

Abstract
Transcriptional regulation is the major regulatory mechanism that controls the spatial and temporal expression of genes during development. This is carried out by transcription factors (TFs), which recognize and bind to their cognate binding sites. Recent studies suggest a modular organization of TF-binding sites, in which clusters of transcription-factor binding sites cooperate in the regulation of downstream gene expression. In this study, we report our computational identification and experimental verification of muscle-specific cis-regulatory modules in Caenorhabditis elegans. We first identified a set of motifs that are correlated with muscle-specific gene expression. We then predicted muscle-specific regulatory modules based on clusters of those motifs with characteristics similar to a collection of well-studied modules in other species. The method correctly identifies 88% of the experimentally characterized modules with a positive predictive value of at least 65%. The prediction accuracy of muscle-specific expression on an independent test set is highly significant (P < 0.0001). We performed in vivo experimental tests of 12 predicted modules, and 10 of those drive muscle-specific gene expression. These results suggest that our method is highly accurate in identifying functional sequences important for muscle-specific gene expression and is a valuable tool for guiding experimental designs.
In metazoans, the gene-regulatory information that directs development is encoded in their genomic DNA sequence. The temporal and spatial expression pattern of genes is controlled by short cis-regulatory elements that act as binding sites for transcription factors. Through interactions with the basal transcription apparatus and other regulatory proteins, transcription factors determine either activation or repression of the target gene at a particular developmental time or within a particular cell or tissue. Therefore, identification of cis-regulatory elements and their binding proteins constitute an important part of deciphering the role of noncoding sequences. However, the individual binding of a transcription factor to a regulatory element is rarely sufficient to confer context-specific expression. Mounting evidence suggests that complex, cooperative protein–protein interactions between transcription factors are required to determine gene expression patterns (Arnone and Davidson 1997; Kamachi et al. 2000; Li et al. 2000; Remenyi et al. 2004). Therefore, identification of all of the component regulatory elements and understanding how they interact with each other are crucial to fully understanding the transcriptional regulatory network.
Given the fast increasing number of genome sequences, our ability to decipher the encoded information lags far behind. For example, Caenorhabditis elegans is the first metazoan organism whose genome was sequenced. However, our understanding of the sequences that control tissue-specific gene expression is still limited. This limited understanding comes mainly from experimental investigation of the regulatory sequences of individual genes, which began almost 20 yr ago (Spieth et al. 1988). In C. elegans, cis-regulation of tissue-specific gene expression is known only for a few genes in some tissues, such as hypodermal cell, excretory cell, vulva, muscle, and neurons (Okkema et al. 1993; Gilleard et al. 1999; Gower et al. 2001; Hwang and Lee 2003; Landmann et al. 2004; Teng et al. 2004; Wang and Chamberlin 2004; Zhao et al. 2005). Progress is limited because of the complexity of the analysis. It involves dissection of all of the sequences around the gene of interest, which could be >10 kb long, to search for functional sequences. To facilitate the study of tissue-specific gene regulation in C. elegans, we use C. elegans muscle-specific gene expression as an example to explore the feasibility of identifying tissue-specific regulatory sequences through a computational approach. In C. elegans, muscle development has been an extensive area of research for a long time. Transcription factors of the basic helix-loop-helix class (hlh-1, Ce-Twist), the NK-2 class (ceh-22), and the T-box family (tbx-2, mls-1) have been shown to be critical for muscle specification and development (Okkema et al. 1993; Chen et al. 1994; Okkema and Fire 1994; Harfe and Fire 1998; Kostas and Fire 2002; Smith and Mango 2007). The promoter regions of several muscle-specific genes (myo-1, myo-2, myo-3, unc-54, hlh-1, and ace-1) have been studied in detail to identify important DNA regulatory sequences using sequence deletions or mutations (Okkema et al. 1993; Chen et al. 1994; Culetto et al. 1999). However, no general rules about the transcriptional regulatory mechanisms that control gene expression in muscle tissue have been identified.
Studies from various organisms have revealed a common theme that transcription factor binding sites tend to be interconnected and function together to confer a particular context-specific expression on the target gene. Those clusters of transcription factor binding sites form a regulatory module that can be located in the upstream, downstream, or intronic sequences and can be moved from their native context and still recapitulate a portion of the native expression pattern independent of their position and orientation to the basal promoter (Arnone and Davidson 1997). Modules have been shown to be very useful in studying temporal and spatial gene expression regulation. Modular structure of regulatory elements is widely present in higher eukaryotes (Kirchhamer et al. 1996; Arnone and Davidson 1997) and has been noted in C. elegans (Jantsch-Plunger and Fire 1994). Due to the time-consuming and labor-intensive nature of experimental approaches, many computational tools have been developed recently to facilitate the identification of regulatory modules. However, the predictive value of most of the methods is either unknown or less than satisfactory.
Here we describe a de novo computational method for accurate identification of regulatory sequences that confer muscle-specific gene expression, as well as experimental tests of the predicted modules. Comparisons of the predicted modules with experimentally characterized modules show high sensitivity and positive predictive value (PPV, defined as True Positives/All Predictions). A totals of 88% (22/25) of experimentally characterized modules are predicted, and 65% (30/46) of our predicted modules are located within experimentally defined regions. The rest of the predicted modules have not been tested for function, so the PPV could be much higher; it is already much higher than currently available algorithms. We developed a scoring system to predict the muscle specificity for any segment of DNA sequence. When applied to the whole genome, this method can help discriminate muscle genes from non-muscle genes. Because no information about known modules was used for the predictions, we expected the new predictions to have the same sensitivity and PPV. To examine this, we experimentally tested the functionality of 12 predicted modules. Of these 12 modules, three are located within known muscle gene promoters and nine are located in the promoters of genes with unknown expression patterns and unknown functions. Ten of the 12 tested modules drive gene expression in muscle tissue, demonstrating that our method is a valuable tool for guiding experimental design. Although we focus on muscle-specific gene expression in this work, we expect the method to be generally applicable to many other context-specific module identification tasks, because our method requires no prior knowledge other than a set of likely coexpressed orthologous genes. C. elegans muscle-specific module prediction tool can be accessed at http://ural.wustl.edu/software.html.
Results
Identification of regulatory motifs
Promoters are commonly defined as the DNA regions located upstream of the transcription start sites that contain the necessary binding elements for proper transcriptional regulation. In C. elegans, 60% of predicted intergenic regions will be fully included within a 2-kb upstream segment (Dupuy et al. 2004). The level of similarity between C. elegans and its relative Caenorhabditis briggsae decreases dramatically 1500 bp upstream of the predicted ATG for most genes with a long intergenic region (Dupuy et al. 2004). Even though some regulatory elements can be located in introns and/or 3′UTRs of genes (Okkema et al. 1993; Jantsch-Plunger and Fire 1994), including those regions in our study, could make computational identification of DNA motifs more difficult, because noise increases with increasing sequence length (Buhler and Tompa 2002; Wang and Stormo 2003). Therefore, we chose to focus on the upstream −2000 to −1 regions. We have used the translation start site (the 0 position) to select the candidate promoter regions because transcriptional start sites have not been determined for most C. elegans genes.
We used the program PhyloCon (Wang and Stormo 2003) for motif identification because comparisons suggest that it outperforms several previous motif-finding programs (Wang and Stormo 2003; MacIsaac et al. 2006). PhyloCon uses position weight matrix-based models (Stormo 2000) to represent ungapped DNA sequence motifs, and conserved motifs identified by this program represent potential regulatory elements. We collected a total of 122 C. elegans genes that are preferentially expressed in muscle tissue (Supplemental Table 1; details given in Methods section), 78 of which have defined C. briggsae orthologs (Supplemental Table 2). PhyloCon was run on the 2-kb upstream sequences of the 78 pairs of muscle genes to predict regulatory motifs, and a total of 18 unique motifs were identified (Table 1).
