRNA transfection support such a hypothesis.
Nevertheless, investigation of how cervical cancer cells could develop a resistant program in the presence of E7 siRNAs for a long term has led us to conclude that suppression of siRNA function in stably transfected CaSki cells at late passages occurs after siRNA duplex unwinding and RISC formation and might involve an siRNA–protein interaction. Our exhaustive approaches further showed that the cells with resistance to endogenous E7 siRNA, in contrast to the cells with no E7 siRNA expression, contain a cytoplasmic protein with a molecular weight of ~50 kDa that specifically and characteristically interacts with E7 siRNAs. More of the same protein appeared as the C-2 cells develop resistance to the siRNA and more in SiHa S-2 cells (16E7 siRNA-resistant) than SiHa S-V cells (16E7 siRNA- susceptible) (Data not shown). Convincingly, this protein preferentially binds to the antisense strand E7 siRNAs. Although the ~50 kDa protein also binds to the E7 siRNA duplexes weakly in the C-2 S100 reactions, this was confirmed to be a result of unwinding activity of the C-2 S100 as reported in HeLa S100 extracts (Martinez et al., 2002). Together with our data showing the presence of >10-fold more siRNA antisense strand than sense strand in the cells with endogenous siRNA expression, our study suggests that the cytoplasmic ~50 kDa protein plays a role in cell development of resistance to E7 siRNA. However, its identity and exact function (s) remain to be determined and are under active investigation. This protein appears not a HPV protein, nor one of cellular ADARs (adenosine deaminases acting on RNA), simply because of its size that is much larger than two viral oncoproteins (E6 and E7), but smaller than ADARs (Yang et al., 2005). We therefore hypothesize that the ~50 kDa protein is a cellular protein. The ~50 kDa protein bound to the unwound antisense E7 siRNA may prevent the antisense siRNA from base-pairing with its target mRNA or prevent the binding of a unknown endonuclease to the double-stranded siRNA:mRNA region. Consequently, interaction of the ~50 kDa protein with the antisense strand siRNA blocks antisense siRNA-guided endonucleolytic cleavage of the mRNA.
Materials and Methods
siRNAs and siRNA expression vectors
Synthetic double strand siRNA 198 duplex, 5′-GCACACACGUAGACAUUCGdTdT-3′, which targets the HPV16 E7 coding region at nt 773–791 in the virus genome, was obtained from Dharmacon, Inc (Lafayette, CO, USA) and its corresponding sense strand siRNA 243, 5′-GCACACACGUAGACAUUCGUU-3′, and antisense strand siRNA 244, 5′-CGAAUGUCUACGUGUGUGCUU-3′, used in Figure 11, were synthesized separately as a single strand siRNA from Integrated DNA Technologies, Inc. (Coralville, IA). Synthetic double strand p53 siRNA (Brummelkamp et al., 2002), GFP siRNA, and non-specific (NS) siRNA with 52% GC content (Cat. #D-001206-08-20) were also purchased from Dharmacon. The siRNA vectors pSUPER and pSUPER-p53 (Brummelkamp et al., 2002) were a generous gift from Dr. Reuven Agami of the Netherlands Cancer Institute. pSUPER was modified by insertion of an eukaryotic selection marker, the blasticidin S deaminase gene, from a Not I and Bam HI fragment of pCMV/Bsd (Invitrogen, Carlsbad, CA). The corresponding E7 siRNA sequence was inserted into the modified pSUPER vector to express a short hairpin (sh) RNA structure from the resulting plasmid, pST66, in mammalian cells as described (Brummelkamp et al., 2002). The same strategy was also applied to prepare plasmid pST83 to express lamin A/C siRNA (5′-GGUGGUGACGAUCUGGGCU-3′, Cat.#D-001050-01-20, Dharmacon) from the modified pSUPER vector. The GFP-E6 and E7 fusion plasmids pZMZ 70 (HPV16 E6 having the E7 coding region as part of its 3′ UTR) and pZMZ74 (HPV16 E7 only) were described in our previous publication (Tao et al., 2003).
