

Abstract
Rad23 proteins function in both DNA repair and protein stability regulation. As ubiquitinated forms of p53 are stabilized after DNA damage in concert with p53 functional activation, and human Rad23 proteins (hHR23A and B) regulate p53 stability in unstressed cells, the role of hHR23B in post-genotoxin regulation of p53 was investigated. Depletion of hHR23B by specific short interfering RNA before genotoxic exposure attenuated p53, p21 and bax induction, abrogated the accumulation of ubiquitinated p53 and suppressed apoptosis. Expression of ubiquitin derivatives with all lysines mutated except K48 or K63 demonstrated that K48-linked p53-ubiquitin conjugates were specifically induced after DNA damage. hHR23B, along with native and ubiquitinated p53, accumulated in chromatin after genotoxic exposure, and the accumulation of ubiquitinated p53 in chromatin was prevented by hHR23B depletion. Chromatin immunoprecipitation analysis demonstrated that hHR23B and p53 both localized to the p21 promoter shortly after DNA damage. hHR23B thus plays a critical role in the activation and function of p53 after specific genotoxic exposures.
Keywords: p53, hHR23, ubiquitin, proteasome, DNA damage, chromatin
Introduction
The Rad23 family of proteins integrate DNA repair with the ubiquitin (Ub)–proteasome system. They participate in global genome-nucleotide excision repair (GG-NER) by interacting with and stabilizing Rad4/XP-C, whereas a conserved ubiquitin-like (UbL) domain recognizes the proteasome and C-terminal ubiquitin-associated (UBA) domains capture polyubiquitin-conjugated proteins (Sweder and Madura, 2002; Ng et al., 2003; Ortolan et al., 2004; Xie et al., 2004; Hsieh et al., 2005). Type II UbL proteins, such as Rad23 and Dsk2, may serve as receptors for proteasome substrate recognition (Verma et al., 2004), but can also interfere with the degradation of ubiquitinated substrates, as well as block deubiquitinases, if in stoichiometric excess (Raasi and Pickart, 2003).
Human Rad23 proteins (hHR23A and B) can specifically modulate p53 abundance and stability. Depletion of hHR23A and B with short interfering RNA (siRNA) results in more rapid degradation of p53 by the proteasome, whereas their overexpression induces the accumulation of ubiquitinated p53 (Glockzin et al., 2003; Brignone et al., 2004). These seemingly paradoxical effects of hHR23 proteins on p53 degradation are consistent with suggestions that the effects of Rad23/hHR23 on degradation are highly sensitive to stoichiometric variation (Raasi and Pickart, 2003; Verma et al., 2004). Additionally, hHR23A and B interact with mouse double minute 2 (MDM2), where MDM2 functions to antagonize the stabilizing function of hHR23 toward p53, directly promoting p53 recognition and degradation by the proteasome (Brignone et al., 2004). This model has been recently bolstered by the finding that MDM2, itself, contacts the 20S core particle of the proteasome (Sdek et al., 2005).
Ubiquitination of p53 is generally considered as an inhibitory negative signal that leads to the protein’s destruction (Harris and Levine, 2005). Although transient increases in polyubiquitinated p53 have been observed after ionizing radiation (Maki and Howley, 1997), the mechanism of accumulation and function, if any, of these species remains unclear.
The present study was initiated to investigate the role of hHR23B in regulating p53 turnover and p53-dependent cellular responses after DNA damage. hHR23B was required for normal kinetics of p53 activation after DNA damage, and its depletion attenuated p53 target gene activation and apoptosis. Ubiquitinated p53 and hHR23B were observed in chromatin after DNA damage, in a manner dependent on physiological levels of hHR23B. hHR23B was colocalized with p53 at the p21 promoter after DNA damage, suggesting that ubiquitinated p53 species, protected by hHR23B, might function in p53 transcriptional responses.
Results and discussion
Physiologic hHR23B levels are required for normal p53 induction after DNA damage
As hHR23 proteins play an important role in both DNA repair and the regulation of p53 stability (Sweder and Madura, 2002; Brignone et al., 2004), the effect of siRNA-mediated knockdown of hHR23B after treatment of cells with different DNA-damaging agents was investigated. U2OS cells were exposed to control or hHR23B siRNA and then mock-treated or treated with doxorubicin (Dox), UV, etoposide, gamma irradiation (IR) or mitomycin-C (MMC) for 3, 6 or 16 h. The doses of each agent (except for IR, which was not cytotoxic for U2OS) were those in the linear range for cytotoxicity (Supplementary Figure S1). For each time point, cell lysates were analysed by immunoblot for levels of hHR23B, p53 and the p53 target genes bax and p21 (Figure 1a–e).
