

Abstract
Bad, a proapoptotic Bcl-2 family protein, plays a critical role in determining cell death/survival. The phosphatidylinositol 3-kinase (PI3-K)/Akt pathway and the c-Jun N-terminal kinase (JNK) pathway are thought to be involved in regulation of Bad. Therefore, the present study was done to clarify the role of Bad as a common target of the PI3-K/Akt and JNK pathways after transient focal cerebral ischemia (tFCI) in rats. We found that Akt activity increased at 3 h and then decreased, whereas JNK activity increased 7 to 24 h in the peripheral area after tFCI. Administration of LY294002, a PI3-K-specific inhibitor, exacerbated DNA fragmentation, whereas administration of SP600125, a JNK-specific inhibitor, attenuated it. Inhibited by LY294002, phospho-Bad (Ser136) expression increased in the peripheral area 3 h after tFCI, with suppression of Akt activity. Furthermore, phospho-Bad (Ser136) and phospho-Akt (Ser473) were colocalized. Decreases in phospho-Bad (Ser136) and Bad/14-3-3 dimerization, and increases in Bcl-XL/Bad or Bcl-2/Bad dimerization observed 7 to 24 h after tFCI, were prevented by SP600125 administration, with inhibition of JNK activity. The present study indicates that signal predominance varies from PI3-K/Akt-mediated survival signaling to JNK-mediated death signaling with the development of neuronal damage in the peripheral area after tFCI. This study also suggests that PI3-K/Akt has a role in Bad inactivation, whereas the JNK pathway is involved in Bad activation. We conclude that Bad may be an integrated checkpoint of PI3-K/Akt-mediated survival signaling and JNK-mediated death signaling and that it contributes to cell fate in the peripheral area after cerebral ischemia.
Keywords: Akt, Bad, cerebral ischemia, JNK, signal balance
Introduction
Cell fate is determined by the balance of survival and death signals under normal and pathological conditions. Survival signals raise the threshold above which apoptotic signals can occur, and a reduction in survival signals renders cells more susceptible to apoptotic signals. The mitochondria-dependent apoptotic pathway plays a critical role in neuronal death after cerebral ischemia (Fujimura et al, 1999; Sugawara et al, 1999).
Akt is a substrate of phosphatidylinositol-3 kinase (PI3-K) and inhibits apoptosis by inactivating proapoptotic proteins such as Bad and by activating anti-apoptotic proteins such as NF-kappaB (Datta et al, 1997; del Peso et al, 1997; Kane et al, 1999; Ozes et al, 1999). A temporal increase in Akt phosphorylation was reported after cerebral ischemia (Ouyang et al, 1999; Noshita et al, 2001; Yano et al, 2001); Akt activation is considered to be neuroprotective.
In contrast, c-Jun N-terminal kinase (JNK) is activated after various cell stress applications (Davis, 2000). In addition to c-Jun-mediated transcription mechanisms, JNK promotes apoptotic cell death by direct regulation of Bcl-2 family members such as Bcl-2, Bim, and Bad (Deng et al, 2001; Donovan et al, 2002; Lei et al, 2002; Bhakar et al, 2003; Putcha et al, 2003; Becker et al, 2004). More recently, studies have shown that JNK triggers the mitochondrial apoptotic pathway via activation of Bax and Bim after cerebral ischemia (Okuno et al, 2004; Gao et al, 2005).
Bad promotes an apoptotic cascade by binding to and inhibiting actions of Bcl-XL and Bcl-2 (Yang et al, 1995; Zha et al, 1997). Akt mediates phosphorylation of Bad (Ser136), which binds with 14-3-3 and reduces the attachment of Bad to Bcl-XL (Zha et al, 1996; Datta et al, 1997; del Peso et al, 1997). In contrast, JNK reduces the affinity of Bad for 14-3-3 and promotes the association of Bad with Bcl-XL or Bcl-2 via phosphorylation of Bad (Ser128) or 14-3-3 (Donovan et al, 2002; Sunayama et al, 2005). Bad has been reported to be involved in cell death after brain ischemia (Saito et al, 2003, Abe et al, 2004, D³użniewska et al, 2005).
Many neuroprotective agents that target only cell death pathways have been failures (Chan, 2004). A key to regulating cell death/survival is to clarify the molecular mechanisms by which survival and apoptotic signals integrate. Bad may be a molecular switch for both survival and apoptotic signals and may contribute to cell fate. Therefore, the present study was performed to clarify the role of Bad as an integrated checkpoint of survival and death signals in the peripheral area after brain ischemia.
Materials and methods
Animal Model
Adult male Sprague-Dawley rats (250 to 280 g) were used in the present study. The animals were anesthetized with an intraperitoneal injection of pentobarbital (40 mg/kg). A burr hole (2-mm diameter) was carefully made in the skull for measurement of cerebral blood flow (CBF), with dura matter preserved at this time. The location of the burr hole was 3 mm dorsal and 5 mm lateral to the left from the bregma, which is located in the upper part of the middle cerebral artery (MCA) territory.
The next day, the animals were anesthetized with a nitrous oxide/oxygen/isoflurane mixture (69%/30%/2%) during surgical preparation. After a midline skin incision, the left external carotid artery was exposed and its branches were electrocoagulated. A 22.0-mm 3-0 surgical monofilament nylon suture, blunted at the end, was introduced into the left internal carotid artery through the external carotid artery stump, according to our previous report (Okuno et al, 2004). Body temperature was maintained at 37 ± 0.5°C, using a heating pad, during the surgical procedure for MCA occlusion (MCAO). After 90 min of MCAO, CBF was restored by removal of the nylon thread. Blood samples were collected from the tail artery before and during MCAO, and just after reperfusion for measurement of pH, PO2, and PCO2. Regional CBF (rCBF) of the left frontoparietal cortex region was measured before and after MCAO and after reperfusion using a laser blood flowmeter (LASERFLO; Vasamedics, St. Paul, MN, USA). Sham control animals were treated in the same way without MCAO. After the incision was closed, the animals recovered and were allowed free access to water and food at ambient temperature until sampling. The experimental protocol and procedures were in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals and were approved by the Administrative Panel on Laboratory Animal Care of Stanford University.
Drug Injection
To investigate the role of the PI3-K/Akt pathway and the JNK pathway after transient focal cerebral ischemia (tFCI), a highly selective PI3-K inhibitor and a specific JNK inhibitor were injected, according to our previous reports (Noshita et al, 2001; Okuno et al, 2004; Yu et al, 2005). The PI3-K inhibitor, LY294002 (Calbiochem, San Diego, CA, USA), and the JNK inhibitor, SP600125 (Calbiochem), were dissolved in dimethyl sulfoxide (DMSO) and phosphate-buffered saline (PBS), respectively. The scalp was incised on the midline, and the skull was exposed. LY294002 (5 mmol/L in 25% DMSO in PBS), SP600125 (1.0 mg/kg in 25% DMSO in PBS) and the vehicle (25% DMSO in PBS) were injected intracerebroventricularly (10 μl, bregma; 1.4 mm lateral, 0.8 mm posterior, 3.6 mm deep) 30 min before MCAO.
Histology
One, 3, 7, and 24 h after reperfusion (n = 5 per each time point), the rats were deeply anesthetized with pentobarbital and perfused through the heart with 200 ml of ice cold heparinized saline followed by 300 ml of 4% paraformaldehyde in 0.1 M phosphate buffer (pH 7.4). Their brains were removed and postfixed by immersion in the same fixative (1 day, 4°C) and rapidly frozen after cryoprotection. For histochemistry, frozen coronal sections were cut from the same region encompassing the striatum (bregma +0.70 mm) at 10 μm on a cryostat at −25°C and collected on glass slides. Brain sections from each animal were stained with cresyl violet for assessment of brain injury. Infarction was recognized by the loss of staining and the abundance of neurons with typical ischemic cell changes, i.e., shrunken and darkly stained cell bodies with condensed, often triangular and darkly stained nuclei (Garcia, 1992). To investigate DNA fragmentation, we performed a terminal deoxynucleotidyl transferase-mediated dUTP nick end labeling (TUNEL) study using a commercial kit (NeuroTACS II in situ apoptosis detection kit, model 4823-30-K; Trevigen, Gaithersburg, MD, USA). Briefly, after pretreatment with NeuroPore (kit component), the sections were incubated with a mixture of terminal deoxynucleotidyl transferase (TdT) dNTP mix, TdT enzyme, and TdT Mn2+ (kit components) in 1 × TdT-labeling buffer for 1 h at 37°C. The sections were then incubated with streptavidin-horseradish peroxidase, and staining was developed with diaminobenzidine.
