

Abstract
The cytokine interleukin (IL) 25 has been implicated in the initiation of type 2 immunity by driving the expression of type 2 cytokines such as IL-5 and IL-13, although its role in the regulation of immunity and infection-induced inflammation is unknown. Here, we identify a dual function for IL-25: first, in promoting type 2 cytokine-dependent immunity to gastrointestinal helminth infection and, second, in limiting proinflammatory cytokine production and chronic intestinal inflammation. Treatment of genetically susceptible mice with exogenous IL-25 promoted type 2 cytokine responses and immunity to Trichuris. IL-25 was constitutively expressed by CD4+ and CD8+ T cells in the gut of mouse strains that are resistant to Trichuris, and IL-25–deficient mice on a genetically resistant background failed to develop a type 2 immune response or eradicate infection. Furthermore, chronically infected IL-25−/− mice developed severe infection-induced intestinal inflammation associated with heightened expression of interferon-γ and IL-17, identifying a role for IL-25 in limiting pathologic inflammation at mucosal sites. Therefore, IL-25 is not only a critical mediator of type 2 immunity, but is also required for the regulation of inflammation in the gastrointestinal tract.
CD4+ T helper 2 (TH2) cells that produce the cytokines IL-4, IL-5, IL-9, and IL-13 mediate type 2 immunity by activating innate and adaptive components of the immune response (1–3). Type 2 immune responses are thought to have evolved primarily to control pathogen invasion at mucosal sites and are required for the development of resistance to helminth infections. However, dysregulated type 2 responses to environmental antigens can result in severe pathological states such as asthma and allergic inflammation, conditions that affect an estimated 15–30% of the United States population (4, 5). Understanding the factors that regulate type 2 immunity and inflammation is a critical aspect of developing novel approaches to treat these debilitating infections and inflammatory diseases.
IL-25 (IL-17E) is a recently described member of the IL-17 cytokine gene family (6–8). IL-25 is produced by activated TH2 cells and mast cells and signals through the IL-25 R (IL-17Rh1 or IL-17BR), a single pass transmembrane receptor that is expressed in the kidneys, liver, intestine, and on dendritic cells activated in the presence of TH2 cytokines (9–13). Administration of exogenous IL-25 has been shown to induce the expression of IL-5 and IL-13 by an accessory cell population and has been suggested to be involved in the initiation and amplification of type 2 responses (11). However, whether IL-25 plays any role in the regulation of immunity and inflammation in vivo is unknown.
Infection of mice with the helminth Trichuris muris is a well-defined model system to study the cellular and molecular requirements of type 2 immunity in the gastrointestinal (GI) tract. Resistance to infection (e.g., in BALB/c or C57BL/6 mice) is associated with the hallmarks of type 2 immunity: outgrowth of antigen-specific CD4+ TH2 cells that produce IL-4 and IL-13, mastocytosis, IgG1 and IgE production, goblet cell hyperplasia, and production of RELMβ, which is a novel goblet cell-specific protein associated with immunity to helminth parasites (14–18). In contrast, CD4+ TH1 cells that produce IFN-γ promote parasite persistence, resulting in chronic infection (e.g., in AKR mice). Most susceptible strains of mice that harbor persistent parasites exhibit moderate infection-induced pathology, whereas strains deficient in IL-10 or NFκB1 develop severe intestinal inflammation that can lead to death (19, 20). Thus, infection with Trichuris provides a tractable system to study immunity and inflammation in the GI tract. Here, we provide evidence that IL-25 is a critical cytokine in both promoting type 2 cytokine-dependent resistance to Trichuris infection and in inhibiting destructive intestinal inflammation.
RESULTS AND DISCUSSION
To test whether IL-25 could induce a protective type 2 response and confer immunity to Trichuris in a normally susceptible strain, infected AKR mice were treated with recombinant IL-25 (rIL-25). Although control-treated mice exhibited a polarized Trichuris-specific IFN-γ response in the absence of detectable IL-4 (Fig. 1 A), administration of rIL-25 resulted in the induction of an antigen-specific IL-4 response and extinguished IFN-γ production (Fig. 1 A). In addition, rIL-25–treated AKR mice exhibited the hallmarks of a protective TH2 cell response, including reduced levels of Trichuris-specific IgG2a (Fig. S1, available at http://www.jem.org/cgi/content/full/jem.20051496/DC1), an increase in the numbers of goblet cells in the cecum (Fig. 1 B) and increased expression of RELMβ (Fig. 1 C). Consistent with the development of a Trichuris-specific type 2 response, rIL-25 treatment conferred resistance to infection, as untreated AKR mice had a high parasite burden, whereas treated AKR mice achieved sterile cure (Fig. 1 D). IL-25–induced immunity was lymphocyte dependent, as infected SCID mice treated with rIL-25 did not exhibit goblet cell responses (Fig. 1 B) and were unable to eradicate infection (Fig. 1 D). Thus, exogenous IL-25 can promote protective lymphocyte-dependent type 2 responses and act as an effective therapeutic treatment to enhance type 2 immunity in the GI tract.