Cell transfection and Western blot
The efficiency of each siRNA described above was assayed in HPV16+ cervical carcinoma cells or HPV− human 293 cells by transient transfection using Oligofectamine (Invitrogen). Transfection efficiencies were established by transfection of HPV 16+ cervical cancer cell lines, CaSki and SiHa cells, with 40 nM of FITC-siRNA 198 at the same transfection conditions. Cell growth curves were determined by cell counting. Cell cycle analysis was performed by FACS in the presence of Vindelov’s propidium iodide buffer (Vindelov, 1977). Apoptotic cells were determined using annexin V-FITC detection kit I (Cat. #556547, BD PharMingen, San Diego, CA) according to the manufacturer’s instruction. All cervical cancer cell lines used for both cell cycle and apoptosis analysis were prepared at 48 h for SiHa and 72 h for CaSki after transfection with 40 nM of siRNA 198.
For stable transfection, each pSUPER-derived construct was linearized by Xmn I before cell transfection was performed using Lipofectamine 2000 (Invitrogen). The transfected cells were selected in the presence of blasticidin S (10 μg/ml) in Dulbecco’s modified Eagle’s medium containing 10% fetal bovine serum. Single cells from a cell population with stable siRNA transfection were further isolated by serial dilution in 96-well plates and grown in the same selection medium as described above. After the stable cells grew up and became confluent in the selection medium, the cells with stable siRNA transfection were subcultured in the same selection medium by cell passage every 4–5 days.
Protein extracts and total cell RNA were prepared at various time points after transfection, as described in each Figure. For Western blot analyses, protein samples (approximately 30 μg each) were resolved by NuPAGE Bis-Tris gel (Invitrogen) electrophoresis before being transferred onto a nitrocellulose membrane. The membranes were then blotted with different antibodies, including anti-p21 (6B6, BD PharMingen), anti-p53 (Ab-6, Oncogene, Cambridge, MA), anti-HPV16 E7 (SC-6981, Santa Cruz, CA), anti-cyclin A (SC-751, Santa Cruz), anti-lamin A/C (SC-7292, Santa Cruz), and anti-β tubulin (5H1, BD PharMingen)
RNase protection assays (RPA) and Northern blot
Total cell RNA was prepared using TRIzol as recommended by the manufacturer (Invitrogen). An 16E6E7-specific antisense RNA probe was synthesized by in vitro transcription and used for RPA for detection of the E6E7 RNAs, as described (Zheng et al., 2004). Antisense RNA multiprobes were prepared from a hCC-2 human cell cycle multi-probe template set (BD PharMingen, Cat.# 556160) by in vitro transcription and used for RPA for detection of various cell cycle-related RNA transcripts.
Expression and processing of the shRNAs from stable siRNA cell lines were examined by Northern blot using a 32P-labeled sense oligo (oZMZ379, 5′-GCACACACGTAGACATTCG-3′) or a 32P-labeled anti-sense oligo (oZMZ380, 5′-CGAATGTCTACGTGTGTGC-3′) for 16E7 siRNAs or a 32P-labeled sense oligo (oST211, 5′-GACTCCAGTGGTAATCTAC-3′ ) or a 32P-labeled antisense oligo (oST212, 5′-GTAGATTACCACTGGAGTC-3′) for p53 siRNAs. Briefly, 30 μg of total RNA was resolved by electrophoresis in a 12% denaturing PAGE gel, transferred to a Hybond N membrane (NEN Life Science Products, Inc., Boston, MA), prehybridized in hybridization buffer (Sigma, St Louis, MO) for 1 hour at 41°C, and then hybridized overnight with the 32P-labeled oligo at 41°C. The membrane was washed twice, once in 2X SSC, 1% SDS at 37°C, and once in 0.1X SSC, 0.5% SDS in 40°C, for 20 min each wash. The radiographic data on the membrane were captured using a Molecular Dynamic PhosphorImager Storm 860 and analyzed with Image Quant Software. For an RNA loading control, the membrane was deprobed with 0.1X SSC, 0.5% SDS at 90°C for 10 min and then reprobed with a 32P-labeled U6 small nuclear (sn)RNA-specific oligo (oST197, 5′-AAAATATGGAACGCTTCACGA-3′).
Cloning and sequencing of siRNA expressed from siRNA-resistant cell lines
Total cell RNA (~600 μg) extracted from CaSki-derived stable siRNA cell line C-2 at passage 40 was resolved on a denaturing 15% PAGE gel and the RNAs with sizes of 18–30 nts were collected, reverse transcribed, PCR amplified, and cloned into pCRII-TOPO vector using the TOPO TA cloning kit (Invitrogen) based on published protocols (Elbashir et al., 2001; Pfeffer et al., 2003). The inserts were determined by DNA sequencing.