Immunohistochemistry
The brain sections were prepared as those for the histological studies. They were incubated in 0.3% hydrogen peroxide in methanol for 30 min to quench endogenous peroxide. After washing in Tris-buffered saline (TBS), nonspecific binding was blocked with 5% normal bovine serum in TBS. The brain sections were incubated overnight at 4°C with a rabbit polyclonal anti-phospho-Akt (p-Akt) (Ser473) antibody (1:100; Cell Signaling Technology, Beverly, MA, USA), a mouse monoclonal anti-phospho-JNK (p-JNK) (Thr183/Tyr185) antibody (1:100; Santa Cruz Biotechnology, Santa Cruz, CA, USA), and a rabbit polyclonal anti-phospho-Bad (p-Bad) (Ser136) antibody (1:50; Oncogene Research Products, San Diego, CA, USA). The sections were then incubated with biotinylated anti-rabbit immunoglobulin G (IgG) or anti-mouse IgG at a 1:200 dilution, followed by incubation with avidin-biotin-peroxidase complex (Vector Laboratories, Burlingame, CA, USA) for 30 min. Diaminobenzidine was used as a color substrate. A set of sections were stained in a similar way without the primary antibodies to confirm the specificity of the primary antibodies. The sections were examined with a microscope (Axioplan 2; Carl Zeiss Jena GmbH, Jena, Germany). The region of interest (ROI) was set in the medial region of the corpus callosum, according to a rat brain atlas (Paxinos and Watson, 1996), as shown in Figure 1C. The location of these ROI (ROI1) was determined based on the results of cresyl violet staining and a TUNEL study, as described below. The area of loss of cresyl violet staining and TUNEL-positive cells expanded to the medial part of the striatum and some parts of the cortex between 7 and 24 h. Therefore, we defined the medial striatal region as the peripheral area. We investigated the temporal profile of interesting proteins in this region while confirming the spatial relationship between the proteins and the area of loss of cresyl violet staining in the current study.
Figure 1.
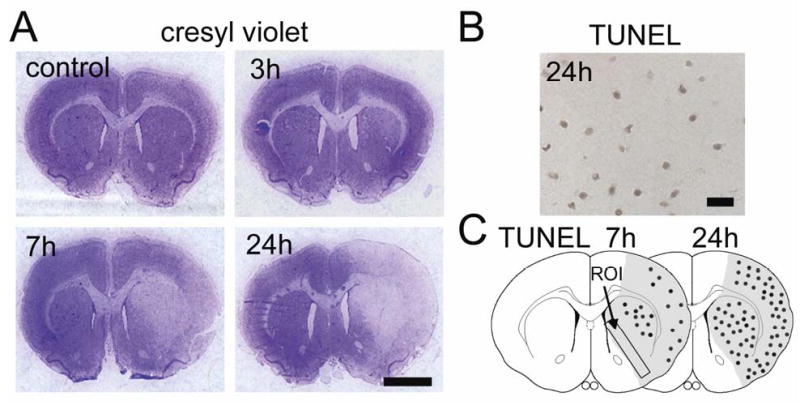
(A) Representative low-magnification photographs of cresyl violet staining. A pale, weakly stained area appeared in the lateral part of the striatum 3 h after tFCI. The loss of staining expanded to the medial part of the striatum and cortex between 7 and 24 h. Scale bar represents 2.5 mm. (B) Representative photomicrograph of TUNEL staining in the medial striatal region 24 h after tFCI, when TUNEL-positive cells were observed in the entire ischemic area. Scale bar represents 50 μm. (C) Illustration of the distribution of TUNEL-positive cells 7 and 24 h after tFCI. These cells were observed in the lateral part of the striatum at 7 h with some scattered positive cells in the cortex, and increased markedly in the entire ischemic area at 24 h. ROI was set in the coronal brain slice 0.7 mm anterior to bregma based on the rat brain atlas by Paxinos and Watson (1996); ROI1 indicates the medial striatal region. The shaded areas represent the ischemic area in the MCA territory based on the results of cresyl violet staining 24 h after tFCI.
Immunofluorescent Double Labeling
For the immunofluorescence detection of either p-Akt (Ser473) or p-Bad (Ser136) and neuron-specific nuclear protein (NeuN), the sections were incubated overnight at 4°C together with the above-mentioned anti-p-Akt (Ser473) (1:100) or anti-p-Bad (Ser136) antibodies (1:50), and a mouse monoclonal anti-NeuN antibody (1:500; Chemicon, Temecula, CA, USA). The sections were then incubated with Alexa 546-labeled goat anti-rabbit IgG and Alexa 488-labeled donkey anti-mouse IgG (1:250; Molecular Probes, Eugene, OR, USA) for 1 h at room temperature.
For the immunofluorescence detection of p-JNK (Thr183/Tyr185) and NeuN, the sections were incubated overnight at 4°C with the above-mentioned anti-p-JNK (Thr183/Tyr185) antibody (1:100). The sections were then incubated with Alexa 546 goat anti-mouse IgG (1:250; Molecular Probes) for 1 h at room temperature. After washing in TBS, the sections were incubated with Alexa 488-conjugated mouse anti-NeuN monoclonal antibody (1:500; Chemicon).
For the immunofluorescence detection of p-Akt (Ser473) and p-JNK (Thr183/Tyr185), the sections were incubated together with the above-mentioned anti-p-JNK (Thr183/Tyr185) (1:100) and anti-p-Akt (Ser473) (1:100) antibodies. The sections were then incubated with Alexa 546-labeled goat anti-mouse IgG and Alexa 488-labeled donkey anti-rabbit IgG (1:250; Molecular Probes) for 1 h at room temperature.
For the immunofluorescence detection of p-Bad (Ser136) and p-Akt (Ser473), the sections were incubated together with the above-mentioned anti-p-Bad (Ser136) (1:50) and mouse monoclonal anti-p-Akt (Ser473) (1:100) antibodies (Cell Signaling Technology). The sections were then incubated with Alexa 546-labeled goat anti-mouse IgG and Alexa 488-labeled donkey anti-rabbit IgG (1:250; Molecular Probes) for 1 h at room temperature.
A set of sections was stained in a similar way without the primary antibodies. Immunofluorescence was evaluated using the above-mentioned microscope. Fluorescence of Alexa 488 was observed at excitation of 495 nm and emission of > 515 nm and fluorescence of Alexa 546 was observed at excitation of 510 nm and emission of > 580 nm.
Western Blot Analysis
For Western blot analysis, four sham-operated rats and 16 rats with ischemia (1, 3, 7, and 24 h after 90 min of MCAO; n = 4 at each time point) were studied. Samples were obtained from the entire MCA territory on the ischemic side. After decapitation, the brains were quickly removed and cut into pieces. For whole-cell protein extraction, the tissue was sonicated with about seven volumes of protein extraction buffer (20 mmol/L HEPES potassium hydroxide [pH 7.5], 10 mmol/L potassium chloride, 1.5 mmol/L magnesium chloride, 1 mmol/L EDTA, 1 mmol/L EGTA, 0.7% protease inhibitor cocktail [p8340; Sigma-Aldrich, St. Louis, MO, USA], and 1% phosphatase inhibitor cocktails [p2850 and p5726; Sigma]). The homogenate was centrifuged at 10,000 × g for 15 min and the supernatant was used for this study. Assays to determine the protein concentration were performed by comparison with a known concentration of bovine serum albumin using a kit (23227; Pierce, Rockford, IL, USA).
Sodium dodecyl sulfate-polyacrylamide gel electrophoresis was performed according to our previous report (Noshita et al, 2001). In brief, the lysate equivalent of 40 μg of protein from each brain was run on the gel at 120 V together with a size marker (RPN800; Amersham, Piscataway, NJ, USA). The protein on the gel was subsequently transferred to a polyvinylidene fluoride transfer membrane (LC2002; Invitrogen, Carlsbad, CA, USA) in a buffer containing methanol, glycine, Tris base, and sodium dodecyl sulfate. After the transfer, the membrane was placed in 5% powdered milk in TBS with 0.1% Tween 20 to block nonspecific binding and was then incubated with primary antibodies for 12 h at 4°C. After washing, membranes were treated with horseradish peroxidase-conjugated secondary antibodies and then with enhanced chemiluminescence Western blotting detection reagents (RPN2132; Amersham). The same membranes were subsequently used for β-actin immunodetection and equal protein loading was ensured.
Coimmunoprecipitation
The procedure for precipitation was performed as described previously with some modification (Springer et al, 2000). For coimmunoprecipitation, four sham-operated rats and 16 rats with ischemia (1, 3, 7, and 24 h after 90 min of MCAO; n = 4 at each time point) were studied. Samples were prepared as described in the Western blot analysis. Three hundred micrograms of protein were used for coimmunoprecipitation. The protein samples were incubated with 50% slurry of protein G-Sepharose (Amersham) for 1 h at 4°C, and mixed samples were centrifuged at 14,000 × g for 2 min. The supernatant was incubated with 2 μg of antibodies and 20 μl of protein G-Sepharose at 4°C overnight. The negative control was prepared with protein G-Sepharose without an antibody. The 14,000 × g pellets were washed four times. After adding the same volume of Tris-glycine sodium dodecyl sulfate sample buffer (Invitrogen) to the samples, we boiled these samples to remove the Sepharose beads. After centrifugation at 14,000 × g for 1 min, the supernatant was immunoblotted as described in the Western blot analysis. Whole-cell extracts from sham control brains (40 μg) were also immunoblotted as a positive control.