Figure 1.

Exogenous IL-25 promotes immunity to Trichuris in genetically susceptible AKR mice. AKR mice were infected with Trichuris and treated with PBS or rIL-25 intraperitoneally every other day from days 8–18. (A) IL-25 induces a protective TH2 cytokine response. Antigen-specific IFN-γ and IL-4 production from restimulated mLN cells was determined by ELISA. White bars, medium; black bars, Trichuris antigen. (B) IL-25 promotes lymphocyte-dependent goblet cell responses. Cecal sections from day 20 infected AKR or SCID mice with or without rIL-25 treatment were stained for goblet cells. Bar, 50 μm. (C) IL-25 induces RELMβ production after infection with Trichuris. RELMβ levels were determined by immunoblotting of protein isolated from fecal pellets isolated from infected mice at days 14 and 17 after infection. (D) IL-25 promotes resistance to infection with Trichuris. Worm burdens from infected AKR or SCID mice with or without rIL-25 treatment were determined at day 20 after infection. Results are presented as mean ± SEM and are representative of three independent experiments with three to four mice per group. ND, not detected. *, P < 0.01.
We next tested whether early differences in expression of IL-25 was a predictor of infection outcome in genetically resistant versus susceptible strains. Although no infection-induced increases in IL-25 mRNA were observed after infection with Trichuris (unpublished data), we found that resistant BALB/c mice expressed constitutively higher levels of IL-25 mRNA in the cecum but not the mesenteric lymph node (mLN) compared with susceptible AKR mice, whereas both strains expressed equivalent but low levels of IL-25 mRNA in the mLN (Fig. 2 A). Furthermore, IL-25 receptor (IL-25R) mRNA expression by BALB/c mice was elevated in both compartments (Fig. 2 A). To determine the cellular source of IL-25, we examined the expression of a lacZ reporter construct inserted into the IL-25 locus. This reporter system allows for the amplification of the endogenous IL-25 response by using the fluorescent β-galactosidase substrate, FDG. Upon examination of the mLN and cecal patch (CP) (the murine equivalent of the appendix) for expression of the lacZ/IL-25, we identified that populations of CD4+ and CD8+ T cells residing specifically in the CP of naive mice expressed β-galactosidase activity (Fig. 2 B). We observed no FDG staining in CD19+, FcɛRI+, CCR3+, NK1.1+, CD11c+, or CD11b+ cells from the mLN or CP (unpublished data). Thus, IL-25 is constitutively expressed by a subset of T cells that are found in the CP of resistant mice.
Figure 2.

IL-25 is expressed constitutively in the cecal patch of mice resistant to Trichuris. (A) Increased expression of IL-25 and IL-25R mRNA in the cecum of resistant BALB/c (B/c) mice. IL-25 and IL-25R mRNA levels were determined by real-time PCR from the cecum or mLN of naive BALB/c or AKR mice. (B) CD4+ and CD8+ T cells in the cecal patch express lacZ/IL-25. Cells from the mLN or cecal patch of IL-25−/− mice were stained with FDG and CD4 or CD8. Numbers represent frequency of FDG+ CD4+ or CD8+ T cells. Data are representative of three to four independent experiments with three to four mice per group.
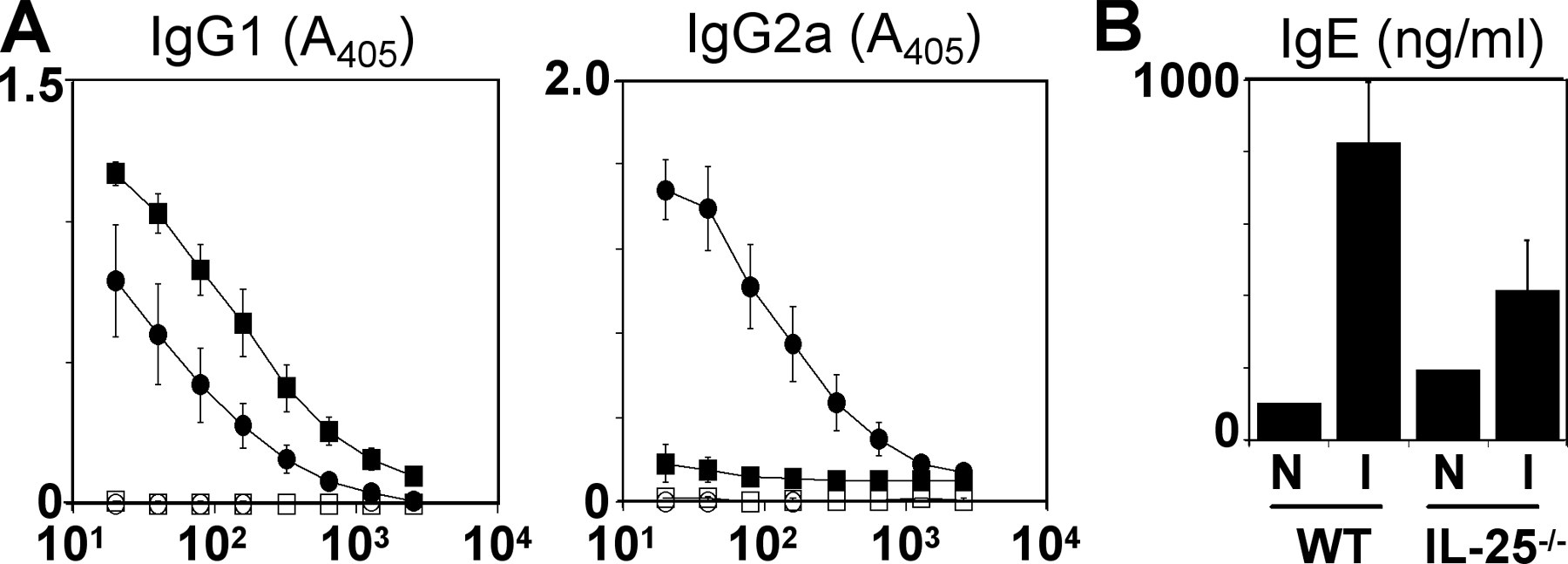
