

Abstract
An improved protocol for the large scale production of 1-benzenesulfinyl piperidine and other sulfinamides is described. It is demonstrated that 1-benzenesulfinyl pyrrolidine and N,N-diethyl benzenesulfinamide function analogously to 1-benzenesulfinyl piperidine in the trifluoromethanesulfonic anhydride-mediated activation of thioglycosides, and that their less crystalline nature enables them to be used at −78 °C as opposed to the −60 °C required to keep 1-benzenesulfinyl piperidine in solution. N,N-Dicyclohexyl benzenesulfinamide does not activate thioglycosides in combination with trifluoromethanesulfonic anhydride which is attributed to its greater steric bulk.
INTRODUCTION
We recently introduced the combination of 1-benzenesulfinyl piperidine (BSP, 1) and trifluoromethanesulfonic anhydride (Tf2O) as a powerful means of activation of both armed and disarmed thioglycosides.[1,2] In designing the sulfinamide activating system we set several criteria, the most important of which was the need for a stable, crystalline reagent capable, in combination with triflic anhydride, of rapidly activating a wide range of armed and disarmed thioglycosides at low temperature.[1] BSP met these criteria admirably and activates most thioglycosides for coupling in a matter of minutes at −60 °C as demonstrated in a series of subsequent synthetic endeavors from this[3–7] and other laboratories.[8–13] The highly crystalline nature of BSP, however, limits its solubility below −60 °C, which explains the choice of this temperature for coupling reactions as opposed to the more convenient −78 °C achieved with dry ice/acetone cooling baths. This minor inconvenience and, more importantly, the recognition that liquid analogs of BSP might ultimately prove preferable in automated oligosaccharide synthesis applications requiring robotic dispensation prompted the synthesis and evaluation of other sulfinamides as described here.
We began with a refinement to our synthesis of BSP, the original version of which was based on the production of benzenesulfinyl chloride from diphenyl disulfide, sulfuryl chloride, and acetic anhydride,[1] which itself is a more convenient version of the protocol of Douglass and Norton employing sulfuryl chloride instead of chlorine gas.[14] While adequate this protocol requires careful control of temperature in the evaporation of the byproduct, acetyl chloride, if a high quality sulfinyl chloride is to be obtained. The use of lower quality sulfinyl chloride results in lower yields of BSP due to the formation of contaminants which hinder the direct isolation by crystallization. We have now found that a modification of a protocol described by Craig[15,16] affords higher quality benzenesulfinyl chloride and thereby facilitates the isolation of the ensuing sulfinamides. In this method sodium benzenesulfinate is suspended in toluene in the presence of catalytic tetrabutylammonium bromide and is treated with thionyl chloride. Removal of the volatiles below 25 °C then affords crude benzenesulfinyl chloride in admixture with sodium chloride. This mixture is used immediately in the derivatization of secondary amines. In this manner we were able to prepare high quality benzenesulfinyl piperidine in 86% yield on a 55 g scale. Similarly prepared on multigram scales were 1-benzenesulfinyl pyrrolidine (2),[17] N,N-diethyl benzenesulfinamide (3),[18] and N,N-dicyclohexyl benzenesulfinamide (4). Two of these sulfinamides (2 and 4) were crystalline with melting points bracketing that of BSP, while a third (3) was a free-flowing, distillable liquid. All were found to be soluble in dichloromethane at −78 °C.
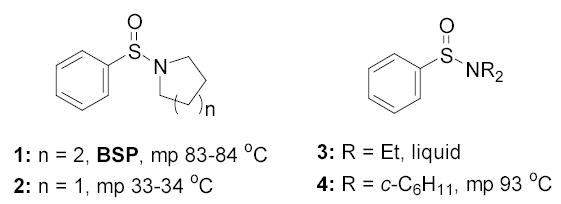
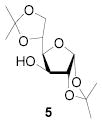
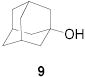
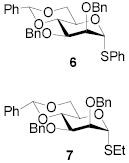
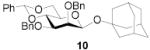
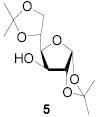
All three compounds (2–4) were assayed for their ability to activate to a standard mannosyl donor 6,[19] in combination with triflic anhydride and the hindered base 2,4,6-tri-tert-butylpyrimidine (TTBP),[20] and to effect its coupling to 1,2;5,6-diacetone glucofuranose 5. As reported in Table 1, with 2 and 3 the results were qualitatively the same as those previously obtained with BSP originally at −60 °C.[1] Reagent 4, on the other hand, did not affect activation of 6 under these conditions. We attributed this failure to the greater steric bulk in the 4-Tf2O adduct effectively preventing approach to the thioglycoside and elected not to pursue this particular sulfinamide further. A number of other couplings were conducted by means of thioglycoside activation with 2 and/or 3 in combination with Tf2O, each of which demonstrated comparable results to those obtained with BSP (Table 1).
