


Abstract
We present the novel prokaryotic gene finder GISMO, which combines searches for protein family domains with composition-based classification based on a support vector machine. GISMO is highly accurate; exhibiting high sensitivity and specificity in gene identification. We found that it performs well for complete prokaryotic chromosomes, irrespective of their GC content, and also for plasmids as short as 10 kb, short genes and for genes with atypical sequence composition. Using GISMO, we found several thousand new predictions for the published genomes that are supported by extrinsic evidence, which strongly suggest that these are very likely biologically active genes. The source code for GISMO is freely available under the GPL license.
INTRODUCTION
Since the mid-1990s, automated gene finders for prokaryotic genome sequences have become available that allow the unsupervised discovery of genes from raw genomic sequence (1–9). This accomplishment, accompanied by impressive values of accuracy, has made prokaryotic gene prediction one of the showcases of computational biology. Subsequent developments have focused mostly on the introduction of novel techniques to more accurately capture sequence composition (4), modeling of the gene structure (7,10) and development of models that allow the unsupervised discovery of multiple gene classes (8,11).
Because of the high accuracy initially reported for most programs, some might consider prokaryotic gene prediction solved, but from the point of a practitioner, this is not quite the case yet. For some programs the predictive accuracy is uncertain, as they have not been re-evaluated since the original evaluation on a handful of genomes. The recent development of techniques that improve predictions by combining the output of multiple programs (6,12,13) shows that accuracy can be increased. Another issue is that some programs are only accessible via a web interface, which for genome projects—due to the confidentiality of the data—is frequently not an option.
Here we describe our novel gene finder GISMO (Gene Identification using a Support Vector Machine for ORF classification), which is freely available under the GPL license. GISMO has high classification accuracy: it is very sensitive, meaning that it identifies most known genes, and specific, i.e. it produces reliable predictions. Our program combines a hidden Markov model (HMM)-based search for protein domains with a support vector machine (SVM) to identify coding regions based on sequence composition. An advantage of the HMM-based search for protein domains compared with pair-wise sequence searches is the higher accuracy in discriminating between signal and noise for protein family members (14). Also, genes with new orderings of known protein domains can be detected easily. An SVM classifier is constructed for composition-based identification of protein-encoding genes. The SVM is a machine learning technique with a strong theoretical foundation (15,16) that has been used to improve classification accuracy in biological applications such as the detection of protein family members (17–19), RNA and DNA binding proteins (20), and the functional classification of gene expression data (21). The SVM is a maximum margin classifier that can solve non-linear classification problems by learning an optimally separating hyperplane in a higher-dimensional feature space. By use of non-linear kernel functions such as a Gaussian kernel, complex and non-linear decision functions can be learned by the SVM. Even if items of one class are clustered in multiple separate sub-regions in the input space they can be clearly separated from the other class (Figure 1). The learnt hyperplane allows accurate discrimination between classes that cannot be separated linearly in the input space, as may be the case when phenomena such as horizontal gene transfer, translational selection and leading/lagging strand biases influence the sequence composition of genes (22–24).
Figure 1.

Class boundaries learned by the SVM with different kernel functions. Circles and crosses represent instances of a toy example training set. Colored regions indicate the two classes learned by three example SVM applications. (A) A linear decision function learned with a linear kernel. (B) A polynomial kernel allows realization of a polynomial separating surface. (C) With a Gaussian kernel the SVM can learn disjoint decision functions that surround a multitude of ‘islands’ of items from the same class (30).
GISMO was evaluated with 165 prokaryotic chromosomes and 223 plasmid sequences. For the chromosomal sequences, GISMO identified 94.3% of the genes (98.9% for genes with annotated function), and 94.3% of its predictions corresponded to annotated genes. Several thousand of the new predictions for the published genomes are supported by extrinsic evidence, suggesting that these very probably are biologically active genes that are missing in the annotations. We also address some of the most challenging problems for prokaryotic gene finders, including the correct identification of short genes (7,25) and of genes with atypical sequence composition and the prediction of genes when only little sequence material is available, as in the case of extrachromosomal replicons. The composition-based SVM, which uses vectors of sequence composition in the (low-dimensional) space of codon usage, is well suited for these tasks and achieved the highest classification accuracy for all cases when compared with two other popular, freely available programs. GISMO predictions for the 165 genomic sequences are available for download in GFF at http://www.CeBiTec.Uni-Bielefeld.DE/groups/brf/software/gismo.
MATERIALS AND METHODS
Datasets
The annotation and genomic sequence of 165 bacterial and archaeal chromosomes were downloaded from EMBL (26), and 223 plasmids longer than 10 kb were downloaded from NCBI (http://www.ncbi.nlm.nih.gov/). Annotated genes tagged as pseudogenes or not corresponding to an open reading frame (beginning with a start codon, ending with an in-frame stop codon, no internal stop codon) were excluded from the reference set of annotated genes. Sets of function-known genes were created based on the gene product description. All genes supported by evidence, such as an annotated function or gene product, noted sequence conservation, or experimental support, were included in these sets. Short genes were defined as genes with <300 bp of sequence. Putative horizontally transferred genes were obtained for 57 genomes from HGT-DB (27), all having >100 genes predicted horizontally transferred. The sequences used in this study as well as tables with evaluation details are available at http://www.CeBiTec.Uni-Bielefeld.DE/groups/brf/software/gismo.
Gene-finding algorithm
GISMO proceeds in three phases: (i) an initial search for extrinsic support with HMM profiles of protein domains, (ii) the training and application of an SVM-based intrinsic classifier, and (iii) the merger of the different sources of evidence and prediction of optimal start sites.
In the first phase, the forward and reverse strand of the DNA sequence are translated in all three reading frames, and the translations are searched for protein domains contained in the Pfam-A database (28). Significant hits to the protein domain models (e-value <0.01) are mapped onto the open reading frames (ORFs) at the appropriate position in the genomic sequence. These ORFs constitute the initial set of domain-supported genes.
