


Abstract
Most ab initio pseudoknot predicting methods provide very few folding scenarios for a given RNA sequence and have low sensitivities. RNA researchers, in many cases, would rather sacrifice the specificity for a much higher sensitivity for pseudoknot detection. In this study, we introduce the Pseudoknot Local Motif Model and Dynamic Partner Sequence Stacking (PLMM_DPSS) algorithm which predicts all PLM model pseudoknots within an RNA sequence in a neighboring-region-interference-free fashion. The PLM model is derived from the existing Pseudobase entries. The innovative DPSS approach calculates the optimally lowest stacking energy between two partner sequences. Combined with the Mfold, PLMM_DPSS can also be used in predicting complicated pseudoknots. The test results of PLMM_DPSS, PKNOTS, iterated loop matching, pknotsRG and HotKnots with Pseudobase sequences have shown that PLMM_DPSS is the most sensitive among the five methods. PLMM_DPSS also provides manageable pseudoknot folding scenarios for further structure determination.
INTRODUCTION
Most RNA secondary structure prediction approaches are thermodynamic energy minimization methods (1,2), such as Mfold and Vienna RNA packages implemented with Zuker's dynamic programming algorithm based on the thermodynamic model (3,4). These methods do not predict pseudoknots. Pseudoknots are RNA structure elements formed upon standard base-pairing of a loop region with residues outside that loop (Figure 1) (5). The pseudoknot database Pseudobase has two types of pseudoknots (6): the H-type which has only two cross-pairing stems and the complicated type which contains recursively cross-pairing (more than two) stems. In the September 2005 Pseudobase, 229 out of 238 unique entries are H-type pseudoknots. Even though pseudoknots are involved in <5% of overall RNA base-pairing, they have various important biological functions (5). Among many important functions, pseudoknots are essential for some virus ribosome entry sites (7), are crucial in promoting frameshifting (8–14) and are critically related to some diseases (15).
Figure 1.
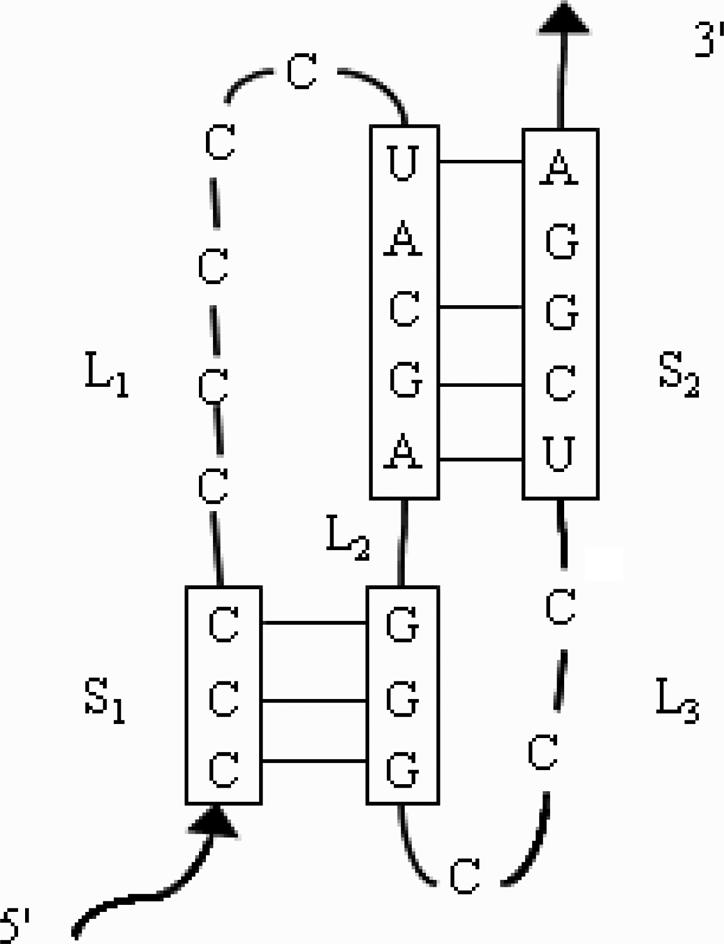
A typical H-type pseudoknot. S1 and S2 denote stem 1 and stem 2; L1, L2 and L3 denote loop 1, loop 2 and loop 3.
