

Abstract
Vibrational microscopy and imaging offer several advantages for a variety of dermatological applications, ranging from studies of isolated single cells (corneocytes) to characterization of endogenous components in intact tissue. Two applications are described to illustrate the power of these techniques for skin research. First, the feasibility of tracking structural alterations in the components of individual corneocytes is demonstrated. Two solvents, DMSO and chloroform/methanol, commonly used in dermatological research, are shown to induce large reversible alterations (α-helix to β-sheet) in the secondary structure of keratin in isolated corneocytes. Second, factor analysis of image planes acquired with confocal Raman microscopy to a depth of 70 μm in intact pigskin, demonstrates the delineation of specific skin regions. Two particular components that are difficult to identify by other means were observed in the epidermis. One small region was formed from a conformationally ordered lipid phase containing cholesterol. In addition, the presence of nucleated cells in the tissue (most likely keratinocytes) was revealed by the spectral signatures of the phosphodiester and cytosine moieties of cellular DNA.
Keywords: Confocal Raman, Infrared imaging, Corneocytes, Epidermis, Protein secondary structure
Introduction
Technological developments in infrared and Raman microscopic imaging have permitted several new types of biomedical applications. As demonstrated elsewhere in this volume, the diagnosis of pathological states in a wide range of tissues is now feasible, and is being pursued vigorously in laboratories around the world. The vast volume of spectral data generated from imaging experiments permits sophisticated multivariate statistical analysis of data and enables the experimentalist to discern tissue regions where spectral signatures from pathological states differ from those of healthy tissue.
An important attribute of vibrational spectroscopy is the availability of spectra–structure correlations from many tissue components. This extensive background information can provide a useful supplement for biomedical diagnostics. By way of example, Boskey and her collaborators have characterized a new form of brittle bone disease (osteoporosis) based on structural changes in the collagen component of mineralized tissue [1]. This pathological state differs from the more widely known forms of osteoporosis, which are characterized by altered hydroxyapatite levels/structure. The origin of the spectral changes in the collagen component of pathological tissue biopsies was traced to increased levels of collagen cross links and alterations in their spatial distribution. Evidently, the availability of such information facilitates the development of strategies for tracing the origin (i.e., genetic predisposition, external factors, etc.) of the pathology.
Skin is another tissue where the availability of spectra–structure correlations as well as the ability to detect spectral signals from exogenous constituents opens up new possibilities for vibrational microscopic imaging experiments relevant to pharmacology, physiology, and biochemistry [2–4]. In this article, we provide two examples depicting the interplay of vibrational microscopic imaging with spectra/structure correlations.
The first example uses both IR and Raman microscopy to evaluate structural changes in the keratin of single cells (corneocytes) induced by treatment with dimethyl sulfoxide (DMSO) or chloroform/methanol (C/M). These solvents have been widely used in dermatology for studies of permeation enhancement and for extracting lipid from the stratum corneum (SC), respectively. The characterization of DMSO-induced structural changes in keratin will aid in the determination of DMSO permeation pathways and in the evaluation of SC barrier recovery. Assessing C/M’s effect on keratin in isolated corneocytes including the reversibility of potential structural changes, will facilitate the delineation of solvent treatment effects from that of lipid removal.
In the second example, we demonstrate that confocal Raman microscopy along with multivariate (factor) analysis provides the detection of particular skin regions (stratum corneum, epidermis, dermis) as well as endogenous sub-areas (keratinocytes, lipid inclusions) in the epidermis. The availability of spectra–structure correlations permits molecular structure information to be deduced from either the spectra or from the factor loadings.
Materials and methods
Preparation of corneocytes
The Rutgers University Internal Review Board has approved all protocols. Corneocytes were collected by tape stripping (Sellotape, 3M Scotchguard) human forearm skin. The forearm was flushed with water for several minutes prior to tape stripping. The first two strips were discarded and corneocytes from the third tape strip were isolated as follows. Corneocytes were flushed from the tape with HPLC-grade hexane. The hexane/corneocyte suspension was sonicated for approximately 5 min to break up desmosomes and lipid cohesion between individual corneocytes. Corneocytes were then isolated using nitrocellulose membrane filters with a pore size of 0.22 μm (GE Osmonics Labstore, Minnetonka, MN) and resuspended with fresh hexane. This procedure was repeated three times. A small aliquot of this suspension was separately deposited on either CaF2 windows for transmission IR or gold-coated silicon substrates for Raman measurements. Samples were dried overnight under house vacuum prior to spectroscopic measurements. The remaining corneocytes were dried under N2 gas prior to treatment with DMSO or C/M.