In the next phase, a composition-based SVM classifier is trained and applied for gene identification. All genes carrying a strongly supported domain motif (e-value <10−40) are used as training instances for the CDS (coding sequence) class, and ORFs located in the ‘shadow’ of these genes are used as the training items for the non-coding ORF (nORF) class. More specifically, shadow ORFs are used that are located on another frame with an overlap of ≥90 bp with a domain-supported gene. As input to the SVM classifier, all ORFs are represented as vectors of sequence composition features. We evaluated 10 feature types for their suitability as input: oligonucleotides of length 3–9, amino acids and di-amino acids, and a combination of codons and amino acids.
Vectors of sequence composition features are composed from different sequence features F. In the case of oligonucleotide features each F is the list of all words of one chosen length k over the alphabet of all nucleotides {a,c,g,t} (for k = 3: F = aaa,aac,..,ttt; for k = 4: F = aaaa,aaac, … , tttt). For the amino acid and di-amino acid feature type each F is defined in an analogous way: Here, F is the list of all amino acids and di-amino acids, respectively (F = Ala,Arg, … , Val for the amino acid feature type; F = AlaAla, AlaArg, … , ValVal for the di-amino acid feature type). Now, let fi be the feature at position i in one F. To represent each ORF x by a vector v = (v1, … , vc) of sequence composition features, we evaluate the frequencies of all fi in x.
For the oligonucleotide feature type, only ‘in-frame’ oligonucleotides, i.e. oligonucleotides beginning at positions 1,4,7,… of x, are considered to account for the 3-periodicity of the genetic code. vi is the in-frame frequency of oligonucleotide fi in x, divided by the normalization factor r:
The normalization factor r for an ORF of length n is r =n/3 for k = 3, r = n/3−1 for k ∈ {4,5,6,} and r =n/3−2 for k ∈ {7,8,9}.
For the (di-) amino acid feature type, vi are defined as:
where fi is the (di-) amino acid at position i in F, and the normalization factor r for an ORF of length n is r = n/3 for amino acids and r = n/3−1 for di-amino acids.
We found that 64-dimensional vectors of relative codon frequencies (i.e. in-frame oligonucleotide trimers) allow the most accurate discrimination between genes and nORFs and are particularly well suited for the identification of short genes and the training of accurate classifiers for plasmid sequences. The SVM classifier is trained with a Gaussian kernel function (30). Therefore, all CDS and nORFs from the training set are implicitly mapped from the input space of sequence composition to the feature space determined by the Gaussian kernel. In this feature space a hyperplane is learned by the SVM that optimally separates all training ORFs from the two classes. A suitable Gaussian kernel parameter γ and SVM parameter C (see the following section ‘support vector machine algorithm’) are determined in a grid search of the parameter space by fivefold cross-validation on the training set: The training set is partitioned into five subsamples. In five steps one sample is retained as the validation set, and an SVM is trained on all remaining samples. In each step the validation set is classified with the trained SVM, and the achieved classification accuracy is measured. The cross-validation process is repeated in a grid search for different values for the Gaussian kernel parameter γ and for the SVM parameter C. Finally, values for γ and C that result in the best classification accuracy are chosen.
Subsequently, all ORFs longer than a specified minimum length (set by the user) are extracted from the genomic sequence and represented by a sequence composition vector. These sequence composition vectors are mapped to the feature space determined by the Gaussian kernel. A class is assigned to each vector depending on its relative location with respect to the learned separating hyperplane. Based on the distance to the learned hyperplane, an additional score, called the SVM-score, can be calculated and used to classify a novel item in one of the two classes (see the SVM algorithm below).
In the third phase, domain- and composition-supported CDSs are combined into one set. Gene starts are adjusted from the ‘longest possible coding sequence’ to alternative positions. All predictions supported by strong evidence for the existence of a protein domain (e-value <0.01) or characteristic CDS sequence composition (SVM-score >−0.1) are kept. CDS candidates supported by weaker evidence are removed if they overlap more than 50 bp with a reliable candidate.
SVM algorithm
The SVM (15,16) is a supervised learning algorithm with a strong theoretical foundation and high classification accuracy for many applications. SVMs can learn accurate classifiers for data sets that cannot be linearly separated in the input space (30). This is achieved by the choice of a suitable kernel function to transform the input data into another feature space where it is easier to compute an accurate classification (Figure 1). By learning the optimal separating hyperplane in this feature space, a non-linear classifier can be learned in the original input space. In the case of GISMO, each item of the training set (CDSs and nORFs) is represented by a vector v of its sequence composition features. Given a training set of m vectors vj = (v1, … , vc)j (1≤j≤m) with known class labels (+1 for CDS, −1 for nORF), the SVM in training learns a hyperplane (w, b) that optimally separates the items of the two classes. The vector w that is learned by an SVM is defined as
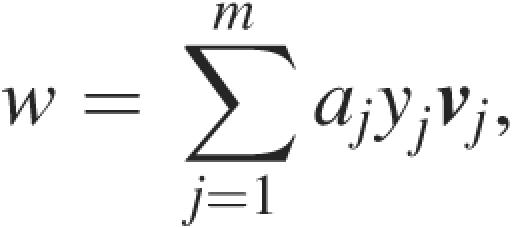 |
where aj are weights that are assigned to each vj during training. b is a scalar (29). With a learned hyperplane (w, b), a query vector v (an ORF represented by its vector of sequence composition) can be classified based on the decision value (the svm-score):
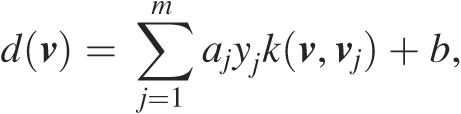 |
where k(v, vj) is a kernel function (29). In the case of GISMO, k (v, vj) is the Gaussian kernel: k(v, vj) = e−γ‖v-vj‖2.
In other words: To calculate the decision value d(v), v is compared with the sequence composition vj of each training ORF using the Gaussian kernel function. If v is more ‘similar’ to the CDSs from the training set a positive score is obtained, otherwise d(v) is negative. Depending on whether d(v) is larger than or smaller than 0, items are usually classified into one of the two classes by the SVM. To increase the sensitivity of GISMO, a relaxed cut-off is used—an ORF is classified as CDS if d(v) ≥ −0.6; otherwise it is classified as nORF.