The two main approaches for pseudoknot prediction are comparative methods and ab initio methods. Comparative methods in general are more accurate, but they require a high level of homologous sequence similarities; thus they are not applicable for many novice sequence structure predictions (16,17). The ab initio pseudoknot prediction methods can be applied to all RNA sequences and are important tools for RNA research. The ab initio pseudoknots-included RNA structure prediction methods, mainly various modifications of Zuker's algorithm, predict pseudoknots in the context of the entire RNA sequence; therefore these methods are often time consuming. PKNOTS, with the gap matrix approach and time complexity O(n6.8), searches for the pseudoknot-included RNA secondary structure with the optimally lowest energy (18); the NUPACK partition function algorithm with time complexity O(n5) considers only the H-type pseudoknots (19).
Currently, there are two efficient pseudoknot predicting methods, pknotsRG and iterated loop matching (ILM). These two methods use heuristic approaches and simple pseudoknot models to reduce time cost (20,21). The pknotsRG (the pknotsRG-enf version) with O(n4) time complexity uses a simple pseudoknot model with three canonical rules and prioritizes the pseudoknot stem (pseudo-stems) detection. ILM with O(n3) time complexity uses the ILM method that heuristically picks a stem-forming region pair according to the local optima.
All of these methods provide just one folding scenario per sequence, and they tune the prediction performance by balancing the sensitivity and the specificity. RNA researchers, in many cases, would rather compromise specificity for higher sensitivity. They would like to be given a group of folding scenarios per sequence with a high confidence that a structure of interest (e.g. a pseudoknot) would be included if theoretically possible. Once an exhaustive range of pseudoknot possibilities has been provided, the determination of a pseudoknot could then be conducted through filtration with knowledge, chemical probing, mutagenesis and other means. HotKnots is the most recent method that uses a heuristic approach in exploring alternative pseudoknot-included secondary structures for a given sequence (22). HotKnots provides multiple folding possibilities, but the choices are not exhaustive and it might miss the actual pseudoknot structures.
Despite several efforts to estimate free energy parameters for pseudoknot loops (23–26), there is still no solid understanding of the pseudoknot folding mechanism (27). Recently, Aalbert et al. (25) have reported that pseudoknots have very specific local sequence restrictions and that short loop 2 in H-type pseudoknots play an important role in stabilizing the pseudoknot structure. Cao et al. (26) have studied the pseudoknot loop thermodynamics through the experimentally determined atomic coordinates and have suggested that pseudoknot stem lengths are closely related to the loop lengths. Based on the above results, we believe that pseudoknots can be searched through a local motif model.
Short loop 2 size (l2size) is not only favorable in stabilizing pseudoknot structure (25), but also is the key to forming the pseudoknot-specific knot-like structure. In Pseudobase, >90% of pseudoknots have l2size ≤ 2. Because short l2sizes are crucial to ensure the sequence pattern specificity for the Pseudoknot local motif (PLM) model construction, we have focused only on pseudoknots with l2sizes ≤ 2, and will ignore those pseudoknots with l2size > 2 until we have better understanding about their sequence pattern specificities.
In this study, based on our previously designed Similar enRiched Parikh Vector Searching (SRPVS) algorithm for the protein intrasequence inversed repeats study (28), we have developed the Pseudoknot Local Motif Model and Dynamic Partner Sequence Stacking (PLMM_DPSS) algorithm for pseudoknot prediction. The purpose of PLMM_DPSS is to allow biologists to identify interactions which can be tested experimentally. The PLM model is constructed based on 182 H-type pseudoknots in the Pseudobase. DPSS, a modification of the dynamic programming for sequence alignment algorithm (29), calculates the lowest stacking energy between two potential stem-forming regions. Even though the current PLM model is derived only from H-type pseudoknots, it can also be applied to complicated pseudoknots searching if we view a complicated pseudoknot as a composition of a core H-type pseudoknot and some other stems further crossing the pseudoknot stem (PK-stem) regions. With a comprehensive PLM model, PLMM_DPSS is guaranteed to be highly sensitive because it simply searches pseudoknot stems without any interference from the neighboring regions.
The PLMM_DPSS web server and the Supplementary Data including the executables, data sets and other related information are available at http://bioinformatics.ist.unomaha.edu:8080/x/PLMM_DPSS.html.
MATERIALS AND METHODS
PLMM_DPSS first decomposes the input sequence into subsequences and then calculates the stacking energy value between each subsequence pair. The subsequence pairs that have met the PLM model size and energy requirements will be considered as potential PK-stems and will then be paired and assembled. Finally, PLMM_DPSS will output those PLM-model-compatible stem pairs as predicted H-type pseudoknots.
Pseudoknot local motif model
The PLM model was constructed from the Pseudobase in September 2005 (6). Out of a total of 238 unique entries, we have excluded the redundant pseudoknots (>85% similarity), unnatural SELEX pseudoknots (30), the pseudoknots with l2size > 2 and pseudoknots with loop 1 or loop 3 sizes >800. The PLM model is then built based on 182 H-type pseudoknots.
