

Abstract
In neurons, proper distribution of mitochondria in axons and at synapses is critical for neurotransmission, synaptic plasticity, and axonal outgrowth. However, mechanisms underlying mitochondrial trafficking throughout the long neuronal processes have remained elusive. Here, we report that syntabulin plays a critical role in mitochondrial trafficking in neurons. Syntabulin is a peripheral membrane-associated protein that targets to mitochondria through its carboxyl-terminal tail. Using real-time imaging in living cultured neurons, we demonstrate that a significant fraction of syntabulin colocalizes and co-migrates with mitochondria along neuronal processes. Knockdown of syntabulin expression with targeted small interfering RNA or interference with the syntabulin–kinesin-1 heavy chain interaction reduces mitochondrial density within axonal processes by impairing anterograde movement of mitochondria. These findings collectively suggest that syntabulin acts as a linker molecule that is capable of attaching mitochondrial organelles to the microtubule-based motor kinesin-1, and in turn, contributes to anterograde trafficking of mitochondria to neuronal processes.
Introduction
The proper intracellular distribution of mitochondria is crucial for the normal physiology of neurons. In addition to playing an essential role in the aerobic production of ATP, mitochondria regulate local Ca2+ concentrations in some nerve terminals (Werth and Thayer, 1994); this mitochondria-mediated Ca2+ buffering at synapses has been implicated in certain forms of synaptic plasticity (Zucker, 1999). Disruption of normal mitochondrial function is believed to be responsible for excitotoxic injury and many neurodegenerative diseases (Raha and Robinson, 2000; Sawa, 2001; Swerdlow and Kish, 2002). Mitochondria accumulate near active growth cones of developing neurons (Morris and Hollenbeck, 1993), and invariably are present within the synaptic terminals (Shepherd and Harris, 1998; Rowland et al., 2000). The loss of mitochondria from axon terminals in the Drosophila milton mutant resulted in defective synaptic transmission (Stowers et al., 2002). Dendritically distributed mitochondria play an essential role for the support of synapse density and plasticity (Li et al., 2004). Mitochondria in the cell bodies of neurons are transported down the neuronal processes in response to changes in the local energy state and metabolic demand (Hollenbeck, 1996). Because of their extreme polarity, neurons require specialized mechanisms to regulate the transport, targeting, and retention of mitochondria at specific subcellular locations. Thus, efficient control of mitochondrial distribution and transport in response to cellular processes and stimuli is essential for neuronal development and synaptic function.
Mitochondria undergo saltatory and bidirectional movements through a combination of dynamic events, which inevitably results in slow net movement at instantaneous velocities of 0.3–2.0 μm sec–1 (Allen et al., 1982; Hollenbeck, 1996; Ligon and Steward, 2000). Disruption of axonal transport, which can occur when specific motor proteins are disrupted and which is found in some neurodegenerative diseases, such as Alzheimer's and Huntington's diseases (Hurd and Saxton, 1996; Gunawardena and Goldstein, 2001; Gunawardena et al., 2003), results in a nonuniform and low-density distribution of mitochondria within the axon. The kinesin family of molecular motors is responsible for anterograde transport of axonal mitochondria, whereas members of the cytoplasmic dynein family are the driving force behind retrograde movement (Hollenbeck, 1996; Ligon and Steward, 2000). Although the need for multiple kinesins in axonal transport of mitochondria is unclear, KIF5B (kinesin-1 heavy chain; KHC) is believed to be a key molecular motor for driving anterograde mitochondrial movement in neurons. However, how these motors interact dynamically with mitochondrial membranes, and how trafficking components, including molecular motors, linkers (or adaptors), and receptors of mitochondria, are assembled into transport machinery are poorly understood.
Syntabulin is a newly identified syntaxin-binding protein that links syntaxin-containing vesicles to KHC, and thus, mediates the transport of syntaxin to the neuronal processes (Su et al., 2004). Here, we report that syntabulin also associates with mitochondria in vivo and links these organelles to KIF5B. This association mediates mitochondrial trafficking along axonal processes, and consequently, contributes to proper distribution of mitochondria in neurons.
