

Abstract
Expression of the Salmonella enterica serovar Typhimurium pathogenicity island 2 (SPI-2) type III secretion system is controlled by the two-component regulatory system SsrA-SsrB. We used a transcriptomic approach to help define the SsrA-SsrB regulon. We identified a gene encoding an uncharacterized effector (SseL) whose translocation into host cells depends on the SPI-2 secretion system. SseL has similarities to cysteine proteases with deubiquitinating activity. A GST-SseL fusion protein specifically hydrolyzed mono- and polyubiquitin substrates in vitro with a preference for K63-linked ubiquitin chains. Ubiquitin-modified proteins accumulated in macrophages infected with Salmonella sseL mutant strains but to a lesser extent when infected with bacteria expressing active protein, demonstrating that SseL functions as a deubiquitinase in vivo. Salmonella sseL mutant strains did not show a replication defect or induce altered levels of cytokine production upon infection of macrophages but were defective for a delayed cytotoxic effect and were attenuated for virulence in mice.
Keywords: cytotoxicity, SPI-2, deubiquitinating enzyme
Salmonella enterica serovar Typhimurium (S. Typhimurium) causes gastroenteritis in humans and a systemic disease in certain mouse strains, which is used as an experimental model of typhoid fever. S. Typhimurium invades and replicates within a variety of host cell types (1, 2). After uptake, bacteria reside within a membrane-bound compartment, the Salmonella-containing vacuole (SCV). Intracellular replication and growth in mice requires many virulence proteins, including several encoded by the Salmonella pathogenicity island (SPI) -2 type III secretion system (T3SS) (3). This T3SS is induced intracellularly (4) and translocates several effectors into the vacuolar membrane and host cell cytosol (3). Many effectors are encoded by genes located at different sites in the bacterial chromosome (3, 5, 6).
The full repertoire of effector proteins of the SPI-2 T3SS is unknown. The expression of SPI-2 genes and effector genes located outside the pathogenicity island requires the SPI-2-encoded two-component regulatory system SsrA-SsrB (4). In this work, we used a DNA microarray of S. Typhimurium to compare the levels of mRNAs in wild-type and ssrA mutant bacteria grown in conditions that result in strong expression of the SsrA-SsrB regulon. This led to the identification of a new gene (STM2287) whose expression depends on SsrA. We characterized the product of STM2287 (SseL) and found that it is a deubiquitinase.
Results
Identification of STM2287.
We used an S. Typhimurium LT2 DNA microarray (7) to identify bacterial genes regulated by SsrA-B. RNA was extracted from wild-type or ssrA mutant strains grown in conditions that induce the expression of SPI-2 genes (4), as well as effectors encoded within and outside SPI-2 (8, 9). RNAs were fluorescently labeled and separately hybridized to LT2 DNA microarrays. The 50 genes showing the highest fold change in expression between the two strains are shown in supporting information (SI) Table 1. Many are known or are predicted to encode components of the secretion apparatus, translocon components, chaperones, and effectors.
One of the hypothetical genes, STM2287, was chosen for further analysis, and as a result of experiments described below, STM2287 was designated sseL (Salmonella secreted effector L), in accordance with accepted nomenclature (10). Its chromosomal organization (see SI Fig. 5A) and work described below indicate that it is monocistronic. The G+C content of sseL is 39%, significantly lower than the genome average of 52.2%. Genes encoding nearly identical proteins are present in the genomes of Salmonella enterica serovars Typhi, Paratyphi A, and Choleraesuis, but sseL is absent from the genome of Salmonella bongori, which lacks the SPI-2 locus. The sseL promoter was activated in an ssrA-dependent manner ≈6 h after uptake by J774 macrophages (SI Fig. 5B) and HeLa cells (data not shown), confirming that sseL is part of the SsrA-B regulon.
Translocation and Localization of SseL.
To determine whether SseL is translocated by intracellular bacteria, an epitope-tagged version (SseL-2HA) was expressed from the chromosome in the wild-type strain and a strain carrying a mutation in ssaV [encoding an essential component of the SPI-2 T3SS (11)]. Macrophages were infected for 6–8 h with these strains carrying a plasmid constitutively expressing GFP. Immunofluorescence microscopy showed that SseL is translocated across the vacuolar membrane in a SPI-2-dependent manner (SI Fig. 6 A and B). To localize translocated SseL, infected macrophages and HeLa cells were labeled for HA, Salmonella, and LAMP-1, a lysosomal membrane glycoprotein enriched on the SCV membrane and tubules (Sifs) that emanate from SCVs in epithelial cells (12). There was extensive colocalization between SCV-associated LAMP-1 and SseL-2HA in macrophages (SI Fig. 6C Upper, arrows) and SCV- and Sif-associated LAMP-1 and SseL-2HA in HeLa cells (SI Fig. 6C Lower, arrow). Membrane localization of SseL-2HA was confirmed by fractionation of infected macrophages, followed by SDS/PAGE and immunoblotting (SI Fig. 6D).
SseL Has Deubiquitinating Activity in Vitro.
SseL is predicted to encode a protein of 317 aa, originally annotated as a sulfatase/phosphatase (13). InterProScan searches showed that a region of the primary amino acid sequence (residues 195–267) is similar to cysteine proteases. This region was then compared with a variety of cysteine proteases by using the ClustalW program (14) and MEROPS database. These comparisons indicate that SseL is related to cysteine proteases of clan CE based on the order of the residues important for active site maintenance and catalysis (His, Glu/Asp/Asn, Gln, Cys) (Fig. 1). This clan contains a yeast protease, Ulp1, with desumoylating activity (15). It also contains the adenovirus L3 23K proteinase (Avp), two Chlamydia trachomatis enzymes, and the Yersinia T3SS effector YopJ/P, all of which have been shown to have deubiquitinating activity in vitro (16–18).