Antibodies
The following antibodies were used for Western blot analysis: rabbit polyclonal anti-p-Akt (Ser473), rabbit polyclonal anti-Akt, rabbit monoclonal anti-JNK (56G8) (all from Cell Signaling Technology), rabbit polyclonal p-Bad (Ser136) (Oncogene Research Products), rabbit polyclonal p-Bad (Ser128) (Novus Biologicals, Littleton, CO, USA), mouse monoclonal anti-Bad, mouse monoclonal anti-Bcl-2 (BD PharMingen, San Jose, CA, USA), mouse monoclonal anti-14-3-3, mouse monoclonal anti-Bcl-XL (Santa Cruz Biotechnology), and mouse monoclonal anti-β-actin (Sigma-Aldrich).
The following antibodies were used for coimmunoprecipitation: rabbit polyclonal anti-14-3-3 (Santa Cruz Biotechnology) and rabbit polyclonal anti-Bad (Cell Signaling Technology).
Akt and JNK Activity Assays
JNK and Akt activity was measured with immune complex protein kinase assays, according to the manufacturer’s protocol (Cell Signaling Technology). Samples were prepared as described in the Western blot analysis. Equal volumes of cell lysate (300 μg) were incubated with immobilized Akt (1G1) monoclonal antibody or c-Jun fusion protein beads for Akt or JNK, respectively, at 4°C overnight. After centrifugation, the pellets were suspended by kinase buffer (25 mmol/L Tris [pH 7.5], 5 mmol/L glycerol phosphate, 2 mmol/L dithiothreitol, 0.1 mmol/L Na3VO4, 10 mmol/L MgCl2, and 200 μM adenosine triphosphate) and then were subjected to kinase assays with glycogen synthase kinase (GSK)-3 fusion protein or c-Jun fusion protein as a specific substrate, respectively, at 30°C for 30 min. The activity for Akt or JNK was then measured by Western blot with a 1:1000 dilution of primary antibodies (rabbit polyclonal phospho-GSK3α/β [Ser21/9] antibody or rabbit polyclonal phospho-c-Jun [Ser63]) as described in the Western Blot analysis.
Cell Death Assay
For quantification of apoptosis-related DNA fragmentation, we used a commercial enzyme immunoassay to determine cytoplasmic histone-associated DNA fragments (Roche Diagnostics, Penzberg, Germany). This assay detects apoptotic but not necrotic cell death (Bonfoco et al, 1995). Samples were obtained from the entire MCA territory. Fresh brain tissue was cut into pieces after 24 h of reperfusion, homogenized with a Teflon homogenizer in lysis buffer (50 mmol/L KH2PO4 and 0.1 mmol/L EDTA, pH 7.8), and spun for 10 min at 750 × g . The supernatant was collected and the protein concentration was determined. A cytosolic fraction containing 20 μg of protein was used for the enzyme-linked immunosorbent assay, according to the manufacturer’s protocol.
Quantification and Statistical Analysis
To evaluate the results of the Western blot and coimmunoprecipitation studies, we scanned the film with an imaging densitometer (GS-700; Bio-Rad Laboratories, Hercules, CA, USA) and quantified the optical density with Multi-Analyst software (Bio-Rad Laboratories) as previously reported (Hayashi et al, 2004). The data are expressed as mean ± SD. Statistical evaluation of each sample was carried out by one-way analysis of variance, followed by the Bonferroni/Dunn post hoc test (StatView, version 5.01; SAS Institute Inc., Cary, NC, USA). P < 0.05 was considered a change to a statistically significant degree. Comparison between vehicle- and drug-treated groups was achieved using analysis of variance for repeated measure with Fisher’s protected least-significant difference test post hoc analysis (Stat View). Significance was accepted with P < 0.05.
Results
Physiological Parameters and Histology
There was no significant difference in physiological parameters such as pH, PO2, and PCO2 between the vehicle- and drug-treated groups before or during MCAO or after reperfusion (data not shown). Similarly, each group had the same body temperatures within 37 ± 0.5°C during the surgical procedure for MCAO. The mean rCBF at the left frontoparietal cortex was 14.4 ± 3.7% just after MCAO and 92.0 ± 6.9% just after reperfusion (data represent percentage of before MCAO as mean ± SD). There was also no significant difference in rCBF between the vehicle- and drug-injected groups (data not shown). A cresyl violet staining study showed no neuronal degeneration until 1 h, but a pale, slightly stained area appeared in the lateral part of the striatum 3 h after tFCI (Figure 1A). The damaged area expanded to the medial part of the striatum and the cortex at 7 h, and almost the entire MCA territory became infarcted at 24 h (Figure 1A). A TUNEL study showed that positively-labeled cells were observed in the lateral part of the striatum with some minor labeling in the cortex 7 h after tFCI (Figure 1C). Massive TUNEL labeling was distributed over almost all of the MCA territory 24 h after tFCI (Figure 1B and 1C).
Signal Predominance Varied from Akt Phosphorylation to JNK Phosphorylation with the Development of Cellular Damage after tFCI
p-Akt (Ser473) and Akt were visualized as 60-kDa bands in the whole-cell fraction from the rat brains (Figure 2A). p-Akt (Ser473) was constitutively expressed in the entire region of the sham control brains (Figure 2A and 2C). There was no change in total Akt protein expression after reperfusion in the ischemic brains (Figure 2A). The immunoreactivity of p-Akt (Ser473) increased significantly at 3 h and returned to the level of the sham controls 7 h after tFCI (Figure 2A and 2C). p-Akt (Ser473) immunoreactivity was observed mainly in the cytoplasm and localized mainly in NeuN-positive cells in the medial striatal region at 3 h (Figure 2C and 2D). p-Akt (Ser473) immunoreactivity was not detected in the infarction core area at any time point after tFCI. Akt activity also increased markedly at 3 h and then decreased to the basal level at 24 h (Figure 2B).
Figure 2.
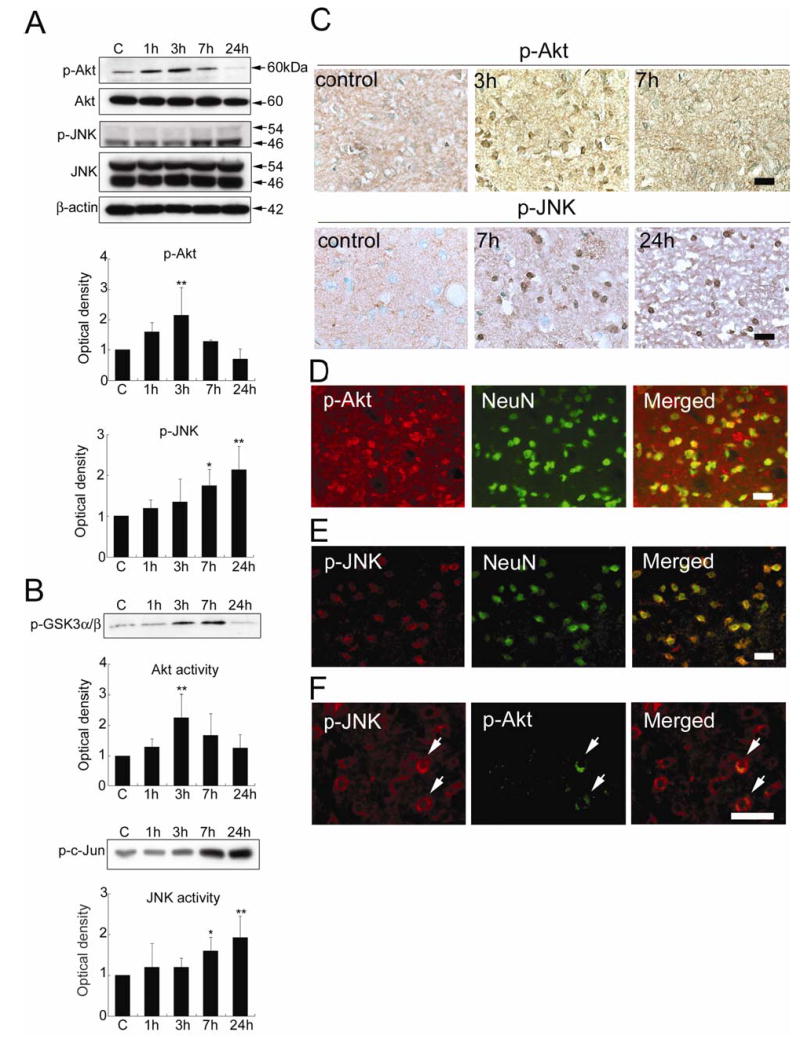
(A) Western blot analysis of phospho-Akt (Ser473) (p-Akt), Akt, phospho-JNK (Thr183/Tyr185) (p-JNK), and JNK in the whole-cell fraction from the MCA territory in rat brains. Expression of p-Akt increased 3 h after tFCI (**P < 0.01). In contrast, p-JNK expression increased at 7 h (*P < 0.05) and further increased up to 24 h (**P < 0.01) after tFCI. Akt and JNK did not significantly change at any time point after tFCI. β-actin was used as an internal control. (B) Akt activity assay with GSK-3 fusion protein as the kinase substrate (p-GSK3α/β) and JNK activity assay with c-Jun fusion protein as the kinase substrate (p-c-Jun) in the whole-cell fraction from the MCA territory of the rat brains. Akt activity increased significantly at 3 h (**P < 0.01), whereas JNK activity increased markedly 7 h (*P < 0.05) and 24 h (**P < 0.01) after tFCI. (C) Representative photomicrographs of immunostaining for p-Akt or p-JNK in the medial striatal region (ROI1). Staining of p-Akt was constitutively observed in the sham control brains. Expression of p-Akt increased at 3 h and returned to the basal level 7 h after tFCI. Expression of p-Akt was detected mainly in the cytoplasm. In contrast, p-JNK was weakly stained in the sham control brains. Immunoreactivity of p-JNK increased at 7 h and was maintained at 24 h after tFCI. p-JNK was expressed mainly in the cytoplasm at 7 h and the staining became more distinct in the perinuclear cytoplasm at 24 h. Scale bars represent 50 μm. (D) Representative photomicrographs of double staining for p-Akt and NeuN in the medial striatal region (ROI1) 3 h after tFCI. Immunoreactivity of p-Akt was mainly detected in the NeuN-positive cells. Scale bar represents 50 μm. (E) Representative photomicrographs of double staining for p-JNK and NeuN in the medial striatal region (ROI1) 7 h after tFCI. Almost all of the p-JNK staining was colocalized with NeuN immunoreactivity. Scale bar represents 50 μm. (F) Representative photomicrographs of double staining for p-JNK and p-Akt in the peripheral area of the MCA territory 7 h after tFCI. Only a few p-JNK-positive cells were double positive for p-Akt (arrows). Scale bar represents 50 μm.