Table 1.
Predicted motifs ranked by over representation index (ORI)

ORI is calculated as described in the Methods. It reflects how more probable it is to find a motif in the muscle-specific promoter set than in the background set. The higher the ORI, the more enriched the motif is in muscle gene promoters.
Muscle specificity of identified motifs
To identify motifs that are enriched in muscle gene promoters we calculated the Over Representation Index (ORI) (Bajic et al. 2004) for each motif (see Methods) using the rest of the genome as a background gene set. ORI takes into account not only the number of patterns found in sequences, but also the proportion of sequences in which the pattern is found. It reflects how much more probable it is to find a particular motif in the muscle-specific promoter set than in the background set. We define motifs that have an ORI >1.2 as muscle-specific motifs, and they are used later in module score calculations. From our catalog of 18 motifs, eight are designated as muscle-specific.
The top four motifs, ranked by ORI (Table 1), are similar to previously identified muscle-specific regulatory motifs (GuhaThakurta et al. 2002, 2004; Ao et al. 2004). Motif 1 (CTCTCTCTCTC) has almost the same consensus sequence as the binding site of transcription-factor TFII-I (currently known as GTF2I) in vertebrates, which binds to 5′-CTCACTCTCT-3′ (Clark et al. 1998). TFII-I family proteins play an important role in regulating muscle gene expression in humans (Polly et al. 2003). However, no C. elegans homolog was identified by BLAST. Motif 3 (CGCCRCCGCCKCC) is similar to the binding site of Drosophila melanogaster transcription factor Adf-1 (CCGCYGCYG YNGCCGV) in the TRANSFAC database (Matys et al. 2003). Homology search identified three genes in C. elegans that have significant similarity to and belong in the same conserved orthologous groups (COG) as Adf-1. All of them have a MADF domain that directs sequence-specific DNA binding. Motif 6 (WCTTTGM) matches several similar matrices that belong to TCF/LEF family transcription factors that are a subfamily of HMG domain proteins that bind to WWCAAWG consensus sequences. It occurs at a similar level in the muscle gene promoters as in the background gene promoters. Therefore, our motif identification step recovered both known muscle-specific motifs as well as binding sites for common transcription factors.
Identification of muscle-specific regulatory modules in C. elegans promoter sequences
Currently, we do not have a good understanding on how motifs are organized to form modules. Modules may vary in the type of motifs, in the total number and the order of binding sites for each type of motif they contain. However, modules usually contain clusters of motifs, and this property has been used in various algorithms to identify regulatory modules (Wagner 1999; Berman et al. 2002; Markstein et al. 2002). In this study, we developed and tested a simple algorithm that is based on motif clustering and takes into account the general properties of well-studied regulatory modules in higher organisms. First, from many cases of well-studied regulatory modules in various organisms, regulatory modules usually consist of two to eight different regulatory motifs (Arnone and Davidson 1997). Therefore, we require that a regulatory module have at least two different motifs. Secondly, Wasserman and Fickett (1998) collected 18 well-characterized regulatory modules from human muscle genes. Most of the modules have at least two muscle-specific motif sites, which can be the sites of the same motif or of different motifs. Based on this information, we require that a regulatory module have at least two muscle-specific motif sites in order to be a muscle-specific regulatory module. Third, we require the distance between any two adjacent sites within a cluster to be ≤40 bp. Although this choice is somewhat arbitrary, the results are fairly insensitive to several reasonable choices of spacing between motifs (see Discussion). In summary, our definition of a muscle-specific regulatory module is a fragment of sequence that consists of clusters of motifs with intersite spaces ≤40 bp, and in which there are at least two different motifs and at least two muscle-specific binding sites (for details of the algorithm, see Methods).
Because some genes have alternative promoters, there are 138 different muscle gene promoters for the 122 muscle-specific genes. We applied this method on the 138 muscle gene promoters and identified 373 modules, an average of 2.7 modules per gene. The size of the modules ranges from 28 to 516 bp with a mean of 144 bp. Kirchhamer et al. (1996) collected 68 experimentally defined modules from Drosophila and mouse. Their size ranges from 40 bp to 8 kb, but they noted that the listed size was the length of DNA fragments used in gene transfer experiments and the actual size of the modules could be much smaller. The number of motifs in our predicted modules ranges from two to 12 with a mean of six. Well-studied modules have two to eight motifs with a mean of five (Arnone and Davidson 1997). Thus, our predicted modules share some general features with those well-studied modules.
Verification of regulatory modules
To evaluate the accuracy of the predicted modules we identified a total of 27 experimentally characterized modules in 16 gene promoters (Table 2). Of those 27 modules, one is located >2 kb upstream of the translation start site, outside the range of our predictions. Two of the modules overlap by >70% of their length (−370 to −686, −458 to −764 in gene T18D3.4) and it has not been tested whether the minimal overlapping region is sufficient for functionality, so they are treated as one module (−370 to −764) when calculating sensitivity and PPV. Therefore, there are a total of 25 experimentally characterized modules located in the regions we studied.
Table 2.
Performance on muscle gene promoters

(NP) Not predicted. (NCP) Not correctly predicted.
A comparison of our predicted modules to those experimentally characterized modules shows that they match closely. For example, T18D3.4 encodes Myo-2, a pharyngeal-specific myosin heavy chain. The −17 to −239 region is defined as the minimal promoter that can drive reporter gene expression in pharyngeal muscles, while two overlapping 0.3-kb fragments (−370 to −686 and −458 to −764) are sufficient for pharyngeal muscle-specific enhancer activity (Okkema et al. 1993). We predicted three modules in the T18D3.4 2-kb upstream sequences that are located at −60 to −263, −430 to −515, and −562 to −733 upstream of the ATG start codon. Therefore, all three predicted modules are located within the experimentally defined regions (Fig. 1). In summary, for the 25 experimentally defined modules, our method correctly detected 88% (22/25). The definition of correct prediction is that the predicted modules overlap at least 50% with reported modules. In those 16 genes, our method predicted a total of 46 modules, of which 30 overlap with experimentally verified modules. Only one is located within a region shown not to be functional in muscle expression. Because the rest have not been tested, we cannot calculate the specificity of the prediction. However, the PPV of the prediction is at least 65% and could be as high as 98% if the rest of the predicted modules are all true positives. Supplemental Figure 2 shows the location of predicted and experimentally characterized modules for the entire set of genes. Also worth noting is that there are 15 experimentally defined modules with a length ≤ 500 bp. The distance from the ends of predicted modules to the ends of experimentally characterized modules ranges from 5 to 182 bp, and the average is 69 bp. These results demonstrate that the predicted regulatory modules are highly correlated with experimentally determined enhancers that direct gene expression in muscle tissues.
Figure 1.

Two examples of comparison between predicted modules and experimentally defined modules. The lines below the scale represent the experimentally defined modules with start and end positions labeled. The black filled box represents the DNA sequence of corresponding gene. The end at the right side is −1 position of the gene. The small filled boxes below represent predicted modules with start and end positions labeled below. Black filled triangles indicate translational start sites. (A) For C09D1.1a, −1 to −588 bp upstream of ATG is an experimentally defined module. Our method predicted three modules and two are located within the first 588 bp (−66 to −209, −235 to −280). (B) T18D3.4 has two experimentally defined modules, −17 to −239 and −370 to −764. We predicted three modules, and all three are located within experimentally defined regions.