In order to ensure the high sensitivity, every parameter in the PLM model is defined to be within a fully inclusive acceptance interval formed by the highest and the lowest values from the data. Table 1 shows some summary data obtained from the 182 Pseudobase H-type pseudoknots. Table 2 illustrates the PLM model parameters based on the data in Table 1 and reflects the relationships among stem 1 5′ region sizes, stem 1 energy values and longest loop 2 sizes in the PLM model. For example, according to Table 1, all pseudoknots with stem 1 5′ region size 4 have the highest stacking energy −6.4 if the longest loop 2 size is 1 and −7.6 if the longest loop 2 size is 2; thus for a stem 1 with 5′ region size 4 and energy value −6.8, its PLM model longest loop 2 size equals 1. The PK-stem energy-sum restriction is also included in the PLM model. An example of PK-stem energy-sum calculation is presented in the Supplementary Figure 1.
Table 1.
The longest loop 2 sizes and other related data in 182 Pseudobase H-type pseudoknots (data for longer stem 1 5′ regions is omitted here)
| Stem 1 5′ region size | Maximum stem 1 energy (kcal/mole) | Longest loop 2 size |
|---|---|---|
| 2 | −3.3 | 0 |
| 3 | −0.2 | 0 |
| 3 | −4.7 | 1 |
| 4 | −3.2 | 0 |
| 4 | −6.4 | 1 |
| 4 | −7.6 | 2 |
Table 2.
The PLM model parameters derived from the data in Table1
| Stem 1 5′ region size | Stem 1 energy lower bound (kcal/mole) | Stem 1 energy upper bound (kcal/mole) | Loop 2 size upper Bound |
|---|---|---|---|
| 2 | −∞ | −3.3 | 0 |
| 3 | −4.7 | −0.2 | 0 |
| 3 | −∞ | −4.7 | 1 |
| 4 | −6.4 | −3.2 | 0 |
| 4 | −7.6 | −6.4 | 1 |
| 4 | −∞ | −7.6 | 2 |
The shortest loop 2 size in the PLM model is always 0
Once a pair of regions is considered as potential PLM stem-forming regions according to their sizes, flanking region sizes, and matched base number, DPSS will calculate the lowest Turner's stacking energy value (31) of the pair and then decide whether this pair is able to form an acceptable PLM PK-stem.
Dynamic Partner Sequence Stacking (DPSS) Algorithm
DPSS is modified from the Dynamic Programming for Sequence Alignment (DPSA) algorithm (20). In a DPSS matrix, each cell has four elements (Table 3). The DPSS algorithm is deployed in Figure 2. An example of the DPSS energy calculation process is presented in Supplementary Figure 2. The DPSS traceback step is similar to the traceback step in the DPSA. Given two subsequences of size v and w, assuming v > w, the DPSS energy calculation has the time complexity of O (v2w) and the space complexity of O (vw). Since the PLM stem-forming region sizes are from 2 to 21, DPSS time and space costs could be considered as constant.
Table 3.
The matrix cell components of DPSA and DPSS, given two sequences Seqx and Seqy (i and j are base locations in Seqx and Seqy)
| DPSA | DPSS |
|---|---|
| F (i, j): score | E(i, j): optimally lowest energy score from (1,1) to (i, j) |
| D(i, j): ↖, ←, ↑ | D(i, j): 1 [ ↖, (i, j) paired], 2 [←, (i, j) paired ), 3 (↑, (i, j) paired], 4 [↖, (i, j) not paired], 5 [←, (i, j) not paired ), 6 (↑, (i, j) not paired] |
| P (i, j): P(i, j) = 1 if (i, j) forms a WC or GU base pair; otherwise P(i, j) = 0 | |
| [Sx (i, j), Sy (i, j)]: the 5′ side base pair that is closest to (i, j) |
‘↖’ denotes that the path from the current cell is directed towards its upper-left cell, ‘←’ denotes that the path is directed towards the left cell, and ‘↑’ denotes that the path is directed towards the upper cell
Figure 2.

Summary of DPSS algorithm. Given Seqx of size v and Seqy of size w (in reversed order), Seqx (a, i) is the region in Seqx flanking locations a and i, and Seqy (b, j) is defined similarly. E (a, b) = 9.00 indicates that no stacking is allowed at (a, b) and the TE function returns the Turner's energy between two nearest base pairs. The stacking energy for 5′  3′ is TE (U, A, ‘A’, ‘G’, C, G) = 1.1 (kcal/mole).
3′ is TE (U, A, ‘A’, ‘G’, C, G) = 1.1 (kcal/mole).