In contrast, bands of p-JNK (Thr183/Tyr185) and JNK were observed at 46 kDa and 54 kDa (Figure 2A). p-JNK (Thr183/Tyr185) was weakly stained in the entire region of the sham control brains (Figure 2C). Total JNK immunoreactivity did not change significantly after tFCI (Figure 2A). Western blot analysis showed that p-JNK (Thr183/Tyr185) immunoreactivity increased significantly at 7 h and reached a peak 24 h after tFCI (Figure 2A). Immunopositive cells for p-JNK (Thr183/Thy185) increased at 7 and 24 h in the medial striatal region after tFCI (Figure 2C). Immunoreactivity for p-JNK (Thr183/Thy185) was detectable mainly in the cytoplasm at 7 h and became stronger in the peri-nuclear cytoplasm at 24 h (Figure 2C). Immunoreactivity for p-JNK (Thr183/Thy185) completely localized in NeuN-positive cells at 7 h (Figure 2E) and at 24 h, but the nuclei were not obviously stained. Immunoreactivity of p-JNK (Thr183/Thy185) was not detected in the infarction core area at any time point after tFCI. The level of JNK activity also notably increased 7 and 24 h after tFCI (Figure 2B).
p-Akt (Ser473) immunoreactivity was observed in only a few phospho-JNK-positive cells, and almost none of the p-JNK (Thr183/Tyr185)-positive cells were double positive for p-Akt (Ser473) at 7 h in the medial striatal region after tFCI (Figure 2F). Although both p-Akt (Ser473) and p-JNK (Thr183/Thy185) increased in the same area after tFCI (Figure 2C), the temporal profiles of p-Akt (Ser473) and Akt activation were different from those of p-JNK (Thr183/Thy185) and JNK activation. The immunostainings without primary antibodies showed no immunoreactivity (data not shown).
The PI3-K Inhibitor (LY294002) Promoted Apoptosis-Related DNA Fragmentation, whereas the JNK Inhibitor (SP600125) Attenuated it after tFCI
In the LY294002-treated group, the level of p-Akt (Ser473) and Akt activity did not change significantly 3 h after tFCI compared with the sham controls. Administration of LY294002 significantly attenuated Akt phosphorylation and Akt activation in the sham control brains and ischemic brains 3 h after tFCI compared with vehicle treatment (**P < 0.01) (Figure 3A). LY294002 administration also significantly exacerbated apoptosis-related DNA fragmentation 24 h after tFCI compared with vehicle treatment (*P < 0.05) (Figure 3C).
Figure 3.
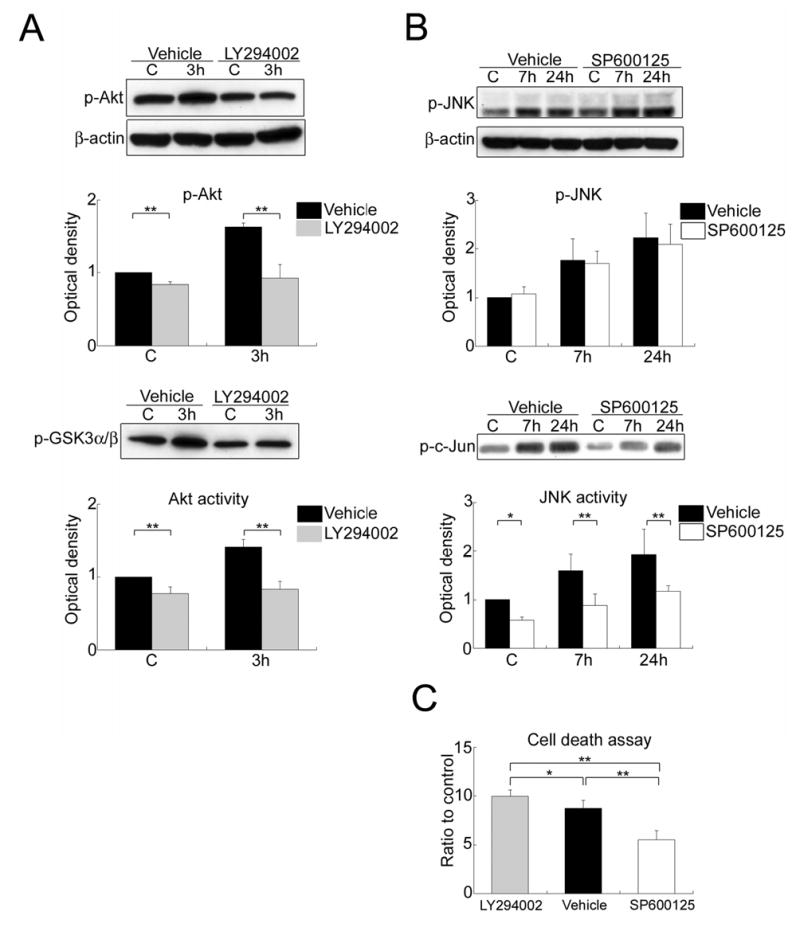
(A) Western blot analysis of phospho-Akt (Ser473) (p-Akt) and Akt activity assay with GSK-3 fusion protein as the kinase substrate (p-GSK3α/β) in the whole-cell fraction from the MCA territory after treatment with the vehicle or LY294002. Administration of LY294002 decreased the level of p-Akt and Akt activity in the sham control brains (C) and the brains with 3 h of reperfusion after tFCI compared with vehicle treatment (**P < 0.01). β-actin was used as an internal control. (B) Western blot analysis of phospho-JNK (Thr183/Tyr185) (p-JNK) and JNK activity assay with c-Jun fusion protein (p-c-Jun) as the kinase substrate in the whole-cell fraction from the MCA territory after treatment with the vehicle or SP600125. Administration of SP600125 caused a significant reduction in JNK activity in the sham control brains and the brains with 7 or 24 h of reperfusion after tFCI compared with vehicle treatment (*P < 0.05, **P < 0.01) without affecting JNK phosphorylation. (C) Apoptotic cell death assay in the cytosolic fraction from the MCA territory after treatment with the vehicle, LY294002, or SP600125. Treatment with LY294002 promoted a marked increase in apoptosis-related DNA fragmentation compared with vehicle or SP600125 treatment (*P < 0.05, **P < 0.01). In contrast, SP600125 attenuated the increase in apoptosis-related DNA fragmentation compared with the vehicle (**P < 0.01).
Administration of the JNK-specific inhibitor (SP600125) significantly reduced the level of JNK activity in the sham control and ischemic brains 7 and 24 h after tFCI compared with the vehicle-injected group (*P < 0.05, **P < 0.01), without inhibition of JNK phosphorylation (Figure 3B). Furthermore, SP600125 administration markedly attenuated apoptosis-related DNA fragmentation 24 h after tFCI compared with the vehicle (**P < 0.01) (Figure 3C). Apoptosis-related DNA fragmentation in the SP600125-treated rats was also less than in the LY294002-treated rats 24 h after tFCI (**P < 0.01) (Figure 3C).
The Spatiotemporal Profile of p-Bad (Ser136) was Consistent with that of p-Akt (Ser473) after tFCI
As shown in Figure 4A, the bands of p-Bad (Ser136) and Bad were observed at 23 kDa in the whole-cell fraction from the rat brains. p-Bad (Ser136) was weakly stained in the entire region of the sham control brains (Figure 4B). Immunoreactivity of total Bad did not change significantly among any animal group (Figure 4A). A significant increase in p-Bad (Ser136) expression was observed in the medial striatal region 3 h after tFCI (Figure 4A and 4B), parallel to the increase in p-Akt (Ser473) and Akt activity. Immunoreactivity for p-Bad (Ser136) was mainly observed in the cytoplasm (Figure 4B and 4C). Almost all of the p-Bad (Ser136)-positive cells were double positive for NeuN at 3 h (Figure 4C). Furthermore, almost all of the p-Bad (Ser136)-positive cells were double positive for p-Akt (Ser473) 3 h after tFCI (Figure 4D). Immunostaining without primary antibodies showed no immunoreactivity (data not shown).