Corneocytes were placed on substrates that were directly dipped in pure DMSO for about 1 min. Separate aliquots of harvested corneocytes were transferred to glass vials using 5 mL C/M (2:1 v/v). Non-covalently bound lipids were extracted for 68 h while stirring at room temperature in sealed vials. Corneocytes, collected by centrifugation, were further washed 3 times using fresh C/M before the final suspension was placed on the appropriate substrate. Samples were dried overnight under vacuum prior to spectroscopic measurements. No vibrational bands from residual solvent were observed in corneocyte spectra. Subsequently, corneocytes from both DMSO and C/M treatment were rehydrated on the solid substrates by exposure to 100% relative humidity overnight before a final set of spectra were acquired.
Intact porcine skin preparation
Skin biopsies from Yucatan white, hairless pigs were purchased from Sinclair Research Center, Inc (Columbia, MO). The stratum corneum of the biopsies was washed with water and samples were cut to a size of approximately 1 × 1 cm2, 0.5-cm-thick sections. Sections were placed, SC side up, into a milled brass cell, covered, and sealed with a microscope coverslip for confocal Raman measurements.
IR microspectroscopy of corneocytes
IR spectra were acquired with the PerkinElmer Spotlight system described elsewhere in detail [4]. The instrument has imaging capabilities, but in this study the point mode was used. Since the typical diameter of corneocytes is about 40–60 μm, an aperture size of 40 × 40 μm2 was chosen to optimize the spectral quality. A total of 256 scans was acquired for each spectrum using 4 cm−1 spectral resolution. One level of zero-filling yielded data encoded every 2 cm−1. Approximately 50 corneocytes were examined from each sample preparation.
Raman microspectroscopy
Raman spectra were acquired with a Kaiser Optical Systems Raman Microprobe. The instrument has been described in detail elsewhere [5, 6]. A 785-nm diode laser generates approximately 7–12 mW of single mode laser power at the sample with a spot size of about 2 μm using a ×100 objective. The backscattered radiation illuminates a near-IR CCD (ANDOR Technology, Model DU 401-BR-DD). Spectral coverage is from 100 to 3,450 cm−1 at spectral resolution of 4 cm−1. Following linearization, data are encoded every 0.3 cm−1. Spectra were acquired from 10–15 corneocytes from each sample preparation using an 80-s exposure time, 4 accumulations, and cosmic ray correction. Optical images of corneocytes were acquired with a Leica microscope (Model DMLP) coupled to the Kaiser system with a ×100 objective.
Confocal Raman spectra of intact pigskin were acquired using a 60-s exposure time, 2 accumulations, and cosmic ray correction. A ×100 oil immersion objective was used for the intact pigskin samples. To evaluate axial characteristics of the optical set-up used in these experiments, the following observations were made with the oil immersion lens using protocols previously published [5]. The test sample consisted of five layers of a polymer laminate which incorporated two chemically identical layers (out of the five), each with a thickness of 7.5 μm separated by an intermediate layer of thickness 15 μm. This provides a center-to-center distance of 22.5 μm between the identical layers. Raman intensity profiles show a baseline-resolved width for each of the chemically identical layers of approximately 12–13 μm, a half-width of the intensity profile for each peak of about 8 μm, and a peak-to-peak distance of about 25 μm, in good agreement with the known test sample physical characteristics. The actual axial spatial resolution in the skin samples cannot be directly ascertained.
Data analysis
Grams/32 AI software version 6.0 (Thermo Galactic, Salem, NH) was used for processing individual Raman and IR spectra. Raman spectra were Fourier smoothed (80–90%) and 2-point baselines were applied to spectral regions of interest. This degree of Fourier smoothing could be applied without inducing spectral lineshape distortions. Confocal Raman spectra were analyzed (e.g., factor analysis) and Raman image planes were generated using ISys software version 3.1 (Spectral Dimensions, Inc. Olney, MD). Factor analysis was performed using the ISys score segregation routine. The analysis seeks to detect simple patterns in the relationships between observed variables in order to reduce the dimensionality of the data. Score segregation begins by normalizing PCA scores which may then be sharpened by raising them to a power specified by the acceleration parameter. The default value of 10 was used. Factor loadings are then calculated according to (S′S)−1*S′*X, unless S′S is not invertible, in which case iteration stops, and a factor is deleted before continuing.
Results and discussion
Solvent-induced conformational changes in the keratin of single cells
Although its mechanism of action is not well understood, DMSO has often been utilized as an efficient permeation enhancer for a wide variety of drugs, including antiviral agents, steroids, and antibiotics [7]. It has been suggested to alter protein conformation in the stratum corneum (SC), the outermost layer of skin and the main barrier to permeability. Anigbogu et al. [8] monitored the protein (keratin) α-helix to β-sheet transition in hydrated SC. In addition, Caspers et al. [3] monitored protein structure following DMSO penetration into human SC in vivo by confocal Raman spectroscopy. A broadening of the Amide I contour was noted, although major new spectral features were not resolved. It is noted that DMSO concentrations differed in the two experiments.