The weights aj that are learned during training may be bounded by a finite value C. Therefore, the SVM parameter C influences the generalization ability of the learned classifier. If C is set to a small value, outlying training items are misclassified (29); this approach can be used to reduce overfitting in the case of small training sets. If C has a finite value, the resulting classifier is called a ‘soft margin SVM’ (29).
With a Gaussian kernel disjoint decision functions can be realized (30). The Gaussian kernel parameter γ influences the local behavior of the learned decision boundary. Setting a value for γ is a tradeoff between a well-fitted or more generalized decision boundary. A large value for γ results in irregular and noisy decision boundaries that are well fit to the training data with more disjoint clusters. A small value for γ, on the other hand, results in smooth and stable boundaries that avoid overfitting and are more robust (30). With a polynomial kernel a polynomial separating surface is learned in the input space (Figure 1).
Measures of accuracy
By comparing the predicted genes with the annotated genes, one can determine the number of correct gene predictions (tp), the number of false gene predictions (fp), the number of genes that were not found (fn), and the number of correctly classified nORFs (tn).
Classification accuracy is measured by the sensitivity (percentage of correctly identified genes) and specificity (percentage of correct predictions). The correlation coefficient describes the agreement of predictions and annotation with a single value in the range of [−1,1], where N = tp + fp + tn + fn. Only predictions and annotated CDSs with >90 bp were included in the analysis. The accuracy for predicting translation start sites was not evaluated. To predict translation start sites GISMO uses GS-Finder, which already has been found to be very accurate (31).
The receiver operating characteristic (ROC) (32) was used to evaluate the suitability of different kernel functions for gene identification with the sequence composition-based SVM classifier. Calculation of the ROC allows a comparison of different methods independent of an individual threshold setting used to discriminate between items of two classes. The ROC value corresponds to the area under a curve of the sensitivity versus the false positive prediction rate [fp/(fp + tn)] across the range of threshold settings. We here use the ROC0.1, which corresponds to the area under the ROC curve up to a false positive prediction rate of 10%.
Accuracy of different kernels for different types of genomes
The SVM for the composition-based identification of genes can be combined with a number of kernel functions that learn different types of discriminatory functions in the input space of sequence composition (Figure 1). We evaluated the classification accuracy achievable with different kernel functions for the genomes of four organisms in detail. For each of these organisms, different properties are most pronounced in genomic sequence composition. The 5.5 Mb genome of Escherichia coli O157:H7 contains a 1.4 Mb large O157:H7-specific region, which has mostly been acquired by lateral transfer (33). Half of this region corresponds to 24 prophages and prophage-like elements. The codon usage of E.coli is also influenced by translational selection. In the space of codon usage, E.coli genes separate into three classes, containing horizontally acquired, typical, or highly expressed genes (23). The genome of Treponema pallidum has a strong strand-specific bias in codon usage, shows little evidence of translation selection (22), and contains 76 horizontally transferred genes according to HGT-DB. The codon usage of Chlamydia trachomatis genes reflects a complex mixture of influences, the strongest being leading/lagging strand differences and translational selection (34). Its pronounced synteny to the C. pneumonia genome is considered evidence of a minimal foreign gene uptake (35). The codon usage of Buchnera aphidicola is generally very uniform, although a slight leading/lagging strand bias is detectable (36). B.aphidicola is a close relative of E.coli but has a reduced genome that contains only a subset of 564 of the E.coli genes (37). It does not contain horizontally acquired genes, according to HGT-DB.
For all genomes the highest classification accuracy is achieved with one of the non-linear kernel functions (Table 1). Only for E.coli O157:H7 are the ROC0.1 values obtained with the linear and polynomial kernel the same. The most accurate classification for E.coli O157:H7 is achieved with the Gaussian kernel. This shows that the Gaussian kernel is well suited for gene prediction in genomes with distinct gene classes. For the prediction of the genes most strongly influenced by the leading/lagging strand bias of the T.pallidum genome, both the polynomial and the Gaussian kernel allow a more accurate prediction. Even for the very homogeneous B.aphidicola genome where there is little variation in codon usage, the classification accuracy improves with the non-linear kernels. These results show how a non-linear model for genes in the codon usage space can improve the classification accuracy compared to a linear classifier.
Table 1.
ROC analysis of the classification accuracy achieved with different kernel functions
| Organism | Accession no. | Linear | Polynomial | Gaussian | Δbest-linear |
|---|---|---|---|---|---|
| E.coli O157:H7 | BA000007 | 0.960 | 0.960 | 0.968 | 0.008 |
| T.pallidum subsp. Pallidum str. Nichols | AE000520 | 0.920 | 0.930 | 0.929 | 0.010 |
| C.trachomatis D/UW-3/CX | AE001273 | 0.976 | 0.982 | 0.987 | 0.009 |
| B.aphidicola str. APS | BA000003 | 0.986 | 0.991 | 0.989 | 0.005 |
The ROC0.1 measures the discriminatory power of the SVM in gene identification based on sequence composition with a linear, polynomial or Gaussian kernel function.
Overall the obtained differences in ROC values between linear and non-linear kernels are low. Yet, owing to the high number of ORFs, even small differences in ROC values may indicate a considerable change in accuracy. ROC values can be interpreted as the probability that when randomly picking one positive and one negative item, the classifier will assign a higher score to the positive item than to the negative. In the optimal case, with a ROC value of 1, in 100% of cases a higher score will be assigned to the positive item. For an average genome with 3 Mb and ∼3000 ORFs, a change in ROC of, say, 0.01 reflects a difference of 30 ORFs that are correctly classified.