We performed simulations to estimate the statistical significance of obtaining the same sensitivity and PPV, given the promoter sequences and the known regulatory modules. We simulate the distribution of predicted modules in the promoters by randomly picking a start position for each module. The length and number of modules in each gene is kept the same as the predicted modules in that gene. The simulation is repeated 100,000 times and the sensitivity and PPV are calculated for each one. The average sensitivity is 48.8% with standard deviation of 7.8. The average PPV is 35.5% with standard deviation of 5.5. Therefore, the P-values of getting 88% sensitivity and 65% PPV are both much less than 0.001.
Detection of muscle genes on a genome scale
Another test of the accuracy of our module definitions is to use them to predict additional muscle genes. We developed a scoring system to measure the muscle specificity for each module using only the muscle-specific motif sites (see Methods). We expect that the higher the score, the more likely it is to be a muscle-specific module. By ranking all promoters by their scores we should be able to enrich for muscle genes. One difficulty of this assessment is that the expression pattern for most C. elegans genes is unknown. WormBase contains information about the tissue-expression pattern of 2576 genes. There are undoubtedly some omissions in these annotations, where some genes are expressed in tissues beside those listed, but it is likely to be largely correct and is the best data available for this assessment. For these 2576 genes, 1562 are either ubiquitously expressed or expressed in tissues other than muscle. We use these 1562 genes as the negative set. The set of well-characterized muscle genes that were not included in the training set, because we could not identify orthologs in C. briggsae, were used as a test set. We present the results using a Receiver Operator Characteristic (ROC) curve (Fig. 2) (Gribskov and Robinson 1996). For every possible choice of cutoff score, the Y-axis shows the fraction of true positives (known muscle genes) exceeding the cutoff, and the X-axis shows the fraction of false positives (known non-muscle-specific genes) exceeding the threshold. Any form of random predictions would result in points along the diagonal. The result demonstrates that our prediction is well above random, especially for the highest-scoring subset. For example, 30% of the muscle genes are detected at a threshold exceeded by <5% of the negative set, and 50% of the muscle genes are detected at a threshold exceeded by only 12% of the negative set. We use the area under the ROC curve (AUC) to measure how significant our prediction accuracy is. The AUC derived from the ROC curve is 0.7506, which is significantly different from 0.5 with P << 0.0001, indicating that our predictions are highly significant. However, there remain some well-characterized muscle genes that are not well predicted; their scores are not higher than the majority of the negative set genes. This means that we do not yet have a complete model that allows us to predict all muscle-specific gene expression. For some of these genes there is evidence that the muscle module occurs outside of the 2-kb promoter region we have used for scoring, but for others, we have to assume we are still missing some important features.
Figure 2.

ROC curves of muscle gene prediction. Genomic genes are ranked by their muscle-specificity score. We plotted the ROC curves of the set of well-characterized muscle-specific genes that were not used for motif identification (44 test set). The diagonal line represents the result of random guessing. The Y-axis is the fraction of true positives exceeding the cutoff for every cutoff value. The X-axis is the fraction of true negatives that exceed the same cutoff.
Will prior information help?
Our module predictions did not rely on any knowledge about experimentally defined modules, such as which genes contained them, where they were located, or which motifs they contained. We next examined whether the use of prior information about experimentally defined modules can identify a reduced set of motifs that is indispensable for module identification and can improve predictive performance.
First, we tested the performance of module prediction using only muscle-specific motifs. We first noticed that the sensitivity is greatly reduced compared with the prediction made with the full set of motifs. Varying the distance parameter from 20 to 100 bp, the sensitivity ranges from 52% to 72%, while using the full set of motifs has a sensitivity range from 80% to 96%. Secondly, the PPV (from 61.8% to 74.3%) is comparable to the prediction made with the full set of motifs (from 60.5% to 77.4%). Using this motif set to perform genomic predictions does not improve the performance, as determined by the ROC curve of the 44 test set muscle-specific genes (Supplemental Fig. 3). This suggests that some of the non-muscle-specific motifs are important components of muscle-specific modules. We next performed experiments to find a subset of motifs to regain the prediction sensitivity with the same or higher level of PPV. By adding back combinations of one, two, or three non-muscle-specific motifs and using various distance parameters ranging from 20 to 100 bp, we find that there are six cases in which we can obtain both higher sensitivity and higher PPV (Supplemental Table 3). In all cases, motif 6 (WCTTTGM) is included in the motif set. We used three motif sets that give the highest sensitivity and PPV to perform genomic prediction, and plotted the ROC curve of the 44 test set muscle genes. The results suggest that the predictive performances are all comparable to, or worse than, the original set of motifs (Supplemental Fig. 3). Therefore, training on known modules can improve the performance on the training set, but this must be due to overfitting, because it does not improve the genomic predictions in any significant way. These results demonstrate that (1) our method for module identification does not need prior information in order to make high quality predictions; (2) our method is robust; (3) the initial step of motif prediction and redundant motif elimination effectively identifies motifs that are important for regulating muscle-specific gene expression.
Experimental verification of predicted modules
All of the statistical analyses suggest that our method generated high-quality predictions. To test the predictive value of the method on unknown modules and the usefulness in guiding experimental designs, we performed four different types of experiments.
First, we tested our predictive powers by locating the regulatory regions of three genes that are known to be muscle-specific genes, but whose promoters have not been subjected to comprehensive functional analyses. Our results confirmed that our predictions are correct in all three cases. C02D4.2 (ser-2) has at least three alternative promoters that drive C02D4.2 expression in a set of neurons, as well as pharyngeal cells and head muscles (Tsalik et al. 2003). We predicted three modules in C02D4.2a 2-kb upstream region (−91 to −382, −1557 to −1716, and −1769 to −1882). We verified the function of the first predicted module by determining that the first 512 bp upstream of the ATG is sufficient to drive gfp expression only in the head muscle cells (data not shown). Similarly, DNA sequences encompassing the first predicted modules of C33G3.1a (dyc-1) and F08B6.2 (gpc-2) both drive reporter gene expression in the corresponding muscle cells (Table 3; data not shown).
Table 3.
Experimental validation of predicted modules

Second, we tested whether our predictions help to identify muscle-expressing genes in the genome. We randomly picked eight genes of unknown function and unknown expression pattern from the top-ranking predicted muscle genes (ranked from 1 to 198 in the genomic ranking, Table 3). For each gene we assayed whether the minimal upstream sequences encompassing the first predicted modules could drive gene expression in the muscle tissue. Table 3 shows the list of genes tested, as well as the genomic rank of the genes, the location of the predicted modules, and the observed expression patterns. C01B7.3 and C01B7.1 share the 2.6-kb intergenic sequences. C01B7.3 is a predicted gene with no RNAi phenotype and no hit in a BLASTP search in the genome of C. briggsae, Caenorhabditis remanei, Anopheles gambiae, D. melanogaster, Rattus norvegicus, Homo sapiens, C. elegans, and Saccharomyces cerevisiae (WormBase http://www.wormbase.org/.). In our experiment, the 553-bp C01B7.3 promoter did not give any expression pattern. Therefore, C01B7.3 is likely to be a falsely annotated gene. For the remaining seven genes, six are muscle genes, while the minimal promoter region of C10G11.7 drives reporter gene expression exclusively in the neurons (Fig. 3A–L). It is known that muscle genes and neuronal genes share some regulatory elements (Wasserman and Fickett 1998) and 45% of our muscle-specific genes are also expressed in neurons. If we include neuronal genes as positive, 87.5% of our genes are correctly predicted.
Figure 3.