Table 1.
Glycosidic Bond Forming Reactions
| Activator
|
|||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 (BSP) | 2 | 3 | |||||||||
| Acceptor | Donor | Product | Yield (%) | α:β ratio | Yield (%) | α:β ratio | Yield (%) | α:β ratio | |||
|
|
|
77 | 1:>9 | 90 | 1:>9 | 87 | 1:>9 | |||
| - | - | 91 | 1:>9 | 89 | 1:>9 | ||||||
|
|
|
88 | 1:>9 | 88 | 1:>9 | - | - | |||
| - | - | 94 | 1:>9 | 99 | 1:>9 | ||||||
|
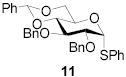
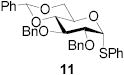
|
|
74 | >9:1 | 89 | 8.3:1 | 82 | 7.3:1 | |||
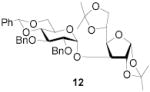
|
|
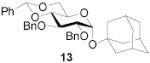
|
72 | >9:1 | 91a | 7.0:1 | 86a | 6.2:1 | |||
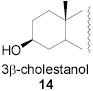
|
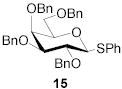
|
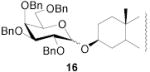
|
78 | 1:1 | 87 | 1:3.0 | 87 | 1:3.9 | |||
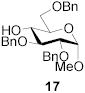
|
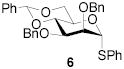
|
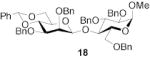
|
- | - | 83 | 1:>9 | - | - | |||
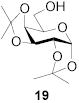
|
|
|
78b | 1:>9 | 85 | 1:>9 | 80 | 1:>9 | |||
|
|
|
70b | 1:>9 | 76b | 1:>9 | 73b | 1:>9 | |||
Reaction was performed under dilute conditions (0.01 M donor dichloromethane) as this led to enhanced α:β ratio;
Reaction was conducted at −40 °C in order to achieve rapid activation of this disarmed donor.
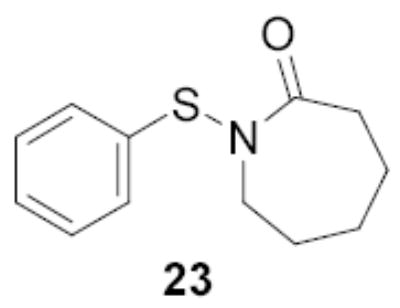
Following the work of Gin on the activation of 1-hydroxy sugars (hemiacetals),[2,21] van Boom and co-workers introduced the combination of diphenyl sulfoxide and triflic anhydride for the activation of thioglycosides and find this reagent combination to be somewhat comparable to the BSP/Tf2O couple.[2,10,22,23] While no actual comparisons between the BSP and Ph2SO methods of thioglycoside activation have been carried out in this study, other work from our laboratory leads us to agree with the conclusion of van Boom and co-workers.[24–27] We anticipate that between BSP, its analogs introduced here, the recent modification of Wong (23),[28] and diphenyl sulfoxide, a reagent will be found to activate almost all classes of thioglycoside, in conjunction with trifluoromethanesulfonic anhydride, under milder conditions than have hitherto been possible.[29–31]
EXPERIMENTAL
General methods
Unless otherwise stated 1H (300 MHz) and 13C (75 MHz) NMR spectra were recorded in CDCl3 solution. Optical rotations were recorded in CHCl3 solution, unless otherwise stated. All solvents were dried and distilled by standard protocols. All reactions were conducted under a blanket of dry nitrogen. All organic extracts were dried over sodium sulfate and concentrated under aspirator vacuum. Chromatographic purifications were carried out over silica gel. All glycosyl donors and acceptors were prepared by the literature methods or were commercial samples. With the exception of the compounds reported below all disaccharides physical characterisitics and spectral data consitent with the literature.1
1-Benzenesulfinyl piperidine (1)