To directly investigate the functional significance of endogenous IL-25 in the development of protective type 2 cytokine responses in the GI tract, IL-25−/− mice were generated by homologous recombination and infected with Trichuris. IL-25−/− mice on a C57BL/6 genetic background were born in expected Mendelian ratios, developed normally, and exhibited no defects in lymphocyte development in the thymus and periphery (unpublished data). After infection, WT mice exhibited enhanced expression of IL-4 and IL-13 mRNA in the draining mLN and elevated IL-4 mRNA levels in the cecum at day 20 after infection with a negligible increase in IFN-γ mRNA expression at both sites (Fig. 3 A). In contrast, IL-4 and IL-13 mRNA levels were significantly reduced in infected IL-25−/− mice with a coincident elevation of IFN-γ mRNA levels (Fig. 3 A). Levels of secreted cytokine protein after antigen restimulation of mLN cells were consistent with ex vivo mRNA levels (unpublished data). The mRNA of other cytokines implicated in resistance to Trichuris, including TNF-α (21) and IL-1β (22), were not expressed at significantly different levels in the mLN between WT and IL-25−/− mice (unpublished data). IL-25−/− mice also exhibited lower serum levels of Trichuris-specific IgG1 (Fig. S2 A, available at http://www.jem.org/cgi/content/full/jem.20051496/DC1) and total IgE (Fig. S2 B), and elevated levels of Trichuris-specific IgG2a (Fig. S2 A), further reflecting a shift from a protective type 2 response to a nonprotective type 1 response in the absence of IL-25. Infection of WT mice with Trichuris resulted in increased numbers of goblet cells in the cecum (Fig. 3 B) and elevated secretion of RELMβ into the GI lumen (Fig. 3 C). In contrast, infected IL-25−/− mice failed to exhibit any significant goblet cell hyperplasia (Fig. 3 B) or detectable secretion of RELMβ (Fig. 3 C). Interestingly, mucosal mast cell responses (numbers of mast cells in the gut and serum levels of mouse mast cell protease-1) that are also associated with type 2 responses at mucosal sites (14, 23) were unimpaired in the absence of IL-25 (Fig. S3, A and B, available at http://www.jem.org/cgi/content/full/jem.20051496/DC1), demonstrating that IL-25 differentially regulates specific components of type 2 cytokine-dependent inflammation in the GI tract. Nevertheless, consistent with a defective type 2 immune response, IL-25−/− mice were unable to eradicate infection and were as susceptible as RAG-deficient mice, whereas WT mice were resistant to infection, clearing worms by day 20 after infection (Fig. 3 D). Thus, endogenous IL-25 is a critical component of type 2 cytokine-dependent immunity in the GI microenvironment.
Figure 3.

Endogenous IL-25 is required for resistance to Trichuris. (A) IL-25 is required for optimal production of TH2 cytokines after infection. IL-4, IL-13, and IFN-γ expression in the mLN and cecum in WT and IL-25−/− mice at day 18 after infection as measured by real-time PCR. (B) Defective goblet cell responses in IL-25−/− mice after infection with Trichuris. Cecal sections from day 18 infected WT or IL-25−/− mice were stained for goblet cells. Bar, 50 μm. (C) IL-25−/− mice fail to up-regulate RELMβ after infection. Protein isolated from fecal pellets on various days after infection was analyzed by SDS-PAGE and immunoblotted for RELMβ. (D) IL-25 is required for resistance to infection with Trichuris. Worm burdens in WT, IL-25−/− and RAG−/− mice at day 30 after infection. Data are presented as mean ± SEM and are representative of four independent experiments with three to four mice per group. N, naive; I, infected.