Figure 4.
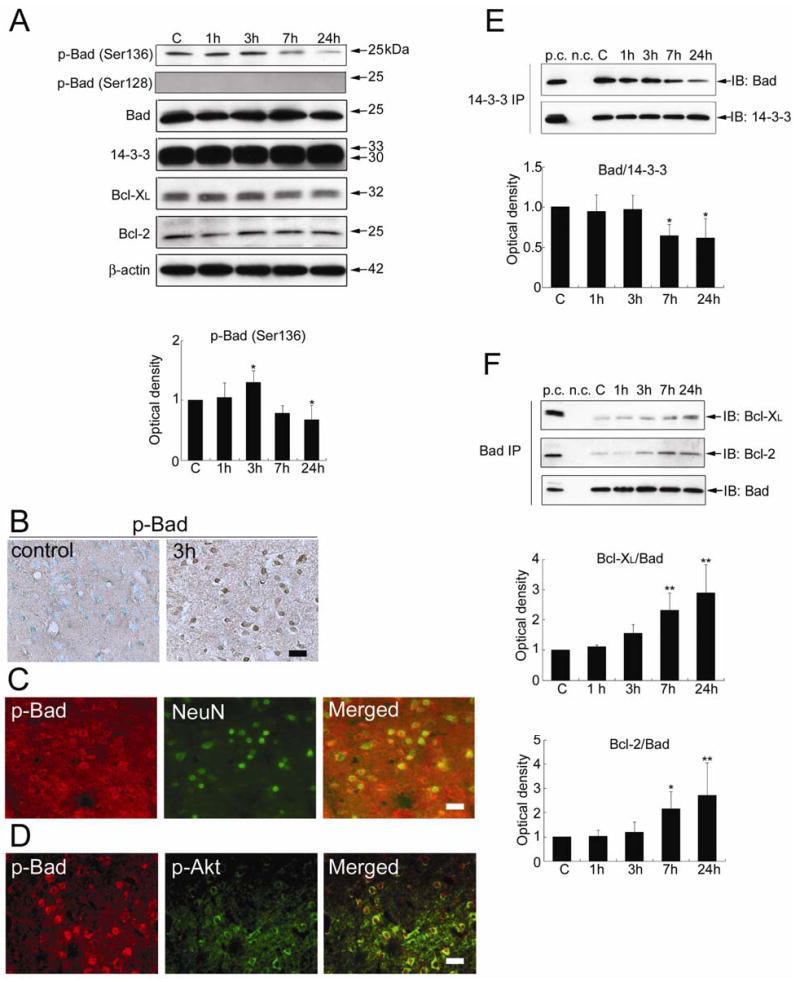
(A) Western blot analysis of phospho-Bad (p-Bad) (Ser136), p-Bad (Ser128), Bad, 14-3-3, Bcl-XL, and Bcl-2 in the whole-cell fraction from the MCA territory. Immunoreactivity of p-Bad (Ser136) increased significantly at 3 h (*P < 0.05) and decreased remarkably 24 h (*P < 0.05) after tFCI. Immunoreactivity of p-Bad (Ser128) was not detected at any time point. Bad, 14-3-3, Bcl-XL, and Bcl-2 immunoreactivity did not change significantly at any time point after tFCI. C, control. β-actin was used as an internal control. (B) Representative photomicrographs of immunostaining for phospho-Bad (Ser136) (p-Bad) in the medial striatal region (ROI1). p-Bad was weakly stained in the sham control brains. Immunoreactivity of p-Bad was observed mainly in the cytoplasm and increased 3 h after tFCI. Scale bar represents 50 μm. (C) Representative photomicrographs of double immunofluorescence of phospho-Bad (Ser136) (p-Bad) and NeuN in the medial striatal region (ROI1) 3 h after tFCI. Almost all of the p-Bad immunoreactivity was localized in the NeuN-positive cells. Scale bar represents 50 μm. (D) Representative photomicrographs of double immunofluorescence of phospho-Bad (Ser136) (p-Bad) and phospho-Akt (Ser473) (p-Akt) in the medial striatal region (ROI1) 3 h after tFCI. Almost all of the p-Bad-positive cells were double positive for p-Akt. Scale bar represents 50 μm. (E) Coimmunoprecipitation of Bad/14-3-3 in the whole-cell fraction from the MCA territory. Bad/14-3-3 dimerization did not change until 3 h and decreased significantly 7 and 24 h after tFCI (*P < 0.05). 14-3-3 was used to show equal precipitation. p.c., positive control; n.c., negative control; IP, immunoprecipitation; IB, immunoblot. (F) Coimmunoprecipitation of Bad and Bcl-XL or Bcl-2 in the whole-cell fraction from the MCA territory. The dimerization of Bad with Bcl-XL or Bcl-2 did not change until 3 h, increased significantly at 7 h, and stayed at the increased level up to 24 h after tFCI (*P < 0.05, **P < 0.01). Bad was used to show equal precipitation.
Dephosphorylation of Bad at Ser136, the Dissociation of Bad from 14-3-3, and the Dimerization of Bad with Bcl-XL or Bcl-2 were Accompanied by JNK Activation after tFCI
The level of p-Bad (Ser136) started decreasing at 7 h and decreased significantly 24 h after tFCI (*P < 0.05) (Figure 4A). Dephosphorylation of Bad (Ser136) was accompanied by the increase in JNK phosphorylation and JNK activity. Immunostaining of p-Bad (Ser136) returned to the basal expression at 7 h and was not detected 24 h after tFCI. Immunoreactivity of p-Bad (Ser128) was not detected in the Western blot analysis (Figure 4A) or immunohistochemistry at any time point after tFCI.
Coimmunoprecipitation of Bad with 14-3-3 showed that the level of Bad/14-3-3 dimerization did not change significantly until 3 h and decreased significantly 7 and 24 h after tFCI (*P < 0.05) (Figure 4E). The decrease in Bad/14-3-3 dimerization was also accompanied by an increase in JNK phosphorylation and JNK activity. In contrast, coimmunoprecipitation of Bcl-XL or Bcl-2 with Bad showed that Bcl-XL/Bad or Bcl-2/Bad dimerization did not change until 3 h, increased markedly at 7 h, and further increased 24 h after tFCI (*P < 0.05, **P < 0.01) (Figure 4F). The increase in dimerization of Bad with Bcl-XL or Bcl-2 was parallel to the increase in JNK phosphorylation and JNK activity. 14-3-3, Bcl-XL, and Bcl-2 were expressed in the rat brains and this expression did not change significantly at any time point after tFCI (Figure 4A).
The PI3-K Inhibitor (LY294002) Reduced the Level of Bad Phosphorylation and the Association of Bad with 14-3-3 without Changing the Level of JNK Activity after tFCI
Administration of LY294002 did not affect the level of p-JNK (Thr183/Tyr185) or JNK activity in the sham control brains or the brains with 3 h of reperfusion after tFCI (data not shown). In the LY294002-injected rats, the level of p-Bad (Ser136) and Bad/14-3-3 dimerization did not change 3 h after tFCI compared with the sham control rats. Treatment with LY294002 significantly attenuated the phosphorylation of Bad (Ser136) in both the sham-operated rats and the rats with 3 h of reperfusion after tFCI compared with vehicle treatment (**P < 0.01) (Figure 5A). Similarly, administration of LY294002 reduced the level of Bad/14-3-3 dimerization in the sham control rats and the rats with 3 h of reperfusion after tFCI compared with the vehicle-treated animals (**P < 0.01) (Figure 5B).
Figure 5.
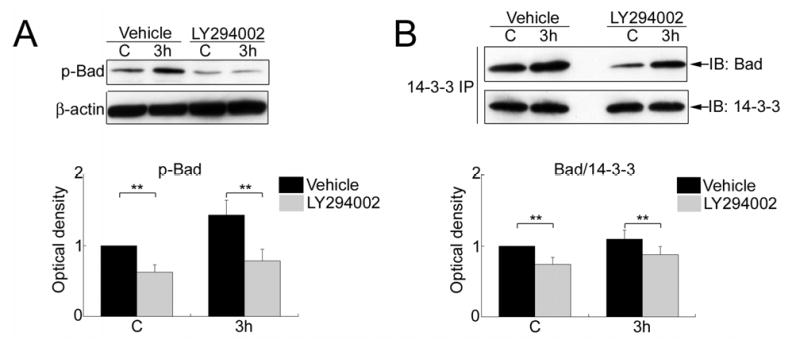
(A) Western blot analysis of phospho-Bad (Ser136) (p-Bad) in the whole-cell fraction from the MCA territory after treatment with the vehicle or LY294002. Administration of LY294002 reduced the level of p-Bad in the sham control brains and the brains with 3 h of reperfusion after tFCI compared with vehicle treatment (**P < 0.01). (B) Coimmunoprecipitation of Bad/14-3-3 in the whole-cell fraction from the MCA territory after treatment with the vehicle or LY294002. Administration of LY294002 decreased Bad/14-3-3 dimerization in the sham control brains and the brains with 3 h of reperfusion after tFCI compared with vehicle treatment. 14-3-3 was used to show equal precipitation. IP, immunoprecipitation; IB, immunoblot.