To an ice-cooled suspension of PhSO2Na (50.0 g, 0.31 mol) and Bu4NBr (4.90 g, 15.2 mmol) in dry toluene (200 mL) was added thionyl chloride (89.6 mL, 1.2 mol) dropwise over 30 min. After the addition, the reaction mixture was stirred at 0 °C for 30 min, then warmed up to room temperature and stirred for a further 2 h. The reaction mixture was then concentrated under reduced pressure keeping the temperature of the water bath below 25 °C after which the residue was diluted with dry toluene (500 mL) and then treated with pyridine (24.6 mL, 0.31 mol) in one portion. The reaction mixture was cooled to 0 °C, followed by dropwise addition of piperidine (60.8 mL, 0.61 mol) over 1 h. After the addition, the reaction mixture was stirred in an ice bath for 2 h, and then warmed up to room temperature and stirred for a further 1 h. The reaction mixture was poured into a vigorously stirred mixture of ice and water (1 L) and NaHCO3 (100 g, 1.2 mol). The organic layer was separated and washed with brine (500 mL), then the aqueous layer was extracted with toluene (2 × 200 mL). The combined organic layer was dried and then concentrated. The residue was purified by recrystallization from ethanol to provide 1 as white crystals (55.0 g, 86%). Mp: 83–84 °C, lit. mp 83–84 °C;[32] 1H NMR δ: 7.62–7.65 (m, 2H), 7.43–7.50 (m, 3H), 3.05–3.12 (m, 2H), 2.88–2.95 (m, 2H), 1.47–1.62 (m, 6H); 13C NMR δ: 143.3, 130.6, 128.7, 126.1, 46.9, 26.1, and 23.8.
1-Benzenesulfinyl pyrrolidine (2).[17]
This compound was prepared analogously to 1 in 73% yield on a scale of 60g. Colorless crystals were obtained by recrystallization from hexane and ethyl acetate. Mp: 33–34 °C, 1H NMR δ: 7.66 (m, 2H), δ 7.49 (m, 3H), δ 3.33 (m, 2H), δ 3.00 (m, 2H), δ 1.83 (m, 4H). 13C NMR δ: 130.6, 128.9, 125.9, 46.2, 26.1.
N,N-Diethyl benzenesulfinamide (3).[18]
This compound was prepared analogously to 1 in 78% on a 55g scale. The crude reaction mixture was purified by distillation under reduced pressure (110 °C, 0.1 mm Hg) to yield the sulfinamide as colorless oil. 1H NMR δ: 7.61 (dd, J = 8.1, 2.4 Hz, 2H), δ 7.42 (m, 3H), δ3.07 (q, J = 7.5 Hz, 4H) 1.11 (t, J = 7.2 Hz, 6H); 13C NMR δ: 130.7, 128.8, 126.4, 42.1, 14.5.
N,N-Dicyclohexyl benzenesulfinamide (4)
This compound was prepared analogously to 1 in 78% on a 16 g scale. Recrystallization from ethyl acetate and hexane gave the sulfonamide as colorless crystals. Mp 93 °C; 1H NMRδ: 7.67-7.64 (m, 2H), 7.49-7.41 (m, 3H), 3.11-3.02 (m, 2H), 2.08-2.03 (m, 2H), 1.81-1.44 (m, 12H), 1.30-1.04 (m, 6H); 13C NMR δ: 145.0, 130.2, 128.6, 126.7, 55.5, 34.9, 26.4, 25.5. Anal. Calcd for C18H27NOS: C, 70.77; H, 8.91. Found: C, 70.81; H, 8.98.
Typical Glycosylation Protocol
A stirred solution of substrate, BSP (1.1 equiv.), TTBP (2.0 equiv.) and 3 Å sieves in CH2Cl2 (0.03 M in substrate) was kept at −78 °C for 15 min. Then Tf2O (1.2 equiv.) was added and after 5 min. the acceptor (1.5 equiv.) in CH2Cl2 (2.0 M) was added. Stirring was continued at −78 °C for 0.5 h and then the reaction mixture was allowed to warm to room temperature over a period of 2 h before it was filtered, washed with a saturated solution of NaHCO3, and brine, dried and concentrated. Chromatographic purification (eluting with mixtures of ethyl acetate in hexane) afforded the coupled products.