Although the cytokines that drive TH1 cell differentiation and type 1 inflammation are well characterized, the factors that promote the development of type 2 responses in vivo remain elusive (24). Based on higher levels of IL-25/IL-25R mRNA in genetically resistant mouse strains, coupled with the results demonstrating that exogenous IL-25 can drive expression of TH2 cytokines, whereas type 2 immunity is severely impaired in IL-25−/− mice, we hypothesized that this cytokine may be the critical factor required for the development of TH2 cells and type 2 responses in vivo. To test whether IL-25 expression was essential for immunity to Trichuris, infected IL-25−/− mice were treated with a combination of anti–IL-12 and anti–IFN-γ monoclonal antibodies. Previous studies have shown that in vivo depletion of type 1 cytokines can promote type 2 cytokine-dependent resistance to Trichuris in normally susceptible hosts (16, 19). Blockade of type 1 cytokines in infected IL-25−/− mice was associated with elevated production of Trichuris-specific IL-4, IL-5, and IL-13 in the draining mLN (Fig. 4 A), enhanced serum IgE responses (Fig. S4 A, available at http://www.jem.org/cgi/content/full/jem.20051496/DC1), and a recovery of intestinal goblet cell responses (Fig. 4 B). Furthermore, anti–IL-12/anti–IFN-γ treated IL-25−/− mice exhibited reduced Trichuris-specific IFN-γ production (Fig. 4 A) and lower serum IgG2a levels (Fig. S4 B). Although control-treated IL-25−/− mice displayed high worm burdens, inhibition of type 1 responses resulted in successful expulsion of Trichuris in IL-25−/− mice (Fig. 4 C). Contrary to the hypothesis that IL-25 is essential for type 2 responses, these results show that expression of IL-4, IL-5, and IL-13 was recovered in the absence of IL-25 if endogenous type 1 cytokines were blocked. Therefore, these data show that the requirement for IL-25 in the development of type 2 immunity is conditional and suggest that, in addition to promoting type 2 responses, IL-25 may function to actively inhibit expression of type 1 cytokines after helminth infection.
Figure 4.

Type 2 inflammation is IL-25 independent when endogenous type 1 responses are inhibited. Infected IL-25−/− mice were treated with anti–IL-12 and anti–IFN-γ every 4 d between days 4 and 20. (A) Blockade of type 1 cytokines in infected IL-25−/− mice recovers TH2 cytokine responses. Antigen-specific IL-4, IL-5, IL-13, and IFN-γ responses by restimulated mLN cells from control (Ig) or αIL-12/αIFN-γ–treated (α12/γ) IL-25−/− mice determined by cytometric bead array (CBA; IL-4, IL-5, and IFNγ) or ELISA (IL-13). (B) Anti–IL-12/anti–IFN-γ treatment recovers goblet cell responses in infected IL-25−/− mice. Goblet cell responses in the cecum of infected WT and control or αIL-12/αIFN-γ–treated IL-25−/− mice on day 20 after infection. Bar, 50 μm. (C) IL-25 is dispensable for resistance to infection with Trichuris when type 1 responses are neutralized. Worm burdens in infected WT and control or αIL-12/αIFN-γ–treated IL-25−/− mice were determined at day 20 after infection. Results are presented as mean ± SEM and represent two independent experiments with three to four mice per group. *, P < 0.01.
In support of an antiinflammatory function of IL-25, we observed a critical role for this cytokine in limiting chronic infection-induced intestinal inflammation. Genetically susceptible strains of mice, such as AKR, as well as mice deficient in IL-4Rα signaling, develop persistent chronic infections but display moderate infection-induced inflammation characterized by minimal crypt hyperplasia and no loss in gross tissue morphology (references 17, 21, 25 and unpublished data). Although we observed no differences in steady-state intestinal structure between AKR, WT, or IL-25−/− mice (Fig. 5 A, top), chronically infected IL-25−/− mice developed severe intestinal inflammation characterized by a transmural infiltrate of lymphocytes, crypt elongation, disrupted intestinal architecture, and a complete absence of goblet cells (Fig. 5 A, bottom). Consistent with the hypothesis that IL-25 may act to limit inflammation, the infection-induced pathology in IL-25−/− mice was associated with heightened levels of the proinflammatory cytokines IFN-γ and IL-17 in the mLN and cecum (Fig. 5, B and C). IL-10−/− mice also develop severe Trichuris-induced intestinal inflammation that can be fatal (20). However, the destructive inflammation observed in IL-25−/− mice was not associated with defective expression of IL-10 in the mLN (Fig. 5 D), suggesting a novel IL-10–independent, IL-25–dependent pathway in the regulation of GI inflammation. Interestingly, mice deficient in NF-κB1 (p50) develop severe intestinal inflammation after infection with Trichuris (19). IL-25 has been shown to activate NF-κB (12), suggesting that IL-25– induced NF-κB1 activation may be a critical component in ameliorating inflammation and maintaining the integrity of the mucosal barrier.
Figure 5.