GFP expression pattern driven by DNA sequences encompassing the predicted modules. (A) C01B7.1::gfp expression in the pharyngeal muscle. (B) C10G11.7::gfp expression in neurons. F27D4.2::gfp expression in pharyngeal muscle (C) and body wall muscles (D, arrow). F45D3.2::gfp expression in the body wall muscle (E, arrows) and neurons (F). T28B8.1::gfp expression in the pharyngeal muscle (G), vulva muscle (H, arrow), and neurons (I). W06H8.6::gfp expression in the body wall muscle (J), vulva muscle (K), pharyngeal muscle (L), and H cells (data not shown). (M) A merge of DIC image and fluorescent image taken for the same worm showing W06H8.6::Δpes10::gfp expression in the pharyngeal muscle cells. (N) K10G6.3::gfp expression in the pharyngeal muscle (arrow) and neurons.
Third, we tested the functionality of modules located further upstream by deletion analysis. The first two predicted modules in K10G6.3 are clustered at −378 to −847. A DNA fragment containing this region drives gfp expression mainly in neurons and occasionally in the pharyngeal muscles (Fig. 3N). Deletion of this region results in complete loss of gfp expression. The first predicted module in F27D4.2 is located at −491 to −1041. A DNA fragment including the predicted module drives reporter gene expression in the pharyngeal muscle (Fig. 3C), body wall muscle (Fig. 3D), and intestine, whereas deletion of the predicted module from the DNA results in loss of gfp expression.
Fourth, we tested the enhancer activity of a predicted module. W06H8.6 is a gene with unknown function and unknown expression pattern that has an upstream sequence >7 kb. In the W06H8.6 2-kb promoter sequence, six modules were predicted. The first one is located at −256 to −591 and the first 675 bp upstream of ATG drives reporter gene expression in body wall muscle (Fig. 3J), vulva muscle (Fig. 3K), and pharyngeal muscle (Fig. 3L), as reported above. Another three are located between −764 and −1183 with intermodule distance of around 40 bp. We tested the functionality of this cluster of modules by introducing the DNA fragment upstream of a minimal pes-10 promoter (Fire et al. 1990) and examined its ability to activate reporter gene expression. The tested DNA fragment drives reporter gene expression only in the pharyngeal muscle cells (Fig. 3M). Although both the promoter and the more upstream module of W06H8.6 directed gene expression in muscle cells, each is expressed in a unique subset of muscle cells.
In summary, we tested the functionality of 12 predicted modules. Ten of them drive gene expression in muscle tissues and one of them is involved in gene expression in neuronal cells. The remaining one showed no expression and may not even correspond to a true gene. This gives a positive predictive value of 83%, and 92%, if we count neuronal regulatory modules as positive. Generally, it takes many similar experiments to dissect the long promoter sequences to identify the functional sequences of a single gene. For the genes we tested, several of them have very long upstream sequences. For example, the upstream sequence of F45D3.2, W06H8.6, and F27D4.2 are 9, 7, and 11 kb, respectively. These results demonstrate that our method is able to both predict unknown genes that are expressed in muscle cells and to reduce the important functional domains, which contain the essential modules, to much smaller regions.
Discussion
The accurate identification of regulatory modules within a genomic sequence would be very useful for the study of gene regulation. However, identifying modules experimentally is a time-consuming and labor-intensive process. We developed a computational approach to predict muscle-specific cis-regulatory modules in C. elegans and performed experimental evaluations of their accuracy. Analysis of the in vivo activity of 12 predicted modules, of which 10 showed the predicted activity, demonstrates the utility of our approach.
We chose muscle genes for this study because muscle has been a fertile ground for molecular genetics studies with C. elegans for three decades. Most of the work focused on the organization, structure, and function of muscle fibers and muscle cells (Moerman and Fire 1997; Moerman and Williams 2006). Recent work identified two genes that are involved in muscle cell fate specification (Kostas and Fire 2002; Smith and Mango 2007). However, the molecular mechanisms that control muscle cell fate specification and differentiation remain unclear. Here we demonstrate a computational approach that can identify motifs and their combinations into regulatory modules, which is very useful in identifying muscle-expressing genes. We tested eight genes of unknown expression pattern and unknown function, which we predicted to have modules for muscle expression. Six of those modules did, indeed, cause expression in muscle cells, while one drove expression in neurons and another showed no expression pattern. In total, we tested 12 predicted modules, with 10 showing activity in regulating muscle gene expression, which gives a PPV of 83%. Many of those were in segments directly upstream of the gene, consistent with C. elegans regulatory regions being compact. But in two cases, K10G6.3 and F27D4.2, we showed that the immediate upstream region was not sufficient for muscle expression, but that inclusion of a predicted module further upstream was. In another case, W06H8.6, we showed that two predicted modules, one immediately upstream and another more distant one, were each sufficient to drive muscle expression, but with different expression patterns.
Although this study focused on modules for muscle expression, we did not use any muscle-specific characteristics, and we expect that our method would work equally well for other tissue-specific expression patterns. The approach is quite simple and requires very little prior information, including no initial information about motifs. The input is merely a set of C. elegans genes known to share a particular expression pattern and their orthologs in another Caenorhabditis genome, so that the program PhyloCon could identify significant motifs. We then used the promoters of non-muscle genes to identify which motifs were muscle specific and which were general. The set of motifs were then combined into predicted modules based on characteristics of a few well-characterized modules found in human, mouse, rat, fly, and sea urchin (Arnone and Davidson 1997; Wasserman and Fickett 1998), namely, that there should be at least two different motifs within the module and at least two occurrences of muscle-specific motifs. The one parameter we explored was the spacing between motifs within a module, but we found the results to be quite consistent over ranges from 20 to 75 bp (Supplemental Table 4); longer spacing often predicted entire upstream regions to be a single module, which is not very useful. We do not specify a particular window size for a module, and they can vary considerably in length. We also do not specify a minimum score, although the score, which is based only on the content of the muscle-specific motifs, is useful for ranking the predicted modules, and the results show that the highest-scoring promoters are the most enriched in muscle-specific genes (Fig. 2). In fact, the score appears to reflect the strength of the module in driving muscle gene expression based on the few experimentally determined modules with quantitative comparisons of activity. For example, Okkema et al. (1993) identified two modules in myo-1 gene (R06C7.10): a strong proximal enhancer located at −123 to −500 and a weak distal enhancer located at −646 to −1752. We predicted three modules. The highest-scoring module (score 42.1) is located near the start site, corresponding to the proximal enhancer, and the two lower-scoring modules (score 18.9 and 11.7) are located distally, corresponding to the weaker enhancer (Supplemental Fig. 2). In F29F11.5, three modules located between −1436 and −1922 upstream of ATG were characterized (DE1, DE2, and DE3) with DE3 showing the strongest enhancer activity (Vilimas et al. 2004). Modules were correctly predicted for DE2 and DE3, with the latter having a higher score (Supplemental Fig. 2). Low-score modules could be functional modules as exampled in Supplemental Figure 2.
While these results demonstrate the utility of our approach, we are still far from having a precise and completely accurate predictor of muscle expression patterns. Two of the 12 predicted modules we tested were not correct. From the ROC curve (Fig. 2) it can be seen that high-scoring promoters are highly enriched in muscle-specific genes, but there are a few non-muscle genes that also have high scores, and there are several muscle-specific genes with only low-scoring modules that do not distinguish well from non-muscle genes. Furthermore, we have only attempted to predict muscle expression in general, rather than for specific classes of muscles. Among the tested modules we see several distinct patterns that include specific subsets of muscles from the head, body wall, vulva, and pharynx, as well as some that also cause expression in subsets of neurons. More work is needed before we can fully model more specific expression patterns. For example, in this study, we have not considered possible modules occurring within introns or downstream of the genes, even though we know of such examples (Jantsch-Plunger and Fire 1994). Nor have we considered the phenomenon that clusters of nearby genes may all be activated coordinately, perhaps through the modification of local chromatin domains (Roy et al. 2002). The recent release of the C. remanei genome sequence (ftp://ftp.wormbase.org/pub/wormbase/genomes/remanei/) will increase our power to detect conserved regulatory motifs using methods such as PhyloCon and PhyloNet (Wang and Stormo 2003, 2005). In addition, comprehensive analysis is now ongoing to determine the complete repertoire of C. elegans TFs and their binding-site motifs (Reece-Hoyes et al. 2005). Together, we expect that these additional data will allow for more comprehensive characterization of regulatory interactions and aid in the determination of the complete regulatory network of a model metazoan.