Methyl 2,3,6-tri-O-benzyl-4-O-(2,3-O-dibenzyl-β-d-mannopyranosyl)-α-d-glucopyranoside (18)
This compound was prepared by the standard protocol and had the following characteristics: [α]D20 -17.1 (c, 0.75); 1H NMR (500 MHz)δ: 7.50-7.48 (m, 2H), 7.42-7.17 (m, 28H), 5.52 (s, 1H), 5.08-5.04 (d, J = 11.0 Hz, 1H), 4.86-4.79 (m, 3H), 4.77 (s, 1H), 4.73 (s, 1H), 4.66-4.56 (m, 4H), 4.36 (s, 1H), 4.30-4.26 (d, J = 12.0 Hz, 1H), 4.14-4.02 (m, 2H), 3.93-3.83 (m, 2H), 3.64-3.58 (m, 2H), 3.54-3.47 (m, 3H), 3.44-3.43 (d, J = 3.0 Hz, 1H), 3.41 (s, 3H), 3.45-3.31 (dd, J = 3.3, 9.6 Hz, 1H), 3.09-3.01 (m, 1H); 13C NMR (125 MHz) δ: 139.9, 139.1, 139.0, 138.8, 138.1, 138.0, 129.3, 129.0, 128.8, 128.7, 128.6, 128.5, 128.2, 128.1, 128.0, 127.9, 127.8, 126.7, 101.9, 101.7, 98.8, 80.7, 79.4, 79.1, 78.7, 78.1, 75.7, 75.4, 74.1, 72.5, 70.0, 69.0, 68.8, 67.7, 55.8; ESI-HRMS Calcd for C55H58O11Na [M + Na]+ : 917.3877. Found 917.3871.
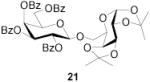
1,2:3,4-Di-O-isopropylidene-3-O-(2,3,4,6-tetra-O-benzoyl-β-d-galactopyranosyl)-α-d-galactopyranose (21)
This compound was prepared by the standard protocol and had the following characterisitcs: [α]D26 +34.7° (c, 3.0), lit.[33] [α]D = +45° (CHCl3); 1H NMR (500 MHz) δ: 8.09 (d, J = 7.6 Hz, 2H), 8.03 (d, J = 7.7 Hz, 2H), 7.98 (d, J = 7.7 Hz, 2H), 7.79 (d, J = 7.7 Hz, 2H), 7.23–7.49 (m, 12H), 6.00 (d, J = 2.8 Hz, 1H), 5.82 (t, J = 10.0 Hz, 1H), 5.62 (dd, J = 3.2, 10.4 Hz, 1H), 5.42 (d, J = 4.9 Hz, 1H), 5.03 (d, J = 8.0 Hz, 1H), 4.68 (dd, J = 6.6, 11.2 Hz, 1H), 4.41–4.45 (m, 2H), 4.35 (t, J = 6.5 Hz, 1H), 4.22 (d, J = 2.7 Hz, 1H), 4.05–4.12 (m, 2H), 3.90–3.93 (m, 2H), 1.40 (s, 3H), 1.24 (s, 3H), 1.22 (s, 3H), 1.20 (s, 3H); 13C NMR (125 MHz)δ: 166.0, 165.6, 165.3, 133.6, 133.2, 133.0, 130.02, 129.98, 129.79, 129.4, 129.3, 129.0, 128.8, 128.6, 128.4, 128.3, 128.2, 124.8, 109.3, 108.4, 101.7, 96.2, 71.8, 71.3, 71.0, 70.5, 70.3, 69.7, 68.4, 68.2, 67.4, 62.4, 62.0, 25.9, 25.7, 24.8, 24.2.