PLMM_DPSS algorithm
PLMM_DPSS uses two steps to predict pseudoknots for an input RNA sequence seq of size n: the Stem Finding step that locates all the potential stems within the sequence, and the Pseudoknot Assembly step that predicts whole pseudoknots based on the results obtained from the Stem Finding step (Figure 3).
Figure 3.

The pseudo-codes of the Stem Finding and the Pseudoknot Assembly steps. Each stemList [x] in the stemLists contains stems with the 5′ region size x and with the 5′ region starting (also ending) base position in nondecreasing order.
An example illustrating how Pseudoknot Assembly assembles the pseudo-stems into pseudoknots is presented in the Supplementary Figure 3. The PLMM_DPSS algorithm provides all PLM-model-compatible pseudoknots for a given sequence, while the PLMM_DPSS_lowest version provides only one pseudoknot with the lowest PK-stem energy-sum if there are several possible pseudoknots overlapping a certain region.
In the worst case, the stem lists contain n2 stems and each stem pairs with a constant c other stems to form c pseudoknots. In this study, the PLM PK-stem region sizes are from 2 to 21 and loop 2 sizes are from 0 to 2, so c = (21−2+1) × 3 = 60. Since the stem 2 5′ region ending position is searched in sorted stem lists through binary search, the PLMM_DPSS algorithm has the time complexity of O (Cn2logn). The constant C = dc, where d is the DPSS energy calculation time cost for each sequence region pair. The stem list size and pseudoknot amount in any real sequence are always far smaller. The PLMM_DPSS space complexity is O(n2). PLMM_DPSS is currently implemented in JAVA and is run under the Linux system with the Intel(R) Xeon(TM) 1700 MHz processor and 256 KB cache size.
RESULTS AND DISCUSSION
In this study, we have tested three data sets containing Pseudobase entries with sequence sizes <140: the pk168 contains 168 H-type entries with loop 2 sizes ≤2; the pkCmplt contains six complicated pseudoknots; and the pkLL2 contains nine H-type pseudoknots with loop 2 size >2. Both pkCmplt and pkLL2 contain pseudoknots not involved in the PLM model building. Because the long loop 2-sized pseudoknots are not typical structures and the Pseudobase has accepted pseudoknots with different levels of structure confirmation, we have selected into pkLL2 only those entries with some forms of experimental confirmation or those entries resulting from the sequence comparisons with experimentally confirmed sequences. More details of pkLL2 sequence selection are in the Supplementary Table 1.
PLMM_DPSS algorithm performance
Figure 4 shows the average numbers of PLM model stems and pseudoknots detected by PLMM_DPSS for all sequences in pk168, pkCmplt and pkLL2 within 12 sequence size ranges (e.g. in the lower graph of Figure 4, the average number of pseudoknots found in the sequence length range 70–79 is 60). Although not very clear due to the lack of sequences, the upward trends of both stem and pseudoknot numbers in Figure 4 are close to linear. Also in Figure 4, in order to provide only the distinguished stems and pseudoknots, a stem will not be counted if it is a substem of a bigger stem and has higher energy value than that of the bigger stem; and a pseudoknot will be ignored if its energy value is higher than another pseudoknot and each of their four PK-stem regions has an overlapping rate ≥80%. The highest pseudoknot amount is <200 for all sequences tested in Figure 4 (see Supplementary Data), so PLMM_DPSS provides a manageable amount of pseudoknot candidates even with the fully inclusive PLM model and exhaustive searching.
Figure 4.

The average numbers of stems (upper graph) and pseudoknots (lower graph) detected by PLMM_DPSS at 12 sequence size ranges. The sequence data involves all sequences in pk168, pkCmplt and pkLL2.
PLMM_DPSS has time complexity of O (Cn2logn). Because the constant in the PLMM_DPSS time complexity is big, the Figure 5 time cost comparisons with pk168, pkCmplt and pkLL2 sequences have shown that PLMM_DPSS is slower than ILM and pknotsRG; and PLMM_DPSS is much more efficient than PKNOTS. One would think that the exhaustive PLMM_DPSS should be slower than the heuristic HotKnots, but since PLMM_DPSS detects only potential pseudoknots and ignores all other folding types, PLMM_DPSS and HotKnots have similar time costs, and the longest time cost of PLMM_DPSS is <2 min (Figure 5).
Figure 5.

Summary of the average time costs of five methods at 12 sequence size ranges. The data involves all sequences in pk168, pkCmplt and pkLL2.