Methods
Identification of C. elegans muscle genes and orthologs in C. briggsae
In this study, we define muscle-specific genes as those that are only expressed in the muscle tissue or expressed in at most two other tissues. We identified a total of 122 C. elegans muscle-specific genes from searching the WormBase (Chen et al. 2005) expression pattern database (http://www.wormbase.org/) and from previous work (GuhaThakurta et al. 2002). C. briggsae orthologs for 78 of the 122 genes were obtained from WormBase. The C. elegans and C. briggsae chromosomal sequence and the gene structures were downloaded from the WormBase ftp-site (ftp://ftp.wormbase.org/pub/wormbase/genomes/, WS123). These were then used to obtain −2000 to −1 upstream regions of muscle-specific genes, as well as an upstream region of all C. elegans genes (22,247).
Identification of putative regulatory motifs and elimination of redundant motifs
PhyloCon (Wang and Stormo 2003) program was run on the upstream sequences (−2000 to −1) of the 78 pairs of C. elegans and C. briggsae orthologous muscle genes. We took the best matrix from each run of PhyloCon, masked all of the incidences of the identified motif in the input file, and repeated until no additional significant motifs were identified. The experiments were performed using various parameters (Wang and Stormo 2003), and motifs identified in all experiments were pooled together. To determine whether any two-position weight matrices were similar, we tested whether two motifs overlap significantly in promoter sequences, as determined by a χ2 test on simulated data. If two motifs overlap significantly, they were considered redundant motifs, and the one with lower information content was removed.
Calculation of over-representation index
Given a weight matrix, the Patser program calculates the probability of observing a sequence with a particular score or greater (Staden 1989; Hertz and Stormo 1999) and determines the default cutoff score based on that P-value. Therefore, a “site” corresponding to a particular motif (weight matrix) is a subsequence that is identified by the Patser program using the cutoff appropriate for each motif.
We adopted the concept of over-representation of a particular pattern in one group of sequences with regard to another group of sequences from Bajic et al. (2004). They define it as:
 |
where Mi is the ith motif. Densityspecific(Mi) is the density at which this motif is found in muscle-specific promoter sequences, and Densitynonspecific(Mi) is the density at which this motif is found in nonspecific sequences. Density is the number of sites of motif i in a sequence of unit length. Proportionspecific is the proportion of muscle-specific promoters that has the motif i. Proportionnonspecific is the proportion of nonspecific promoters that has the motif i. This can be rewritten as:
 |
NumSites is the number of sites of motif i found in muscle-specific promoter sequences, while NumSitens is the number of sites of motif i found in nonspecific sequences; TotalLengthspecific is the total length of muscle-specific promoter sequences and TotalLengthnonspecific is the total length of nonspecific sequences; Ns is the number of muscle-specific promoter sequences where motif i is found, Nns is the number of nonspecific sequences where motif i is found; TotalPromoterspecific is the total number of muscle-specific promoter and TotalPromoternonspecific is the total number of nonspecific sequences, respectively. We use all C. elegans genes other than the 138 muscle gene promoters as nonspecific background sequences.
Searching for cis-regulatory modules
To search for clusters of motifs, we first identify all of the sites for all of the motifs using Patser. Then, we scan the sequence from 5′ to the 3′ end starting from the first site in the sequence. If the next site is less than the cutoff distance away, it is considered to be in the same cluster as the first site. Then, the third site is considered and the distance between it and the second site is calculated. This processes continues until a site is encountered that is too far away from the previous site (exceeds the distance cutoff). This cluster of motifs is a putative regulatory module. Then, we check whether this cluster fits the criteria of muscle-specific module (having at least two types of motifs and two muscle-specific sites). If it fits, it is kept as a muscle-specific regulatory module.
Calculation of module score and promoter score
For a given DNA sequence, the combined probability–proportionality value of multiple motifs is calculated as described (GuhaThakurta et al. 2004). It measures the likelihood that each TF binds at least one of its binding sites in the given sequence. We apply this calculation on each predicted module rather than the whole sequence to calculate the combined probability–proportionality value for each module:
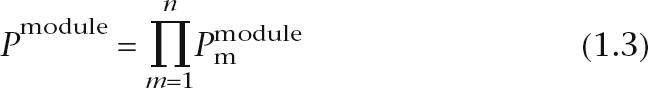 |
where m denotes all of the motifs that exist in the module and n is the total number of different motifs. 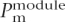 is the probability–proportionality value for motif m in a given module module calculated as described (GuhaThakurta et al. 2004). This treatment is likely oversimplified given the known cooperative binding of transcription factors to promoter elements. However, this does not affect module prediction, it only affects the ranking of genes when we try to discriminate muscle genes from non-muscle genes, and this simplified approach has produced meaningful results. The score for a regulatory module is calculated as log of the combined probability.
is the probability–proportionality value for motif m in a given module module calculated as described (GuhaThakurta et al. 2004). This treatment is likely oversimplified given the known cooperative binding of transcription factors to promoter elements. However, this does not affect module prediction, it only affects the ranking of genes when we try to discriminate muscle genes from non-muscle genes, and this simplified approach has produced meaningful results. The score for a regulatory module is calculated as log of the combined probability.
If a promoter region has more than one identified regulatory module, the muscle-specificity score for the promoter is the sum of the score of all the modules it has
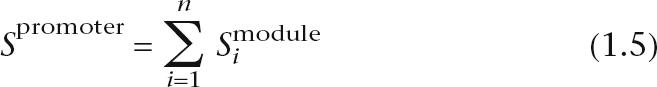 |
where n is the total number of modules in the promoter.
Genome-wide searches
We retrieved 2 kb of upstream sequences from all of the genes in the C. elegans genome (22,247). A muscle-specificity score is calculated for each gene promoter as described above. The promoters were then ranked by the score. If a gene has multiple promoters, we take the highest score and ranking of that gene.
Construction of plasmids and GFP expression analysis
To test the predicted modules close to translational start codons, gene-specific primers were used to amplify the corresponding sequences from fosmid DNAs (Geneservice Ltd). PCR products were cloned into a promoterless GFP vector pLS43 (GuhaThakurta et al. 2004) with nuclear localization signals. Transgenic C. elegans were made as described (Mello et al. 1991) using the collagen gene rol-6 as a coinjection marker. Rolling GFP-expressing progeny were isolated and studied for in vivo GFP expression.
To test the enhancer activity of more distant predicted modules, PCR products were cloned into pPD107.94 (Δpes-10 minimal promoter, a gift from Andrew Fire, Stanford University School of Medicine) (Fire et al. 1990). The construct is used to make transgenic animals for GFP expression study.
Acknowledgments
We thank Ting Wang for assistance with the PhyloCon program and helpful discussions. We also thank Michael L. Nonet, Andrew Fire, and Susan E. Mango for providing reagents used in this work, and Dr. Frank E. Harrell Jr. for helping with statistical analysis of the predictions. This work was supported by NIH grants HG00249, and G.Z. was supported by NIH institutional training grant 5 T32 HG000045-08 and National Institute of General Medical Sciences NRSA service award 1 F32 GM73444-01.
Footnotes
[Supplemental material is available online at www.genome.org.]