Fig. 1.

Amino acid alignment of SseL with a selection of cysteine proteases from clan CE. The conserved catalytic residues (H, D/E/N, Q, and C) are highlighted in gray. ASFV indicates African swine fever virus.
To test whether SseL can cleave ubiquitinated or sumoylated substrates in vitro, SseL and SseLC262A (in which the putative catalytic residue Cys-262 was replaced by Ala) were purified as GST fusion proteins (SI Fig. 7A), and incubated with ubiquitin (Ub)- or SUMO-1-amido methyl coumarin (AMC). Isopeptidase T, a Ub-specific protease, was used as a positive control for Ub cleavage. GST-SseL hydrolyzed Ub-AMC (Fig. 2A) but not SUMO-1-AMC (SI Fig. 7B). The exchange of Cys-262 to Ala abolished the activity of SseL on Ub-AMC (Fig. 2A). The Km of GST-SseL on Ub-AMC was 1.5 μM (Fig. 2B), similar to that of other deubiquitinating enzymes (DUBs) (18, 19).
Fig. 2.

Enzymatic activity of GST-fusion proteins. (A) Purified proteins or isopeptidase T (a Ub-specific protease) were incubated with Ub-AMC, and their activity was determined by release of fluorescent AMC. (B) Michaelis–Menten plot to determine kinetic constants for GST-SseL (Km = 1.5 μM; Vmax = 2354 pM s−1). Cleavage of branched K48- (C) and K63- (D) linked multiUb chains by GST-SseL. Purified proteins (5 μM) were incubated with substrate with (+) or without (−) inhibitor (Ub aldehyde) for 1 h at 37°C and fractioned by SDS/PAGE. Proteins were transferred to PVDF membrane and labeled with an anti-Ub antibody (Zymed). Purified proteins of increasing concentration (0–5.0 μM) were incubated for 30 min at 37°C with equimolar amounts of either K48- (E) or K63- (F) linked multiUb chains and subjected to SDS/PAGE and immunoblotted with an anti-Ub antibody (Zymed). Bands correspond to different Ub oligomers (indicated on the left). GST-SseL in each sample was visualized by Coomassie blue staining.
To examine the ability of SseL to cleave branched polyUb chains, GST-SseL, GST-SseLC262A, or GST were mixed with equimolar amounts of K48-linked or K63-linked multiUb chains. Samples were then subjected to PAGE and immunoblotted by using an anti-Ub antibody. GST-SseL, but not GST-SseLC262A, cleaved polyUb into monomers (Fig. 2 C and D). Ub aldehyde, a specific inhibitor of Ub C-terminal hydrolysis, significantly reduced cleavage of K48-linked Ub chains but inhibited less efficiently the activity of SseL on K63-linked chains (Fig. 2D). When increasing amounts (0–5 μM) of GST-SseL was mixed with either K48-linked Ub chains (Fig. 2E) or K63-linked Ub chains (Fig. 2F), a preference of GST-SseL for K63-linked Ub-chains was evident.
Lack of SseL Leads to an Accumulation of Ubiquitinated Proteins in Infected Cells, and SseL Binds Ub Directly.
To investigate the effects of SseL on ubiquitinated proteins after infection of macrophages, J774 cells were infected for various periods of time with a ΔsseL strain of S. Typhimurium expressing one of two epitope-tagged versions of SseL (SseL-2HA or SseLC262A-2HA) from a plasmid. Infected macrophages were lysed and proteins subjected to PAGE and immunoblotting with an anti-Ub antibody. In uninfected and cells infected with either strain, Ub-modified proteins of low relative molecular mass (Mr) were detected (Fig. 3A). In cells infected with the ΔsseL strain expressing either SseL-2HA or SseLC262A-2HA from a plasmid, Ub-modified proteins of Mr >≈50 kDa accumulated gradually between 8 and 16 h after uptake (Fig. 3A Left). The signals from Ub-modified proteins were very weak in lysates from macrophages infected with S. Typhimurium expressing SseL-2HA but were stronger in macrophages exposed to SseLC262A-2HA, particularly at the 16-h time point (Fig. 3A). The same samples were immunoblotted by using an anti-HA antibody. This showed an equivalent, gradual increase in effector protein levels from both strains (Fig. 3A Left). To examine the levels of ubiquitinated proteins in uninfected cells and cells infected with the ΔsseL strain, the experiment was repeated, and macrophages were lysed at the 16-h time point. The amount of ubiquitinated proteins was lower in uninfected than infected cells, and there was a greater accumulation of ubiquitinated proteins in cells infected with the sseL mutant strain than in cells infected with the wild-type strain. (Fig. 3A Right). The accumulation of ubiquitinated proteins caused by infection with the null mutant was similar to that caused by the strain expressing the cysteine point mutant.
Fig. 3.