PLMM_DPSS prediction of pk168
The PLMM_DPSS algorithm is constructed to meet the requirements of RNA researchers who have asked for a highly sensitive pseudoknot searching tool. In this study, PLMM_DPSS prediction results of pk168 have been compared with those of HotKnots, pknotsRG-enf, ILM and PKNOTS. Since PLMM_DPSS detects only the base pairs that form PK-stems and ignores noncrossing stems, we have used the pk168 prediction results of the PLMM_DPSS_Mfold and PLMM_DPSS_lowest_Mfold packages to ensure a fair result comparison. PLMM_DPSS_Mfold is an integration of PLMM_DPSS and Mfold methods. PLMM_DPSS first predicts H-type pseudoknots for a given sequence, then will feed the sequence file and the corresponding constraint file masking the base pairs already predicted to the Mfold, and then will output the base pair prediction results of both methods (see Supplementary Table 2). The PLMM_DPSS_lowest_Mfold package is built in the same way.
Since both PLMM_DPSS and HotKnots provide multiple folding scenarios for each sequence, we have summed up base pairs from the best predicted scenarios (the highest correctly predicted base pair number scenario per sequence) of both PLMM_DPSS_Mfold and HotKnots (Table 4). Even though both methods provide alternative folding scenarios, PLMM_DPSS_Mfold is more sensitive than HotKnots for pk168 because PLMM_DPSS provides exhaustive PLM pseudoknot folding scenarios, while HotKnots is a heuristic approach that may miss real pseudoknot structures. PLMM_DPSS_Mfold results with the best prediction scenario per pk168 entry are the most sensitive among all listed methods in Table 4.
Table 4.
Summary of pk168 base pair prediction results and evaluations of five methods
| TP | Sensitivity (%) | TP + FP | Specificity (%) | |
|---|---|---|---|---|
| PLMM_DPSS_Mfold | 1839 | 96.2 | 2301 | 79.9 |
| HotKnots (the best scenario) | 1703 | 89.1 | 2094 | 81.3 |
| PLMM_DPSS_lowest_Mfold | 1612 | 84.4 | 2308 | 69.8 |
| HotKnots (the first scenario) | 1334 | 69.8 | 1926 | 69.3 |
| pknotsRG-enf | 1449 | 75.8 | 2031 | 71.3 |
| ILM | 1244 | 65.1 | 2084 | 59.7 |
| PKNOTS | 1398 | 73.2 | 1935 | 72.3 |
PLMM_DPSS_Mfold has two results: the PLMM_DPSS_lowest_Mfold result and the base pair sum result from the best predicted scenario per entry with PLMM_DPSS_Mfold; HotKnots also has two results: the base pair sum from each first result per entry which has the lowest overall energy and the base pair sum result from the best predicted scenario per entry. The total pk168 base pair number is 1911. TP = number of correctly predicted base pairs; sensitivity = TP/(the total base pair number); FP = number of wrongly predicted base pairs; specificity = TP/(TP + FP).
If a pseudoknot is considered as correctly predicted when each of four PK-stem regions is at least partially included, the pk168 test results have also shown that PLMM_DPSS has included all actual pseudoknot structures within its prediction results while HotKnots has missed 17 pseudoknots (see the details at http://bioinformatics.ist.unomaha.edu:8080/x/PLMM_DPSS.html).
Among all the methods that provide only one result per entry (for HotKnots, the first folding scenario per sequence which has the lowest overall energy is selected), PLMM_DPSS_lowest_Mfold, which predicts only one pseudoknot structure with the lowest PK-stem energy-sum per sequence, has the highest sensitivity (Table 4). Because PLMM_DPSS is not designed to provide just one ‘most likely’ pseudoknot folding scenario per sequence, the highly sensitive and precise PLMM_DPSS_lowest_Mfold result for pk168 is an unexpected encouraging bonus and suggest that most ‘typical’ (H-type with loop 2 size <3) pseudoknots are local-folding and are preferred structures of the natural RNA folding mechanism.
In order to validate the PLM model, we have separated the 182 PLM pseudoknots into two data sets: pktest and pktrain. The test data set pktest contains all 30 entries with sequence sizes <140 and the Pseudobase identification numbers >199; and the training data set pktrain contains the remaining 152 entries to build the PLMtrain model. Table 5 has shown that the prediction results of PLMtrain_DPSS_Mfold, PLMtrain_DPSS_lowest_Mfold and the other four methods are consistent with the results in Table 4. Therefore, the PLM model is a robust PLM model, and PLMM_DPSS is a highly sensitive and precisely accurate approach for short loop 2 H-type pseudoknot prediction.
Table 5.