Figure 1.
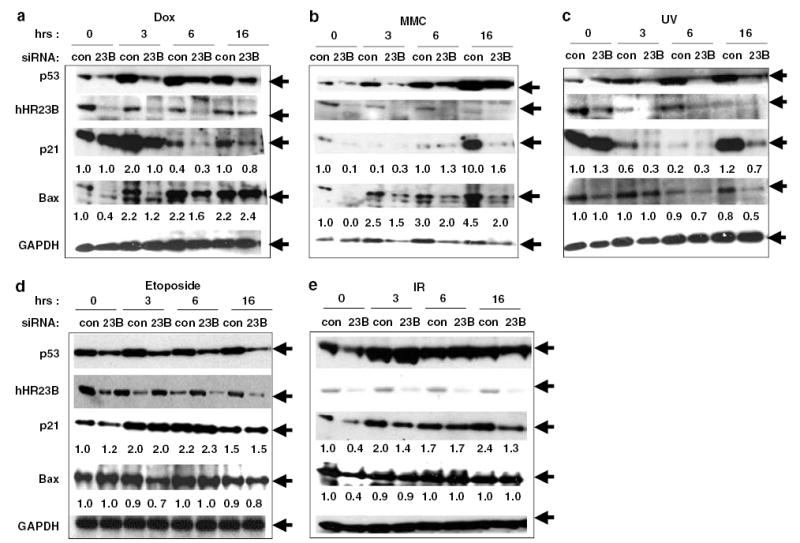
hHR23B depletion attenuates p53, p21 and bax induction by specific genotoxins. U2OS cells were transfected (Oligofectamine, Invitrogen, Carlsbad, CA, USA; 600 pmol siRNA/100 mm dish) with control (IX, Dharmacon, Lafayette, CO, USA) or hHR23B-specific siRNA (AAAGUCAGGCUGUGGUUGACC) for 30 h, followed by treatment with indicated agents and preparation of cell lysates (Brignone et al., 2004) at the indicated times. Lysates were immunoblotted for p53 (DO-1, Santa Cruz, CA, USA), hHR23B (BD, San Jose, CA, USA), p21 (C-19, Santa Cruz), bax (P-19, Santa Cruz) and GAPDH (Immunochemical, Long Beach, CA, USA) as indicated. (a) Dox (2 μM); (b) MMC (8 μg/ml); (c) UV irradiation (50 J/M2); (d) etoposide (1 μM); (e) IR (15 Gy). p21 and bax levels were quantitated by densitometry (values below the corresponding blot) and values normalized to GAPDH for lane loading and the band intensity at t = 0; control siRNA set to 1.0.
In all samples, hHR23B knockdown was evident (Figure 1a–e), although at late time points after MMC or UV treatment, the control levels of hHR23B also reproducibly decreased, as a likely normal cellular response to these particular stresses (M Kaur and S Grossman, unpublished observations). In those samples, hHR23B knockdown was still seen, but to a lesser degree owing to the lower baseline amount (Figure 1b and c). In control siRNA-treated cells, p53 abundance increased 4–12 h before bax or p21 induction was noted (Figure 1a–c), except for the rapid induction of both p53 and p21 after etoposide and IR (Figure 1d and e).
Notably, in certain cultures, levels of p21 were high at baseline, but then decreased early after DNA damage, followed by an expected increase, usually at 16 h after treatment (Figure 1a and c). This finding provided a useful control for the effect of hHR23B depletion on p21 levels independent of p53 induction. As can be seen in Figure 1a and c, hHR23B depletion resulted in no significant change in p21 levels at baseline. As for the transient decrease in p21 seen immediately (3–6 h) after only Dox, MMC and UV (Figure 1a–c), and not etoposide or IR (Figure 1d and e), this has been extensively reported, and likely because of transient destabilization of p21 protein (Wang et al., 1999; Itoh and Linn, 2005; Soria et al., 2006).