The JNK Inhibitor (SP600125) Attenuated Bad Dephosphorylation, the Release of Bad from 14-3-3, and the Association of Bad with Bcl-XL or Bcl-2 without Changing the Level of Akt Activity after tFCI
Administration of SP600125 did not affect the level of p-Akt (Ser473) or Akt activity in the sham controls or the rat brains with 7 or 24 h of reperfusion after tFCI (data not shown). Treatment with SP600125 also had no effect on the level of p-Bad (Ser136) and the dimerization of Bad with 14-3-3, Bcl-XL, or Bcl-2 in the sham control brains compared with vehicle treatment (Figure 6A to 6C).
Figure 6.
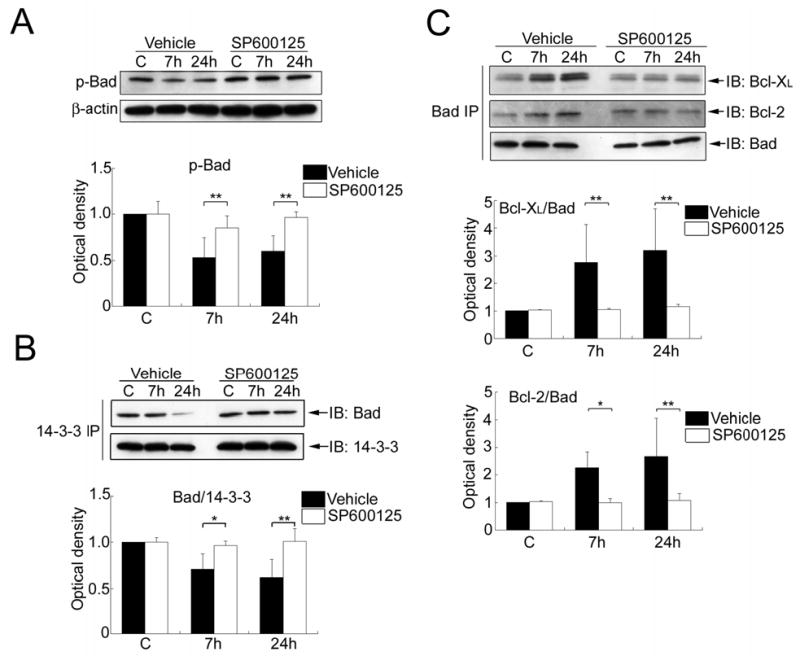
(A) Western blot analysis of phospho-Bad (Ser136) (p-Bad) in the whole-cell fraction from the MCA territory after treatment with the vehicle or SP600125. Administration of SP600125 significantly prevented dephosphorylation of Bad (Ser136) at 7 and 24 h compared with vehicle treatment (**P < 0.01). (B) Coimmunoprecipitation of Bad/14-3-3 in the whole-cell fraction from the MCA territory after treatment with the vehicle or SP600125. Administration of SP600125 significantly attenuated the decrease in Bad/14-3-3 dimerization 7 and 24 h after tFCI compared with vehicle treatment (*P < 0.05,**P < 0.01). 14-3-3 was used to show equal precipitation. IP, immunoprecipitation; IB, immunoblot. (C) Coimmunoprecipitation of Bad with Bcl-XL or Bcl-2 in the whole-cell fraction from the MCA territory after treatment with the vehicle or SP600125. Administration of SP600125 significantly attenuated the increase in Bcl-XL/Bad or Bcl-2/Bad dimerization 7 and 24 h after tFCI compared with vehicle treatment (*P < 0.05, **P < 0.01). Bad was used to show equal precipitation.
In the SP600125-treated rats, the level of p-Bad (Ser136) did not decrease 7 or 24 h after tFCI compared with the sham control animals. Treatment with SP600125 significantly prevented the decrease in p-Bad (Ser136) at 7 and 24 h compared with vehicle treatment (**P < 0.01) (Figure 6A). Similarly, in the group of rats treated with SP600125, the level of Bad/14-3-3 dimerization did not change 7 or 24 h after tFCI compared with the sham control animals. Administration of SP600125 remarkably attenuated the decrease in Bad/14-3-3 dimerization at 7 and 24 h compared with vehicle treatment (*P < 0.05, **P < 0.01) (Figure 6B). Moreover, in the SP600125-injected rats, the level of Bcl-XL/Bad or Bcl-2/Bad dimerization did not remarkably change 7 or 24 h after tFCI compared with the sham controls. Administration of SP600125 prevented the increase in Bcl-XL/Bad or Bcl-2/Bad dimerization 7 or 24 h after tFCI compared with vehicle treatment (*P < 0.05, **P < 0.01) (Figure 6C).
Discussion
In this study, we have shown three major findings: (1) In neurons of the medial striatal region, expression of p-Akt (Ser473) increased 3 h after tFCI and then decreased, whereas expression of p-JNK (Thr183/Thy185) increased at 7 h and further increased up to 24 h. (2) Phospho-Bad (Ser136) expression increased 3 h after tFCI in the neurons of the medial striatal region, but was inhibited by LY294002 administration, with suppression of Akt activity. (3) The decrease in p-Bad (Ser136) and Bad/14-3-3 dimerization and the increase in Bcl-XL/Bad or Bcl-2/Bad dimerization observed 7 to 24 h after tFCI were prevented by SP600125 treatment, with inhibition of JNK activity.
The first set of findings suggests that the predominant signal varies from PI3-K/Akt signaling to JNK signaling in neurons of the medial striatal region after brain ischemia. Furthermore, the present study demonstrated that LY294002 treatment increased DNA damage after tFCI with suppression of Akt activation, while treatment with SP600125 prevented DNA damage with inhibition of JNK activation. Our results of the cresyl violet staining and TUNEL studies showed that cellular damage extended to the medial striatal region, accompanied by a decrease in Akt activity and an increase in JNK activity after 7 h of reperfusion. A cell death factor in the later phase overcomes the early activation of a survival factor in both the CA1 subregion after global ischemia (Ouyang et al, 1999) and in the penumbra area after focal ischemia (Jin et al, 2003). Taken together, these findings may lend support to the view that the balance between PI3-K/Akt-mediated survival signaling and JNK-mediated death signaling can contribute to cell fate in the peripheral area after brain ischemia.
The second group of findings suggests that the PI3-K/Akt pathway is involved in the phosphorylation of Bad at Ser136 in the neurons of the medial striatal region 3 h after tFCI. Akt has been shown to promote cell survival by its ability to phosphorylate Bad at Ser136 (Datta et al, 1997; del Peso et al, 1997). These findings may indicate that Akt activation after brain ischemia contributes to cell survival in the peripheral area via Bad phosphorylation at Ser136. Our results of coimmunoprecipitation demonstrated that dimerization of Bad/14-3-3, Bcl-XL/Bad, or Bcl-2/Bad did not change until 3 h after tFCI in accord with our previous report (Saito et al, 2003). In the sham control brains, LY294002 treatment caused a decrease in p-Bad (Ser136) and Bad/14-3-3 dimerization with a reduction in p-Akt (Ser473) and Akt activity, which supports the view that the PI3-K/Akt pathway is involved in Bad phosphorylation (Ser136) and subsequent dimerization of Bad with 14-3-3 under normal conditions. In contrast, our study demonstrated that after tFCI, an injection of LY294002 also caused a significant reduction in Bad/14-3-3 dimerization at 3 h. However, we cannot rule out the possibility that this reduction might be caused by suppression of the basal level of p-Akt, because Bad/14-3-3 dimerization was not encouraged 3 h after tFCI despite the increase in Akt activity. Phosphorylation of Bad at Ser136 or Ser112 or both culminated in the binding to 14-3-3 in an in vitro study (Zha et al, 1996). The extracellular signal-regulated kinase pathway promotes cell survival through phosphorylation of Bad at Ser112 (Bonni et al, 1999). It would be useful to further investigate whether not only phosphorylation of Bad at Ser136 but also phosphorylation of Bad at Ser112 is required for cell survival via Bad/14-3-3 dimerization after brain ischemia.