Summary of pktest base pair prediction results and evaluations of five methods
| TP | Sensitivity (%) | TP + FP | Specificity (%) | |
|---|---|---|---|---|
| PLMtrain_DPSS_Mfold | 446 | 92.1 | 560 | 79.6 |
| HotKnots (the best scenario) | 420 | 86.8 | 502 | 83.7 |
| PLMtrain_DPSS_lowest_Mfold | 401 | 82.9 | 541 | 74.1 |
| HotKnots (the first scenario) | 373 | 77.1 | 496 | 75.2 |
| pknotsRG-enf | 291 | 60.1 | 475 | 61.3 |
| ILM | 300 | 62.0 | 541 | 55.5 |
| PKNOTS | 339 | 70.0 | 504 | 67.3 |
As in Table 4, PLMtrain_DPSS_Mfold and HotKnots each have two results. The total pktest base pair number is 484.
PLMM_DPSS prediction of pkCmplt
The pkCmplt prediction results have shown that the PLMM_DPSS_Mfold results with the highest correctly predicted number of base pairs per entry are the most sensitive among all listed methods (Table 6), so PLMM_DPSS is also the most sensitive tool for complicated pseudoknot prediction. An illustration of how PLMM_DPSS_Mfold detect a complicated pseudoknot in pkCmplt is presented in the Supplementary Table 2. The PLMM_DPSS_lowest_Mfold result, however, is the least accurate among all methods. We have expected such a result since complicated pseudoknots involve more stems; so picking the most stable H-type pseudoknot scenario alone may miss the actual folding in many cases. The results have also shown that ILM is the second most accurate among all listed methods and is the most accurate among all one-scenario-per-entry methods.
Table 6.
Summary of pkCmplt base pair prediction results and evaluations of five methods
| TP | Sensitivity (%) | TP + FP | Specificity (%) | |
|---|---|---|---|---|
| PLMM_DPSS_Mfold | 111 | 84.1 | 155 | 71.6 |
| HotKnots (the best scenario) | 105 | 79.6 | 155 | 67.7 |
| PLMM_DPSS_lowest_Mfold | 45 | 34.1 | 147 | 30.6 |
| HotKnots (the first scenario) | 69 | 52.3 | 147 | 46.9 |
| pknotsRG-enf | 97 | 73.5 | 155 | 62.6 |
| ILM | 101 | 76.5 | 166 | 60.8 |
| PKNOTS | 65 | 49.2 | 178 | 36.5 |
As in Table4, PLMM_DPSS_Mfold and HotKnots each have two results. The total pkCmplt base pair number is 132.
PLMM_DPSS prediction of pkLL2
In theory, PLMM_DPSS will miss all pseudoknots with loop 2 size >2. However, the pkLL2 prediction results have shown that the PLMM_DPSS_Mfold results (with the highest correctly predicted base pair number per pkLL2 entry) are the most sensitive among all listed methods (Table 7). PLMM_DPSS_MFold is able to detect long loop 2 pseudoknots because some pkLL2 pseudoknots have possible PK-stem base pairs that involve bases in the loop 2 region, a pseudoknot with short loop 2 predicted by PLMM_DPSS may include the PK-stem base pairs of the actual long loop 2 pseudoknot. In general, all applied methods have low sensitivities and specificities. The pkLL2 results in Table 7 have suggested that PLMM_DPSS is a broadly applicable pseudoknot searching tool with high sensitivity.
Table 7.
Summary of pkLL2 base pair prediction results and evaluations of five methods
| TP | Sensitivity (%) | TP + FP | Specificity (%) | |
|---|---|---|---|---|
| PLMM_DPSS_Mfold | 154 | 64.2 | 232 | 66.4 |
| HotKnots (the best scenario) | 141 | 58.8 | 178 | 79.2 |
| PLMM_DPSS_lowest_Mfold | 119 | 49.6 | 271 | 43.9 |
| HotKnots (the first scenario) | 107 | 44.6 | 238 | 45.0 |
| pknotsRG-enf | 119 | 49.6 | 225 | 52.9 |
| ILM | 139 | 57.9 | 280 | 49.6 |
| PKNOTS | 124 | 51.7 | 253 | 49.0 |
As in Table 4, PLMM_DPSS_Mfold and HotKnots each have two results. The total pkLL2 base pair number is 240.
Of all methods that predict one unique folding scenario per sequence, the PLMM_DPSS_lowest_Mfold has fewer correctly predicted base pairs than ILM and PKNOTS, has the same result as pknotsRG-enf and has better results than HotKnots.
In this study, PLMM_DPSS has been shown to be the most sensitive pseudoknot prediction method for all pseudoknots among all tested methods, and the PLMM_DPSS_lowest is also the most precisely sensitive tool for predicting ‘typical’ (H-type with loop 2 size ≤ 2) pseudoknots.