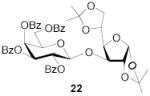
1,2:5,6-Di-O-isopropylidene-3-O-(2,3,4,6-tetra-O-benzoyl-β-d-galactopyranosyl)-α-d-glucofuranose (22)
[34] This compound was prepared by the standard protocol and had the following characterisitcs: [α]D24 +46.1 (c, 1.8); 1H NMR (500 MHz) δ: 8.10-8.08 (d, J = 7.5 Hz, 2H), 8.04-8.02 (d, J = 8.0 Hz, 2H), 8.00-7.94 (d, J = 8.0 Hz, 2H), 7.79-7.77 (d, J = 7.5 Hz, 2H), 7.63-7.60 (t, J = 7.5 Hz, 1H), 7.58-7.55 (t, J = 7.5 Hz, 1H), 7.51-7.41 (m, 6H), 7.38-7.35 (t, J = 7.5 Hz, 2H), 7.26-7.22 (t, J = 8.0 Hz, 2H), 5.99 (d, J = 2.5 Hz, 1H), 5.90 (d, J = 3.5 Hz, 1H), 5.83-5.80 (t, J = 8.0 Hz, 1H), 5.61-5.59 (dd, J = 3.5, 10.5 Hz, 1H), 4.93-4.92 (d, J = 8.0 Hz, 1H), 4.69-4.65 (dd, J = 6.5, 11.0 Hz, 1H), 4.50-4.49 (d, J = 3.5 Hz, 1H), 4.44-4.41 (dd, J = 6.5, 11.0 Hz, 1H), 4.34-4.31 (t, J = 6.5 Hz, 1H), 4.17-4.14 (m, 2H), 4.08-4.07 (d, J = 3.5 Hz, 1H), 3.76-3.71 (m, 2H), 1.40 (s, 3H), 1.29 (s, 3H), 1.13 (s, 3H), 1.09 (s, 3H); 13C NMR (125 MHz) δ: 166.5, 166.0, 165.9, 165.5, 134.0, 133.7, 133.6, 130.5, 130.3, 130.2, 129.85, 129.81, 129.4, 129.2, 129.0, 128.9, 128.7, 128.6, 112.6, 106.8, 102.6, 101.3, 84.4, 79.6, 75.3, 72.0, 71.7, 70.9, 70.1, 68.5, 62.4, 27.5, 26.9, 24.1, 23.1; ESI-HRMS Calcd for C46H46O15Na [M + Na]+ : 861.2734. Found 861.2704.
Acknowledgments
We thank the NIH (GM 62160) for support of this work.
Footnotes
Dedicated with respect to the memory of Professor Jacques H. van Boom
References
- 1.Crich D, Smith M. J Am Chem Soc. 2001;123:9015–9020. doi: 10.1021/ja0111481. [DOI] [PubMed] [Google Scholar]
- 2.Crich D, Lim LBL. Org React. 2004;64:115–251. [Google Scholar]
- 3.Crich D, Li H. J Org Chem. 2002;67:4640–4646. doi: 10.1021/jo0108818. [DOI] [PubMed] [Google Scholar]
- 4.Crich D, de la Mora MA, Cruz R. Tetrahedron. 2002;58:35–44. [Google Scholar]
- 5.Dudkin VY, Crich D. Tetrahedron Lett. 2003;44:1787–1789. [Google Scholar]
- 6.Crich D, Yao Q. J Am Chem Soc. 2004;126:8232–8236. doi: 10.1021/ja048070j. [DOI] [PubMed] [Google Scholar]
- 7.Crich D, Banerjee A, Yao Q. J Am Chem Soc. 2004;126:14930–14934. doi: 10.1021/ja047194t. [DOI] [PubMed] [Google Scholar]
- 8.Mong TKK, Lee HK, Duron SG, Wong CH. Proc Natl Acad Sci USA. 2003;100:797–802. doi: 10.1073/pnas.0337590100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wang Y, Huang X, Zhang LH, Ye XS. Org Lett. 2004;6:4415–4417. doi: 10.1021/ol0483246. [DOI] [PubMed] [Google Scholar]
- 10.Codée JDC, van den Bos LJ, Litjens REJN, Overkleeft HS, van Boeckel CAA, van Boom JH, van der Marel GA. Tetrahedron. 2004;60:1057–1064. [Google Scholar]
- 11.Gadikota RR, Callam CS, Wagner T, Del Fraino B, Lowary TL. J Am Chem Soc. 2003;125:4155–4165. doi: 10.1021/ja029302m. [DOI] [PubMed] [Google Scholar]
- 12.Yamago S, Yamada H, Maruyama T, Yoshida J-i. Angew Chem Int Ed Engl. 2004;43:2145–2148. doi: 10.1002/anie.200353552. [DOI] [PubMed] [Google Scholar]