IL-25 is required to limit infection-induced intestinal inflammation. WT and IL-25−/− mice were infected with Trichuris and allowed to progress to the chronic phase of infection (day 30 after infection). (A) IL-25 is required to inhibit infection-induced pathology. Cecal sections from naive or infected (day 30) AKR, B6 WT, or IL-25−/− mice were stained with hematoxylin and eosin. Bar, 50 μm. (B and C) IL-25 limits expression of proinflammatory cytokines in the draining mLN and gut after infection with Trichuris. (B) Increased Trichuris-specific IFN-γ and IL-17 produced by mLN cells from WT or IL-25−/− mice at day 18 after infection as measured by CBA (IFN-γ) or ELISA (IL-17). White bars, medium; black bars, Trichuris antigen. (C) Expression of mRNA for IFN-γ and IL-17 in the mLN and cecum of WT and IL-25−/− mice at day 30 after infection. (D) IL-10 expression in the mLN is intact in infected IL-25−/− mice. mLN cells from WT or IL-25−/− mice at day 18 after infection were restimulated with (black bars) or without (white bars) Trichuris antigen and IL-10 levels were determined by CBA. IL-10 mRNA levels in the mLN at day 30 after infection were determined by real-time PCR. Results are presented as mean ± SEM and represent three independent experiments with three to four mice per group. ND, not detected.
These results demonstrate that IL-25 plays a dual role in both the development of immunity and in limiting inflammation in the GI tract. As IL-25 is produced constitutively by T cells present in the CP of naive mice, this cytokine is most likely operating early after infection by driving expression of TH2 cytokines and simultaneously limiting the production of proinflammatory cytokines. Consistent with this, IL-25−/− mice fail to develop a polarized type 2 immune response and are unable to clear infection unless endogenous type 1 cytokine responses are blocked. This ability to limit proinflammatory responses is critical, as chronically infected IL-25−/− mice develop severe intestinal pathology characterized by high levels of IFN-γ and IL-17. IFN-γ and IL-17 have been implicated in the pathogenesis of inflammatory bowel disease (26, 27). However, whether the intestinal pathology observed in Trichuris-infected IL-25−/− mice is mediated by dysregulated IL-12–dependent, IFN-γ–producing TH1 cells or by an IL-23–dependent, IL-17–producing T cell population is still not known. It is possible that IL-25 directly inhibits the production of IFN-γ or IL-17 from T cells, or alternatively limits their expression through influencing expression of IL-12/IL-23. We observed no significant difference in the expression levels of the mRNA for the IL-12 and IL-23 subunits p19, p35, and p40 between WT and IL-25−/− mice in the mLN after infection with Trichuris (unpublished data). Therefore, it is unclear at present how IL-25 is limiting inflammation.
Recent studies have shown that IL-17–producing CD4+ T cells mediate chronic inflammation in the central nervous system of animals that develop experimental autoimmune encephalitis (EAE), an animal model of multiple sclerosis that is characterized by chronic inflammatory responses and demyelination of neurons (28, 29). Supporting a role for IL-25 in limiting production of proinflammatory cytokines and destructive tissue inflammation, IL-25−/− mice are highly susceptible to EAE, exhibiting reduced expression of TH2 cytokines, increased numbers of IL-17pos CD4+ T cells in the central nervous system and rapid onset and severity of disease (unpublished data). Together with results presented here, these studies implicate IL-25 as a global inhibitor of chronic inflammatory responses. Therefore, manipulation of IL-25/IL-25R signaling offers the potential to promote or block type 2 responses associated with infection, asthma, or allergic inflammation and may provide a novel therapeutic approach for the treatment of multiple diseases associated with chronic inflammation.
MATERIALS AND METHODS
Animals, parasites, antigens, and infections.
IL-25−/− mice were generated by homologous recombination (HR). After a screen of a C57BL/6J genomic BAC library (Genome Systems) with a full-length mIL-25 cDNA probe, a clone was identified and used to generate a shotgun plasmid library (SeqWright). Using a genomic subclone (F6), a BglII site was introduced in exon1 at the ATG via site-directed mutagenesis. This clone was digested with BglII and a 5,442-bp 5′ region of homology was cloned into the unique BglII site of pbgal-Basic (CLONTECH Laboratories, Inc.). A 1,049-bp 3′ region of homology was generated by PCR using a genomic subclone (C6), incorporating unique SpeI and NotI restriction sites, and cloned into pBS-lox-Neo-lox. This plasmid was digested with SalI and the lox-Neo-lox-3′ region of homology was ligated into the unique SalI site of the 5′ arm-pbgal-Basic, yielding the final targeting vector. The NheI linearized targeting vector was electroporated into C57BL/6 ES cells and Neo-resistant clones were analyzed for HR using a PCR based screening strategy followed by Southern blot confirmation with 5′ and 3′ probes. A confirmed HR clone was electroporated with a Cre recombinase expression containing plasmid and several Neo-sensitive clones were screened using Southern blot to identify the Neo-flipped HR IL-25-LacZ ES line. This line was injected into C57BL/6 blastocysts and IL-25-LacZ-knockin (KI)−/− progeny were obtained by intercrossing IL-25–LacZ-KI+/− animals. A PCR-based genotyping strategy was developed to track the IL-25 WT and KI alleles as follows: IL-25 targeted allele, sense 5′-GCTGACTCTCAACATTCTACTCC TCC-3′, antisense 5′-CCTGCTGCTTCAGGTAGGGCTTTG-3′; wild-type IL-25 allele, sense 5′-CTACAGACAGGCTCCCACATGGACC-3′, antisense 5′-CCTGCTGCTTCAGGTAGGGCTTTG-3′.