Article published online before print. Article and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.5989907
References
- Anyanful A., Sakube Y., Takuwa K., Kagawa H., Sakube Y., Takuwa K., Kagawa H., Takuwa K., Kagawa H., Kagawa H. The third and fourth tropomyosin isoforms of Caenorhabditis elegans are expressed in the pharynx and intestines and are essential for development and morphology. J. Mol. Biol. 2001;313:525–537. doi: 10.1006/jmbi.2001.5052. [DOI] [PubMed] [Google Scholar]
- Ao W., Gaudet J., Kent W.J., Muttumu S., Mango S.E., Gaudet J., Kent W.J., Muttumu S., Mango S.E., Kent W.J., Muttumu S., Mango S.E., Muttumu S., Mango S.E., Mango S.E. Environmentally induced foregut remodeling by PHA-4/FoxA and DAF-12/NHR. Science. 2004;305:1743–1746. doi: 10.1126/science.1102216. [DOI] [PubMed] [Google Scholar]
- Arnone M.I., Davidson E.H., Davidson E.H. The hardwiring of development: Organization and function of genomic regulatory systems. Development. 1997;124:1851–1864. doi: 10.1242/dev.124.10.1851. [DOI] [PubMed] [Google Scholar]
- Bajic V.B., Choudhary V., Hock C.K., Choudhary V., Hock C.K., Hock C.K. Content analysis of the core promoter region of human genes. In Silico Biol. 2004;4:109–125. [PubMed] [Google Scholar]
- Berman B.P., Nibu Y., Pfeiffer B.D., Tomancak P., Celniker S.E., Levine M., Rubin G.M., Eisen M.B., Nibu Y., Pfeiffer B.D., Tomancak P., Celniker S.E., Levine M., Rubin G.M., Eisen M.B., Pfeiffer B.D., Tomancak P., Celniker S.E., Levine M., Rubin G.M., Eisen M.B., Tomancak P., Celniker S.E., Levine M., Rubin G.M., Eisen M.B., Celniker S.E., Levine M., Rubin G.M., Eisen M.B., Levine M., Rubin G.M., Eisen M.B., Rubin G.M., Eisen M.B., Eisen M.B. Exploiting transcription factor binding site clustering to identify cis-regulatory modules involved in pattern formation in the Drosophila genome. Proc. Natl. Acad. Sci. 2002;99:757–762. doi: 10.1073/pnas.231608898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buhler J., Tompa M., Tompa M. Finding motifs using random projections. J. Comput. Biol. 2002;9:225–242. doi: 10.1089/10665270252935430. [DOI] [PubMed] [Google Scholar]
- Chen L., Krause M., Sepanski M., Fire A., Krause M., Sepanski M., Fire A., Sepanski M., Fire A., Fire A. The Caenorhabditis elegans MYOD homologue HLH-1 is essential for proper muscle function and complete morphogenesis. Development. 1994;120:1631–1641. doi: 10.1242/dev.120.6.1631. [DOI] [PubMed] [Google Scholar]
- Chen N., Harris T.W., Antoshechkin I., Bastiani C., Bieri T., Blasiar D., Bradnam K., Canaran P., Chan J., Chen C.K., Harris T.W., Antoshechkin I., Bastiani C., Bieri T., Blasiar D., Bradnam K., Canaran P., Chan J., Chen C.K., Antoshechkin I., Bastiani C., Bieri T., Blasiar D., Bradnam K., Canaran P., Chan J., Chen C.K., Bastiani C., Bieri T., Blasiar D., Bradnam K., Canaran P., Chan J., Chen C.K., Bieri T., Blasiar D., Bradnam K., Canaran P., Chan J., Chen C.K., Blasiar D., Bradnam K., Canaran P., Chan J., Chen C.K., Bradnam K., Canaran P., Chan J., Chen C.K., Canaran P., Chan J., Chen C.K., Chan J., Chen C.K., Chen C.K., et al. WormBase: A comprehensive data resource for Caenorhabditis biology and genomics. Nucleic Acids Res. 2005;33:D383–D389. doi: 10.1093/nar/gki066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho J.H., Eom S.H., Ahnn J., Eom S.H., Ahnn J., Ahnn J. Analysis of calsequestrin gene expression using green fluorescent protein in Caenorhabditis elegans. Mol. Cells. 1999;9:230–234. [PubMed] [Google Scholar]
- Clark M.P., Chow C.W., Rinaldo J.E., Chalkley R., Chow C.W., Rinaldo J.E., Chalkley R., Rinaldo J.E., Chalkley R., Chalkley R. Multiple domains for initiator binding proteins TFII-I and YY-1 are present in the initiator and upstream regions of the rat XDH/XO TATA-less promoter. Nucleic Acids Res. 1998;26:2813–2820. doi: 10.1093/nar/26.11.2813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Culetto E., Combes D., Fedon Y., Roig A., Toutant J.P., Arpagaus M., Combes D., Fedon Y., Roig A., Toutant J.P., Arpagaus M., Fedon Y., Roig A., Toutant J.P., Arpagaus M., Roig A., Toutant J.P., Arpagaus M., Toutant J.P., Arpagaus M., Arpagaus M. Structure and promoter activity of the 5′ flanking region of ace-1, the gene encoding acetylcholinesterase of class A in Caenorhabditis elegans. J. Mol. Biol. 1999;290:951–966. doi: 10.1006/jmbi.1999.2937. [DOI] [PubMed] [Google Scholar]
- Dupuy D., Li Q.R., Deplancke B., Boxem M., Hao T., Lamesch P., Sequerra R., Bosak S., Doucette-Stamm L., Hope I.A., Li Q.R., Deplancke B., Boxem M., Hao T., Lamesch P., Sequerra R., Bosak S., Doucette-Stamm L., Hope I.A., Deplancke B., Boxem M., Hao T., Lamesch P., Sequerra R., Bosak S., Doucette-Stamm L., Hope I.A., Boxem M., Hao T., Lamesch P., Sequerra R., Bosak S., Doucette-Stamm L., Hope I.A., Hao T., Lamesch P., Sequerra R., Bosak S., Doucette-Stamm L., Hope I.A., Lamesch P., Sequerra R., Bosak S., Doucette-Stamm L., Hope I.A., Sequerra R., Bosak S., Doucette-Stamm L., Hope I.A., Bosak S., Doucette-Stamm L., Hope I.A., Doucette-Stamm L., Hope I.A., Hope I.A., et al. A first version of the Caenorhabditis elegans Promoterome. Genome Res. 2004;14:2169–2175. doi: 10.1101/gr.2497604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fire A., Harrison S.W., Dixon D., Harrison S.W., Dixon D., Dixon D. A modular set of lacZ fusion vectors for studying gene expression in Caenorhabditis elegans. Gene. 1990;93:189–198. doi: 10.1016/0378-1119(90)90224-f. [DOI] [PubMed] [Google Scholar]
- Gilleard J.S., Shafi Y., Barry J.D., McGhee J.D., Shafi Y., Barry J.D., McGhee J.D., Barry J.D., McGhee J.D., McGhee J.D. ELT-3: A Caenorhabditis elegans GATA factor expressed in the embryonic epidermis during morphogenesis. Dev. Biol. 1999;208:265–280. doi: 10.1006/dbio.1999.9202. [DOI] [PubMed] [Google Scholar]
- Gower N.J., Temple G.R., Schein J.E., Marra M., Walker D.S., Baylis H.A., Temple G.R., Schein J.E., Marra M., Walker D.S., Baylis H.A., Schein J.E., Marra M., Walker D.S., Baylis H.A., Marra M., Walker D.S., Baylis H.A., Walker D.S., Baylis H.A., Baylis H.A. Dissection of the promoter region of the inositol 1,4,5-trisphosphate receptor gene, itr-1, in C. elegans: A molecular basis for cell-specific expression of IP3R isoforms. J. Mol. Biol. 2001;306:145–157. doi: 10.1006/jmbi.2000.4388. [DOI] [PubMed] [Google Scholar]