Validating novel genes by comparative analysis
To validate the novel genes predicted by GISMO for the published chromosomes, we tested for conservation of sequence, location and functional context, which is one of the strongest indicators of a biologically valid prediction (38). The chromosomal arrangement of the predictions and their surrounding genes was compared to the arrangement of homologous genes in other microbial genomes. Information about gene clusters in different organisms was obtained from the SEED, which is a manually curated comparative genomic database (39). Novel predictions homologous to non-hypothetical entries in this database (80% of the query sequence aligned using BLAST), and located in gene clusters conserved between two or more organisms (with at least two of the three adjacent upstream and downstream genes also found in the neighborhood of the homolog) were categorized as ‘probably coding’ (Figure 2).
Figure 2.

New candidates for the thiS gene of thiamin biosynthesis in the genomes of seven organisms. Homologous genes of the different organisms are drawn in the same color. The newly predicted thiS genes (hatched arrows) and homologs of the adjacent genes occur in conserved gene clusters. Genes with no sequence similarity to any of the displayed genes are colored grey. Overlapping genes are drawn below the continuous line. The displayed annotation is part of the thiamin biosynthesis pathway annotation of the SEED system, which is maintained and manually curated by human experts (36).
Implementation
GISMO is implemented in Perl using an object-oriented approach. From the HMMER package (40), hmmpfam is used to search the Pfam-A database. The parallel execution of hmmpfam on a high performance computing resource is facilitated by the use of a DRMAA-compliant interface (http://www.drmaa.org/). The results of the domain searches with hmmpfam are parsed with the BioPerl library (41). For the SVM-based classification the LIBSVM library and python scripts are utilized (http://www.csie.ntu.edu.tw/~cjlin/libsvm), which perform the scaling of the input data, cross validation for model selection, training of the SVM and the SVM-based classification. The GS-Finder software is used to identify translation start sites.
RESULTS
Accuracy for prokaryotic chromosomes
The accuracy of GISMO was evaluated with 165 publicly available complete prokaryotic chromosomes. Overall, GISMO is both highly sensitive and specific in predicting prokaryotic genes, with a value of 94.3% for both measurements. For the function-known genes, which are annotated with either a functional description or experimental evidence, the sensitivity of GISMO is even 98.9%. We also found that 4336 (16.4%) of the novel GISMO predictions that are not contained in the annotations are supported either by a significant protein domain motif (2423) or by the presence of homologs and a conserved genomic context found in the genomes of other organisms (4329, see ‘Identification of novel genes in the published genomes’ below).
Compared to two other popular gene finders that are freely available, GISMO is the most accurate (Table 2). Figure 3 shows a Venn diagram of the sets of genes predicted by GISMO and the gene finders Glimmer and CRITICA. We point out the high specificity of our program, which predicts ∼26 000 additional genes for the 165 chromosomes in addition to the currently annotated ones, compared with ∼115 000 additional predictions for Glimmer. Compared to CRITICA, which is very specific and produces reliable assignments, GISMO is more sensitive. GISMO's gene-finding accuracy does not seem significantly affected by the genomic GC content: For 42 genomes with a GC content >56%, the sensitivity is 93.5% and the specificity 92.9% (Table 2), <1% (2%) different from the overall accuracy achieved. For the function-known genes of the 42 GC-rich genomes, the sensitivity of GISMO is not reduced (99%).
Table 2.
Gene-finding accuracy for 165 prokaryotic chromosomes
| Gene finder | Cor | Sn (%) | Sp (%) |
|---|---|---|---|
| GISMO | 0.94 (0.93) | 94.3 (93.5) | 94.3 (92.9) |
| Glimmer | 0.87 (0.77) | 94.0 (89.8) | 83.3 (70.0) |
| CRITICA | 0.92 (0.91) | 88.8 (87.1) | 97.1 (96.2) |
The overall agreement of annotation and predictions (Cor), the sensitivity, and the specificity for the gene finders GISMO, Glimmer and CRITICA are shown. The values in parentheses are for the subset of GC-rich genomes (GC content > 56%) in the data set.
Figure 3.

Comparison of the genes predicted by GISMO and two other gene finders (Glimmer and CRITICA) for 165 prokaryotic chromosomes (with 471 884 annotated genes and 333 259 function-known genes in total). (A) Sensitivity (percentage of identified genes) in predicting genes, whereby the numbers in the overlapping areas specify the fractions of genes identified by more than one program. The absolute numbers are given in parentheses. (B) Specificity (percentage of correct predictions) of predictions made by one or more of the programs. The absolute numbers of false predictions are given in parentheses. (C) Sensitivity for the function-known genes. The number of correct predictions is given in parentheses.
Accuracy for short genes
The knowledge of the short genes of an organism is crucial because many proteins with important cellular functions are encoded by genes with <300 bp (e.g., regulatory or ribosomal proteins). Short genes are generally more difficult to identify than longer genes because their sequence carries less information that can be evaluated for classification. Figure 4 shows a comparison of the gene-finding accuracy for GISMO, Glimmer, and CRITICA for different minimum gene lengths. The results clearly show that the classification accuracy decreases with decreasing gene length. For short genes (<300 bp) GISMO has the highest overall prediction accuracy of the three programs, with a sensitivity and specificity of 63% and 69%, respectively (Table 3). CRITICA makes the most reliable predictions but identifies only 46% of the genes. Glimmer is the most sensitive (72%), but 56% of the predictions are false. Statistics suggest that a considerable fraction of the short annotated genes might, in fact, not be genes (42,43) (also, a large fraction of short genes are annotated as ‘hypothetical protein’), which makes an evaluation with more reliable gene sets especially important. For the function-known genes of the short genes, GISMO is also the most sensitive program, whereby sensitivity increases by >23% to 86.4%. GISMO thus has the highest overall classification accuracy and is the most sensitive program for detection of function-known short genes.
Figure 4.

Sensitivity and specificity of predictions for different gene lengths. The values for GISMO, CRITICA, and Glimmer are depicted in green, red, and blue, respectively. (A) Relation of the gene length to the sensitivity and specificity of the three programs. The sensitivity is displayed as a solid line, the specificity as a dashed line. (B) Relation of the gene length to the sensitivity for function-known genes.
Table 3.