We believe that evaluations of the five methods require more than just comparing their results in Table 4 through Table 7. For a fair comparison, all other methods have considered the nonpseudoknot scenarios for each given sequence, while PLMM_DPSS considers only whether pseudoknot structures could possibly exist for a given sequence. The PLMM_DPSS is different from other structure prediction tools: it answers the question ‘Is there any possibility for this region to contain pseudoknots, and if so, what are these possible pseudoknots for future structure experiments?’ rather than ‘What is the most likely structure for this region?’
CONCLUSIONS
The PLMM_DPSS algorithm predicts pseudoknots by employing an innovative neighboring-region-interference-free pseudoknot-stem searching approach. This method allows PLMM_DPSS to provide all possible pseudoknots that conform to the PLM model for a given sequence. PLMM_DPSS has been proven to be more sensitive than four leading pseudoknots prediction tools used for comparison. The test results have also shown that for most H-type short loop 2 Pseudobase entries, the true pseudoknots are those with the lowest PK-stem energy-sum. PLMM_DPSS can also be integrated with Mfold to predict both H-type and complicated pseudoknots. We expect that in the future, the PLM model will be more comprehensive and PLMM_DPSS will take into account the noncrossing stem regions and be evolved into an efficient, reliable and specific tool for predicting pseudoknot-included RNA secondary structures.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
Acknowledgments
The authors would like to thank Dr Nora Chapman, Dr William Tapprich and Dr Eric Haas for helpful discussions and critical reading of the manuscript. Funding to support the work and to pay the Open Access publication charges for this article was provided by the NIH grant number P20 RR16469 from the INBRE program of National Center for Research Resource.
Conflict of interest statement. None declared.
REFERENCES
- 1.Zuker M., Stiegler P. Optimal computer folding of large RNA sequences using thermodynamics and auxiliary information. Nucleic Acids Res. 1981;9:133–148. doi: 10.1093/nar/9.1.133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Clote P. An efficient algorithm to compute the landscape of locally optimal RNA secondary structures with respect to the Nussinov-Jacobson energy model. J. Comput. Biol. 2005;12:83–101. doi: 10.1089/cmb.2005.12.83. [DOI] [PubMed] [Google Scholar]
- 3.Zuker M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 2003;31:3406–3415. doi: 10.1093/nar/gkg595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hofacker I.L. Vienna RNA secondary structure server. Nucleic Acids Res. 2003;31:3429–3431. doi: 10.1093/nar/gkg599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dam E., Pleij K., Draper D. Structural and functional aspects of RNA pseudoknots. Biochemistry. 1992;31:11665–11676. doi: 10.1021/bi00162a001. [DOI] [PubMed] [Google Scholar]
- 6.Batenburg F., Gultyaev A.P., Pleij C., Ng J., Oliehoek J. PseudoBase: a database with RNA pseudoknots. Nucleic Acids Res. 2000;28:201–204. doi: 10.1093/nar/28.1.201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wang C., Le S.Y., Ali N., Siddiqui A. An RNA pseudoknot is an essential structural element of the internal ribosome entry site located within the hepatitis C virus 5′ noncoding region. RNA. 1995;1:526–537. [PMC free article] [PubMed] [Google Scholar]
- 8.Gesteland R.F., Atkins J.F. Recoding: dynamic reprogramming of translation. Annu. Rev. Biochem. 1996;65:741–768. doi: 10.1146/annurev.bi.65.070196.003521. [DOI] [PubMed] [Google Scholar]
- 9.Liphardt J., Napthine S., Kontos H., Brierley I. Evidence for an RNA pseudoknot loop–helix interaction essential for efficient-1 ribosomal frameshifting. J. Mol. Biol. 1999;288:321–335. doi: 10.1006/jmbi.1999.2689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Plant E.P., Dinman J.D. Torsional restraint: a new twist on frameshifting pseudoknots. Nucleic Acids Res. 2005;33:1825–1833. doi: 10.1093/nar/gki329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Plant E.P., Jacobs K.L., Harger J.W., Meskauskas A., Jacobs J.L., Baxter J.L., Petrov A.N., Dinman J.D. The 9-A solution: how mRNA pseudoknots promote efficient programmed -1 ribosomal frameshifting. RNA. 2003;9:168–174. doi: 10.1261/rna.2132503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Su L., Chen L., Egli M., Berger J.M., Rich A. Minor groove RNA triplex in the crystal structure of a