Ubiquitination of proteins in infected macrophages. (A Left) J774 cells were infected with the ΔsseL mutant strain expressing either SseL-2HA or SseLC262A-2HA. At different time points, cells were lysed and proteins subjected to SDS/PAGE, followed by transfer to PVDF membranes and labeling with an anti-Ub antibody (Zymed). The blot was then stripped and reprobed with anti-tubulin and anti-HA antibodies to provide a loading control for host proteins and to demonstrate equivalent expression of epitope-tagged SseL in cells infected with the two strains. (A Right) J774 cells were uninfected or infected for 16 h with wild-type, ΔsseL or ΔsseL mutant strain expressing either SseL-2HA or SseLC262A-2HA. Cell lysates were analyzed as described above, without anti-HA immunoblot. (B) Coimmune precipitation of HA-tagged proteins from macrophages infected for 16 h with the same strains as in A Left). Macrophage lysates were incubated with anti-HA antibody. Antibody-bound proteins were recovered by using protein G-coated beads and immunoblotted (Left) with an anti-HA antibody. HC and LC indicate heavy and light antibody chains. HA labeling indicates equivalent recovery from the two samples. Recovered fractions were probed with antibodies recognizing polyUb (FK1) or both monoUb and polyUb (FK2) (Right). Positions of size markers are indicated on the left of blots.
To determine whether SseLC262A-2HA interacts with ubiquitinated proteins in infected cells, an anti-HA antibody was used to immunoprecipitate SseL-2HA or SseLC262A-2HA from lysates of infected macrophages (Fig. 3B Left) or HeLa cells (data not shown). Immunoblot analysis revealed that SseLC262A-2HA and, to a much lesser extent, SseL-2HA coimmunoprecipitated both mono- and polyubiquitinated proteins from both cell types: the antibody FK1 recognizes polyubiquitinated proteins, whereas FK2 recognizes both forms; the majority of Ub-modified proteins had a relative molecular mass >≈50 kDa (Fig. 3B Right). A prominent band of ≈70 kDa was detected in immunoprecipitates from cells infected with bacteria expressing SseLC262A-2HA (Fig. 3B Right). However, this was not consistently observed in replicate experiments. Together, these results indicate that infection of macrophages with S. Typhimurium results in the accumulation of Ub-modified proteins that are subsequently deubiquitinated by SseL, after translocation of this effector from bacteria across the vacuolar membrane.
A direct interaction between SseL and Ub was confirmed by a yeast two-hybrid screen with a HeLa cell cDNA library (see SI Methods for further details.
SseL Is Required for Salmonella-Induced Delayed Cytotoxicity of Macrophages.
We next compared the intracellular growth of the sseL mutant with that of to the wild-type strain and an ssaV mutant. Replication assays were performed in HeLa cells, J774 macrophages, and in elicited murine peritoneal macrophages. At 10 and 20 h after uptake in each cell-type, the growth of the sseL mutant was indistinguishable from that of the wild-type strain, whereas the ssaV mutant displayed a strong replication defect (data not shown).
The Yersinia DUB YopJ/P inhibits the inflammatory cytokine NF-κB pathway (20–23). We therefore investigated whether SseL suppresses the NF-κB pathway in three independent assays involving activation and degradation of IκBα and release of TNF from macrophages infected with either the wild-type or the ΔsseL strain up to 22 h after uptake. Activation of IκBα was investigated by using a phospho-specific IκBα antibody, and degradation of IκBα was analyzed in samples by using an anti-IκBα antibody. No difference in activation or degradation of IκBα was observed between macrophages infected with the two strains at any time point (data not shown). Release of TNF was consistent with these results, with no significant difference between the two strains at any time point (Fig. 4A).
Fig. 4.

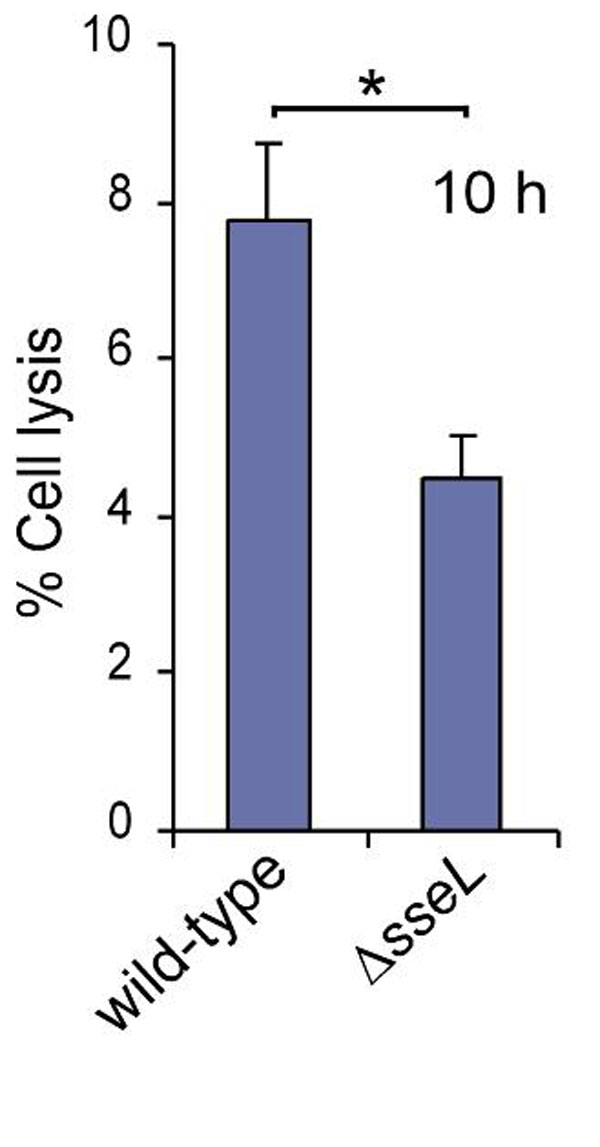
SseL is required for a delayed cytotoxic effect in infected macrophages. (A) TNF release from infected macrophages. J774 cells were infected for different periods of time with the wild-type or ΔsseL strain, and supernatants were recovered and analyzed for TNF by sandwich ELISA. TNF release from macrophages exposed to LPS (1 μg/ml) for 16 h was used as a positive control. (B) Cytotoxicity assay of macrophages after infection with various bacterial strains. Cells were infected for 10 or 20 h and then incubated with PI for 20 min and analyzed by flow cytometry. The levels of PI labeling of uninfected cells were subtracted from those of infected cells at the two time points. In each case, the percentages of PI-positive cells are from a total of 20,000 cells. Samples were analyzed in triplicate, and the standard errors of the mean are shown. The results shown are representative of four independent experiments. (C) Macrophages were infected as in B with the wild-type or ΔsseL strain carrying a GFP-expressing plasmid. At 20 h after uptake, 20,000 GFP-positive macrophages were analyzed, and the number of PI-positive cells was counted. Asterisks indicate P < 0.05.