Induction of p53, p21 and bax was delayed or abrogated by hHR23B siRNA after UV, Dox or MMC (Figure 1a–c). However, hHR23B siRNA had little effect on induction of p21 after etoposide, although p53 levels were generally lower (Figure 1d). After IR, hHR23B siRNA modestly attenuated early p21 induction (30% decrease at 3 h), but not p53 induction, and bax was not induced after either IR or etoposide (Figure 1d and e). These results show that knockdown of hHR23B blocks both p53 stabilization and corresponding activation of at least two p53 target genes, specifically after UV, MMC and Dox. A less apparent impact of hHR23B depletion on either p53 stabilization or p21 induction is noted after etoposide or IR.
hHR23B-dependent accumulation of ubiquitinated p53 after DNA damage
Given the requirement for hHR23B during physiologic p53 induction after DNA damage, along with its known protective effect on p53 degradation in unstressed cells (Brignone et al., 2004), we investigated the consequences of hHR23B depletion on DNA damage-induced changes in p53 ubiquitination. p53–Ub conjugates accumulate transiently in cells exposed to gamma irradiation, although the mechanism of accumulation and function of the conjugates has not been determined (Maki and Howley, 1997). Similarly, high-molecular-weight p53-immunoreactive species accumulated transiently (peak = 6–16 h) after treatment of control siRNA-transfected U2OS cells with Dox (Figure 2a) and MMC (data not shown). Strikingly, hHR23B depletion abrogated the accumulation of ubiquitinated p53 in both MMC- (data not shown) and Dox-treated cells (Figure 2a). Thus, the accumulation of ubiquitinated p53 species after genotoxic exposure is specifically dependent on the presence of physiologic levels of hHR23B.
Figure 2.

hHR23B-dependent accumulation of p53–Ub conjugates after DNA damage. (a) hHR23B-dependent high-molecular-weight forms of p53 transiently accumulate after Dox exposure. U2OS cells were transfected with control or hHR23B-specific siRNA (30 h) followed by Dox treatment (2 μM) for indicated times and immunoblot analysis of cell lysates for p53, hHR23B and GAPDH. (b) K48-linked p53–Ub conjugates transiently accumulate after DNA damage. U2OS cells (100 mm dishes) were transfected (FuGene, Roche) with 5 μg of wild type, K48o or K63o HA-Ub expression plasmids (Wertz et al., 2004) and exposed to 1 μM Dox (0–24 h) 30 h after transfection. (upper panel) At each time point, cell lysates were immunonoprecipitated (IP’d) with anti-p53 Ab (FL-393, Santa Cruz) and IPs analysed by anti-HA (F-7, Santa Cruz) immunoblot. (lower panel) Total cell lysates were separately immunoblotted with anti-HA antibody.
To verify that high-molecular-weight ubiquitinated p53 species do accumulate after DNA damage, and to determine the lysine linkages of the observed conjugates, U2OS cells expressing HA-tagged Ub (Ub-wt), Ub-K48-only (Ub-K48o) and UB-K63-only (Ub-K63o) alleles were exposed to Dox, and lysates harvested at 0, 3, 6, 16 and 24 h were analysed by p53 immunoprecipitation (IP) and HA immunoblot. High-molecular-weight p53 conjugates containing Ub-wt accumulated and decayed with the same kinetics as the endogenous high-molecular-weight p53 species noted in Figure 2a (Figure 2b). Notably, K48-linked p53–Ub conjugates, which are of the type recognized by the hHR23 UBA domains (Raasi et al., 2004), exhibited the same kinetics of post-DNA damage induction as Ub-wt-linked p53 conjugates, whereas K63-linked p53–Ub conjugates were present in untreated cells but strongly suppressed within 6 h after damage (Figure 2b).
Requirement for hHR23B in genotoxin-mediated apoptosis
To address whether hHR23B depletion might, in addition to attenuating p53 activation, also alter genotoxin-mediated apoptosis, hHR23B-depleted or control cells were treated with Dox and then analysed for sub-G1 DNA content (Figure 3a). No significant changes in cell cycle profile were observed in hHR23B siRNA-treated cells, as compared with control cells after mock treatment (Figure 3a). hHR23B depletion, however, markedly reduced the apoptotic fraction at 32 h after Dox treatment (5%), as compared to the apoptotic fraction observed in control siRNA-treated cells (21%) (Figure 3a). These results indicate that Dox-mediated apoptosis requires physiologic levels of hHR23B.
Figure 3.