C57BL/6J, AKR, SCID (CBySmn.CB17-Prkdcscid/J), and RAG−/− (B6.129S7-Rag1tm1Mom/J) mice were obtained from The Jackson Laboratory. Animals were maintained in a specific-pathogen free environment and tested negative for pathogens in routine screening. All experiments were performed following the guidelines of the University of Pennsylvania Institutional Animal Care and Use Committee. Trichuris muris was maintained in genetically susceptible animals. Isolation of Trichuris excretory-secretory antigen and eggs was performed as described previously (14). Mice were infected on day 0 with 150–200 embryonated eggs, and parasite burdens were assessed on various days after infection.
Cytokines and monoclonal antibodies.
Monoclonal antibodies against IL-12 (C17.8) and IFN-γ (XMG-6) were purified from ascites by ammonium sulfate precipitation and extensively dialyzed against PBS. Mice were treated i.p. with 500 μg of each antibody every 4 d between days 4 and 20 after infection. rIL-25 was synthesized and purified as described previously (11). Mice were treated i.p. with 10 μg of rIL-25 every other day between days 8 and 18 after infection.
mRNA analysis.
mRNA expression levels were analyzed using real-time PCR. Total RNA from tissue was isolated, cDNA was prepared, and 10–25 ng of cDNA was used to analyze gene expression in either the SYBR green real-time PCR assay or using predesigned TaqMan probe sets for IL-25, IL-25R, IL-4, IL-13, IL-10, IL-17, IFN-γ, ubiquitin, and β-actin (Applied Biosystems). Reactions were run on the GeneAmp 5700 Sequence Detection System (Applied Biosystems), and expression was normalized to the housekeeping genes ubiquitin or β-actin.
Cell culture and cytokine and lacZ analysis.
At necropsy, the mesenteric LN was harvested, and single cell suspensions were prepared in DMEM supplemented with 10% heat-inactivated FBS, 2 mM glutamine, 100 U/ml penicillin, 100 μg/ml streptomycin, 25 mM Hepes, and 5 × 10−5 M 2-ME. Cells were plated in medium alone or in the presence of Trichuris muris ES Ag (50 μg/ml). Cell-free supernatants were harvested after 48 h, and analyzed for cytokine secretion by sandwich ELISA or cytometric bead array (mouse Th1/Th2 cytokine CBA; BD Biosciences). For determination of lacZ expression, the fluorescent β-galactosidase substrate fluorescein di-β-d-galactopyranoside (FDG; Invitrogen) was added to cells isolated from the mLN or CP of naive or infected IL-25−/− mice that express lacZ in place of IL-25. After 20 min on ice, cells were surface stained with fluorescently labeled antibodies against CD4, CD8, CCR3, CD11b, CD11c, CD19, FcɛRI, and NK1.1 (eBioscience) and analyzed by flow cytometry on a FACSCalibur using CELLQuest Pro software (BD Biosciences).
Serum immunoglobulin analysis.
Serum was analyzed by ELISA for Trichuris-specific IgG1 and IgG2a as described previously (19). Total serum IgE was analyzed with an OptEIA IgE ELISA kit following the manufacturer's recommendations (BD Biosciences).
Goblet cell, RELMβ, and mucosal mast cell responses.
Segments of cecum were removed and fixed in 4% paraformaldehyde. For detection of intestinal goblet cells, 5-μm paraffin-embedded sections were cut and stained with Alcian blue-periodic acid Schiff's reagent or with hematoxylin and eosin. Isolation of proteins from stool samples was performed as described previously (15). Equal amounts of protein were analyzed by SDS-PAGE and immunoblotted for RELMβ with a polyclonal rabbit α-murine RELMβ antibody. Intestinal mast cells were identified by immunohistochemistry using rat anti–mouse mast cell protease-1 (mMCP-1) antibody as described previously (30). Concentrations of mMCP-1 in serum were quantitated as described previously (30).
Statistics.
Results represent the mean ± SEM unless otherwise stated. Statistical significance was determined by Student's t test.
Online supplemental material.
Trichuris-specific serum IgG2a response in AKR mice after infection with Trichuris with or without exogenous rIL-25 is shown in Fig. S1. Fig. S2 depicts the serum immunoglobulin responses (IgG1, IgG2a, and IgE) of wild-type and IL-25−/− mice after infection with Trichuris. Fig. S3 shows the local and systemic mucosal mast cell responses in wild-type and IL-25−/− mice after infection with Trichuris. Fig. S4 shows that blocking IL-12 and IFN-γ in IL-25−/− mice after infection with Trichuris results in decreased serum IgG2a and increased IgG1 and IgE responses. Online supplemental material is available at http://www.jem.org/cgi/content/full/jem.20051496/DC1.