Results
Syntabulin and mitochondria colocalize and co-migrate along neuronal processes
We previously identified syntabulin as a linker molecule that attaches syntaxin-1 cargoes to KHC, which enables the transport of syntaxin-1 to neuronal processes. Our immunocytochemical studies demonstrated that the staining pattern of endogenous syntabulin appeared as vesicular-tubular shapes of different sizes along the processes of cultured hippocampal neurons (Su et al., 2004). This suggested that syntabulin might play a general role in connecting the motor protein KIF5B to its transport cargoes or organelles. Mitochondria represent the major class of organelles transported by KIF5B (Tanaka et al., 1998), and therefore, we asked if syntabulin functions as an adaptor for mitochondrial trafficking in neurons. To address this question, we examined their localizations in cultured hippocampal neurons at day in vitro (DIV) 10. Immunostaining with antibodies against syntabulin and the mitochondrial marker, cytochrome c, showed that most mitochondria colocalized precisely with syntabulin (Fig. 1); this indicated an association of syntabulin with mitochondria in vivo. In addition, many syntabulin vesicular and vesicular-tubular structures are not associated with mitochondria, which suggests that syntabulin also is present in other classes of transport vesicles; possibilities include those carrying syntaxin cargoes within neuronal processes (Su et al., 2004). To determine whether syntabulin associates with mitochondrial movements, we used time-lapse confocal microscopy in living hippocampal neurons that were cotransfected with EGFP-STB (syntabulin) and DsRed-mitotracker. EGFP-STB structures were colocalized with mitochondria along axonal processes, which together, migrated in the anterograde direction toward extending growth cones (Fig. 2).
Figure 1.

Endogenous syntabulin colocalizes with mitochondria in the somata and processes of cultured hippocampal neurons. (A–C) Low-density hippocampal cultures at DIV10 were coimmunostained with affinity-purified antibodies against mitochondrial marker cytochrome c (red, B) and syntabulin (green, C). (A) The image is shown in a merged differential interference contrast. (A′–C′) Close-up of the boxed region from A–C. Syntabulin staining appeared as elongated vesicular-tubular structures and many of these colocalized with mitochondria in neuronal somata and neuritic processes (white arrows). Bar in (A′), 10 μm.
Figure 2.
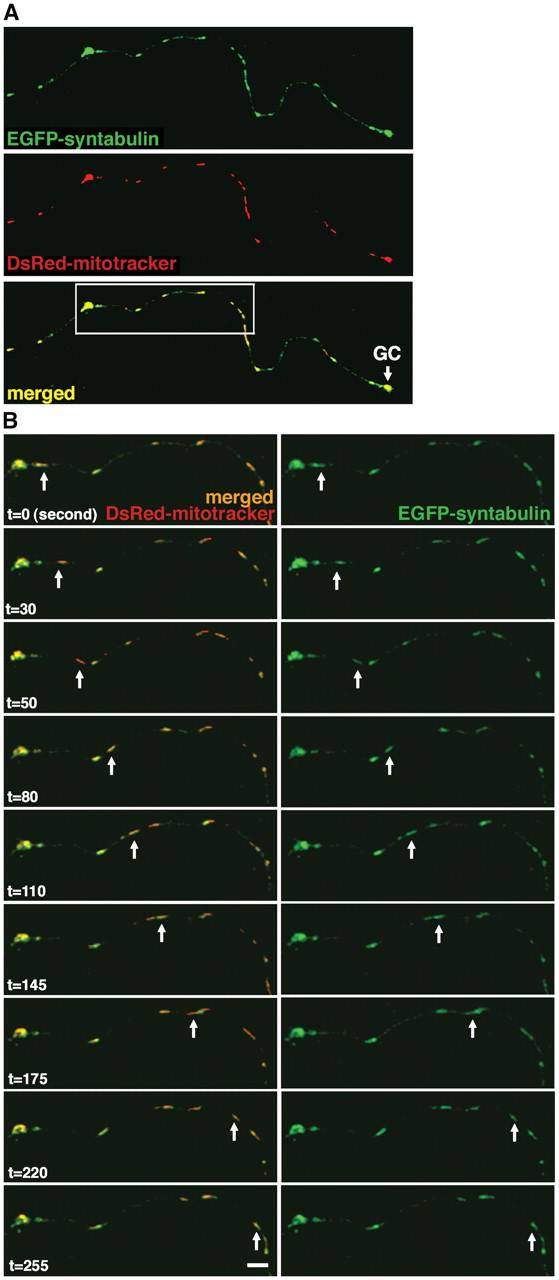
Syntabulin and mitochondria co-migrate along neuronal processes. (A) Full-length STB partially colocalizes with mitochondria along axonal process and within the growth cone (GC). Cultured hippocampal neurons (DIV6) were cotransfected with EGFP-STB (green) and DsRed-mitotracker (DsRed2-tagged mitochondrial targeting sequence of cytochrome c oxidase, red), and the selected axonal process was imaged. (B) STB-associated mitochondria move along the axonal processes. The selected axonal process (white box in panel A) from a live hippocampal neuron was examined by time-lapse imaging 18 h after transfection. The arrows point to a moving mitochondrion associated with EGFP-STB, which migrated anterogradely along the axon toward the growth cone at an average velocity of ∼0.3–0.4 μm s−1. Images were collected every 5 s. Bar in (B), 10 μm.