IR (1,200–3,600 cm−1) and Raman (800–1,720 cm−1) spectra, shown in Fig. 1a and b, respectively, demonstrate the quality of data obtained from untreated human corneocytes. Several strong bands characteristic of the protein and lipid constituents are observed in each spectrum. The Amide I and Amide II bands observed at approximately 1,650 and 1,550 cm−1 in the IR spectra, respectively, are of particular interest. The Amide I band is also one of the strongest bands in the Raman spectrum. In both types of spectra, the measured Amide I frequency reflects the predominantly helical secondary structure found in epidermal keratin. Absorbance in the Amide I region due to ceramides and other components, some containing amide groups and others C=C, is expected to be low compared to keratin absorbance in corneocytes isolated from the topmost layers of the stratum corneum [6]. Raman spectral quality is very good considering the thickness of an individual corneocyte (0.5–0.8 μm) and is comparable to spectra obtained from much thicker stratum corneum samples and intact skin [2, 5, 8]. It is likely that the observed signal has been enhanced due to a surface-enhanced Raman scattering (SERS) effect between the corneocytes and gold-coated substrate. Raman band assignments pertinent to stratum corneum samples have been listed previously [5, 9, 10].
Fig. 1.
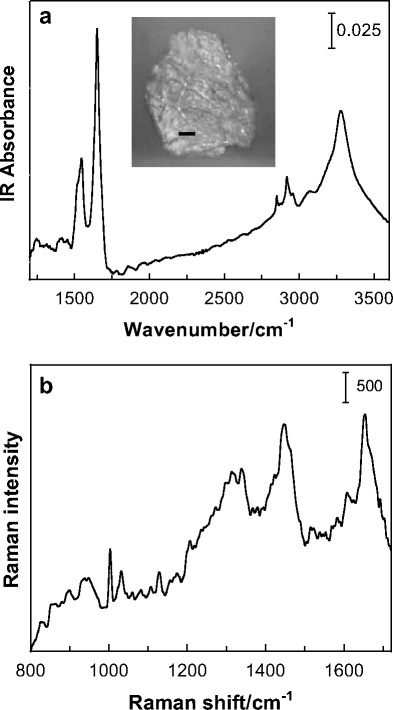
Vibrational microscopy of human corneocytes. a IR point mode spectrum (1,150–3,600 cm−1 region) acquired using a 40 μm2 aperture of a single corneocyte isolated from the third sequential tape strip applied to human forearm skin. The inset shows the optical image of the corneocyte (bar = 10 μm). b Raman spectrum (800–1,720 cm−1 region) of a similarly isolated corneocyte displaying bands characteristic of the lipid and protein (keratin) components
IR and Raman spectra of isolated corneocytes prior to and following immersion in DMSO, and after an overnight rehydration period (top to bottom trace) are displayed in Fig. 2a and b, respectively. A similar series of IR and Raman spectra acquired from corneocytes treated with C/M are shown in Fig. 2c and d. Both solvents induce several alterations in the spectra. The most significant change in the IR spectra is the appearance of an intense low frequency Amide I component observed for the DMSO-treated sample at 1,626 cm−1 in conjunction with a weak high frequency shoulder at approximately 1,695 cm−1. This pattern is diagnostic for formation of antiparallel β-sheet structure [11]. A redistribution of the maximum intensity in the IR Amide II band from 1,550 to 1,517 cm−1 for the same two spectra is also consistent with β-sheet formation [11].
Fig. 2.
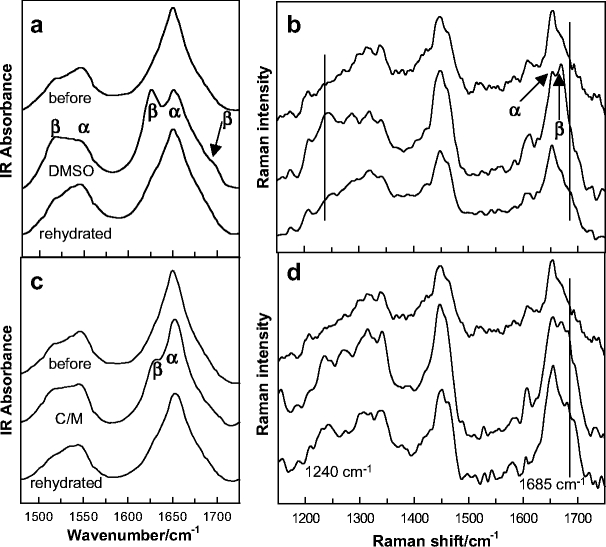
Solvent-induced conformational changes in the keratin of single corneocytes. a and b IR and Raman spectra, respectively, of an isolated corneocyte prior to and following immersion in DMSO, and after an overnight rehydration period (top to bottom, respectively). c and d IR and Raman spectra, respectively, of a corneocyte prior to and following C/M treatment, and after an overnight rehydration period (top to bottom, respectively). In the IR spectra (a and c), α-helical and β-sheet components of the Amide I (ca. 1,650 cm−1) and II (ca. 1,500–1,550 cm−1) bands are noted. In the Raman spectra (b and d), α-helical and β-sheet components of the Amide I are marked along with disordered constituents at 1,685 cm−1. One component of the Amide III band is marked at 1,240 cm−1
Helix:sheet transitions are well documented for different types of keratin, and have been detected by IR spectroscopy. In particular, Oertel [12] used IR to observe similar DMSO-induced changes in human SC. The current work extends observation of this conformational transition to isolated human corneocytes. It is also noted that, based on the IR Amide I and II band contours, a fair amount of helical structure remains subsequent to DMSO treatment.