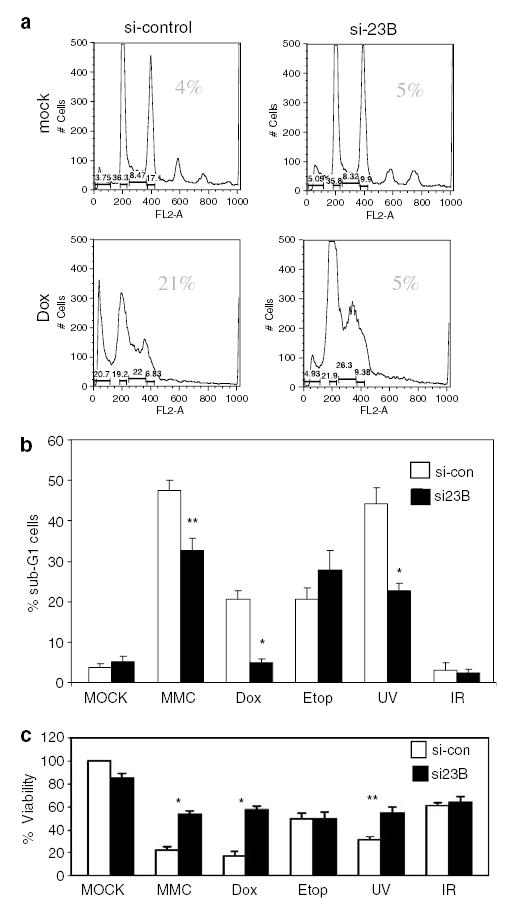
Genotoxin-mediated apoptosis requires hHR23B. (a) Dox-induced apoptosis of U2OS cells requires hHR23B. U2OS cells transfected with control or hHR23B-specific siRNA (30 h) were treated with Dox (2 μM) for 32 h, followed by propidium iodide staining and FACS analysis. The percentage of cells with sub-G1 DNA content is indicated in the upper right of each panel. Results of a representative experiment are shown. (b,c) Depletion of hHR23B attenuates genotoxin-induced apoptosis and cytotoxicity. U2OS cells were transfected with control (open bars) or hHR23B-specific siRNA (black bars), followed (after 30 h) by treatment with Dox (2 μM), MMC (8 μg/ml), etoposide (1 μM), UV (50 J/M2) or IR (15 Gy). At 32 h after initial genotoxic exposure, cells were stained with propidium iodide and analysed by FACS for sub-G1 content (b), or analysed for viability by 3-(4,5-dimethylthia-zol-2-yl)-2,5-diphenyl-2H-tetrazolium bromide (MTT) assay (c). In (c), the value obtained for mock and control siRNA-treated cells was set at 100%. Error bars represent 1.0 s.d. * P < 0.01 and ** P < 0.05.
To investigate whether hHR23B contributes to apoptosis caused by other genotoxins, the effect of hHR23B depletion on apoptosis induction and cell viability after treatment with UV, MMC, etoposide, IR, as well as Dox, was determined (Figure 3b and c). Additionally, the effect of XP-C knockdown on genotoxin-induced apoptosis was also determined to help distinguish the contribution to cell death from hHR23B-dependent NER (as part of the XP-C complex) vs its other functions in protein stability regulation (Supplementary Figure S2).
hHR23B depletion before UV, MMC or Dox treatment resulted in a statistically significant reduction (relative reduction 28% MMC, 47% UV, 77% Dox) in apoptosis and increase in viability (relative increase 160% UV, 250% MMC, 300% Dox; Figure 3b and c). IR did not cause apoptosis or loss of viability in U2OS cells, and hHR23B siRNA thus had no impact (Figure 3b and c). hHR23B depletion also had no significant effect on apoptotic fraction or cell viability after etoposide treatment (Figure 3b and c). The lack of a negative effect of hHR23B siRNA on etoposide-induced apoptosis also controlled for any non-specific effects of hHR23B siRNA on the apoptosis pathway, or of hHR23B itself on downstream events in apoptosis.
Robust XP-C depletion (Supplementary Figure S2a), in comparison to the effects of hHR23B depletion, had no impact (etoposide, UV) or promoted (MMC, Dox) genotoxin-induced apoptosis (Supplementary Figure S2b), suggesting that the inhibition of genotoxin-induced apoptosis and cytotoxicity due to hHR23B depletion could not be accounted for by a reduction in DNA repair efficiency. Thus, consistent with defective p53 and p53 target gene induction, apoptosis after Dox, MMC and UV was hindered after depletion of hHR23B, suggesting that hHR23B plays an important role in agent-specific genotoxin-mediated apoptosis that may depend on signals generated by specific types of lesions, such as the covalent adducts found especially after treatment with UV, MMC and Dox (Cutts et al., 2003; Pfeifer et al., 2005; Lee et al., 2006).