Syntabulin is a membrane-associated protein that targets to mitochondria through its carboxyl-terminal tail
To test further whether syntabulin associates with mitochondria, we prepared mitochondria-enriched fractions from rat forebrain using Percoll gradient centrifugation. Immunoblotting of the solubilized mitochondrial fraction demonstrated that syntabulin and the mitochondrial markers, cytochrome c and TOM20, were enriched in mitochondrial fractions relative to whole brain homogenates (Fig. 3 A). However, enrichment of syntabulin is low compared with mitochondrial markers, which is consistent with our findings that syntabulin also is present in other classes of transport vesicles, including syntaxin cargoes (Su et al., 2004). In contrast, there were no detectable protein markers of nonmitochondrial organelles and membrane compartments, including synaptophysin (synaptic vesicles), syntaxin-1 and SNAP-25 (synaptic plasma membrane), p115 (Golgi), EEA1 (endosomes), and Grp78 (ER) in the preparation; this was an indication of the purity of our mitochondrial preparation. To confirm further that syntabulin is associated with mitochondria, we immunoisolated mitochondrial organelles from the mitochondrial-enriched fraction by magnetic beads (Dynabeads) that were coated with the antibody against TOM20, a mitochondrial outer membrane protein. Our results demonstrated that syntabulin is associated specifically with these purified mitochondria (Fig. 3 B).
Figure 3.

Syntabulin is a peripheral membrane-associated protein of mitochondria. (A) Syntabulin is enriched in the mitochondrial fraction. Neuronal mitochondria fraction (mito) was prepared with Percoll-gradient centrifugation from rat forebrain. The relative enrichment of mitochondria was compared with equal total protein (20 μg/lane) of brain homogenates (BH) by detecting cytochrome c (the mitochondrial protein resident in the intermembrane space) and TOM20 (the mitochondrial outer membrane protein). Relative purity of the mitochondrial fraction was assessed by sequential immunoblotting of nonmitochondrial markers, including synaptophysin (synaptic vesicles), SNAP-25 and syntaxin-1 (synaptic plasma membrane), p115 (Golgi), EEA1 (endosomes), and Grp78 (ER). (B) Immunoisolation of mitochondrial organelles. The mitochondrial fraction was incubated with magnetic beads that were coated with the antibody against TOM20, a mitochondrial outer membrane protein, or normal IgG control. The beads-bound fractions were subjected to immunoblotting. (C) Syntabulin is a membrane peripheral protein of mitochondria. Mitochondria-enriched fraction was subjected to Triton X-114 (TX-114) phase partitioning. In: input of the starting material; A and D: the aqueous or detergent phase of Triton X-114 phase partitioning, respectively.
To define the nature of syntabulin's association with mitochondria, we performed Triton X-114 phase partitioning of syntabulin from mitochondrial-enriched membrane fractions. As shown in Fig. 3 C, syntabulin, along with the peripherally associated cytochrome c, was present exclusively in the aqueous phase after Triton X-114 phase separation. In contrast, TOM20, an integral membrane protein of mitochondria, was retained in the detergent phase following Triton X-114 phase partitioning. Our biochemical data—taken together with the evidence of the colocalization of endogenous syntabulin with mitochondria in neurons (Fig. 1), mitochondrial-targeting of exogenously expressed EGFP-STB (Fig. 2), and the exclusive presence of syntabulin in the aqueous phase after Triton X-114 phase partitioning—led us to conclude that syntabulin is a peripheral membrane-associated protein of mitochondria.