The Raman spectrum (Fig. 2b, middle trace) corroborates the β-sheet assignment with the appearance of a high frequency Amide I component at about 1,669 cm−1 and a strong Amide III component at approximately 1,240 cm−1. Both features are within frequency ranges assigned to antiparallel β-sheets [13, 14]. The observed changes in the Amide I region of the Raman spectrum are consistent with a previous report detailing the effects of a series of aqueous DMSO solutions on human SC [8]. Comparisons between individual cells and the SC in the Amide III region are difficult due to overlapped bands from lipid components in the SC sample. In the current work, a second slightly weaker band is also observed for the DMSO-treated cells at approximately 1,270 cm−1. Bands in this region are usually assigned to α-helical structure [13, 14]. Generally, caution is required in associating characteristic frequency ranges with conformation for the Amide III mode given that the NH in-plane bend internal coordinate contributes to a number of modes in the 1,200–1,400 cm−1 region [15] and secondary-structure-dependent coupling occurs between the Amide III and the Cα–H bending vibration [16, 17]. This complexity, in addition to the inherent heterogeneity of corneocytes and variable lipid content of the samples examined herein, precludes an in-depth analysis of the Amide III contour in the current context.
The majority of the DMSO-induced secondary structure change is reversed upon rehydration of the corneocytes as demonstrated by comparison of the top and bottom spectra shown in Fig. 2a and b. Some residual broadening remains on the low frequency side of the Amide I band in the IR spectrum. The average full width at half height for the Amide I band in the IR spectra increased from 44 cm−1 for untreated corneocytes to 52 cm−1 following DMSO treatment and rehydration, indicating that some residual sheet structure may be present. An accurate measure of the Amide I bandwidth in the Raman spectra is precluded by the presence of a weak side chain vibration at approximately 1,608 cm−1. The general reversibility of the conformational change upon hydration was also observed by Oertel [12] for isolated SC.
C/M solutions are commonly used to extract lipids from isolated SC and intact skin samples, but solvent effects on the structure of the remaining constituents have not been detailed. The utility of vibrational spectroscopy for evaluation of keratin secondary structure after lipid removal is somewhat hampered by the native ceramide contribution to the Amide I and II band prior to solvent exposure. In SC samples it is therefore difficult to accurately access the contribution ceramides make to Amide I and II band intensities and position. In the current work, this concern is mostly overcome by acquiring spectra of isolated corneocytes in which the ceramide contribution to the Amide modes is substantially reduced.
Several changes are apparent in the IR and Raman spectra of corneocytes upon exposure to C/M (Fig. 2c and d, compare top to middle traces). Some of the solvent-induced alterations are similar to those just discussed for the DMSO-treated cells, e.g., the low (high) frequency Amide I component in the IR (Raman) spectrum at 1,626 cm−1 (1,670 cm−1) and the appearance of an Amide III band at 1,240 cm−1 in the Raman spectrum. These features provide strong evidence that C/M also induces antiparallel β-sheet formation. Differences are also evident when spectra from the two treatments are compared. Broadening on the high frequency side of the Amide I band in the Raman spectra is much more intense after C/M treatment vs. DMSO (compare the middle trace in Fig. 2d to b). Amide I Raman intensity in this region (ca. 1,685 cm−1) has been assigned to non-hydrogen-bonded disordered structures [13]. The broadening in the Raman Amide I band is not completely reversible upon rehydration of the sample that had been treated with C/M, even though the intensity of the β-sheet component at about 1,670 cm−1 has diminished (Fig. 2d, bottom trace). In addition, upon rehydration, intensity remains in the Amide III band at approximately 1,240 cm−1, although this band also broadens. Finally, measurements of the Amide I bandwidth in IR spectra upon rehydration of several C/M-treated corneocytes are essentially the same (±2 cm−1) as widths measured for untreated cells. The close proximity of Amide I bands arising from helical and random coil structures makes it difficult to differentiate between these two forms. Considering both the Raman and IR spectral data after rehydration, it seems safe to conclude that the majority of keratin exists in a helical form with some disordered structure remaining.