S. Typhimurium has been shown to have a delayed cytotoxic activity against macrophages in vitro. This begins ≈12 h after infection and depends on the SPI-2 T3SS (24), although the effector(s) responsible has not been identified. We therefore investigated the plasma membrane integrity of J774 macrophages at 10 or 20 h after infection with the wild-type strain or various mutant strains by measuring the uptake of propidium iodide (PI), a membrane-impermeable dye. In a first set of experiments, 20,000 macrophages were quantified for each strain at both time points. The wild-type strain consistently induced significantly greater cytotoxicity than the ssaV mutant strain, the ΔsseL strain, and the ΔsseL strain expressing SseLC262A-2HA (Fig. 4B). In a further experiment involving only the GFP-expressing wild-type and ΔsseL strains, 20,000 GFP-positive macrophages were analyzed for PI uptake. The number of PI-positive macrophages infected with wild-type bacteria was approximately double that of macrophages infected with the ΔsseL strain (Fig. 4C). The reduced cytotoxicity of the ΔsseL strain can be attributed solely to a loss of SseL, because a plasmid expressing SseL-2HA restored cytotoxicity to a level similar to that caused by the wild-type strain (Fig. 4B). Release of cytosolic lactate dehydrogenase (LDH) was additionally used to monitor death of macrophages infected with the five strains used in Fig. 4B. Significantly greater LDH was released from macrophages infected with the wild-type and psseL-2HA-complemented mutant strain than from cells infected with the mutant strains (data not shown). LDH release was also measured from elicited macrophages infected with wild-type or ΔsseL strain. A significant difference in cytotoxicity was observed at 10 h after uptake (SI Fig. 8).
SseL Is a Salmonella Virulence Factor.
Competitive index (CI) (25) tests were used to assess the importance of SseL in virulence. The CIs for the wild-type strain versus the ΔsseL mutant strain were 0.66 ± 0.075 after i.p. inoculation and 0.37 ± 0.057 after oral inoculation of mice. The CI of the ΔsseL strain expressing SseL-2HA from a plasmid versus the ΔsseL strain was 0.68 ± 0.05 after i.p. inoculation. The CI of the wild-type strain versus the ΔsseL strain expressing SseLC262A-2HA from a plasmid was 0.50 ± 0.029 after i.p. inoculation. Therefore, the null and point-mutant strains display virulence attenuation during the systemic phase of infection, and this can be attributed directly to the mutation in sseL rather than a polar effect or secondary mutation in this strain. Taken together, these data show that the deubiquitinating and cytotoxic activities of SseL are necessary for Salmonella virulence.
Discussion
In this work, we describe the characterization of sseL, a previously uncharacterized member of the SsrA-SsrB regulon encoding an effector of the SPI-2 T3SS. SseL is translocated onto the vacuolar surface in infected host cells, where it targets the host cell Ub pathway by acting as a DUB. It is not required for intracellular bacterial replication but is required for SPI-2-dependent cytotoxicity of macrophages and Salmonella virulence in the mouse model of typhoid fever.
In mammalian cells, ubiquitination proceeds via three enzymatic steps comprising a Ub-activating enzyme (E1), transfer of Ub to one of several E2-conjugating enzymes, and then transfer of the activated Ub to a lysine residue in the target protein by one of a large family of E3 ligases. Ub can itself be further ubiquitinated, generating one or more branched chains conjugated through Lys 48; these modifications target the protein to the proteasome for degradation. Alternatively monoubiquitinated proteins, or proteins modified by Ub chains linked through Lys 63, influence processes such as membrane trafficking, transcription, and other signaling pathways (26).
Ub processing is also controlled by a large family of DUBs, which are mainly cysteine proteases. These can rescue proteins from proteasome degradation, or down-regulate signaling or trafficking events (27). A small number of viral and bacterial DUBs have been discovered (16, 17, 28); these include two recently identified Chlamydia trachomatis proteins, whose physiological functions and requirement for virulence are unknown (18), and YopJ/P, a Yersinia T3SS effector that suppresses the NF-κB pathway involved in inflammatory responses (29). Normally, activation of the pathway leads to proteasomal degradation of ubiquitinated IκBα, enabling the nuclear translocation of NF-κB and the transcription of cytokine genes. However, in macrophages infected with Yersinia pseudotuberculosis, the deubiquitinating (17) and/or acetylating (23) activities of YopJ/P prevent the degradation of IκBα. YopJ/P is also required for rapid apoptosis of macrophages (30, 31). However, the mechanism by which its suppressive effect on the NF-κB pathway is linked to its proapoptotic activity, and the relative importance of these two activities in virulence, are not well understood (32).