To determine the sequence that is required for syntabulin targeting to mitochondria, we constructed truncated mutants of syntabulin and cotransfected these mutants with DsRed-mitotracker into cultured hippocampal neurons. EGFP-STB (600–663), which encodes the carboxyl-terminal tail of syntabulin, precisely colocalized with DsRed-mitotracker–labeled mitochondria (Fig. S1 A; available at http://www.jcb.org/cgi/content/full/jcb.200506042/DC1) and was sufficient to target to mobile mitochondria along processes of living neurons (Video 1; available at http://www.jcb.org/cgi/content/full/jcb.200506042/DC1). However, EGFP-STB (1–604), which lacks the carboxyl-terminal sequence, appeared as small vesicular structures, most of which did not associate with mitochondria within neuronal processes (Fig. S1 B). Furthermore, EGFP-STB (1–604) colocalized with endogenous Bassoon and DsRed–syntaxin-1 vesicles in cultured hippocampal neurons (unpublished data). This suggested that although the carboxyl-terminal sequence is not required for syntabulin to target to syntaxin cargoes, it is critical for mitochondrial targeting. Altogether, our biochemical and cell biologic results support our assumption that syntabulin attaches to different membrane organelles and transport cargoes by way of distinct targeting sequences.
Knockdown of syntabulin reduces mitochondrial distribution in neuronal processes
To address whether syntabulin is involved in the distribution of mitochondria within neuronal processes, we reduced syntabulin expression with a syntabulin-targeted small interfering siRNA (stb-siRNA). Six 21-nt sequences specific to syntabulin were tested extensively and rigorously under multiple controls in our previous studies. One, which corresponds specifically to amino acid residues 101–107 of syntabulin, was selected for the current study because it was able to knockdown, efficiently and specifically, endogenous syntabulin in hippocampal neurons and exogenously expressed syntabulin in COS-7 cells (Su et al., 2004). Scrambled siRNA that was not homologous to any sequence in GenBank was used as a negative control. Developing neurons at DIV4–5 were cotransfected with DsRed-mitotracker and stb-siRNA or control siRNA. A cGFP marker encoded in the siRNA vector allowed us to trace the transfected neurons and visualize neuronal morphology. Mitochondrial distributions were examined by imaging cGFP fluorescence and DsRed-labeled mitochondria in neurons 4 or 5 d after transfection. At this stage of development, mitochondria were abundant and clustered in soma and large proximal dendrites, but were separated from each other in axons and smaller dendrites. In contrast to the abundant and uniform distribution of mitochondria in processes of cells that were transfected with a control siRNA (Fig. 4 A), knockdown of syntabulin by stb-siRNA resulted in a significant lower density of mitochondria in the distal regions of processes (Fig. 4 B).
Figure 4.
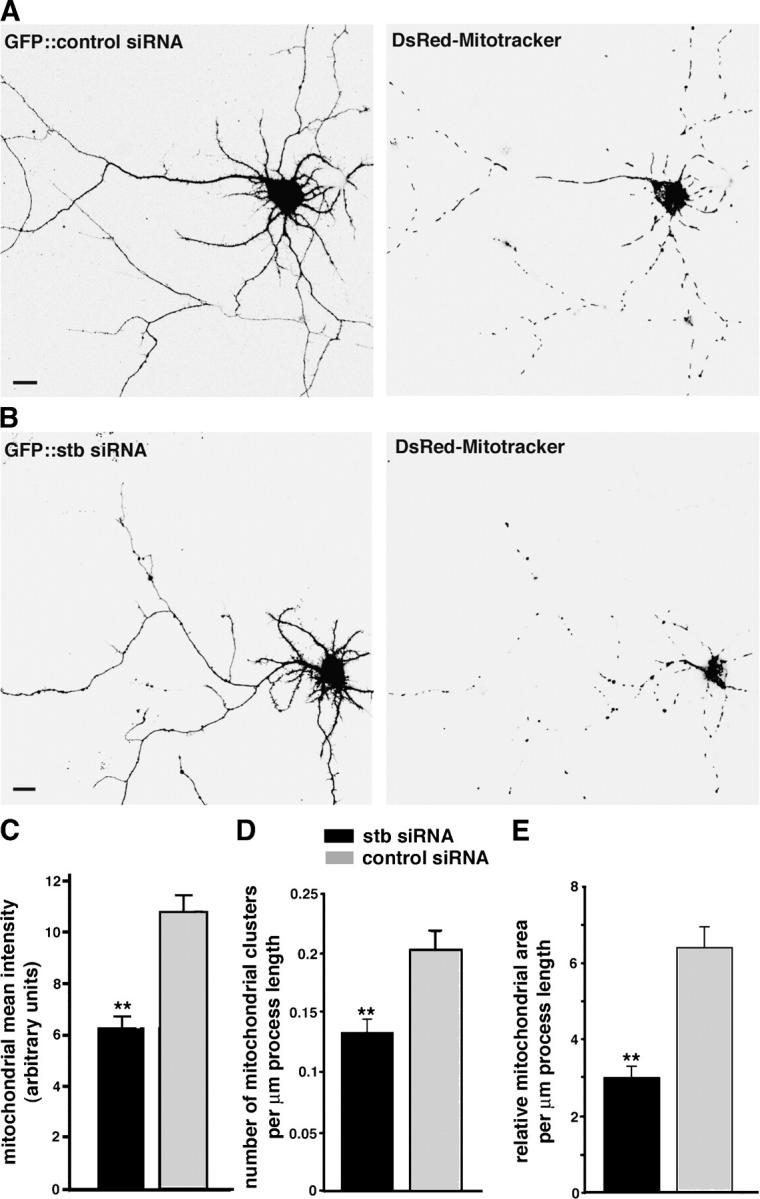
Knockdown of syntabulin reduces mitochondrial density in the neuronal processes of cultured hippocampal neurons. (A, B) Hippocampal neurons (DIV4 or 5) were cotransfected with DsRed-mitotracker and stb-siRNA or control siRNA. Mitochondrial distribution was examined by imaging GFP fluorescence (left panels) and DsRed-mitochondria (right panels) in the neurons 4 or 5 d after transfection. Note that in the processes of the neurons expressing the stb-siRNA (B), an abnormally lower density of mitochondria was observed relative to that of the neurons that were transfected with control siRNA (A). Bars, 10 