The third group of findings suggests that the JNK pathway is involved in dephosphorylation of Bad at Ser136, the release of Bad from 14-3-3, and the association of Bad with Bcl-XL or Bcl-2 after brain ischemia. The JNK pathway induces phosphorylation of Bad at Ser128, which reduces the affinity of Bad for 14-3-3, and then promotes the apoptotic effect of Bad (Donovan et al, 2002; Bhakar et al, 2003). In the present study, we did not detect immunoreactivity for p-Bad (Ser128) in any group in either the Western blot analysis or in immunohistochemistry, which indicates that phosphorylation of Bad at Ser128 may not be involved in Bad activation after brain ischemia. In contrast, phosphorylation of 14-3-3 by JNK releases Bad from 14-3-3 and promotes dephosphorylation of Bad (Ser136) and the association of Bad with Bcl-XL or Bcl-2 (Sunayama et al, 2005). A recent study showed that expression of phosphorylated 14-3-3 increases after tFCI and is diminished in brains treated with SP600125 (Gao et al, 2005). These findings may indicate that JNK activation contributes to Bad activation via 14-3-3 phosphorylation after brain ischemia. Moreover, JNK has been found to regulate expression of some Bcl-2 family proteins, such as DP5 and Bim, via a c-Jun-mediated transcription mechanism (Harris and Johnson, 2001; Putcha et al, 2003). In addition to the cytosolic localization of p-JNK immunoreactivity and no significant change in total Bad expression after tFCI, we also have preliminary data showing that the level of phospho-c-Jun (Ser63) was not changed after tFCI. However, we cannot exclude the possibility that JNK activation could be involved in Bad activation via a transcriptional mechanism. In addition, we cannot rule out the possibility that restoration of p-Bad (Ser136) could be an indirect effect of JNK inhibition, secondary to its neuroprotective effect. More study of the mechanisms of Bad regulation by the JNK pathway is needed.
After apoptotic stimuli, activated Bad can displace Bax binding to Bcl-XL or Bcl-2 (Yang et al, 1995; Zha et al, 1996) and facilitate the action of Bax. Bax-mediated mitochondria-dependent apoptosis plays an important role in ischemic neuronal injury (Cao et al, 2001). Moreover, Bax is essential for JNK-dependent apoptosis (Lei et al, 2002), and the JNK pathway has been shown to mediate Bax translocation after tFCI (Okuno et al, 2004; Gao et al, 2005). These findings may indicate that the JNK pathway is implicated in Bax translocation and subsequent apoptosis partly through Bad activation after brain ischemia.
Increased Akt activity can lead to suppression of the JNK pathway and this cross-talk between Akt and JNK may underlie many of the prosurvival effects of Akt (Cerezo et al, 1998; Kim et al, 2001; Barthwal et al, 2003). In the current study, LY294002 did not reduce the level of p-JNK or JNK activity, which indicates that the temporal increase in Akt activity after brain ischemia may have no effect on JNK activity. Similarly, we demonstrated that SP600125 did not cause any change in Akt phosphorylation or Akt activity. Together, these findings suggest that the PI3-K/Akt and JNK pathways may be involved in regulation of Bad, independent of each other, after brain ischemia.
In summary, the present study indicates that signal predominance varies from PI3-K/Akt-mediated survival signaling to JNK-mediated death signaling in the peripheral area after brain ischemia, with the development of cellular damage. This study also suggests that the PI3-K/Akt pathway has a role in Bad inactivation, whereas the JNK pathway is involved in Bad activation after brain ischemia. This information shows that Bad may be an integrated target of PI3-K/Akt-mediated survival signaling and JNK-mediated death signaling and that it may contribute to cell fate in the peripheral area after brain ischemia (Figure 7). In light of this observation, Bad may be one of the potential therapeutic targets for treatment of ischemic stroke.
Figure 7.
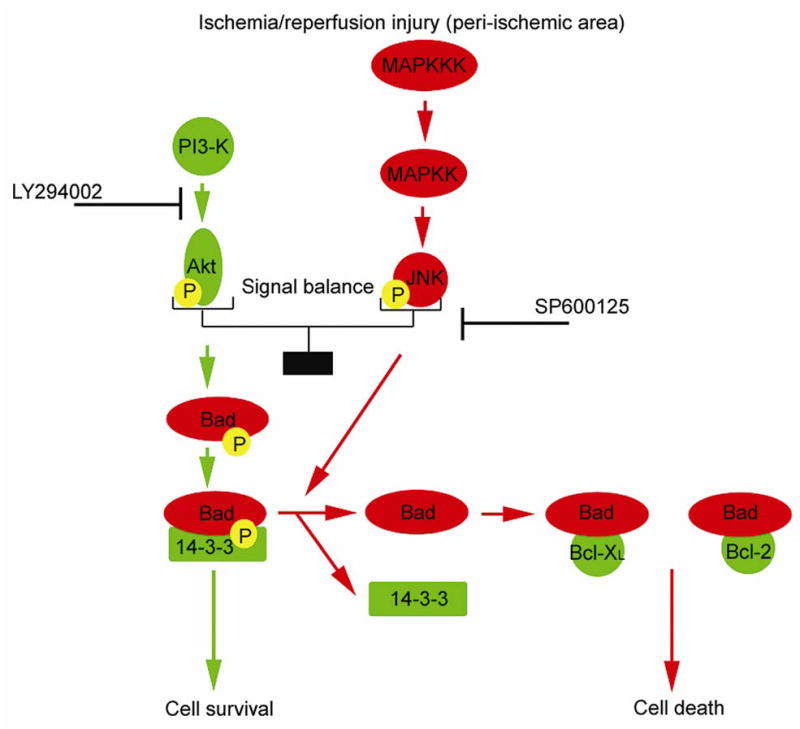
Schematic depicting the effect of the PI3-K/Akt pathway and the JNK pathway on Bad activation in the peripheral area after brain ischemia/reperfusion injury and pharmacological manipulation. The PI3-K/Akt pathway promotes phosphorylation of Bad and the association of Bad with 14-3-3, which mediates cell survival. In contrast, the JNK pathway encourages the dissociation of Bad from 14-3-3, the dephosphorylation of Bad, and the dimerization of Bad with Bcl-XL or Bcl-2, followed by cell death. After ischemia/reperfusion injury, the signal balance between the PI3-K/Akt pathway and the JNK pathway regulates Bad activation. Bad might play a crucial role as a determinant of cell fate after cerebral ischemia. MAPKKK, mitogen-activated protein kinase kinase kinase; MAPKK, mitogen-activated protein kinase kinase; P, phosphate group.
Acknowledgments
We thank Liza Reola and Bernard Calagui for technical assistance, Cheryl Christensen for editorial assistance, and Elizabeth Hoyte for figure preparation.
Footnotes
This work was supported by National Institutes of Health grants P50 NS14543, RO1 NS25372, RO1 NS36147, and RO1 NS38653.
References
- Abe T, Takagi N, Nakano M, Furuya M, Takeo S. Altered Bad localization and interaction between Bad and Bcl-XL in the hippocampus after transient global ischemia. Brain Res. 2004;1009:159–168. doi: 10.1016/j.brainres.2004.03.003. [DOI] [PubMed] [Google Scholar]
- Barthwal MK, Sathyanarayana P, Kundu CN, Rana B, Pradeep A, Sharma C, et al. Negative regulation of mixed lineage kinase 3 by protein kinase B/AKT leads to cell survival. J Biol Chem. 2003;278:3897–3902. doi: 10.1074/jbc.M211598200. [DOI] [PubMed] [Google Scholar]
- Becker EBE, Howell J, Kodama Y, Barker PA, Bonni A. Characterization of the c-Jun N-terminal kinase-BimEL signaling pathway in neuronal apoptosis. J Neurosci. 2004;24:8762–8770. doi: 10.1523/JNEUROSCI.2953-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhakar AL, Howell JL, Paul CE, Salehi AH, Becker EBE, Said F, et al. Apoptosis induced by p75NTR overexpression requires Jun kinase-dependent phosphorylation of Bad. J Neurosci. 2003;23:11373–11381. doi: 10.1523/JNEUROSCI.23-36-11373.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonfoco E, Krainc D, Ankarcrona M, Nicotera P, Lipton SA. Apoptosis and necrosis: two distinct events induced, respectively, by mild and intense insults with N-methyl-D-aspartate or nitric oxide/superoxide in cortical cell cultures. Proc Natl Acad Sci USA. 1995;92:7162–7166. doi: 10.1073/pnas.92.16.7162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonni A, Brunet A, West AE, Datta SR, Takasu MA, Greenberg ME. Cell survival promoted by the Ras-MAPK signaling pathway by transcription-dependent and -independent mechanisms. Science. 1999;286:1358–1362. doi: 10.1126/science.286.5443.1358. [DOI] [PubMed] [Google Scholar]
- Cao G, Minami M, Pei W, Yan C, Chen D, O’Horo C, et al. Intracellular Bax translocation after transient cerebral ischemia: implications for a role of the mitochondrial apoptotic signaling pathway in ischemic neuronal death. J Cereb Blood Flow Metab. 2001;21:321–333. doi: 10.1097/00004647-200104000-00001. [DOI] [PubMed] [Google Scholar]
- Cerezo A, Martínez-A C, Lanzarot D, Fischer S, Franke TF, Rebollo A. Role of Akt and c-Jun N-terminal kinase 2 in apoptosis induced by interleukin-4 deprivation. Mol Biol Cell. 1998;8:3107–3118. doi: 10.1091/mbc.9.11.3107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan PH. Future targets and cascades for neuroprotective strategies. Stroke. 2004;35(Suppl I):2748–2750. doi: 10.1161/01.STR.0000143325.25610.ac. [DOI] [PubMed] [Google Scholar]