S100 preparation and gel mobility shift assays
Cytoplasmic S100 extracts of CaSki- or SiHa-derived cells were prepared based on published methods (Mayeda and Krainer, 1999). Total cell extracts were prepared from the S100 preparation pellet enriched with cell nuclei by sonication in Buffer D five times (5 seconds each on ice) followed by centrifugation at 100,000 x g for 1 hour at 4°C. Each gel mobility shift experiment was conducted with 5 x 105 cpm of the 32P-labeled siRNA duplex incubating with 20 μg of S100 extract in a 20-μl reaction volume containing 1X gel shift binding buffer (Promega, Madison, WI), 1 mM ATP, and 6 μg of tRNA for 20 min at 30°C. The reaction mixture was then incubated on ice for 10 min before directly loading onto a pre-run, 4% native PAGE gel (acrylamide/Bis = 80:1) and run at 150 V at 4°C for 2.5 hours. After electrophoresis, the gel was dried and exposed to X-ray film.
Protein-RNA interaction
Protein-RNA interaction was analyzed by UV cross-linking assay. Briefly, 20 μg of S100 extract or total cell extract was mixed with 5 x 105 cpm of 32P-labeled siRNA in a 30-μl reaction volume containing 1X gel shift binding buffer, 1 mM ATP, and 30 μg heparin in the presence or absence of tRNA or other competitors, as shown in Figure 10 and Figure 11, for 20 min at room temperature, followed by 10 min on ice. The reaction mixture was then irradiated with UV for 10 min in a UV Stratalinker (Stratagene) before being loaded onto a 4%–12% NuPAGE Bis-Tris gel. After electrophoresis, the gel was fixed and stained with Coomassie blue and then dried on a filter paper and exposed to X-ray film.
Acknowledgments
This research was supported by the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research. We thank Reuven Agami for providing pSUPER and pSUPER-p53 vectors, many of our colleagues for their encouragement in the course of these studies, Sohrab Bodaghi in our lab for providing bacterially expressed HPV16 E7 for the Western blot control. We also thank Douglas Lowy, Robert Yarchoan, Bruce Paterson, Eric Sontheimer, and Natasha Caplen for their critical reading of our manuscript.
References
- 1.Aebi U, Cohn J, Buhle L, Gerace L. Nature. 1986;323:560–564. doi: 10.1038/323560a0. [DOI] [PubMed] [Google Scholar]
- 2.Boyer SN, Wazer DE, Band V. Cancer Res. 1996;56:4620–4624. [PubMed] [Google Scholar]
- 3.Bridge AJ, Pebernard S, Ducraux A, Nicoulaz AL, Iggo R. Nat Genet. 2003;34:263–264. doi: 10.1038/ng1173. [DOI] [PubMed] [Google Scholar]
- 4.Brummelkamp TR, Bernards R, Agami R. Science. 2002;296:550–553. doi: 10.1126/science.1068999. [DOI] [PubMed] [Google Scholar]
- 5.Butz K, Ristriani T, Hengstermann A, Denk C, Scheffner M, Hoppe-Seyler F. Oncogene. 2003;22:5938–5945. doi: 10.1038/sj.onc.1206894. [DOI] [PubMed] [Google Scholar]
- 6.Doench JG, Petersen CP, Sharp PA. Genes Dev. 2003;17:438–442. doi: 10.1101/gad.1064703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dykxhoorn DM, Novina CD, Sharp PA. Nat Rev Mol Cell Biol. 2003;4:457–467. doi: 10.1038/nrm1129. [DOI] [PubMed] [Google Scholar]