Accuracy of GISMO, Glimmer and CRITICA in predicting short genes (<300 bp)
| Gene finder | Cor | Sn | Snfk (%) | Sp |
|---|---|---|---|---|
| GISMO | 0.64 | 63.0 | 86.4 | 69.0 |
| Glimmer | 0.54 | 72.0 | 83.7 | 44.0 |
| CRITICA | 0.60 | 46.0 | 67.4 | 84.0 |
Snfk denotes the sensitivity in detecting function-known genes.
Identification of horizontally transferred genes
Genes obtained by horizontal gene transfer can possess an unusual codon usage, base composition, and GC content (44). Therefore, it can be difficult to identify these genes based on the evaluation of intrinsic sequence properties. Generative methods such as Markov chains or hidden Markov models, which create a mean-based model of sequence composition by averaging over the sequence properties of their training collections, can have difficulties with genes that are best described by more than one distribution. This issue has been addressed by the inclusion of an additional model for the genes with ‘atypical’ sequence composition (8). The SVM has the convenient feature that it learns to optimally discriminate the genes from the non-coding ORFs during the training phase. In an unsupervised fashion, it discovers the shape that is most suitable for discrimination—which can be non-linear or even disjoint for genes distributed over multiple clusters in the input space (Figure 1). This property is convenient for gene prediction, as horizontal gene transfer is only one of several forces influencing sequence composition and affecting different genomes to different extents, and for each case the optimally separating boundaries can be found anew.
GISMO predicted 91.1% of the probably horizontally acquired genes with atypical sequence composition that were obtained from HGT-DB for 57 genomes. Of the function-known genes with atypical composition, 98.5% were identified, which is very similar to the overall sensitivity for prokaryotic chromosomes (98.8%), and significantly higher than the sensitivity of the other programs (Table 4).
Table 4.
Sensitivity in the detection of probably horizontally acquired genes with atypical sequence composition
| Gene finder | Sn (%) | Snfk (%) |
|---|---|---|
| GISMO | 91.1 | 98.5 |
| Glimmer | 87.3 | 94.4 |
| CRITICA | 72.8 | 86.3 |
Snfk denotes the sensitivity in detecting function-known genes.
Gene-finding accuracy for plasmids
Short DNA sequences such as plasmids yield only small sets for the training of intrinsic sequence models. For complex models with many parameters this situation can lead to overfitting and reduced prediction accuracy. GISMO is well suited for classification based on small training data sets because of (i) the use of a ‘soft margin,’ which allows the misclassification of outliers during training and avoids overfitting of SVMs, and (ii) the low dimensionality of the input space of codon usage, which we found to be optimal for gene prediction with a composition-based SVM.
For the 223 plasmid sequences >10 kb, GISMO achieves an average sensitivity of 89.1% and specificity of 80.3% and has the highest overall accuracy of the three programs (Table 5). The sensitivity increases to 96.1% for the function-known genes of the plasmids. Although this is lower than for the prokaryotic chromosomes, GISMO is very sensitive and specific, if one considers the size of the training sets available. For example, the two IncQ-like antibiotic resistance plasmids pIE1115 and pIE1130 (44) are the shortest sequences used in this survey. Both are 10 687 bp long, but differ in sequence and gene content. For pIE1115, the positive training set for the SVM consisted of five domain-supported genes, the negative training set of 60 shadow ORFs. Eight of the ten annotated genes were correctly identified, with only three additional predictions. For pIE1130, seven annotated genes were initially identified by their protein domain motifs. The training set for the composition-based classifier consisted of the seven domain-supported genes and 79 shadow ORFs. The SVM then identified two of the remaining four annotated genes, with only one additional prediction. That the classifier is able to accurately distinguish between genes and nORFs is demonstrated by the following numbers: For pIE1115, 120 of the 123 non-coding ORFs longer than 90 bp were correctly assigned, and 125 of 126 for pIE1130.
Table 5.
Gene-finding accuracy for 223 plasmids >10 kb
| Gene finder | Cor | Sn (%) | Snfk(%) | Sp (%) |
|---|---|---|---|---|
| GISMO | 0.82 | 89.1 | 96.1 | 80.3 |
| Glimmer | 0.79 | 89.3 | 94.4 | 74.5 |
| CRITICA | 0.58 | 45.4 | 55.7 | 87.3 |
Snfk denotes the sensitivity in detecting function-known genes.
Comparison with EasyGene and GenemarkS
GISMO was also compared with the HMM-based programs EasyGene and GenemarkS, considered among the most accurate bacterial gene finders (3,7). The accuracy was evaluated on a restricted test set as both programs are only accessible via a public web interface. While GISMO and GenemarkS automatically derive training sets with genome-specific compositional sequence properties, EasyGene can be run only via its Web interface with pretrained models. Pretrained models are available only for a limited number of sequenced genomes. Therefore, the performance of GISMO, EasyGene and GenemarkS was compared for the 25 of the 365 genomic sequences for which a pretrained EasyGene model was available. For these 25 genomic sequences, all three programs display a high accuracy (Table 6). GISMO has the highest overall accuracy, with an average sensitivity and specificity of ∼95%. GenemarkS has the highest average sensitivity for all genes, whereas EasyGene is most reliable. For the function-known genes, both GenemarkS and GISMO identify ∼99% and thus are 3.4% more sensitive than EasyGene. EasyGene is more specific (+2.1%) but less sensitive than GISMO (−4.0%).
Table 6.
Gene-finding accuracy for 25 genomic sequences
| Gene finder | Cor | Sn (%) | Snfk(%) | Sp (%) |
|---|---|---|---|---|
| GISMO | 0.943 | 95.1 | 99.0 | 94.7 |
| EasyGene | 0.930 | 91.1 | 95.5 | 96.8 |
| GenemarkS | 0.938 | 96.0 | 99.1 | 93.0 |
Snfk denotes the sensitivity in detecting function-known genes.