Localization of hHR23B and ubiquitinated p53 in chromatin
To gain further insight into the potential association between hHR23B, ubiquitinated p53 and p53 transcriptional responses, U2OS cells treated with control or hHR23B siRNA were exposed to Dox, and the chromatin fraction probed for p53 and hHR23B (Figure 4a). In control siRNA-treated cells, hHR23B and p53 were both found in chromatin before Dox exposure. Native p53 levels in chromatin were modestly induced (especially at 3 and 6 h) after DNA damage, whereas hHR23B levels were induced at 3 and 6 h and then fell at 16 and 24 h (Figure 4a). Unexpectedly, hHR23B levels in chromatin from hHR23B siRNA-treated cells, although reduced at 0, 6 and 16 h, were not reduced at the 3 and 24 h time points (Figure 4a, middle panel), despite a reduction in level at those time points when assayed in whole-cell lysates from the same cells (Figure 4a, lower panel). This may represent differential stability of hHR23B protein in chromatin vs. other cellular compartments, offsetting the decrease in total cellular hHR23B observed in whole-cell lysates.
Figure 4.
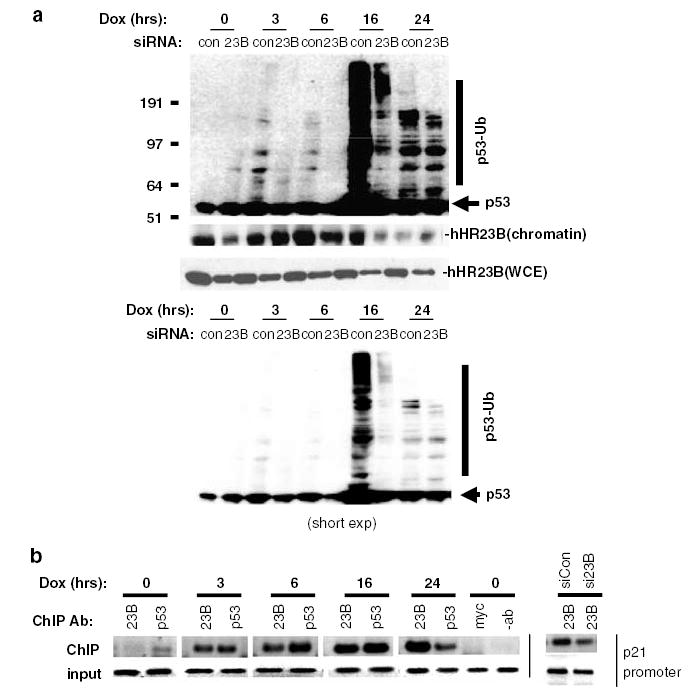
hHR23B, p53 and p53-Ub localization to chromatin and the p21 promoter after DNA damage. (a) hHR23B-dependent high-molecular-weight forms of p53 transiently accumulate in chromatin after Dox exposure. Chromatin isolated as described (Szak et al., 2001) from cells transfected with control or hHR23B siRNA (30 h) and treated with Dox (2 μM) for the indicated times 30 h after siRNA transfection was immunoblotted for p53 and hHR23B. Crosslinks were reversed in the chromatin preparation before immunoblot analysis as described (Szak et al., 2001). (b) hHR23B and p53 are coordinately recruited to the p21 promoter after DNA damage. (left panel) U2OS cells were treated with Dox (2 μM) for the indicated times, and hHR23B and p53 chromatin immunoprecipitations (ChIPs) analysed for the presence of the p21 promoter by PCR amplification of a 230 bp fragment (−2280 to −2050) as described (Szak et al., 2001). Negative control ChIPs were performed with chromatin derived from untreated (time = 0) cells using anti-myc antibody (myc) or protein G-Sepharose beads without antibody (−ab). (right panel) Chromatin prepared from U2OS cells treated with control or hHR23B siRNA for 30 h before treatment with Dox for 3 h was analysed by hHR23B ChIP.