- 13.van den Bos L, Codée JDC, van der Toorn JC, Boltje TJ, van Boom JH, Overkleeft HS, van der Marel GA. Org Lett. 2004;6:2165–2168. doi: 10.1021/ol049380+. [DOI] [PubMed] [Google Scholar]
- 14.Douglass IB, Norton RV. J Org Chem. 1968;33:2104–2106. [Google Scholar]
- 15.Craig D, Daniels K, MacKenzie AR. Tetrahedron. 1993;49:11263–11304. [Google Scholar]
- 16.For an earlier version of this protocol see: Kurzer F.Org Synth Coll Vol 19634937–939. [Google Scholar]
- 17.Bujnicki B, Drabowicz J, Mikolajczyk M, Kolbe A, Stefaniak L. J Org Chem. 1996;61:7593–7596. doi: 10.1021/jo960205j. [DOI] [PubMed] [Google Scholar]
- 18.Matsuo J-i, Iida D, Tatani K, Mukaiyama T. Bull Chem Soc Jpn. 2002;75:223–234. [Google Scholar]
- 19.Crich D, Sun S. Tetrahedron. 1998;54:8321–8348. [Google Scholar]
- 20.Crich D, Smith M, Yao Q, Picione J. Synthesis. 2001:323–326. [Google Scholar]
- 21.Gin, D. Y. In Glycochemistry. Principles, Synthesis, and Applications; Wang, P., Bertozzi, C. R., Eds.; Dekker: New York, 2001, pp 33–52.
- 22.Codée JDC, Litjens REJN, den Heeten R, Overkleeft HS, van Boom JH, van der Marel GA. Org Lett. 2003;5:1519–1522. doi: 10.1021/ol034312t. [DOI] [PubMed] [Google Scholar]
- 23.Codée JDC, van den Bos J, Litjens REJN, Overkleeft HS, van Boom JH, van der Marel GA. Org Lett. 2003;5:1947–1950. doi: 10.1021/ol034528v. [DOI] [PubMed] [Google Scholar]
- 24.Crich D, Vinod AU. J Org Chem. 2005;70:1291–1296. doi: 10.1021/jo0482559. [DOI] [PubMed] [Google Scholar]
- 25.Crich D, de la Mora M, Vinod AU. J Org Chem. 2003:8142–8148. doi: 10.1021/jo0349882. [DOI] [PubMed] [Google Scholar]
- 26.Crich D, Hutton TK, Banerjee A, Jayalath P, Picione J. Tetrahedron: Asymmetry. 2005;16:105–119. [Google Scholar]
- 27.The differing polarity of the byproducts from the sulfonamide method and the diphenyl sulfoxide methods is an advantage and it is sometimes found that switching from one of these activating systems to the other can greatly facilitate purification.
- 28.Durón SG, Polat T, Wong CH. Org Lett. 2004;6:839–841. doi: 10.1021/ol0400084. [DOI] [PubMed] [Google Scholar]
- 29.Norberg, T. In Modern Methods in Carbohydrate Synthesis; Khan, S. H., O'Neill, R. A., Eds.; Harwood Academic Publishers: Amsterdam, 1996, pp 82–106.
- 30.Garegg PJ. Adv Carbohydr Chem Biochem. 1997;52:179–266. doi: 10.1016/s0065-2318(08)60091-8. [DOI] [PubMed] [Google Scholar]
- 31.Oscarson, S. In Carbohydrates in Chemistry and Biology; Ernst, B., Hart, G. W., Sinaÿ, P., Eds.; Wiley-VCH: Weinheim, 2000; Vol. 1, pp 93–116.
- 32.Maricich TJ, Angeletakis CN. J Org Chem. 1984;49:1931–1934. [Google Scholar]
- 33.Garegg P, Norberg T. Acta Chem Scand. 1979;33:116–118. [Google Scholar]
- 34.An erroneous data set was inadvertantly recorded previously1 for this compound. The correct data is given here.