S. Typhimurium can cause macrophage death in several different ways. Two types of cell death that occur in the first few hours after infection of cultured macrophages require the SPI-1 T3SS and its associated translocase/effector protein, SipB. One of these depends on caspase-1 (33), whereas the other is caspase-1-independent and involves mitochondrial disruption and autophagy (34). However, SipB is essential for the translocation of all SPI-1 effectors across the host-cell plasma membrane (35), making it very difficult to assess the biological significance of the different forms of cell death caused by SipB. The SPI-1 T3SS functions during invasion of the gut epithelium but does not contribute significantly to systemic growth of S. Typhimurium in the mouse (36).
A delayed cytotoxic activity of S. Typhimurium has also been reported. This begins ≈12 h after infection of cultured macrophages and depends on the SPI-2 T3SS (24). It also appears to involve caspase-1 (37) and the protein kinase PKR (38), but again its biological significance has not been established, and the effector(s) responsible had not been identified previously. From the results shown in Fig. 4 B and C and SI Fig. 8, we conclude that SseL is an effector that accounts for a proportion of SPI-2-dependent cytotoxicity in macrophages. The cytotoxic effect of the sseL mutant was not as low as that of the ssaV mutant. This could be due to the replication defect of the ssaV mutant; alternatively, other SPI-2 T3SS effector(s) might be involved in the process.
In contrast to YopJ/P, we obtained no evidence that SseL suppresses the NF-κB pathway, and the mechanism by which the deubiquitinating activity of SseL is connected to its requirement for delayed cytotoxicity in macrophages remains to be investigated. A large number of both mono- and polyubiquitinated proteins were immunoprecipitated by SseLC262A-2HA. These proteins could reflect different degrees of ubiquitination of one or a few proteins or mono- and polyubiquitination of many targets. Our work indicates that ubiquitination and deubiquitination of proteins occurs on the cytoplasmic face of the SCV, something that is not evident by immunofluorescence microscopy by using antibodies against Ub [(39), A.R. and D.W.H., unpublished data). Presumably, deubiquitination of these protein(s) by SseL promotes a macrophage-killing pathway that might also involve caspase-1 and PKR. In vitro SseL displayed a clear preference for K63-linked Ub-chains, suggesting that SseL interferes with a signaling pathway rather than inhibiting proteasomal-dependent degradation of its target(s).
The main conclusion to emerge from this work is that the delayed cytotoxic effect of S. Typhimurium in macrophages (24, 37) is likely to be a biologically relevant phenomenon. Apoptotic nuclei are a characteristic of neutrophils and macrophages in tissues of mice infected with S. Typhimurium (40), but it has not been clear whether these dying cells represent a host resistance response to infection or a means by which S. Typhimurium evades host defenses. By showing that SseL contributes to SPI-2-dependent macrophage death and that sseL mutant strains are attenuated for virulence in the systemic phase of infection, we provide evidence that the cytotoxic activity depends on SseL and plays an important role in Salmonella virulence. This might enable the spread of bacteria throughout infected organs by incoming uninfected macrophages engulfing either infected dying cells or bacteria released by them into the extracellular space.
Materials and Methods
Bacterial Strains and Growth Conditions.
All strains and growth conditions are published in SI Methods and SI Table 2.
Microarray Procedures and Analysis.
A detailed description of the microarray screening procedure is published in SI Methods.
Plasmids and Construction of Mutant Strain.
Plasmids used in this study are listed in SI Table 3. Plasmid pFPV25.1, carrying gfpmut3A under the control of a constitutive promoter, was introduced into bacterial strains for fluorescence visualization where indicated (41). The plasmid psseL::gfp contains a 300-bp DNA fragment immediately upstream of the start codon of sseL (amplified by primers STM2287F/R, in SI Table 4) and was inserted into the promoter trap vector pFPV25. The chromosomal deletion of sseL in S. Typhimurium was performed by using the one-step gene-disruption technique (42).
Epitope Tagging, Site-Directed Mutagenesis, and Construction of Expression Vectors.
The influenza virus HA epitope DNA sequence was fused to the chromosomal copy of sseL according to Uzzau et al. (43), by using plasmid pSU315. sseL-2HA was transduced by bacteriophage P22 into the ssaV mutant strain following the method of Davis et al. (44). Site-directed mutagenesis was performed according to manufacturer's recommendations (Quickchange, Stratagene). Cys at residue 262 of SseL was changed to Ala by using primers C262A-F and C262A-R. Primers used for construction of all constructs are listed in SI Table 4.
Antibodies.
All antibodies used are published in SI Methods.
Cell Culture.
HeLa (93021013) and J774 (91051511) cells were obtained from the European Collection of Cell Cultures, Salisbury, U.K. Cells were grown in DMEM (GIBCO, Carlsbad, CA) supplemented with 10% FCS. Cells were grown at 37°C in 5% CO2. Elicited i.p. macrophages were obtained from BALB/c mice challenged with 5 mM sodium periodate 4 days before harvesting. Cells were seeded at a density of 4 × 105 per ml and incubated for 24 h before bacterial challenge.