Supplemental Material
Acknowledgments
The authors thank S. Longworth and J. Northrup for assistance with lacZ/FDG staining and E.J. Pearce and P. Scott for critical reading of the manuscript.
This work was supported by National Institutes of Health grant nos. AI61570 (to D. Artis) and AI42334 (to C.A. Hunter); Molecular Biology Core, Morphology Core and Pilot Feasibility Program of the National Institute of Diabetes and Digestive and Kidney Diseases Center grant no. DK50306 (to D. Artis); the Crohn's and Colitis Foundation of America's William and Shelby Modell Family Foundation Research Award (to D. Artis); and the Irvington Institute for Immunological Research Postdoctoral Fellowship (to C. Zaph). Schering-Plough Biopharma (formerly DNAX Research Inc.) is funded by the Schering-Plough Corporation.
The authors have no conflicting financial interests.
A.M. Owyang and C. Zaph contributed equally to this work.
References
- 1.Murphy, K.M., and S.L. Reiner. 2002. The lineage decisions of helper T cells. Nat. Rev. Immunol. 2:933–944. [DOI] [PubMed] [Google Scholar]
- 2.Mowen, K.A., and L.H. Glimcher. 2004. Signaling pathways in Th2 development. Immunol. Rev. 202:203–222. [DOI] [PubMed] [Google Scholar]
- 3.Stetson, D.B., D. Voehringer, J.L. Grogan, M. Xu, R.L. Reinhardt, S. Scheu, B.L. Kelly, and R.M. Locksley. 2004. Th2 cells: orchestrating barrier immunity. Adv. Immunol. 83:163–189. [DOI] [PubMed] [Google Scholar]
- 4.Cohn, L., J.A. Elias, and G.L. Chupp. 2004. Asthma: mechanisms of disease persistence and progression. Annu. Rev. Immunol. 22:789–815. [DOI] [PubMed] [Google Scholar]
- 5.Umetsu, D.T., J.J. McIntire, O. Akbari, C. Macaubas, and R.H. DeKruyff. 2002. Asthma: an epidemic of dysregulated immunity. Nat. Immunol. 3:715–720. [DOI] [PubMed] [Google Scholar]
- 6.Kawaguchi, M., M. Adachi, N. Oda, F. Kokubu, and S.K. Huang. 2004. IL-17 cytokine family. J. Allergy Clin. Immunol. 114:1265–1273. [DOI] [PubMed] [Google Scholar]
- 7.Kolls, J.K., and A. Linden. 2004. Interleukin-17 family members and inflammation. Immunity. 21:467–476. [DOI] [PubMed] [Google Scholar]
- 8.Moseley, T.A., D.R. Haudenschild, L. Rose, and A.H. Reddi. 2003. Interleukin-17 family and IL-17 receptors. Cytokine Growth Factor Rev. 14:155–174. [DOI] [PubMed] [Google Scholar]
- 9.Hurst, S.D., T. Muchamuel, D.M. Gorman, J.M. Gilbert, T. Clifford, S. Kwan, S. Menon, B. Seymour, C. Jackson, T.T. Kung, et al. 2002. New IL-17 family members promote Th1 or Th2 responses in the lung: in vivo function of the novel cytokine IL-25. J. Immunol. 169:443–453. [DOI] [PubMed] [Google Scholar]
- 10.Ikeda, K., H. Nakajima, K. Suzuki, S. Kagami, K. Hirose, A. Suto, Y. Saito, and I. Iwamoto. 2003. Mast cells produce interleukin-25 upon FcɛRI-mediated activation. Blood. 101:3594–3596. [DOI] [PubMed] [Google Scholar]
- 11.Fort, M.M., J. Cheung, D. Yen, J. Li, S.M. Zurawski, S. Lo, S. Menon, T. Clifford, B. Hunte, R. Lesley, et al. 2001. IL-25 induces IL-4, IL-5, and IL-13 and Th2-associated pathologies in vivo. Immunity. 15:985–995. [DOI] [PubMed] [Google Scholar]
- 12.Lee, J., W.H. Ho, M. Maruoka, R.T. Corpuz, D.T. Baldwin, J.S. Foster, A.D. Goddard, D.G. Yansura, R.L. Vandlen, W.I. Wood, and A.L. Gurney. 2001. IL-17E, a novel proinflammatory ligand for the IL-17 receptor homolog IL-17Rh 1. J. Biol. Chem. 276:1660–1664. [DOI] [PubMed] [Google Scholar]
- 13.Gratchev, A., J. Kzhyshkowska, K. Duperrier, J. Utikal, F.W. Velten, and S. Goerdt. 2004. The receptor for interleukin-17E is induced by Th2 cytokines in antigen-presenting cells. Scand. J. Immunol. 60:233–237. [DOI] [PubMed] [Google Scholar]
- 14.Artis, D., A. Villarino, M. Silverman, W. He, E.M. Thornton, S. Mu, S. Summer, T.M. Covey, E. Huang, H. Yoshida, et al. 2004. The IL-27 receptor (WSX-1) is an inhibitor of innate and adaptive elements of type 2 immunity. J. Immunol. 173:5626–5634. [DOI] [PubMed] [Google Scholar]