- 8.Elbashir SM, Lendeckel W, Tuschl T. Genes Dev. 2001;15:188–200. doi: 10.1101/gad.862301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gonzalez SL, Stremlau M, He X, Basile JR, Munger K. J Virol. 2001;75:7583–7591. doi: 10.1128/JVI.75.16.7583-7591.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Goodwin EC, Yang E, Lee CJ, Lee HW, DiMaio D, Hwang ES. Proc Natl Acad Sci U S A. 2000;97:10978–10983. doi: 10.1073/pnas.97.20.10978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Haley B, Zamore PD. Nat Struct Mol Biol. 2004;11:599–606. doi: 10.1038/nsmb780. [DOI] [PubMed] [Google Scholar]
- 12.Hall AH, Alexander KA. J Virol. 2003;77:6066–6069. doi: 10.1128/JVI.77.10.6066-6069.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hammond SM, Bernstein E, Beach D, Hannon GJ. Nature. 2000;404:293–296. doi: 10.1038/35005107. [DOI] [PubMed] [Google Scholar]
- 14.Howley PM, Lowy DR. Papillomaviruses and their replication. In: Knipe DM, Howley PM, Griffin DE, Lamb RA, Martin MA, Roizman B, Straus SE, editors. Field Virology. Lippincott Williams & Wilkins; Philadelphia: 2001. pp. 2197–2229. [Google Scholar]
- 15.Jiang M, Milner J. Oncogene. 2002;21:6041–6048. doi: 10.1038/sj.onc.1205878. [DOI] [PubMed] [Google Scholar]
- 16.Kasschau KD, Xie Z, Allen E, Llave C, Chapman EJ, Krizan KA, Carrington JC. Dev Cell. 2003;4:205–217. doi: 10.1016/s1534-5807(03)00025-x. [DOI] [PubMed] [Google Scholar]
- 17.Kessis TD, Slebos RJ, Nelson WG, Kastan MB, Plunkett BS, Han SM, Lorincz AT, Hedrick L, Cho KR. Proc Natl Acad Sci U S A. 1993;90:3988–3992. doi: 10.1073/pnas.90.9.3988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Knight SW, Bass BL. Mol Cell. 2002;10:809–817. doi: 10.1016/s1097-2765(02)00649-4. [DOI] [PubMed] [Google Scholar]
- 19.Lecellier CH, Dunoyer P, Arar K, Lehmann-Che J, Eyquem S, Himber C, Saib A, Voinnet O. Science. 2005;308:557–560. doi: 10.1126/science.1108784. [DOI] [PubMed] [Google Scholar]
- 20.Li H, Li WX, Ding SW. Science. 2002;296:1319–1321. doi: 10.1126/science.1070948. [DOI] [PubMed] [Google Scholar]
- 21.Li WX, Li H, Lu R, Li F, Dus M, Atkinson P, Brydon EW, Johnson KL, Garcia-Sastre A, Ball LA, Palese P, Ding SW. Proc Natl Acad Sci U S A. 2004;101:1350–1355. doi: 10.1073/pnas.0308308100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lu S, Cullen BR. J Virol. 2004;78:12868–12876. doi: 10.1128/JVI.78.23.12868-12876.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mallory AC, Reinhart BJ, Bartel D, Vance VB, Bowman LH. Proc Natl Acad Sci U S A. 2002;99:15228–15233. doi: 10.1073/pnas.232434999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Martinez J, Patkaniowska A, Urlaub H, Luhrmann R, Tuschl T. Cell. 2002;110:563–574. doi: 10.1016/s0092-8674(02)00908-x. [DOI] [PubMed] [Google Scholar]
- 25.Mayeda A, Krainer AR. Methods Mol Biol. 1999;118:309–314. doi: 10.1385/1-59259-676-2:309. [DOI] [PubMed] [Google Scholar]
- 26.Nykanen A, Haley B, Zamore PD. Cell. 2001;107:309–321. doi: 10.1016/s0092-8674(01)00547-5. [DOI] [PubMed] [Google Scholar]
- 27.Pfeffer S, Lagos-Quintana M, Tuschl T. Cloning of small RNA molecules. In: Ausubel FM, Brent R, Kingston RE, Moore DD, Seidman JG, Smith JA, Struhl K, editors. Current Protocols in Molecular Biology. John Wiley & Sons, Inc; New York: 2003. pp. 26.4.1–26.4.18. [Google Scholar]
- 28.Pham JW, Pellino JL, Lee YS, Carthew RW, Sontheimer EJ. Cell. 2004;117:83–94. doi: 10.1016/s0092-8674(04)00258-2. [DOI] [PubMed] [Google Scholar]