Bacterial Infection of Epithelial Cells and Macrophages and Immunofluorescence Microscopy.
Infection of HeLa cells and macrophages was performed as described (25, 45). Cells were fixed, permeabilized, and incubated with antibodies as described (25). Labeled cells were analyzed by using a fluorescence microscope (BX50; Olympus, Melville, NY) or a confocal laser scanning microscope (LSM510; Zeiss, Thornwood, NY).
CI Assay.
Female BALB/c mice (B and K Universal, Hull, U.K.) of 18–22 g were used for all infection studies and were challenged either i.p. or by oral gavage (p.o.) with 0.2 ml of bacteria suspended in physiological saline solution. The bacterial inocula used in the experiments were 5 × 104 (i.p.) or 5 × 108 (p.o.) cfus of each strain. At least five mice were inoculated per strain mixture for each experiment. Mice were killed after 48 h (i.p.) or 5 days (p.o.) after inoculation. Each CI value is the mean of three independent experiments.
Yeast Two-Hybrid Analysis.
The Matchmaker Gal4 Two-Hybrid System 3 kit (Clontech) was used for the yeast two-hybrid screen of a pretransformed HeLa cDNA library (Clontech, Mountain View, CA) by following protocols of the manufacturer. (Further details are published in SI Methods).
Protein Purification.
Plasmids for expression of GST-fusion proteins (GST-SseL, GST-SseLC262A) were constructed in the pGEX-4T-2 vector by using the primers listed in SI Table 4. A detailed description of the protein purification is published in SI Methods.
Protease Assays.
GST constructs were used at a concentration of 1 μM. isopeptidase T or SENP1 (Boston Biochem, Cambridge, MA) were used at a concentration of 100 nM. Samples were incubated with 1–7 μM ubiquitin-AMC or SUMO-1-AMC (Boston Biochem) in reaction buffer (50 mM Hepes, pH 8.0, and 1 mM DTT) at a total volume of 40 μl. Liberation of AMC at room temperature was monitored in a fluorimetric microplate reader (Cary Eclipse; Varian, Palo Alto, CA) with excitation at 380 nm and emission at 460 nm. For analysis of enzyme kinetics relative fluorescence, units were converted to pM AMC by using a standard curve of relative fluorescence units versus AMC concentration. Kinetic constants were calculated from Michaelis–Menten plots with nonlinear regression analysis using Graph-Pad software, with the assumption that 100% of the purified protein was active. For the polyubiquitinase assay, Lys-48- or Lys-63-linked polyubiquitin chains (25 ng/μl, Biomol, Plymouth Meeting, PA) were mixed with 5 μM GST-SseL or increasing amounts of GST-SseL (0–5 μM) in Tris-Cl, pH 7.4, and left at 37°C for 30 min to 1 h. Some samples were pretreated with the Ub aldehyde (3 μM; Biomol) for 30 min before being mixed with the polyUb-linked chains. Samples were subsequently fractionated on a 12% acrylamide gel and transferred to Immobilon-P (PVDF) membrane (Millipore, Bedford, MA). The membrane was probed with anti-Ub and anti-mouse HRP antibodies and visualized by using the ECL Western Blotting Analysis system (GE Healthcare, Buckinghamshire, U.K.).
Preparation of Cell Lysates, Coimmunoprecipitation, and Fractionation.
HeLa cells or J774 macrophages (107 cells) were infected for 16 h with an sseL mutant carrying pWSKsseL-2HA or pWSKsseLC262A-2HA, and coimmunoprecipitation was performed by using an HA-antibody (a complete description is published in SI Methods together with a description of cell lysate preparation after infection of macrophages). Fractionation of macrophages after overnight infection was performed as described (46).
TNF Immunoassay and Cytotoxicity Assays.
Supernatants were recovered at different time points from J774 macrophages after challenge with various Salmonella strains or purified LPS (Sigma, St. Louis, MO) at 1 μg/ml. Samples were centrifuged at 14,000 × g and transferred into a clean tube. TNF levels were measured by using sandwich ELISA according to manufacturer's recommendations (Diaclone, Stamford, CT). For flow cytometry, macrophages were seeded at a density of 3 × 105 cells per well in six-well tissue culture plates 24 h before use. Infections were performed as described above. At 10 and 20 h after uptake, infected macrophages were resuspended in 0.5 ml of cold PBS containing 2 μg/ml PI. In each sample, 20,000 cells or 20,000 GFP-positive cells were analyzed on a FACScalibur cytometer (Becton Dickinson, Franklin Lakes, NJ), and data were analyzed by using CellQuest software. LDH assays were performed by using In Situ Death Detection Kit (Roche, Indianapolis, IN).
DNA Sequence Analysis.
Web sites used for sequence analysis are published in SI Methods.
Supplementary Material
Acknowledgments
We acknowledge the technical support of Erika Rosivatz (fluorimetric readings), Matt Rolfe (S. Typhimurium microarrays), Ciaran Mckeown (protein purification), and Paul Evans for advice on DUBs, and we thank Christoph Tang and members of the Holden laboratory for critical review of the manuscript. This work was supported by grants from the European Molecular Biology Oganization (to A.R. and J.G.), the Swedish Research council (to A.R.), the Medical Research Council and Wellcome Trust (U.K.) (to D.W.H.), and Biotechnology and Biological Sciences Research Council Core Strategic Grant (to J.C.D.H.).