Identification of novel genes in the published genomes
Since the current annotations are missing many important genes (38), the novel predictions of GISMO were further investigated. For the 165 genomes used in this survey, 26 454 of the 468 368 GISMO predictions did not match an annotated gene. A strong indicator for a biologically active gene is the presence of a significant motif of a Pfam protein domain. Of the newly predicted genes, 2423 (9.2%) exhibit such motifs with strong statistical support (E-value <10−10) and do not overlap with any annotated gene by >10 amino acids. An additional 4329 (16.4%) new predictions are part of conserved gene clusters found in the same or similar orders in other microbial genomes (see Section ‘Material and Methods’ above). In total, 4336 (16.4%) of the novel GISMO predictions are supported by external sources of evidence (Pfam hit or a conserved cluster) that suggest that these predictions are truly biologically active genes. We describe several interesting examples below:
The thiS gene encodes a sulfur-carrying protein that is involved in the biosynthesis of thiamin (vitamin B1) (45). The thiS gene has been identified in a cluster with the thiE, thiG and thiF genes in a wide range of genomes (46) but is currently unknown for Clostridium tetani E88, Photobacterium profundum SS9, Shigella flexneri 2a 301, Shigella flexneri 2a 2457T, Wigglesworthia glossinidia, Yersinia pestis CO92, and Yersinia pestis KIM. For each of these genomes, GISMO predicted a novel probably-coding gene with significant homology to known thiS orthologs. The novel predictions are strongly supported by their genomic context, which comprises clusters of known genes of thiamin biosynthesis (Figure 2).
GISMO also predicted 99 genes encoding ribosomal proteins that are currently missing from the genome annotations. For example, two novel GISMO predictions for E.coli CFT073 and Wolinella succinogenes DSM 1740 are very similar to the ribosomal protein L32 in E.coli K12 and Helicobacter pylori 26695. The homologs of the two novel predictions and their adjacent genes appear in a conserved order in various organisms (Figure 5). Many of the probably-coding genes are as short as the ribosomal protein-encoding genes, a situation that explains why they were missed before. In summary, our results indicate that a considerable percentage of our additional predictions are novel and currently unknown protein-encoding genes that are missing from the annotations.
Figure 5.

Candidates for missing l32 genes in E.coli CFT073 and W.Succinogenes DSM. Homologous genes are displayed in the same color. The novel predictions (hatched arrows) and homologs of the surrounding genes occur in conserved order in closely related genomes.
CONCLUSIONS
The gene finder GISMO presented in this work uses state-of-the-art techniques from computational biology and machine learning to accurately predict protein-encoding genes for prokaryotic genome sequences. Initially, evidence for genes is compiled by protein-domain searches with profile HMMs, which are a well-known and highly accurate means for finding members of protein families and allow a more accurate discrimination between signal and noise than pair-wise sequence comparisons. They also allow the detection of genes that have protein domains in different order from that of known proteins. An SVM-based classifier is used for gene prediction based on sequence composition. The SVM is a machine learning technique that is well suited for prokaryotic gene prediction because it guarantees the unsupervised discovery of the shape in the space of sequence composition that is best suited for discrimination between genes and non-coding ORFs. The distribution of microbial genes in the space of sequence composition is affected by various influences that are pronounced to different extent for different genomes and thus require a careful and time-consuming analysis (34,36). The SVM allows the program to learn an accurate classifier, even when the distribution of items in this space is influenced by various factors such as gene expression rate, acquisition by horizontal transfer, or leading/lagging strand-related features. Gene identification for genomes in all cases was improved with non-linear classification functions, demonstrating the suitability of this approach.
In our extensive evaluation, we found GISMO to be very accurate. For the prokaryotic chromosomes, GISMO has an overall sensitivity and specificity of 94.3%. For the genes annotated with either a function or experimental evidence, the sensitivity is 98.9%. In comparison with the two popular programs Glimmer and CRITICA, which are freely available for a local installation, we found GISMO to be the most accurate also for finding genes shorter than 300 bp, for identifying genes with atypical sequence composition, and for predicting genes for short genomic sequences such as plasmid sequences. What makes this observation even more significant is the fact that GISMO is the only one of the three programs that was not used for annotating any of the genomes used in the evaluation.
In a comparison of GISMO to EasyGene and GeneMarkS on 25 genomic sequences, all three programs were very accurate, but GISMO slightly outperformed the other two in terms of overall accuracy. Therefore, GISMO presents an open source alternative to these programs for local use and integration into genome annotation pipelines.
For the prediction of translation initiation sites, GISMO uses the GS-Finder software. Since GS-Finder has already been shown to be very accurate (31), the accuracy of GISMO in gene start site prediction was not evaluated in this survey.
For the public genomes, we found several thousand new predictions that are strongly supported by external evidence and very likely correspond to real but unannotated genes. Many of these are short, such as 99 missing ribosomal protein-encoding genes, a fact that might explain why they were not found before.
The low-dimensional input space of codon frequencies that we found to be optimal for gene identification with the SVM-based compositional classifier allows accurate classification for short genes, as well as for short genomic sequences with a low number of available training items. SVMs are also intrinsically well suited for small data sets because they avoid overfitting the learned model by using a ‘soft margin’ in the model optimization step.
GISMO has already been used to predict genes in more than 20 genome annotation projects, for the reannotation of genomes as well as in the international effort to annotate a thousand genomes (39). We hope that our new gene finder will be widely used in microbial genome annotation and reannotation projects and will contribute to the generation of high-quality annotations.
Acknowledgments
The authors thank Michael Dondrup, Ross Overbeek, Gordon Pusch and Gail Pieper for valuable discussions and comments. The authors also thank Santi Garcia-Vallve for supplying information for the genomes contained in HGT-DB. L.K. was supported by the DFG Graduiertenkolleg 635 Bioinformatik. F.M. was supported in part by the U.S. Department of Energy, under Contract W-31-109-Eng-38. Funding to pay the Open Access publication charges for this article was provided by the International NRW Graduate School in Bioinformatics and Genome Research.
Conflict of interest statement. None declared.