- Datta SR, Dudek H, Tao X, Masters S, Fu H, Gotoh Y, et al. Akt phosphorylation of BAD couples survival signals to the cell-intrinsic death machinery. Cell. 1997;91:231–241. doi: 10.1016/s0092-8674(00)80405-5. [DOI] [PubMed] [Google Scholar]
- Davis RJ. Signal transduction by the JNK group of MAP kinases. Cell. 2000;103:239–252. doi: 10.1016/s0092-8674(00)00116-1. [DOI] [PubMed] [Google Scholar]
- del Peso L, González-Garcia M, Page C, Herrera R, Nuñez G. Interleukin-3-induced phosphorylation of BAD through the protein kinase Akt. Science. 1997;278:687–689. doi: 10.1126/science.278.5338.687. [DOI] [PubMed] [Google Scholar]
- Deng X, Xiao L, Lang W, Gao F, Ruvolo P, May WS., Jr Novel role for JNK as a stress-activated Bcl2 kinase. J Biol Chem. 2001;276:23681–23688. doi: 10.1074/jbc.M100279200. [DOI] [PubMed] [Google Scholar]
- D³użniewska J, Berêsewicz B, Wojewódzka U, Gajkowska B, Zab³ocka B. Transient cerebral ischemia induces delayed proapoptotic Bad translocation to mitochondria in CA1 sector of hippocampus. Mol Brain Res. 2005;133:274–280. doi: 10.1016/j.molbrainres.2004.10.013. [DOI] [PubMed] [Google Scholar]
- Donovan N, Becker EBE, Konishi Y, Bonni A. JNK phosphorylation and activation of BAD couples the stress-activated signaling pathway to the cell death machinery. J Biol Chem. 2002;277:40944–40949. doi: 10.1074/jbc.M206113200. [DOI] [PubMed] [Google Scholar]
- Fujimura M, Morita-Fujimura Y, Kawase M, Copin J-C, Calagui B, Epstein CJ, et al. Manganese superoxide dismutase mediates the early release of mitochondrial cytochrome c and subsequent DNA fragmentation after permanent focal cerebral ischemia in mice. J Neurosci. 1999;19:3414–3422. doi: 10.1523/JNEUROSCI.19-09-03414.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao Y, Signore AP, Yin W, Cao G, Yin X-M, Sun F, et al. Neuroprotection against focal ischemic brain injury by inhibition of c-Jun N-terminal kinase and attenuation of the mitochondrial apoptosis-signaling pathway. J Cereb Blood Flow Metab. 2005;25:694–712. doi: 10.1038/sj.jcbfm.9600062. [DOI] [PubMed] [Google Scholar]
- Garcia JH. The evolution of brain infarcts. A review. J Neuropathol Exp Neurol. 1992;51:387–393. doi: 10.1097/00005072-199207000-00001. [DOI] [PubMed] [Google Scholar]
- Harris CA, Johnson EM., Jr BH3-only Bcl-2 family members are coordinately regulated by the JNK pathway and require Bax to induce apoptosis in neurons. J Biol Chem. 2001;276:37754–37760. doi: 10.1074/jbc.M104073200. [DOI] [PubMed] [Google Scholar]
- Hayashi T, Saito A, Okuno S, Ferrand-Drake M, Dodd RL, Chan PH. Oxidative injury to the endoplasmic reticulum in mouse brains after transient focal ischemia. Neurobiol Dis. 2004;15:229–239. doi: 10.1016/j.nbd.2003.10.005. [DOI] [PubMed] [Google Scholar]
- Jin G, Omori N, Li F, Nagano I, Manabe Y, Shoji M, et al. Protection against ischemic brain damage by GDNF affecting cell survival and death signals. Neurol Res. 2003;25:249–253. doi: 10.1179/016164103101201454. [DOI] [PubMed] [Google Scholar]
- Kane LP, Shapiro VS, Stokoe D, Weiss A. Induction of NF-κB by the Akt/PKB kinase. Curr Biol. 1999;9:601–604. doi: 10.1016/s0960-9822(99)80265-6. [DOI] [PubMed] [Google Scholar]
- Kim AH, Khursigara G, Sun X, Franke TF, Chao MV. Akt phosphorylates and negatively regulates apoptosis signal-regulating kinase 1. Mol Cell Biol. 2001;21:893–901. doi: 10.1128/MCB.21.3.893-901.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lei K, Nimnual A, Zong W-X, Kennedy NJ, Flavell RA, Thompson CB, et al. The Bax subfamily of Bcl2-related proteins is essential for apoptotic signal transduction by c-Jun NH2-terminal kinase. Mol Cell Biol. 2002;22:4929–4942. doi: 10.1128/MCB.22.13.4929-4942.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noshita N, Lewén A, Sugawara T, Chan PH. Evidence of phosphorylation of Akt and neuronal survival after transient focal cerebral ischemia in mice. J Cereb Blood Flow Metab. 2001;21:1442–1450. doi: 10.1097/00004647-200112000-00009. [DOI] [PubMed] [Google Scholar]
- Okuno S, Saito A, Hayashi T, Chan PH. The c-Jun N-terminal protein kinase signaling pathway mediates Bax activation and subsequent neuronal apoptosis through interaction with Bim after transient focal cerebral ischemia. J Neurosci. 2004;24:7879–7887. doi: 10.1523/JNEUROSCI.1745-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ouyang Y-B, Tan Y, Comb M, Liu C-L, Martone ME, Siesjö BK, et al. Survival- and death-promoting events after transient cerebral ischemia: phosphorylation of Akt, release of cytochrome c, and activation of caspase-like proteases. J Cereb Blood Flow Metab. 1999;19:1126–1135. doi: 10.1097/00004647-199910000-00009. [DOI] [PubMed] [Google Scholar]
- Ozes ON, Mayo LD, Gustin JA, Pfeffer SR, Pfeffer LM, Donner DB. NF-κB activation by tumour necrosis factor requires the Akt serine-threonine kinase. Nature. 1999;401:82–85. doi: 10.1038/43466. [DOI] [PubMed] [Google Scholar]
- Paxinos GT, Watson C. The rat brain in stereotaxic coordinates. 3rd edn. Academic Press; San Diego: 1996. p. 98. [Google Scholar]
- Putcha GV, Le S, Frank S, Besirli CG, Clark K, Chu B, et al. JNK-mediated BIM phosphorylation potentiates BAX-dependent apoptosis. Neuron. 2003;38:899–914. doi: 10.1016/s0896-6273(03)00355-6. [DOI] [PubMed] [Google Scholar]
- Saito A, Hayashi T, Okuno S, Ferrand-Drake M, Chan PH. Overexpression of copper/zinc superoxide dismutase in transgenic mice protects against neuronal cell death after transient focal ischemia by blocking activation of the Bad cell death signaling pathway. J Neurosci. 2003;23:1710–1718. doi: 10.1523/JNEUROSCI.23-05-01710.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Springer JE, Azbill RD, Nottingham SA, Kennedy SE. Calcineurin-mediated BAD dephosphorylation activates the caspase-3 apoptotic cascade in traumatic spinal cord injury. J Neurosci. 2000;20:7246–7251. doi: 10.1523/JNEUROSCI.20-19-07246.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugawara T, Fujimura M, Morita-Fujimura Y, Kawase M, Chan PH. Mitochondrial release of cytochrome c corresponds to the selective vulnerability of hippocampal CA1 neurons in rats after transient global cerebral ischemia. J Neurosci. 1999;19:1–6. doi: 10.1523/JNEUROSCI.19-22-j0002.1999. RC39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sunayama J, Tsuruta F, Masuyama N, Gotoh Y. JNK antagonizes Akt-mediated survival signals by phosphorylating 14-3-3. J Cell Biol. 2005;170:295–304. doi: 10.1083/jcb.200409117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang E, Zha J, Jockel J, Boise LH, Thompson CB, Korsmeyer SJ. Bad, a heterodimeric partner for Bcl-xL and Bcl-2, displaces Bax and promotes cell death. Cell. 1995;80:285–291. doi: 10.1016/0092-8674(95)90411-5. [DOI] [PubMed] [Google Scholar]
- Yano S, Morioka M, Fukunaga K, Kawano T, Hara T, Kai Y, et al. Activation of Akt/protein kinase B contributes to induction of ischemic tolerance in the CA1 subfield of gerbil hippocampus. J Cereb Blood Flow Metab. 2001;21:351–360. doi: 10.1097/00004647-200104000-00004. [DOI] [PubMed] [Google Scholar]
- Yu F, Sugawara T, Maier CM, Hsieh LB, Chan PH. Akt/Bad signaling and motor neuron survival after spinal cord injury. Neurobiol Dis. 2005;20:491–499. doi: 10.1016/j.nbd.2005.04.004. [DOI] [PubMed] [Google Scholar]
- Zha J, Harada H, Osipov K, Jockel J, Waksman G, Korsmeyer SJ. BH3 domain of BAD is required for heterodimerization with BCL-XL and pro-apoptotic activity. J Biol Chem. 1997;272:24101–24104. doi: 10.1074/jbc.272.39.24101. [DOI] [PubMed] [Google Scholar]
- Zha J, Harada H, Yang E, Jockel J, Korsmeyer SJ. Serine phosphorylation of death agonist BAD in response to survival factor results in binding to 14-3-3 not BCL-XL. Cell. 1996;87:619–628. doi: 10.1016/s0092-8674(00)81382-3. [DOI] [PubMed] [Google Scholar]