Abbreviations
- AMC
amido methyl coumarin
- CI
competitive index
- DUB
deubiquitinating enzyme
- LDH
lactate dehydrogenase
- PI
propidium iodide
- SCV
Salmonella-containing vacuole
- SPI
Salmonella pathogenicity island
- T3ss
type III secretion system
- Ub
ubiquitin.
Note Added in Proof.
While this study was under revision, Coombes et al. (47) reported on the identification of sseL as a gene dependent on SsrB for expression. In agreement with our study, they showed that sseL is a SPI-2 T3SS effector required for virulence in mice.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS direct submission.
This article contains supporting information online at www.pnas.org/cgi/content/full/0610095104/DC1.
References
- 1.Rathman M, Barker LP, Falkow S. Infect Immun. 1997;65:1475–1485. doi: 10.1128/iai.65.4.1475-1485.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Buchmeier NA, Heffron F. Infect Immun. 1991;59:2232–2238. doi: 10.1128/iai.59.7.2232-2238.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Waterman SR, Holden DW. Cell Microbiol. 2003;5:501–511. doi: 10.1046/j.1462-5822.2003.00294.x. [DOI] [PubMed] [Google Scholar]
- 4.Cirillo DM, Valdivia RH, Monack DM, Falkow S. Mol Microbiol. 1998;30:175–188. doi: 10.1046/j.1365-2958.1998.01048.x. [DOI] [PubMed] [Google Scholar]
- 5.Geddes K, Worley M, Niemann G, Heffron F. Infect Immun. 2005;73:6260–6271. doi: 10.1128/IAI.73.10.6260-6271.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kujat Choy SL, Boyle EC, Gal-Mor O, Goode DL, Valdez Y, Vallance BA, Finlay BB. Infect Immun. 2004;72:5115–5125. doi: 10.1128/IAI.72.9.5115-5125.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Eriksson S, Lucchini S, Thompson A, Rhen M, Hinton JC. Mol Microbiol. 2003;47:103–118. doi: 10.1046/j.1365-2958.2003.03313.x. [DOI] [PubMed] [Google Scholar]
- 8.Worley MJ, Ching KH, Heffron F. Mol Microbiol. 2000;36:749–761. doi: 10.1046/j.1365-2958.2000.01902.x. [DOI] [PubMed] [Google Scholar]
- 9.Garmendia J, Beuzon CR, Ruiz-Albert J, Holden DW. Microbiology. 2003;149:2385–2396. doi: 10.1099/mic.0.26397-0. [DOI] [PubMed] [Google Scholar]
- 10.Hensel M, Shea JE, Raupach B, Monack D, Falkow S, Gleeson C, Kubo T, Holden DW. Mol Microbiol. 1997;24:155–167. doi: 10.1046/j.1365-2958.1997.3271699.x. [DOI] [PubMed] [Google Scholar]
- 11.Beuzon CR, Banks G, Deiwick J, Hensel M, Holden DW. Mol Microbiol. 1999;33:806–816. doi: 10.1046/j.1365-2958.1999.01527.x. [DOI] [PubMed] [Google Scholar]
- 12.Garcia-del Portillo F, Zwick MB, Leung KY, Finlay BB. Proc Natl Acad Sci USA. 1993;90:10544–10548. doi: 10.1073/pnas.90.22.10544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.McClelland M, Sanderson KE, Spieth J, Clifton SW, Latreille P, Courtney L, Porwollik S, Ali J, Dante M, Du F, et al. Nature. 2001;413:852–856. doi: 10.1038/35101614. [DOI] [PubMed] [Google Scholar]
- 14.Chenna R, Sugawara H, Koike T, Lopez R, Gibson TJ, Higgins DG, Thompson JD. Nucleic Acids Res. 2003;31:3497–3500. doi: 10.1093/nar/gkg500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Li SJ, Hochstrasser M. Nature. 1999;398:246–251. doi: 10.1038/18457. [DOI] [PubMed] [Google Scholar]
- 16.Balakirev MY, Jaquinod M, Haas AL, Chroboczek J. J Virol. 2002;76:6323–6331. doi: 10.1128/JVI.76.12.6323-6331.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhou H, Monack DM, Kayagaki N, Wertz I, Yin J, Wolf B, Dixit VM. J Exp Med. 2005;202:1327–1332. doi: 10.1084/jem.20051194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Misaghi S, Balsara ZR, Catic A, Spooner E, Ploegh HL, Starnbach MN. Mol Microbiol. 2006;61:142–150. doi: 10.1111/j.1365-2958.2006.05199.x. [DOI] [PubMed] [Google Scholar]
- 19.Nanao MH, Tcherniuk SO, Chroboczek J, Dideberg O, Dessen A, Balakirev MY. EMBO Rep. 2004;5:783–788. doi: 10.1038/sj.embor.7400201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Collier-Hyams LS, Zeng H, Sun J, Tomlinson AD, Bao ZQ, Chen H, Madara JL, Orth K, Neish AS. J Immunol. 2002;169:2846–2850. doi: 10.4049/jimmunol.169.6.2846. [DOI] [PubMed] [Google Scholar]