- Gribskov M., Robinson N.L., Robinson N.L. The use of receiver operating characteristic (ROC) analysis to evaluate sequence matching. Comput. Chem. 1996;20:25–34. doi: 10.1016/s0097-8485(96)80004-0. [DOI] [PubMed] [Google Scholar]
- GuhaThakurta D., Schriefer L.A., Hresko M.C., Waterston R.H., Stormo G.D., Schriefer L.A., Hresko M.C., Waterston R.H., Stormo G.D., Hresko M.C., Waterston R.H., Stormo G.D., Waterston R.H., Stormo G.D., Stormo G.D. Identifying muscle regulatory elements and genes in the nematode Caenorhabditis elegans. Pac. Symp. Biocomput. 2002;7:425–436. doi: 10.1142/9789812799623_0040. [DOI] [PubMed] [Google Scholar]
- GuhaThakurta D., Schriefer L.A., Waterston R.H., Stormo G.D., Schriefer L.A., Waterston R.H., Stormo G.D., Waterston R.H., Stormo G.D., Stormo G.D. Novel transcription regulatory elements in Caenorhabditis elegans muscle genes. Genome Res. 2004;14:2457–2468. doi: 10.1101/gr.2961104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harfe B.D., Fire A., Fire A. Muscle and nerve-specific regulation of a novel NK-2 class homeodomain factor in Caenorhabditis elegans. Development. 1998;125:421–429. doi: 10.1242/dev.125.3.421. [DOI] [PubMed] [Google Scholar]
- Hertz G.Z., Stormo G.D., Stormo G.D. Identifying DNA and protein patterns with statistically significant alignments of multiple sequences. Bioinformatics. 1999;15:563–577. doi: 10.1093/bioinformatics/15.7.563. [DOI] [PubMed] [Google Scholar]
- Hwang S.B., Lee J., Lee J. Neuron cell type-specific SNAP-25 expression driven by multiple regulatory elements in the nematode Caenorhabditis elegans. J. Mol. Biol. 2003;333:237–247. doi: 10.1016/j.jmb.2003.08.055. [DOI] [PubMed] [Google Scholar]
- Jantsch-Plunger V., Fire A., Fire A. Combinatorial structure of a body muscle-specific transcriptional enhancer in Caenorhabditis elegans. J. Biol. Chem. 1994;269:27021–27028. [PubMed] [Google Scholar]
- Kagawa H., Sugimoto K., Matsumoto H., Inoue T., Imadzu H., Takuwa K., Sakube Y., Sugimoto K., Matsumoto H., Inoue T., Imadzu H., Takuwa K., Sakube Y., Matsumoto H., Inoue T., Imadzu H., Takuwa K., Sakube Y., Inoue T., Imadzu H., Takuwa K., Sakube Y., Imadzu H., Takuwa K., Sakube Y., Takuwa K., Sakube Y., Sakube Y. Genome structure, mapping and expression of the tropomyosin gene tmy-1 of Caenorhabditis elegans. J. Mol. Biol. 1995;251:603–613. doi: 10.1006/jmbi.1995.0459. [DOI] [PubMed] [Google Scholar]
- Kamachi Y., Uchikawa M., Kondoh H., Uchikawa M., Kondoh H., Kondoh H. Pairing SOX off: With partners in the regulation of embryonic development. Trends Genet. 2000;16:182–187. doi: 10.1016/s0168-9525(99)01955-1. [DOI] [PubMed] [Google Scholar]
- Kirchhamer C.V., Yuh C.H., Davidson E.H., Yuh C.H., Davidson E.H., Davidson E.H. Modular cis-regulatory organization of developmentally expressed genes: Two genes transcribed territorially in the sea urchin embryo, and additional examples. Proc. Natl. Acad. Sci. 1996;93:9322–9328. doi: 10.1073/pnas.93.18.9322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kostas S.A., Fire A., Fire A. The T-box factor MLS-1 acts as a molecular switch during specification of nonstriated muscle in C. elegans. Genes & Dev. 2002;16:257–269. doi: 10.1101/gad.923102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krause M., Harrison S.W., Xu S.Q., Chen L., Fire A., Harrison S.W., Xu S.Q., Chen L., Fire A., Xu S.Q., Chen L., Fire A., Chen L., Fire A., Fire A. Elements regulating cell- and stage-specific expression of the C. elegans MyoD family homolog hlh-1. Dev. Biol. 1994;166:133–148. doi: 10.1006/dbio.1994.1302. [DOI] [PubMed] [Google Scholar]
- Landmann F., Quintin S., Labouesse M., Quintin S., Labouesse M., Labouesse M. Multiple regulatory elements with spatially and temporally distinct activities control the expression of the epithelial differentiation gene lin-26 in C. elegans. Dev. Biol. 2004;265:478–490. doi: 10.1016/j.ydbio.2003.09.009. [DOI] [PubMed] [Google Scholar]
- Li R., Pei H., Watson D.K., Pei H., Watson D.K., Watson D.K. Regulation of Ets function by protein–protein interactions. Oncogene. 2000;19:6514–6523. doi: 10.1038/sj.onc.1204035. [DOI] [PubMed] [Google Scholar]
- MacIsaac K.D., Wang T., Gordon D.B., Gifford D.K., Stormo G.D., Fraenkel E., Wang T., Gordon D.B., Gifford D.K., Stormo G.D., Fraenkel E., Gordon D.B., Gifford D.K., Stormo G.D., Fraenkel E., Gifford D.K., Stormo G.D., Fraenkel E., Stormo G.D., Fraenkel E., Fraenkel E. An improved map of conserved regulatory sites for Saccharomyces cerevisiae. BMC Bioinformatics. 2006;7:113. doi: 10.1186/1471-2105-7-113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markstein M., Markstein P., Markstein V., Levine M.S., Markstein P., Markstein V., Levine M.S., Markstein V., Levine M.S., Levine M.S. Genome-wide analysis of clustered Dorsal binding sites identifies putative target genes in the Drosophila embryo. Proc. Natl. Acad. Sci. 2002;99:763–768. doi: 10.1073/pnas.012591199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matys V., Fricke E., Geffers R., Gossling E., Haubrock M., Hehl R., Hornischer K., Karas D., Kel A.E., Kel-Margoulis O.V., Fricke E., Geffers R., Gossling E., Haubrock M., Hehl R., Hornischer K., Karas D., Kel A.E., Kel-Margoulis O.V., Geffers R., Gossling E., Haubrock M., Hehl R., Hornischer K., Karas D., Kel A.E., Kel-Margoulis O.V., Gossling E., Haubrock M., Hehl R., Hornischer K., Karas D., Kel A.E., Kel-Margoulis O.V., Haubrock M., Hehl R., Hornischer K., Karas D., Kel A.E., Kel-Margoulis O.V., Hehl R., Hornischer K., Karas D., Kel A.E., Kel-Margoulis O.V., Hornischer K., Karas D., Kel A.E., Kel-Margoulis O.V., Karas D., Kel A.E., Kel-Margoulis O.V., Kel A.E., Kel-Margoulis O.V., Kel-Margoulis O.V., et al. TRANSFAC: Transcriptional regulation, from patterns to profiles. Nucleic Acids Res. 2003;31:374–378. doi: 10.1093/nar/gkg108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mello C.C., Kramer J.M., Stinchcomb D., Ambros V., Kramer J.M., Stinchcomb D., Ambros V., Stinchcomb D., Ambros V., Ambros V. Efficient gene transfer in C.elegans: Extrachromosomal maintenance and integration of transforming sequences. EMBO J. 1991;10:3959–3970. doi: 10.1002/j.1460-2075.1991.tb04966.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moerman D.G., Fire A., Fire A. Muscle: Structure, function, and development. In: Riddle D.L., et al., editors. C. elegans II. Cold Spring Harbor Laboratory Press; Cold Spring Harbor, NY: 1997. pp. 147–184. [PubMed] [Google Scholar]