As seen previously for p53 extracted in whole-cell lysates (Figure 2), high-molecular-weight p53 species consistent with p53–Ub conjugates were found in chromatin after control siRNA treatment and Dox exposure (detectable at 3 and 6 h, peak at 16 h), but at a significantly reduced level (especially at 3, 6 and 16 h), after hHR23B siRNA treatment (Figure 4a, top panel). Interestingly, the abundance of high-molecular-weight p53 species was most noticeably reduced at 16 h after Dox treatment, which was the time point at which maximum depletion of hHR23B in chromatin by siRNA was observed. At 24 h, however, hHR23B levels in chromatin were distinctly lower than those at earlier time points, but equal between control and hHR23B siRNA-treated samples, and the levels of high-molecular-weight p53 species were likewise equivalent. Physiologic levels of hHR23B are thus required for the induction and maintenance of p53–Ub conjugates in chromatin after DNA damage.
hHR23B localizes to the p21 promoter
To determine if hHR23B might serve a specific functional role in chromatin, the p21 promoter was analysed for the presence of hHR23B before and after genotoxic exposure (Figure 4b, left panel), and in the presence or absence of control or hHR23B siRNA (Figure 4b, right panel). Sheared, crosslinked chromatin was immunoprecipitated with anti-hHR23B, negative control anti-myc or positive control anti-p53 antibodies, followed by disruption of crosslinks and p21 promoter-specific PCR reaction. Whereas no antibody and control anti-c-myc chromatin immunoprecipitations (ChIPs) of chromatin prepared from untreated (t = 0) U2OS cells were devoid of the p21 promoter fragment (Figure 4b, left panel, last two lanes), anti-hHR23B and anti-p53 ChIPs captured limited (more for p53) but detectable amounts of the fragment (Figure 4b, left panel; first two lanes). As expected (Szak et al., 2001), p53 abundance at the p21 promoter dramatically increased by 3 h after Dox treatment, peaked between 6 and 16 h and declined at 24 h (Figure 4b, left panel; t = 3–24 h). hHR23B followed a similar pattern, with robust localization to the p21 promoter by 3 h, and continued increasing association through 24 h (Figure 4b, left panel; t = 3–24 h).
The specificity of the hHR23B ChIP for the detection of bona fide hHR23B and not an irrelevant crossreactive protein at the p21 promoter was confirmed by performing a similar ChIP on chromatin obtained from control or hHR23B siRNA-treated U2OS cells 3 h after Dox exposure. As shown in Figure 4b (right panel), substantially less p21 promoter fragment was observed in an anti-hHR23B ChIP of chromatin prepared from hHR23B vs control siRNA-treated cells. Thus, in U2OS cells, both p53 and hHR23B simultaneously load onto the p21 promoter within 3 h of DNA damage –coordinate with the first detection of ubiquitinated p53 species in chromatin and also preceding p21 induction that occurs at 16 h (Figure 1) after Dox treatment.
Of the agents tested in this work, UV, Dox and MMC shared a requirement for hHR23B to induce p53, bax, p21 and apoptosis, and also shared the property of causing covalent modifications of DNA (Cutts et al., 2003; Pfeifer et al., 2005; Lee et al., 2006). Intriguingly, a role for NER (in which hHR23 has a role) has been proposed in the sensing or repair of UV-, MMC- and Dox-induced lesions (Robles et al., 1999; Kruczynski et al., 2004; Lee et al., 2006). However, it does not appear that physiological levels of the hHR23 DNA repair partner protein XP-C were required for cell death, suggesting that hHR23B functions apart from repair were responsible for its impact on genotoxin-induced apoptosis. Moreover, mHR23B knockout mouse cells are not defective for NER, owing to compensation by mHR23A (Ng et al., 2002), suggesting that human cells depleted for hHR23B are likewise competent for NER. Thus, loss of NER is not a likely explanation for the effects of hHR23B depletion observed in this work.
The mechanism by which hHR23B is recruited to and functions in chromatin, if not via repair complexes, especially with regard to p53 ubiquitination and p21 activation, remains unclear. Previous work has suggested that hHR23A can be recruited in a three-way complex to p53 in vivo via bridging by MDM2, which interacts with both p53 and hHR23A/B (Brignone et al., 2004). If true, this model would predict that DNA damage, especially by UV, MMC and Dox, should increase the avidity or abundance of hHR23B/MDM2 complexes as a means of targeting hHR23B to the vicinity of p53 where it could stabilize p53-conjugated K48-linked Ub chain(s). Such an activity of hHR23 has been observed for model substrates (Wang et al., 2003; Raasi et al., 2004).