REFERENCES
- 1.Salzberg S.L., Delcher A.L., Kasif S., White O. Microbial gene identification using interpolated Markov models. Nucleic Acids Res. 1998;26:544–548. doi: 10.1093/nar/26.2.544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Badger J.H., Olsen G.J. CRITICA: coding region identification tool invoking comparative analysis. Mol. Biol. Evol. 1999;16:512–524. doi: 10.1093/oxfordjournals.molbev.a026133. [DOI] [PubMed] [Google Scholar]
- 3.Besemer J., Lomsadze A., Borodovsky M. GeneMarkS: a self-training method for prediction of gene starts in microbial genomes. Implications for finding sequence motifs in regulatory regions. Nucleic Acids Res. 2001;29:2607–2618. doi: 10.1093/nar/29.12.2607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Delcher A.L., Harmon D., Kasif S., White O., Salzberg S.L. Improved microbial gene identification with GLIMMER. Nucleic Acids Res. 1999;27:4636–4641. doi: 10.1093/nar/27.23.4636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Frishman D., Mironov A., Mewes H.W., Gelfand M. Combining diverse evidence for gene recognition in completely sequenced bacterial genomes. Nucleic Acids Res. 1998;26:2941–2947. doi: 10.1093/nar/26.12.2941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Guo F.B., Ou H.Y., Zhang C.T. ZCURVE: a new system for recognizing protein-coding genes in bacterial and archaeal genomes. Nucleic Acids Res. 2003;31:1780–1789. doi: 10.1093/nar/gkg254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Larsen T.S., Krogh A. EasyGene—a prokaryotic gene finder that ranks ORFs by statistical significance. BMC Bioinformatics. 2003;4:21. doi: 10.1186/1471-2105-4-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lukashin A.V., Borodovsky M. GeneMark.hmm: new solutions for gene finding. Nucleic Acids Res. 1998;26:1107–1115. doi: 10.1093/nar/26.4.1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shibuya T., Rigoutsos I. Dictionary-driven prokaryotic gene finding. Nucleic Acids Res. 2002;30:2710–2725. doi: 10.1093/nar/gkf338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lomsadze A., Ter-Hovhannisyan V., Chernoff Y.O., Borodovsky M. Gene identification in novel eukaryotic genomes by self-training algorithm. Nucleic Acids Res. 2005;33:6494–6506. doi: 10.1093/nar/gki937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mahony S., McInerney J.O., Smith T.J., Golden A. Gene prediction using the self-organizing map: automatic generation of multiple gene models. BMC Bioinformatics. 2004;5:23. doi: 10.1186/1471-2105-5-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.McHardy A.C., Goesmann A., Puhler A., Meyer F. Development of joint application strategies for two microbial gene finders. Bioinformatics. 2004;20:1622–1631. doi: 10.1093/bioinformatics/bth137. [DOI] [PubMed] [Google Scholar]
- 13.Tech M., Merkl R. YACOP: Enhanced gene prediction obtained by a combination of existing methods. In Silico Biol. 2003;3:441–451. [PubMed] [Google Scholar]
- 14.Park J., Karplus K., Barrett C., Hughey R., Haussler D., Hubbard T., Chothia C. Sequence comparisons using multiple sequences detect three times as many remote homologues as pairwise methods. J. Mol. Biol. 1998;284:1201–1210. doi: 10.1006/jmbi.1998.2221. [DOI] [PubMed] [Google Scholar]
- 15.Boser B., Guyon I., Vapnik V.N. In: Haussler D., editor. Proceedings of the 5th Annual ACM Workshop on Computational Learning Theory; ACM Press; 1992. pp. 144–152. [Google Scholar]
- 16.Vapnik V.N. The Nature of Statistical Learning Theory. Springer; 1995. [Google Scholar]
- 17.Hou Y., Hsu W., Lee M.L., Bystroff C. Efficient remote homology detection using local structure. Bioinformatics. 2003;19:2294–2301. doi: 10.1093/bioinformatics/btg317. [DOI] [PubMed] [Google Scholar]
- 18.Jaakkola T., Diekhans M., Haussler D. A discriminative framework for detecting remote protein homologies. J. Comput. Biol. 2000;7:95–114. doi: 10.1089/10665270050081405. [DOI] [PubMed] [Google Scholar]
- 19.Leslie C., Eskin E., Noble W.S. The spectrum kernel: a string kernel for SVM protein classification. Pac. Symp. Biocomput. 2002:564–575. [PubMed] [Google Scholar]
- 20.Cai Y.D., Lin S.L. Support vector machines for predicting rRNA-, RNA-, and DNA-binding proteins from amino acid sequence. Biochim. Biophys. Acta. 2003;1648:127–133. doi: 10.1016/s1570-9639(03)00112-2. [DOI] [PubMed] [Google Scholar]
- 21.Brown M.P., Grundy W.N., Lin D., Cristianini N., Sugnet C.W., Furey T.S., Ares M., Jr, Haussler D. Knowledge-based analysis of microarray gene expression data by using support vector machines. Proc. Natl Acad. Sci. USA. 2000;97:262–267. doi: 10.1073/pnas.97.1.262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lafay B., Lloyd A.T., McLean M.J., Devine K.M., Sharp P.M., Wolfe K.H. Proteome composition and codon usage in spirochaetes: species-specific and DNA strand-specific mutational biases. Nucleic Acids Res. 1999;27:1642–1649. doi: 10.1093/nar/27.7.1642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Medigue C., Rouxel T., Vigier P., Henaut A., Danchin A. Evidence for horizontal gene transfer in Escherichia coli speciation. J. Mol. Biol. 1991;222:851–856. doi: 10.1016/0022-2836(91)90575-q. [DOI] [PubMed] [Google Scholar]
- 24.Moszer I., Rocha E.P., Danchin A. Codon usage and lateral gene transfer in Bacillus subtilis. Curr. Opin. Microbiol. 1999;2:524–528. doi: 10.1016/s1369-5274(99)00011-9. [DOI] [PubMed] [Google Scholar]