ribosomal frameshifting viral pseudoknot. Nature Struct. Biol. 1999;6:285–292. doi: 10.1038/6722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kim Y.G., Su L., Maas S., O'Neill A., Rich A. Specific mutations in a viral RNA pseudoknot drastically change ribosomal frameshifting efficiency. Proc. Natl Acad. Sci. USA. 1999;96:14234–14239. doi: 10.1073/pnas.96.25.14234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cornish P.V., Hennig M., Giedroc D.P. A loop 2 cytidine-stem 1 minor groove interaction as a positive determinant for pseudoknot-stimulated -1 ribosomal frameshifting. Proc. Natl Acad. Sci. USA. 2005;102:12694–12699. doi: 10.1073/pnas.0506166102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yingling Y.G., Shapiro B.A. Dynamic behavior of the telomerase RNA hairpin structure and its relationship to dyskeratosis congenita. J. Mol. Biol. 2005;348:27–42. doi: 10.1016/j.jmb.2005.02.015. [DOI] [PubMed] [Google Scholar]
- 16.Tabaska J.E., Cary R.B., Gabow H.N., Stormo G.D. An RNA folding method capable of identifying pseudoknots and base triples. Bioinformatics. 1998;14:691–699. doi: 10.1093/bioinformatics/14.8.691. [DOI] [PubMed] [Google Scholar]
- 17.Gutell R.R., Power A., Hertz G.Z., Putz E.J., Stormo G.D. Identifying constraints on the higher-order structure of RNA: continued development and application of comparative sequence analysis methods. Nucleic Acids Res. 1992;20:5785–5795. doi: 10.1093/nar/20.21.5785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rivas E., Eddy S.R. A dynamic programming algorithm for RNA structure prediction including pseudoknots. J. Mol. Biol. 1999;285:2053–2068. doi: 10.1006/jmbi.1998.2436. [DOI] [PubMed] [Google Scholar]
- 19.Dirks R.M., Pierce N.A. A partition function algorithm for nucleic acid secondary structure including pseudoknots. J. Comp. Chem. 2003;24:1664–1677. doi: 10.1002/jcc.10296. [DOI] [PubMed] [Google Scholar]
- 20.Reeder J., Giegerich R. Design, implementation and evaluation of a practical pseudoknot folding algorithm based on thermodynamics. BMC Bioinformatics. 2004;5:104. doi: 10.1186/1471-2105-5-104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ruan J.R., Stormo G.D., Zhang W. An iterated loop matching approach to the prediction of RNA secondary structures with pseudoknots. Bioinformatics. 2004;20:58–66. doi: 10.1093/bioinformatics/btg373. [DOI] [PubMed] [Google Scholar]
- 22.Ren J., Rastegari B., Condon A., Hoos H.H. HotKnots: heuristic prediction of RNA secondary structures including pseudoknots. RNA. 2005;11:1494–1504. doi: 10.1261/rna.7284905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gluick T.C., Draper D.E. Thermodynamics of folding a pseudoknotted mRNA fragment. J. Mol. Biol. 1994;241:246–326. doi: 10.1006/jmbi.1994.1493. [DOI] [PubMed] [Google Scholar]
- 24.Gultyaev A.P., Batenburg F., Pleij C. An approximation of loop free energy values of RNA H-pseudoknots. RNA. 1999;5:609–617. doi: 10.1017/s135583829998189x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Aalberts D.P., Hodas N.O. Asymmetry in RNA pseudoknots: observation and theory. Nucleic Acids Res. 2005;33:2210–2214. doi: 10.1093/nar/gki508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cao S., Chen S.J. Predicting RNA pseudoknot folding thermodynamics. Nucleic Acids Res. 2006;34:2634–2652. doi: 10.1093/nar/gkl346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tinoco I., Bustamante C. How RNA folds. J. Mol. Biol. 1999;293:271–281. doi: 10.1006/jmbi.1999.3001. [DOI] [PubMed] [Google Scholar]
- 28.Huang X., Ali H., Sadanandam A., Singh R. The fifth IEEE Symposium on BioInformatics and BioEngineering. 2005. Protein motif searching through similar enriched parikh vector identification; pp. 285–289. [Google Scholar]
- 29.Needleman S.B., Wunsch C.D. A general method applicable to the search for similarities in the amino acid sequences of two proteins. J. Mol. Biol. 1970;48:443–453. doi: 10.1016/0022-2836(70)90057-4. [DOI] [PubMed] [Google Scholar]
- 30.Tuerk C., MacDougal S., Gold L. RNA pseudoknots that inhibit human immunodeficiency virus type 1 reverse transcriptase. Proc. Natl Acad. Sci. USA. 1992;89:6988–6992. doi: 10.1073/pnas.89.15.6988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Xia T., SantaLucia J., Jr, Burkard M.E., Kierzek R., Schroeder S.J., Jiao X., Cox C., Turner D.H. Thermodynamic parameters for an expanded nearest-neighbor model for formation of RNA duplexes with Watson–Crick base pairs. Biochemistry. 1998;37:14719–14735. doi: 10.1021/bi9809425. [DOI] [PubMed] [Google Scholar]