The hHR23B-dependent accumulation of K48-linked p53–Ub conjugates after DNA damage correlated with p53 stabilization, p21/bax induction and apoptosis. p53–Ub conjugates could contribute to transcription-dependent functions of p53, which are required for downstream p53 activities (Slee et al., 2004; Schuler and Green, 2005). A role for such ubiquitin-transcription factor conjugates in transcription regulation has been proposed, as the ubiquitination of transactivators and the recruitment of whole (26S) or partial (19S) protea-somes have been associated with RNA polymerase II transactivation (Muratani and Tansey, 2003). Moreover, the discovery of hHR23B loading onto the p21 promoter with p53 suggests that it, or similar Type II UbL proteins, might participate more broadly in activated transcription, either by stabilizing Ub chains, recruiting proteasomes or both.
Supplementary Material
Acknowledgments
We thank V Sharma for assistance with ChIP assays. This work was supported by CA107532 from NCI.
References
- Brignone C, Bradley KE, Kisselev AF, Grossman SR. A post-ubiquitination role for MDM2 and hHR23A in the p53 degradation pathway. Oncogene. 2004;23:4121–4129. doi: 10.1038/sj.onc.1207540. [DOI] [PubMed] [Google Scholar]
- Cutts SM, Swift LP, Rephaeli A, Nudelman A, Phillips DR. Sequence specificity of adriamycin-DNA adducts in human tumor cells. Mol Cancer Ther. 2003;2:661–670. [PubMed] [Google Scholar]
- Glockzin S, Ogi FX, Hengstermann A, Scheffner M, Blattner C. Involvement of the DNA repair protein hHR23 in p53 degradation. Mol Cell Biol. 2003;23:8960–8969. doi: 10.1128/MCB.23.24.8960-8969.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris SL, Levine AJ. The p53 pathway: positive and negative feedback loops. Oncogene. 2005;24:2899–2908. doi: 10.1038/sj.onc.1208615. [DOI] [PubMed] [Google Scholar]
- Hsieh HC, Hsieh YH, Huang YH, Shen FC, Tsai HN, Tsai JH, et al. HHR23A, a human homolog of Saccharo-myces cerevisiae Rad23, regulates xeroderma pigmentosum C protein and is required for nucleotide excision repair. Biochem Biophys Res Commun. 2005;335:181–187. doi: 10.1016/j.bbrc.2005.07.067. [DOI] [PubMed] [Google Scholar]
- Itoh T, Linn S. The fate of p21CDKN1A in cells surviving UV-irradiation. DNA Repair (Amst) 2005;4:1457–1462. doi: 10.1016/j.dnarep.2005.08.008. [DOI] [PubMed] [Google Scholar]
- Kruczynski A, Barret JM, Van Hille B, Chansard N, Astruc J, Menon Y, et al. Decreased nucleotide excision repair activity and alterations of topoisomerase IIalpha are associated with the in vivo resistance of a P388 leukemia subline to F11782, a novel catalytic inhibitor of topoisomerases I and II. Clin Cancer Res. 2004;10:3156–3168. doi: 10.1158/1078-0432.ccr-1305-2. [DOI] [PubMed] [Google Scholar]
- Lee YJ, Park SJ, Ciccone SL, Kim CR, Lee SH. An in vivo analysis of MMC-induced DNA damage and its repair. Carcinogenesis. 2006;27:446–453. doi: 10.1093/carcin/bgi254. [DOI] [PubMed] [Google Scholar]
- Maki CG, Howley PM. Ubiquitination of p53 and p21 is differentially affected by ionizing and UV radiation. Mol Cell Biol. 1997;17:355–363. doi: 10.1128/mcb.17.1.355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muratani M, Tansey WP. How the ubiquitin–protea-some system controls transcription. Nat Rev Mol Cell Biol. 2003;4:192–201. doi: 10.1038/nrm1049. [DOI] [PubMed] [Google Scholar]