- Moerman D.G., Williams B.D., Williams B.D. Sarcomere assembly in C. elegans muscle. In: T.C.e.R. Community,, editor. WormBook. WormBook; 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okkema P.G., Fire A., Fire A. The Caenorhabditis elegans NK-2 class homeoprotein CEH-22 is involved in combinatorial activation of gene expression in pharyngeal muscle. Development. 1994;120:2175–2186. doi: 10.1242/dev.120.8.2175. [DOI] [PubMed] [Google Scholar]
- Okkema P.G., Harrison S.W., Plunger V., Aryana A., Fire A., Harrison S.W., Plunger V., Aryana A., Fire A., Plunger V., Aryana A., Fire A., Aryana A., Fire A., Fire A. Sequence requirements for myosin gene expression and regulation in Caenorhabditis elegans. Genetics. 1993;135:385–404. doi: 10.1093/genetics/135.2.385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polly P., Haddadi L.M., Issa L.L., Subramaniam N., Palmer S.J., Tay E.S., Hardeman E.C., Haddadi L.M., Issa L.L., Subramaniam N., Palmer S.J., Tay E.S., Hardeman E.C., Issa L.L., Subramaniam N., Palmer S.J., Tay E.S., Hardeman E.C., Subramaniam N., Palmer S.J., Tay E.S., Hardeman E.C., Palmer S.J., Tay E.S., Hardeman E.C., Tay E.S., Hardeman E.C., Hardeman E.C. hMusTRD1alpha1 represses MEF2 activation of the troponin I slow enhancer. J. Biol. Chem. 2003;278:36603–36610. doi: 10.1074/jbc.M212814200. [DOI] [PubMed] [Google Scholar]
- Reece-Hoyes J.S., Deplancke B., Shingles J., Grove C.A., Hope I.A., Walhout A.J., Deplancke B., Shingles J., Grove C.A., Hope I.A., Walhout A.J., Shingles J., Grove C.A., Hope I.A., Walhout A.J., Grove C.A., Hope I.A., Walhout A.J., Hope I.A., Walhout A.J., Walhout A.J. A compendium of Caenorhabditis elegans regulatory transcription factors: A resource for mapping transcription regulatory networks. Genome Biol. 2005;6:R110. doi: 10.1186/gb-2005-6-13-r110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Remenyi A., Scholer H.R., Wilmanns M., Scholer H.R., Wilmanns M., Wilmanns M. Combinatorial control of gene expression. Nat. Struct. Mol. Biol. 2004;11:812–815. doi: 10.1038/nsmb820. [DOI] [PubMed] [Google Scholar]
- Roy P.J., Stuart J.M., Lund J., Kim S.K., Stuart J.M., Lund J., Kim S.K., Lund J., Kim S.K., Kim S.K. Chromosomal clustering of muscle-expressed genes in Caenorhabditis elegans. Nature. 2002;418:975–979. doi: 10.1038/nature01012. [DOI] [PubMed] [Google Scholar]
- Smith P.A., Mango S.E., Mango S.E. Role of T-box gene tbx-2 for anterior foregut muscle development in C. elegans. Dev. Biol. 2007;302:25–39. doi: 10.1016/j.ydbio.2006.08.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spieth J., MacMorris M., Broverman S., Greenspoon S., Blumenthal T., MacMorris M., Broverman S., Greenspoon S., Blumenthal T., Broverman S., Greenspoon S., Blumenthal T., Greenspoon S., Blumenthal T., Blumenthal T. Regulated expression of a vitellogenin fusion gene in transgenic nematodes. Dev. Biol. 1988;130:285–293. doi: 10.1016/0012-1606(88)90434-4. [DOI] [PubMed] [Google Scholar]
- Staden R. Methods for calculating the probabilities of finding patterns in sequences. Comput. Appl. Biosci. 1989;5:89–96. doi: 10.1093/bioinformatics/5.2.89. [DOI] [PubMed] [Google Scholar]
- Stormo G.D. DNA binding sites: Representation and discovery. Bioinformatics. 2000;16:16–23. doi: 10.1093/bioinformatics/16.1.16. [DOI] [PubMed] [Google Scholar]
- Teng Y., Girard L., Ferreira H.B., Sternberg P.W., Emmons S.W., Girard L., Ferreira H.B., Sternberg P.W., Emmons S.W., Ferreira H.B., Sternberg P.W., Emmons S.W., Sternberg P.W., Emmons S.W., Emmons S.W. Dissection of cis-regulatory elements in the C. elegans Hox gene egl-5 promoter. Dev. Biol. 2004;276:476–492. doi: 10.1016/j.ydbio.2004.09.012. [DOI] [PubMed] [Google Scholar]
- Tsalik E.L., Niacaris T., Wenick A.S., Pau K., Avery L., Hobert O., Niacaris T., Wenick A.S., Pau K., Avery L., Hobert O., Wenick A.S., Pau K., Avery L., Hobert O., Pau K., Avery L., Hobert O., Avery L., Hobert O., Hobert O. LIM homeobox gene-dependent expression of biogenic amine receptors in restricted regions of the C. elegans nervous system. Dev. Biol. 2003;263:81–102. doi: 10.1016/s0012-1606(03)00447-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vilimas T., Abraham A., Okkema P.G., Abraham A., Okkema P.G., Okkema P.G. An early pharyngeal muscle enhancer from the Caenorhabditis elegans ceh-22 gene is targeted by the Forkhead factor PHA-4. Dev. Biol. 2004;266:388–398. doi: 10.1016/j.ydbio.2003.10.015. [DOI] [PubMed] [Google Scholar]
- Wagner A. Genes regulated cooperatively by one or more transcription factors and their identification in whole eukaryotic genomes. Bioinformatics. 1999;15:776–784. doi: 10.1093/bioinformatics/15.10.776. [DOI] [PubMed] [Google Scholar]
- Wang X., Chamberlin H.M., Chamberlin H.M. Evolutionary innovation of the excretory system in Caenorhabditis elegans. Nat. Genet. 2004;36:231–232. doi: 10.1038/ng1301. [DOI] [PubMed] [Google Scholar]
- Wang T., Stormo G.D., Stormo G.D. Combining phylogenetic data with co-regulated genes to identify regulatory motifs. Bioinformatics. 2003;19:2369–2380. doi: 10.1093/bioinformatics/btg329. [DOI] [PubMed] [Google Scholar]
- Wang T., Stormo G.D., Stormo G.D. Identifying the conserved network of cis-regulatory sites of a eukaryotic genome. Proc. Natl. Acad. Sci. 2005;102:17400–17405. doi: 10.1073/pnas.0505147102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wasserman W.W., Fickett J.W., Fickett J.W. Identification of regulatory regions which confer muscle-specific gene expression. J. Mol. Biol. 1998;278:167–181. doi: 10.1006/jmbi.1998.1700. [DOI] [PubMed] [Google Scholar]
- Zhao Z., Fang L., Chen N., Johnsen R.C., Stein L., Baillie D.L., Fang L., Chen N., Johnsen R.C., Stein L., Baillie D.L., Chen N., Johnsen R.C., Stein L., Baillie D.L., Johnsen R.C., Stein L., Baillie D.L., Stein L., Baillie D.L., Baillie D.L. Distinct regulatory elements mediate similar expression patterns in the excretory cell of Caenorhabditis elegans. J. Biol. Chem. 2005;280:38787–38794. doi: 10.1074/jbc.M505701200. [DOI] [PubMed] [Google Scholar]