The extent of the C/M-induced protein structural changes has been observed to vary among corneocytes. IR spectra of some cells with significant lipid bands (i.e., those characterized by relatively intense methylene stretching modes near 2,850 and 2,918 cm−1) still exhibit notable protein conformation changes (data not shown), while some cells from which the lipid has been essentially completely extracted do not show significant changes in protein secondary structure. It thus appears that the extent of lipid extraction is not directly related to C/M-induced protein conformational changes. Similar results have been noted in IR studies of DMSO-induced protein conformation changes in SC samples [12]. Additional experiments under conditions of controlled hydration are needed to further explore possible origins of these conformational changes. One explanation for the α-helix to β-sheet structure change in keratin is that DMSO displaces bound water necessary for maintaining secondary structure [12]. DMSO is known to form an association complex with water stronger than that formed between water molecules alone [8]. It may be that C/M destabilizes keratin’s native secondary structure in a manner similar to DMSO, since the majority of the native structure is restored after both solvent treatments when the corneocytes are exposed to high relative humidity.
The importance of the current experiments lies in the fact, that in studies of supposedly native protein structures in the SC, C/M is often used to remove lipid components. As shown in the current work, this procedure is not innocuous, but can induce irreversible changes in protein structure in corneocytes.
Confocal Raman microscopy: delineation of skin regions from factor analysis
As demonstrated above, Raman microscopy complements the information available from IR. Although Raman scattering is inherently weaker than IR absorbance, the technique offers two major advantages for tissue studies. First, the spatial resolution of the method is inherently better than IR. For an excitation wavelength of 785 nm, Rayleigh’s rule for spatial resolution (ΔZ = 1.22λ/2(NA)) predicts a spatial resolution of approximately 0.5 μm for a wavelength (λ) of 785 nm and a numerical aperture (NA) of the ×100 objective of 0.95. In practice, as has been discussed in Materials and methods and elsewhere, the apparent depth and related measured optical properties are degraded by refractive index variations in the tissue [5, 18, 19]. Second, and equally important, spectra may be acquired in a confocal manner. Thus, physical sectioning of samples is not required to obtain molecular structure information from within the tissue. This evidently opens up the possibility for in vivo examination of skin, especially at the low laser powers (ca. 10 mW) used in these experiments. Puppels’ group has demonstrated the utility of confocal Raman determination of water levels in vivo in skin [20].
The benefit of using both a multivariate approach and existing spectra–structure correlations to characterize the microanatomy of skin is demonstrated in the next few figures. An optical micrograph of a 5-μm-thick pigskin section, shown in Fig. 3a, allows delineation of different skin regions. The outermost layered stratum corneum (SC) region appears to be ca. 15-μm thick with the underlying epidermis (ca. 50-μm thick) and dermis discernible. The physical sampling used in the confocal Raman measurement precludes the acquisition of a meaningful micrograph from the same sample; however to allow for comparison, confocal Raman spectra were acquired from a similar pigskin biopsy using approximately the same dimensions. Spectra were acquired from an untreated piece of pigskin, in steps of 2.5 μm in the z direction and 5 μm in the x (or y) direction. The area sampled was 75×40 μm so that 240 spectra (30 × 8) were acquired during the experiment. These were analyzed with factor analysis as described in Materials and methods and as applied to liposome-treated pigskin in a previous report [21]. In the current work, five significant factors were determined as shown in Fig. 3b, while score images for each factor are depicted in Fig. 3c. Three of the score images and corresponding factor loadings in Fig. 3 may be directly correlated with the known regions of skin as follows: Factor 1 corresponds to the SC, Factor 4 to the viable epidermis, and Factor 5 to the dermis. Thus, delineation of the skin regions based on the factor score images is wholly consistent with the optical image. The two remaining factor score images (Factors 2 and 3 ) highlight particular features within the viable epidermis. Examination of the factor loadings or averaged spectra corresponding to those pixels where the respective scores are high provides an indication of the molecular origin of the components. In general, factor plots tend to offer somewhat higher S/N ratios, since they represent a substantial condensation of data. However, it is important to remember that factors include all sources of variance in the data. For example, baseline distortions may appear as “features” in the factor loadings, which however are not related to vibrational modes in the sample. Thus, it is useful to routinely compare factors with appropriately selected spectra. In Fig. 4 an average spectrum of the stratum corneum is overlaid with the average localized to the small spatial region highlighted in Fig. 3c, labeled Factor 2. The average spectrum labeled “ordered lipid” has strong bands near 1,058, 1,130, 1,296, 2,880, and 2,850 cm−1 (Fig. 4b and c). These bands are readily assigned [9, 22] to lipid C–C stretching vibrations (1,058, 1,130 cm−1) of a conformationally ordered (all-trans) chain, lipid CH2 twist (1,296 cm−1) and lipid CH2 stretch (2,850 and 2,880 cm−1). In addition, the bands observed at 605 and 700 cm−1 (Fig. 4a) are indicative of cholesterol [23]. Thus, the small pocket in Fig. 3c (Factor 2) likely contains an ordered lipid phase. The fact that the same bands are present in the average stratum corneum spectrum (Fig. 4, top), albeit at lower intensity, strengthens the assignment. Highly ordered lipid phases (ceramides, fatty acids, and cholesterol) comprise the inter-corneocyte matrix of the stratum corneum. In addition, infrared spectroscopic evidence exists (D.J. Moore, unpublished results) for ordered lipid regions with relatively low protein content in appendages (i.e., pores and hair follicles) well below the stratum corneum. It is possible that the confocal Raman image planes shown in Fig. 3c cut through such an appendage.