- 15.Artis, D., M.L. Wang, S.A. Keilbaugh, W. He, M. Brenes, G.P. Swain, P.A. Knight, D.D. Donaldson, M.A. Lazar, H.R.P. Miller, et al. 2004. RELMβ/FIZZ2 is a goblet cell-specific immune-effector molecule in the gastrointestinal tract. Proc. Natl. Acad. Sci. USA. 101:13596–13600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Else, K.J., F.D. Finkelman, C.R. Maliszewski, and R.K. Grencis. 1994. Cytokine-mediated regulation of chronic intestinal helminth infection. J. Exp. Med. 179:347–351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bancroft, A.J., A.N. McKenzie, and R.K. Grencis. 1998. A critical role for IL-13 in resistance to intestinal nematode infection. J. Immunol. 160:3453–3461. [PubMed] [Google Scholar]
- 18.Cliffe, L.J., and R.K. Grencis. 2004. The Trichuris muris system: a paradigm of resistance and susceptibility to intestinal nematode infection. Adv. Parasitol. 57:255–307. [DOI] [PubMed] [Google Scholar]
- 19.Artis, D., S. Shapira, N. Mason, K.M. Speirs, M. Goldschmidt, J. Caamano, H.C. Liou, C.A. Hunter, and P. Scott. 2002. Differential requirement for NF-κB family members in control of helminth infection and intestinal inflammation. J. Immunol. 169:4481–4487. [DOI] [PubMed] [Google Scholar]
- 20.Schopf, L.R., K.F. Hoffmann, A.W. Cheever, J.F. Urban Jr., and T.A. Wynn. 2002. IL-10 is critical for host resistance and survival during gastrointestinal helminth infection. J. Immunol. 168:2383–2392. [DOI] [PubMed] [Google Scholar]
- 21.Artis, D., N.E. Humphreys, A.J. Bancroft, N.J. Rothwell, C.S. Potten, and R.K. Grencis. 1999. Tumor necrosis factor α is a critical component of interleukin 13-mediated protective T helper cell type 2 responses during helminth infection. J. Exp. Med. 190:953–962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Helmby, H., and R.K. Grencis. 2004. Interleukin 1 plays a major role in the development of Th2-mediated immunity. Eur. J. Immunol. 34:3674–3681. [DOI] [PubMed] [Google Scholar]
- 23.Marshall, J.S. 2004. Mast-cell responses to pathogens. Nat. Rev. Immunol. 4:787–799. [DOI] [PubMed] [Google Scholar]
- 24.Trinchieri, G. 2003. Interleukin-12 and the regulation of innate resistance and adaptive immunity. Nat. Rev. Immunol. 3:133–146. [DOI] [PubMed] [Google Scholar]
- 25.Bancroft, A.J., D. Artis, D.D. Donaldson, J.P. Sypek, and R.K. Grencis. 2000. Gastrointestinal nematode expulsion in IL-4 knockout mice is IL-13 dependent. Eur. J. Immunol. 30:2083–2091. [DOI] [PubMed] [Google Scholar]
- 26.Camoglio, L., A.A. Te Velde, A.J. Tigges, P.K. Das, and S.J. Van Deventer. 1998. Altered expression of interferon-γ and interleukin-4 in inflammatory bowel disease. Inflamm. Bowel Dis. 4:285–290. [DOI] [PubMed] [Google Scholar]
- 27.Fujino, S., A. Andoh, S. Bamba, A. Ogawa, K. Hata, Y. Araki, T. Bamba, and Y. Fujiyama. 2003. Increased expression of interleukin 17 in inflammatory bowel disease. Gut. 52:65–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cua, D.J., J. Sherlock, Y. Chen, C.A. Murphy, B. Joyce, B. Seymour, L. Lucian, W. To, S. Kwan, T. Churakova, et al. 2003. Interleukin-23 rather than interleukin-12 is the critical cytokine for autoimmune inflammation of the brain. Nature. 421:744–748. [DOI] [PubMed] [Google Scholar]
- 29.Langrish, C.L., Y. Chen, W.M. Blumenschein, J. Mattson, B. Basham, J.D. Sedgwick, T. McClanahan, R.A. Kastelein, and D.J. Cua. 2005. IL-23 drives a pathogenic T cell population that induces autoimmune inflammation. J. Exp. Med. 201:233–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Scudamore, C.L., L. McMillan, E.M. Thornton, S.H. Wright, G.F. Newlands, and H.R. Miller. 1997. Mast cell heterogeneity in the gastrointestinal tract: variable expression of mouse mast cell protease-1 (mMCP-1) in intraepithelial mucosal mast cells in nematode-infected and normal BALB/c mice. Am. J. Pathol. 150:1661–1672. [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}