- 29.Provost P, Dishart D, Doucet J, Frendewey D, Samuelsson B, Radmark O. EMBO J. 2002;21:5864–5874. doi: 10.1093/emboj/cdf578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Reynolds A, Leake D, Boese Q, Scaringe S, Marshall WS, Khvorova A. Nat Biotechnol. 2004;22:326–330. doi: 10.1038/nbt936. [DOI] [PubMed] [Google Scholar]
- 31.Scadden AD, Smith CW. EMBO Rep. 2001;2:1107–1111. doi: 10.1093/embo-reports/kve244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Scheffner M, Werness BA, Huibregtse JM, Levine AJ, Howley PM. Cell. 1990;63:1129–1136. doi: 10.1016/0092-8674(90)90409-8. [DOI] [PubMed] [Google Scholar]
- 33.Schulze A, Mannhardt B, Zerfass-Thome K, Zwerschke W, Jansen-Durr P. J Virol. 1998;72:2323–2334. doi: 10.1128/jvi.72.3.2323-2334.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Schwarz DS, Hutvagner G, Du T, Xu Z, Aronin N, Zamore PD. Cell. 2003;115:199–208. doi: 10.1016/s0092-8674(03)00759-1. [DOI] [PubMed] [Google Scholar]
- 35.Sledz CA, Holko M, de Veer MJ, Silverman RH, Williams BR. Nat Cell Biol. 2003;5:834–839. doi: 10.1038/ncb1038. [DOI] [PubMed] [Google Scholar]
- 36.Smotkin D, Wettstein FO. Proc Natl Acad Sci U S A. 1986;83:4680–4684. doi: 10.1073/pnas.83.13.4680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Soldan SS, Plassmeyer ML, Matukonis MK, Gonzalez-Scarano F. J Virol. 2005;79:234–244. doi: 10.1128/JVI.79.1.234-244.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tang S, Zheng ZM. J Biol Chem. 2002;277:14547–14556. doi: 10.1074/jbc.M111308200. [DOI] [PubMed] [Google Scholar]
- 39.Tao M, Kruhlak M, Xia S, Androphy E, Zheng ZM. J Virol. 2003;77:13232–13247. doi: 10.1128/JVI.77.24.13232-13247.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tomari Y, Du T, Haley B, Schwarz DS, Bennett R, Cook HA, Koppetsch BS, Theurkauf WE, Zamore PD. Cell. 2004;116:831–841. doi: 10.1016/s0092-8674(04)00218-1. [DOI] [PubMed] [Google Scholar]
- 41.Vindelov LL. Virchows Arch B Cell Pathol. 1977;24:227–242. [PubMed] [Google Scholar]
- 42.Vousden KH. Cell. 2000;103:691–694. doi: 10.1016/s0092-8674(00)00171-9. [DOI] [PubMed] [Google Scholar]
- 43.Yaginuma Y, Westphal H. Cancer Res. 1991;51:6506–6509. [PubMed] [Google Scholar]
- 44.Yang W, Wang Q, Howell KL, Lee JT, Cho DS, Murray JM, Nishikura K. J Biol Chem. 2005;280:3946–3953. doi: 10.1074/jbc.M407876200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yoshinouchi M, Yamada T, Kizaki M, Fen J, Koseki T, Ikeda Y, Nishihara T, Yamato K. Mol Ther. 2003;8:762–768. doi: 10.1016/j.ymthe.2003.08.004. [DOI] [PubMed] [Google Scholar]
- 46.Yu JY, DeRuiter SL, Turner DL. Proc Natl Acad Sci U S A. 2002;99:6047–6052. doi: 10.1073/pnas.092143499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zamore PD, Tuschl T, Sharp PA, Bartel DP. Cell. 2000;101:25–33. doi: 10.1016/S0092-8674(00)80620-0. [DOI] [PubMed] [Google Scholar]
- 48.Zeng Y, Yi R, Cullen BR. Proc Natl Acad Sci U S A. 2003;100:9779–9784. doi: 10.1073/pnas.1630797100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhang H, Kolb FA, Brondani V, Billy E, Filipowicz W. EMBO J. 2002;21:5875–5885. doi: 10.1093/emboj/cdf582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zheng ZM, Tao M, Yamanegi K, Bodaghi S, Xiao W. J Mol Biol. 2004;337:1091–1108. doi: 10.1016/j.jmb.2004.02.023. [DOI] [PubMed] [Google Scholar]
- 51.zur Hausen H. Nat Rev Cancer. 2002;2:342–350. doi: 10.1038/nrc798. [DOI] [PubMed] [Google Scholar]