Fig. 3.
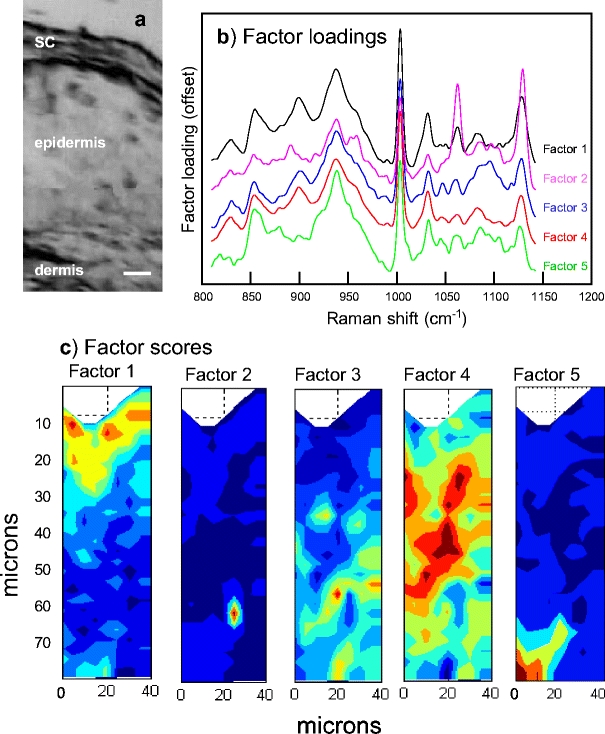
a An optical micrograph of a microtomed 5-μm-thick pigskin section acquired using the PerkinElmer Spotlight (bar = 10 μm). The results of factor analysis in the 800–1,150 cm−1 region conducted on a confocal Raman map acquired from intact pigskin. b The five distinct factor loadings generated by the ISys score segregation algorithm (see Materials and methods) are offset and labeled Factor 1–5. c The spatial distribution of factor scores for each of the loadings as marked. Dark blue indicates the lowest score with green, yellow, orange, and red indicative of progressively higher scores. Factor loadings and score images have been assigned to different micro regions in skin as described in the text
Fig. 4.
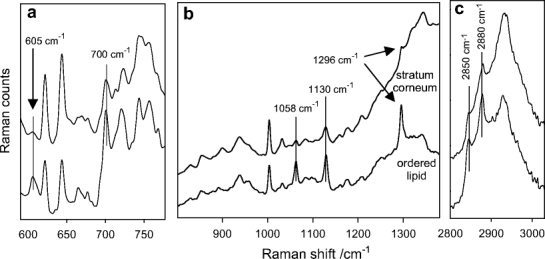
Averaged Raman spectra from within the stratum corneum (top) and lipid (bottom) regions as noted in Fig. 3b and c as Factor 1 and 2, respectively. a The 590–790 cm−1 region with bands assigned to cholesterol marked at 605 and 700 cm−1. b The 800–1,380 cm−1 region displays several bands (1,058, 1,130, and 1,296 cm−1) characteristic for ordered (all-trans) lipid acyl chains. c The CH stretching region (2,800–3,030 cm−1) with methylene symmetric (2,850 cm−1) and asymmetric (2,880 cm−1) stretching modes noted. The frequencies and relative intensity of the bands are consistent with well-ordered lipid acyl chains
Finally, Factor 3 (Fig. 3b), whose score image reveals several small-localized pockets in the epidermis, contains one broad spectral feature centered at approximately 1,090 cm−1 that is not present in the other factor loadings. Based on both the position of this particular band and the spatial distribution of the factor score, the band is tentatively assigned to the phosphodiester mode of DNA [24, 25]. Since the identification of DNA is of interest, as it would reveal the location of nuclei within keratinocytes or other cells in the tissue, an additional strategy was employed to acquire better spectra from the epidermis.