μm. (C–E) Quantification of relative mitochondrial density in the neuronal processes. The transfected neurons were imaged under the same conditions and the same settings below saturation at a resolution of 1,024 × 1,024 pixels (12 bit). One main process (the longest process of each neuron) was traced manually for more than 200 μm in length, starting from the cell body. Normalized mean intensity of mitochondrial fluorescence in neuronal processes was expressed as the mean intensity in arbitrary units per square area of process (C). Alternatively, the relative mitochondrial density was estimated by determining the number of mitochondrial clusters (D) or relative mitochondrial area (E) per μm length of process. All data were obtained from a total of 3708.11 μm of process length of neurons (n = 16) expressing stb-siRNA, and 3786.5 μm of process length of neurons (n = 16) transfected with control siRNA in three independent experiments. Note that the neurons transfected with stb-siRNA resulted in a significant reduction in mitochondrial density within processes by all three of the quantitative methods: mitochondrial mean intensity (C) was 6.29 ± 0.46 (mean ± SEM, P < 0.001); the number of mitochondrial clusters per μm process length (D) was 0.13 ± 0.01 (P < 0.001); and the relative mitochondrial area/μm process length (E) was 2.99 ± 0.30 (P < 0.001), relative to the cells transfected with the control siRNA (10.85 ± 0.63 for mitochondrial mean intensity, 0.20 ± 0.02 clusters/μm process length, and 6.38 ± 0.55 relative mitochondrial area/μm process length). **P < 0.001 relative to control siRNA by t test.
We next estimated the relative mitochondrial density in the main processes (see Discussion) by three quantitative methods. Fluorescence intensity of DsRed-mitotracker in processes was measured as the mean intensity of selected areas, and was expressed in arbitrary units per square unit area of process (Fig. 4 C). The relative density of mitochondria was calculated further by determining the number of mitochondrial clusters (Fig. 4 D) or relative mitochondrial area (Fig. 4 E) per μm length of process; these were automatically calculated and scored based on the fluorescence intensity profile. Transfection of neurons with stb-siRNA revealed a significant reduction in mitochondrial density within processes by all three quantitative methods; this indicated that syntabulin is required for proper mitochondrial distribution in neuronal processes.
The STB–KHC interaction is required for trafficking of mitochondria to neuronal processes
KHC consists of two functional parts: a head with a motor domain and a tail, which interacts with cargo directly or indirectly through light chains (Diefenbach et al., 1998; Gunawardena and Goldstein, 2004). Our previous biochemical studies identified a specific syntabulin interaction with the COOH-terminal tail of KHC in vitro, and expression of the KHC COOH-terminal tail (cargo-binding domain, CBD) impaired trafficking of syntabulin vesicular structures in neuronal processes (Su et al., 2004). To characterize further the interaction of syntabulin with KHC, we performed in vitro binding studies with truncated mutants of syntabulin. Our data showed that a segment from amino acid residues 145–230, which does not overlap with the syntaxin-binding domain (310–417) (Su et al., 2004), is required for binding to KHC-CBD (Fig. S2 A; available at http://www.jcb.org/cgi/content/full/jcb.200506042/DC1). It is able to block formation of the STB-CBD complex in a dose-dependent manner (Fig. S2 B), and thus, was named as the KHC-binding domain (KBD) of syntabulin.