- 25.Linke B., McHardy A.C., Neuweger H., Krause L., Meyer F. REGANOR: a gene prediction server for prokaryotic genomes and a database of high quality gene predictions for prokaryotes. Appl. Bioinformatics. 2006;5:193–198. doi: 10.2165/00822942-200605030-00008. [DOI] [PubMed] [Google Scholar]
- 26.Cochrane G., Aldebert P., Althorpe N., Andersson M., Baker W., Baldwin A., Bates K., Bhattacharyya S., Browne P., van den Broek A., et al. EMBL Nucleotide Sequence Database: developments in 2005. Nucleic Acids Res. 2006;34:D10–D15. doi: 10.1093/nar/gkj130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Garcia-Vallve S., Guzman E., Montero M.A., Romeu A. HGT-DB: a database of putative horizontally transferred genes in prokaryotic complete genomes. Nucleic Acids Res. 2003;31:187–189. doi: 10.1093/nar/gkg004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bateman A., Coin L., Durbin R., Finn R.D., Hollich V., Griffiths-Jones S., Khanna A., Marshall M., Moxon S., Sonnhammer E.L., et al. The Pfam protein families database. Nucleic Acids Res. 2004;32:D138–141. doi: 10.1093/nar/gkh121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schoelkopf A., Schmola J. Learning with Kernels. Cambridge, MA: MIT Press; 2002. [Google Scholar]
- 30.Hastie T., Tibshirani R., Friedman J.H. The Elements Of Statistical Learning. Springer Verlag; 2003. [Google Scholar]
- 31.Ou H.Y., Guo F.B., Zhang C.T. GS-Finder: a program to find bacterial gene start sites with a self-training method. Int. J. Biochem. Cell. Biol. 2004;36:535–544. doi: 10.1016/j.biocel.2003.08.013. [DOI] [PubMed] [Google Scholar]
- 32.Swets J.A. Measuring the accuracy of diagnostic systems. Science. 1988;240:1285–1293. doi: 10.1126/science.3287615. [DOI] [PubMed] [Google Scholar]
- 33.Hayashi T., Makino K., Ohnishi M., Kurokawa K., Ishii K., Yokoyama K., Han C.G., Ohtsubo E., Nakayama K., Murata T., et al. Complete genome sequence of enterohemorrhagic Escherichia coli O157:H7 and genomic comparison with a laboratory strain K-12. DNA Res. 2001;8:11–22. doi: 10.1093/dnares/8.1.11. [DOI] [PubMed] [Google Scholar]
- 34.Romero H., Zavala A., Musto H. Codon usage in Chlamydia trachomatis is the result of strand-specific mutational biases and a complex pattern of selective forces. Nucleic Acids Res. 2000;28:2084–2090. doi: 10.1093/nar/28.10.2084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Read T.D., Brunham R.C., Shen C., Gill S.R., Heidelberg J.F., White O., Hickey E.K., Peterson J., Utterback T., Berry K., et al. Genome sequences of Chlamydia trachomatis MoPn and Chlamydia pneumoniae AR39. Nucleic Acids Res. 2000;28:1397–1406. doi: 10.1093/nar/28.6.1397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rispe C., Delmotte F., van Ham R.C., Moya A. Mutational and selective pressures on codon and amino acid usage in Buchnera, endosymbiotic bacteria of aphids. Genome Res. 2004;14:44–53. doi: 10.1101/gr.1358104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shigenobu S., Watanabe H., Hattori M., Sakaki Y., Ishikawa H. Genome sequence of the endocellular bacterial symbiont of aphids Buchnera sp. APS Nature. 2000;407:81–86. doi: 10.1038/35024074. [DOI] [PubMed] [Google Scholar]
- 38.Osterman A., Overbeek R. Missing genes in metabolic pathways: a comparative genomics approach. Curr. Opin. Chem. Biol. 2003;7:238–251. doi: 10.1016/s1367-5931(03)00027-9. [DOI] [PubMed] [Google Scholar]
- 39.Overbeek R., Begley T., Butler R.M., Choudhuri J.V., Chuang H.Y., Cohoon M., de Crecy-Lagard V., Diaz N., Disz T., Edwards R., et al. The subsystems approach to genome annotation and its use in the project to annotate 1000 genomes. Nucleic Acids Res. 2005;33:5691–5702. doi: 10.1093/nar/gki866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Eddy S.R. Profile hidden Markov models. Bioinformatics. 1998;14:755–763. doi: 10.1093/bioinformatics/14.9.755. [DOI] [PubMed] [Google Scholar]
- 41.Stajich J.E., Block D., Boulez K., Brenner S.E., Chervitz S.A., Dagdigian C., Fuellen G., Gilbert J.G., Korf I., Lapp H., et al. The Bioperl toolkit: Perl modules for the life sciences. Genome Res. 2002;12:1611–1618. doi: 10.1101/gr.361602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Skovgaard M., Jensen L.J., Brunak S., Ussery D., Krogh A. On the total number of genes and their length distribution in complete microbial genomes. Trends Genet. 2001;17:425–428. doi: 10.1016/s0168-9525(01)02372-1. [DOI] [PubMed] [Google Scholar]
- 43.Nielsen P., Krogh A. Large-scale prokaryotic gene prediction and comparison to genome annotation. Bioinformatics. 2005;21:4322–4329. doi: 10.1093/bioinformatics/bti701. [DOI] [PubMed] [Google Scholar]
- 44.Ochman H., Lawrence J.G., Groisman E.A. Lateral gene transfer and the nature of bacterial innovation. Nature. 2000;405:299–304. doi: 10.1038/35012500. [DOI] [PubMed] [Google Scholar]
- 45.Begley T.P., Downs D.M., Ealick S.E., McLafferty F.W., Van Loon A.P., Taylor S., Campobasso N., Chiu H.J., Kinsland C., Reddick J.J., et al. Thiamin biosynthesis in prokaryotes. Arch. Microbiol. 1999;171:293–300. doi: 10.1007/s002030050713. [DOI] [PubMed] [Google Scholar]
- 46.Rodionov D.A., Vitreschak A.G., Mironov A.A., Gelfand M.S. Comparative genomics of thiamin biosynthesis in procaryotes. New genes and regulatory mechanisms. J. Biol. Chem. 2002;277:48949–48959. doi: 10.1074/jbc.M208965200. [DOI] [PubMed] [Google Scholar]