The experiment, whose results are depicted in Fig. 5, was carried out as follows. The top ≈20 μm of a pigskin section, including most of the SC, were removed by tape stripping prior to confocal Raman imaging. This allows for the acquisition of higher quality spectra in deeper regions of the viable epidermis. The Raman experiment was also carried out with more data points/unit area (2-μm step size) providing a closer examination of the tissue compared to the sample presented in Fig. 3. The presence of cell nuclei was revealed by factor analysis conducted over two wavenumber regions, each separate analysis restricted to compute only two factor loadings. An additional DNA band assigned to cytosine is detected in the factor loadings shown in Fig. 5b, which also strengthens the previously identified phosphodiester DNA band assignment at about 1,090 cm−1 [24, 25]. As expected, in the tape stripped sample, the phosphodiester band is again observed (Fig. 5c). The score image shown in Fig. 5a is derived from the factor with the strong cytosine band and is essentially the same as the score image generated from the phosphodiester factor (not shown). Thus, the image shown in Fig. 5a, depicting keratinocyte or other epidermal cell (e.g., melanocyte) nuclei, demonstrate the ability of confocal Raman to detect DNA in intact skin tissue.
Fig. 5.
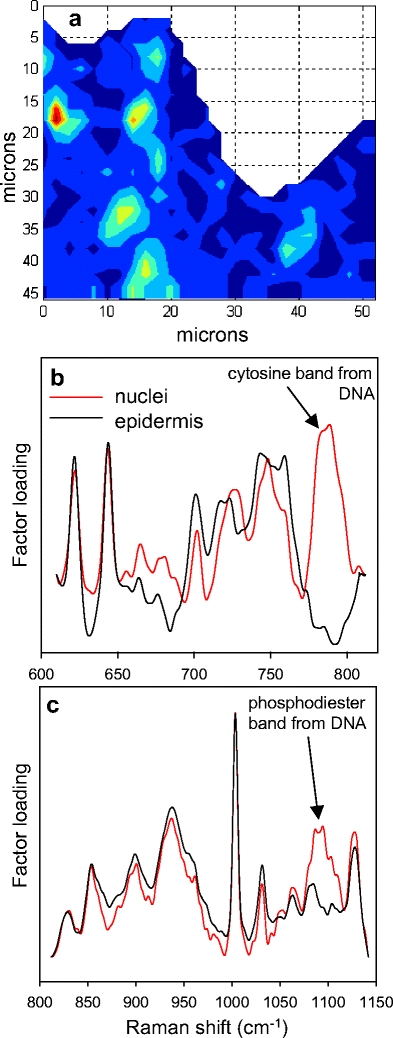
The results of factor analysis conducted in two separate spectral regions for a confocal Raman map of pigskin after stratum corneum removal by tape stripping. Both analyses were performed allowing the generation of only two factors. In each case, one factor contained a spectral feature specific to DNA and the other mapped in a fairly uniform way throughout the epidermal region. a Factor score image for the factor-labeled nuclei shown in (b) with red corresponding to the highest score and dark blue to the lowest. b Factor loadings for the 600–820 cm−1 region with a distinctive vibrational band at approximately 785 cm−1 assigned to cytosine. c Factor loadings for the 800–1,150 −1 region with a second characteristic DNA vibrational band due to the phosphodiester backbone stretching mode, at about 1,090 cm−1 noted
Conclusions
The current experiments highlight some unique advantages of vibrational microscopic imaging. An understanding of the spectroscopy of the various tissue components permits insight into the molecular origins of the images generated from factor analysis. Thus, identification of the lipid pocket in Fig. 3c (Factor 2) and of the cell nuclei (as detected from the DNA spectral signatures) in Fig. 3c (Factor 3) and Fig. 5a were greatly facilitated by the availability of Raman spectra of lipids and of DNA, respectively. In addition, the availability of correlations between lipid spectra and chain conformation, permitted assignment of the lipid inclusions as having arisen from ordered lipid phases. The biological relevance of this observation remains to be determined. From a pharmacological perspective, the detection of cell nuclei (most likely keratinocytes) within intact skin permits co-localization experiments to begin to be designed. For example, for classes of drugs targeted to these cells, confocal Raman measurements will permit determination of whether the putative therapeutic agent reaches its intended target.
In addition, although all microscopic methods can obviously readily detect single cells, the ability of IR- or Raman-based methods to detect conformational changes within a particular component of a single isolated cell is unique. The example we have chosen in Fig. 2 is of practical importance in skin research, since solvents such as DMSO or chloroform/methanol are used for permeation enhancement and lipid extraction, respectively. In each case these solvents were found to be far from innocuous. Each induced large conformational changes in the cellular keratin. This fact had been known previously from spectra of intact SC, but two aspects of the current measurements are novel. The observation of IR data from single cells provides a sharper spectral assignment of the bands. We note for example that ceramides, a major component of the SC, have strong bands arising from the amide bond located in these lipids. These modes contribute significantly to the spectra of the intact SC and overlap protein Amide I and II vibrations; however, they are much reduced in relative intensity in spectra of cells. Thus, studies of the reversibility of the solvent-induced conformational changes benefit from the reduced intensity of ceramide interference.