- 21.Boland A, Cornelis GR. Infect Immun. 1998;66:1878–1884. doi: 10.1128/iai.66.5.1878-1884.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ruckdeschel K, Harb S, Roggenkamp A, Hornef M, Zumbihl R, Kohler S, Heesemann J, Rouot B. J Exp Med. 1998;187:1069–1079. doi: 10.1084/jem.187.7.1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mukherjee S, Keitany G, Li Y, Wang Y, Ball HL, Goldsmith EJ, Orth K. Science. 2006;312:1211–1214. doi: 10.1126/science.1126867. [DOI] [PubMed] [Google Scholar]
- 24.van der Velden AW, Lindgren SW, Worley MJ, Heffron F. Infect Immun. 2000;68:5702–5709. doi: 10.1128/iai.68.10.5702-5709.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Beuzon CR, Meresse S, Unsworth KE, Ruiz-Albert J, Garvis S, Waterman SR, Ryder TA, Boucrot E, Holden DW. EMBO J. 2000;19:3235–3249. doi: 10.1093/emboj/19.13.3235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Haglund K, Dikic I. EMBO J. 2005;24:3353–3359. doi: 10.1038/sj.emboj.7600808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Amerik AY, Hochstrasser M. Biochim Biophys Acta. 2004;1695:189–207. doi: 10.1016/j.bbamcr.2004.10.003. [DOI] [PubMed] [Google Scholar]
- 28.Kattenhorn LM, Korbel GA, Kessler BM, Spooner E, Ploegh HL. Mol Cell. 2005;19:547–557. doi: 10.1016/j.molcel.2005.07.003. [DOI] [PubMed] [Google Scholar]
- 29.Orth K, Xu Z, Mudgett MB, Bao ZQ, Palmer LE, Bliska JB, Mangel WF, Staskawicz B, Dixon JE. Science. 2000;290:1594–1597. doi: 10.1126/science.290.5496.1594. [DOI] [PubMed] [Google Scholar]
- 30.Monack DM, Mecsas J, Ghori N, Falkow S. Proc Natl Acad Sci USA. 1997;94:10385–10390. doi: 10.1073/pnas.94.19.10385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mills SD, Boland A, Sory MP, van der Smissen P, Kerbourch C, Finlay BB, Cornelis GR. Proc Natl Acad Sci USA. 1997;94:12638–12643. doi: 10.1073/pnas.94.23.12638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhang Y, Ting AT, Marcu KB, Bliska JB. J Immunol. 2005;174:7939–7949. doi: 10.4049/jimmunol.174.12.7939. [DOI] [PubMed] [Google Scholar]
- 33.Hersh D, Monack DM, Smith MR, Ghori N, Falkow S, Zychlinsky A. Proc Natl Acad Sci USA. 1999;96:2396–2401. doi: 10.1073/pnas.96.5.2396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hernandez LD, Pypaert M, Flavell RA, Galan JE. J Cell Biol. 2003;163:1123–1131. doi: 10.1083/jcb.200309161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Collazo CM, Galan JE. Mol Microbiol. 1997;24:747–756. doi: 10.1046/j.1365-2958.1997.3781740.x. [DOI] [PubMed] [Google Scholar]
- 36.Galan JE, Curtiss R., III Proc Natl Acad Sci USA. 1989;86:6383–6387. doi: 10.1073/pnas.86.16.6383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Monack DM, Detweiler CS, Falkow S. Cell Microbiol. 2001;3:825–837. doi: 10.1046/j.1462-5822.2001.00162.x. [DOI] [PubMed] [Google Scholar]
- 38.Hsu LC, Park JM, Zhang K, Luo JL, Maeda S, Kaufman RJ, Eckmann L, Guiney DG, Karin M. Nature. 2004;428:341–345. doi: 10.1038/nature02405. [DOI] [PubMed] [Google Scholar]
- 39.Perrin AJ, Jiang X, Birmingham CL, So NS, Brumell JH. Curr Biol. 2004;14:806–811. doi: 10.1016/j.cub.2004.04.033. [DOI] [PubMed] [Google Scholar]
- 40.Richter-Dahlfors A, Buchan AM, Finlay BB. J Exp Med. 1997;186:569–580. doi: 10.1084/jem.186.4.569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Valdivia RH, Falkow S. Science. 1997;277:2007–2011. doi: 10.1126/science.277.5334.2007. [DOI] [PubMed] [Google Scholar]
- 42.Datsenko KA, Wanner BL. Proc Natl Acad Sci USA. 2000;97:6640–6645. doi: 10.1073/pnas.120163297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Uzzau S, Figueroa-Bossi N, Rubino S, Bossi L. Proc Natl Acad Sci USA. 2001;98:15264–15269. doi: 10.1073/pnas.261348198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Davis RH, Botstein D, Roth JR. A Manual for Genetic Engineering: Advanced Bacterial Genetics. Cold Spring Harbor, NY: Cold Spring Harbor Lab Press; 1980. [Google Scholar]
- 45.Ruiz-Albert J, Yu XJ, Beuzon CR, Blakey AN, Galyov EE, Holden DW. Mol Microbiol. 2002;44:645–661. doi: 10.1046/j.1365-2958.2002.02912.x. [DOI] [PubMed] [Google Scholar]
- 46.Yu XJ, Liu M, Holden DW. Mol Microbiol. 2004;54:604–619. doi: 10.1111/j.1365-2958.2004.04297.x. [DOI] [PubMed] [Google Scholar]
- 47.Coombes BK, Lowden MJ, Bishop JL, Wickman ME, Brown NF, Duong N, Osborne S, Gal-Mor O, Finlay BB. Infect Immun. 2007;75:574–580. doi: 10.1128/IAI.00985-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}