- Ng JM, Vermeulen W, van der Horst GT, Bergink S, Sugasawa K, Vrieling H, et al. A novel regulation mechanism of DNA repair by damage-induced and RAD23-dependent stabilization of xeroderma pigmentosum group C protein. Genes Dev. 2003;17:1630–1645. doi: 10.1101/gad.260003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng JM, Vrieling H, Sugasawa K, Ooms MP, Grootegoed JA, Vreeburg JT, et al. Developmental defects and male sterility in mice lacking the ubiquitin-like DNA repair gene mHR23B. Mol Cell Biol. 2002;22:1233–1245. doi: 10.1128/MCB.22.4.1233-1245.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ortolan TG, Chen L, Tongaonkar P, Madura K. Rad23 stabilizes Rad4 from degradation by the Ub/proteasome pathway. Nucleic Acids Res. 2004;32:6490–6500. doi: 10.1093/nar/gkh987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfeifer GP, You YH, Besaratinia A. Mutations induced by ultraviolet light. Mutat Res. 2005;571:19–31. doi: 10.1016/j.mrfmmm.2004.06.057. [DOI] [PubMed] [Google Scholar]
- Raasi S, Orlov I, Fleming KG, Pickart CM. Binding of polyubiquitin chains to ubiquitin-associated (UBA) domains of HHR23A. J Mol Biol. 2004;341:1367–1379. doi: 10.1016/j.jmb.2004.06.057. [DOI] [PubMed] [Google Scholar]
- Raasi S, Pickart CM. Rad23 ubiquitin-associated domains (UBA) inhibit 26 S proteasome-catalysed proteolysis by sequestering lysine 48-linked polyubiquitin chains. J Biol Chem. 2003;278:8951–8959. doi: 10.1074/jbc.m212841200. [DOI] [PubMed] [Google Scholar]
- Robles AI, Wang XW, Harris CC. Drug-induced apoptosis is delayed and reduced in XPD lymphoblastoid cell lines: possible role of TFIIH in p53-mediated apoptotic cell death. Oncogene. 1999;18:4681–4688. doi: 10.1038/sj.onc.1202862. [DOI] [PubMed] [Google Scholar]
- Schuler M, Green DR. Transcription, apoptosis and p53: catch-22. Trends Genet. 2005;21:182–187. doi: 10.1016/j.tig.2005.01.001. [DOI] [PubMed] [Google Scholar]
- Sdek P, Ying H, Chang DL, Qiu W, Zheng H, Touitou R, et al. MDM2 promotes proteasome-dependent ubiquitin-independent degradation of retinoblastoma protein. Mol Cell. 2005;20:699–708. doi: 10.1016/j.molcel.2005.10.017. [DOI] [PubMed] [Google Scholar]
- Slee EA, O’Connor DJ, Lu X. To die or not to die: how does p53 decide? Oncogene. 2004;23:2809–2818. doi: 10.1038/sj.onc.1207516. [DOI] [PubMed] [Google Scholar]
- Soria G, Podhajcer O, Prives C, Gottifredi V. P21Cip1/WAF1 downregulation is required for efficient PCNA ubiquitination after UV irradiation. Oncogene. 2006;25:2829–2838. doi: 10.1038/sj.onc.1209315. [DOI] [PubMed] [Google Scholar]
- Sweder K, Madura K. Regulation of repair by the 26S proteasome. J Biomed Biotechnol. 2002;2:94–105. doi: 10.1155/S1110724302205033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szak ST, Mays D, Pietenpol JA. Kinetics of p53 binding to promoter sites in vivo. Mol Cell Biol. 2001;21:3375–3386. doi: 10.1128/MCB.21.10.3375-3386.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verma R, Oania R, Graumann J, Deshaies RJ. Multiubiquitin chain receptors define a layer of substrate selectivity in the ubiquitin–proteasome system. Cell. 2004;118:99–110. doi: 10.1016/j.cell.2004.06.014. [DOI] [PubMed] [Google Scholar]
- Wang JA, Fan S, Yuan RQ, Ma YX, Meng Q, Goldberg ID, et al. Ultraviolet radiation down-regulates expression of the cell-cycle inhibitor p21WAF1/CIP1 in human cancer cells independently of p53. Int J Radiat Biol. 1999;75:301–316. doi: 10.1080/095530099140483. [DOI] [PubMed] [Google Scholar]
- Wang Q, Goh AM, Howley PM, Walters KJ. Ubiquitin recognition by the DNA repair protein hHR23a. Biochemistry. 2003;42:13529–13535. doi: 10.1021/bi035391j. [DOI] [PubMed] [Google Scholar]
- Wertz IE, O’Rourke KM, Zhou H, Eby M, Aravind L, Seshagiri S, et al. De-ubiquitination and ubiquitin ligase domains of A20 downregulate NF-kappaB signalling. Nature. 2004;430:694–699. doi: 10.1038/nature02794. [DOI] [PubMed] [Google Scholar]
- Xie Z, Liu S, Zhang Y, Wang Z. Roles of Rad23 protein in yeast nucleotide excision repair. Nucleic Acids Res. 2004;32:5981–5990. doi: 10.1093/nar/gkh934. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.