Finally, it is feasible to begin to examine biochemical heterogeneity at a single cellular level. Issues such as the relationship of protein structure (either native or solvent-modified) to hydration levels or to the levels of natural moisturizing factors, in single cells may be profitably probed. In addition, changes in the spectra of cells from pathological states may be examined.
Acknowledgements
This work was generously supported by PHS grant GM 29864-25 to RM. Additional support from Rutgers University toward acquisition of instrumentation is also acknowledged.
Abbreviations
- C/M
chloroform/methanol
- DMSO
dimethyl sulfoxide
- SC
stratum corneum
References
- 1.Paschalis EP, Shane E, Lyritis C, Skarantavos G, Mendelsohn R, Boskey AL (2004) J Bone Miner Res 19:2000–2004 [DOI] [PMC free article] [PubMed]
- 2.Caspers PJ, Lucassen GW, Wolthuis R, Bruining HA, Puppels GJ (1998) Biospectroscopy 4:S31–S39 [DOI] [PubMed]
- 3.Caspers PJ, Williams AC, Carter EA, Edwards HG, Barry BW, Bruining HA, Puppels GJ (2002) Pharm Res 19:1577–1580 [DOI] [PubMed]
- 4.Xiao C, Moore DJ, Rerek ME, Flach CR, Mendelsohn R (2005) J Invest Dermatol 124:622–632 [DOI] [PubMed]
- 5.Xiao C, Flach CR, Marcott C, Mendelsohn R (2004) Appl Spectrosc 58:382–389 [DOI] [PubMed]
- 6.Zhang G, Moore DJ, Mendelsohn R, Flach CR (2006) J Invest Dermatol 126:1088–1094 [DOI] [PubMed]
- 7.Barry BW (2004) Adv Drug Deliv Rev 56:603–618 [DOI] [PubMed]
- 8.Anigbogu ANC, Williams AC, Barry BW, Edwards HGM (1995) Int J Pharm 125:265–282 [DOI]
- 9.Barry BW, Edwards HGM, Williams AC (1992) J Raman Spectrosc 23:641–645 [DOI]
- 10.Frushour BG, Koenig JL (1975) Biopolymers 14:379–391 [DOI] [PubMed]
- 11.Mendelsohn R, Mantsch HH (1986) Fourier transform infrared studies of lipid-protein interaction. In: Watts A, de Pont JJHM (eds) Progress in protein-lipid interaction, vol 2. Elsevier, Amsterdam, pp 103–146
- 12.Oertel RP (1977) Biopolymers 16:2329–2345 [DOI] [PubMed]
- 13.Yager P, Gaber BP (1987) Membranes. In: Spiro TG (ed) Biological applications of Raman spectroscopy. Wiley, New York, pp 203–261
- 14.Peticolas WL (1995) Raman spectroscopy of DNA and proteins. In: Sauer K (ed) Methods enzymol. Academic Press, New York, pp 389–397 [DOI] [PubMed]
- 15.Krimm S, Bandekar J (1986) Adv Protein Chem 38:181–364 [DOI] [PubMed]
- 16.Asher SA, Ianoul A, Mix G, Boyen MN, Karnoup A, Diem M, Schweitzer-Stenner R (2001) J Amer Chem Soc 123:11775–11781 [DOI] [PubMed]
- 17.Oboodi MR, Alva C, Diem M (1984) J Phys Chem 88:501–505 [DOI]
- 18.Everall NJ (2000) Appl Spectrosc 54:1515–1520 [DOI]
- 19.Everall NJ (2000) Appl Spectrosc 54:773–782 [DOI]
- 20.Caspers PJ, Lucassen GW, Carter EA, Bruining HA, Puppels GJ (2001) J Invest Dermatol 116:434–441 [DOI] [PubMed]
- 21.Mendelsohn R, Flach CR, Moore DJ (2006) Biochim Biophys Acta 1758:923–933 [DOI] [PubMed]
- 22.Lippert JL, Peticolas WL (1971) Proc Natl Acad Sci USA 68:1572–1576 [DOI] [PMC free article] [PubMed]
- 23.Baraga JJ, Feld MS, Rava RP (1992) Proc Natl Acad Sci USA 89:3473–3477 [DOI] [PMC free article] [PubMed]
- 24.Deng H, Bloomfield VA, Benevides JM, Thomas GJ (1999) Biopolymers 50:656–666 [DOI] [PubMed]
- 25.Peticolas WL, Kubasek WL, Thomas GA, Tsuboi M (1987) Nucleic acids. In: Spiro TG (ed) Biological applications of Raman spectroscopy. Wiley, New York, pp 81–134