To examine the potential role of STB–KHC interaction in mitochondrial distribution, we interfered with the STB–KHC interaction by expressing the KBD in neurons and observing mitochondrial distribution. Hippocampal neurons at DIV5 were cotransfected with DsRed-mitotracker and GFP-STB-KBD or GFP for 48–72 h followed by imaging assessment. GFP-KHC-CBD was used as a dominant-negative mutant of KHC, because exogenous expression of the GFP-KHC-CBD impaired trafficking of syntabulin-linked vesicles (Su et al., 2004) and glutamate receptor–interacting protein–1 and AMPA receptor in neurons (Setou et al., 2002). GFP-STB-NT (1–80), an amino-terminal domain of syntabulin that is not required for binding to syntaxin or KHC, was expressed as a second negative control. The distribution pattern of mitochondria along processes after expression of GFP alone (Fig. 5 A) or GFP-STB-NT (Fig. 5 B) was similar to that of mitochondria found in control siRNA-transfected neurons (Fig. 4 A). In contrast, expression of GFP-STB-KBD (Fig. 5 C) or GFP-KHC-CBD (Fig. 5 D) resulted in a striking change in mitochondrial distribution; mitochondrial density along the processes was significantly lower relative to that in neurons that were transfected with GFP.
Figure 5.

Expression of the KHC-CBD or the STB-KBD reduces mitochondrial density in neuronal processes. (A–D) Cultured hippocampal neurons (DIV5) were transfected with DsRed-mitotracker and expression vector encoding GFP control (A), GFP-STB-NT (B), GFP-STB-KBD (C), or GFP-KHC-CBD (D). Mitochondrial distribution in the neuronal processes was examined by imaging GFP fluorescence (top panels) and DsRed-mitochondria (bottom panels) 48–72 h after transfection. Neurons transfected with GFP-KHC-CBD or GFP-STB-KBD showed a marked reduction in the density of mitochondria along the neuronal processes relative to that of neurons transfected with GFP or GFP-STB-NT. Bars, 10 μm. (E–G) Quantification of the relative mitochondrial density in neuronal processes. Relative mitochondrial density was expressed as the fluorescence mean intensity of DsRed-mitotracker per square unit area of process of transfected neurons (E), number of mitochondrial clusters (F), or relative mitochondrial area (G) per μm of process length of transfected neurons. All images were collected under the same settings below saturation and from 15 neurons for each group in three independent experiments. Total process length measured was 4111.7 μm (GFP), 4148 μm (GFP-STB-NT), 3963.74 μm (GFP-STB-KBD), or 3053.97 μm (GFP-KHC-CBD). Histograms indicate mean ± SEM. *P <0.01; **P < 0.002; ***P < 0.001 relative to GFP controls by t test.
To evaluate the relative distribution of mitochondria, we compared the mean fluorescence intensity of DsRed-mitotracker in the processes of transfected neurons. Expression of the CBD or the KBD significantly reduced the mean intensity of DsRed-mitotracker in neuronal processes (CBD: 5.69 ± 0.43, P < 0.001; KBD: 6.26 ± 0.67, P < 0.002) compared with that from GFP-transfected neurons (10.629 ± 1.01) (Fig. 5 D). We assessed mitochondrial density further in terms of the number of mitochondrial clusters (Fig. 5 E) and relative mitochondrial area (Fig. 5 F) per μm length process. Exogenous expression of the KBD or the CBD markedly reduced the number of mitochondrial clusters/μm length of process (CBD: 0.12 ± 0.02, P < 0.01; KBD: 0.11 ± 0.01, P < 0.001; GFP: 0.21 ± 0.03) and relative mitochondrial area/μm length of process (CBD: 2.50 ± 0.33, P < 0.001; KBD: 2.29 ± 0.25, P < 0.001; EGFP: 5.44 ± 0.62). However, relative distribution of mitochondria within processes after expression of GFP-STB-NT (1–80) showed no significant difference compared with that of mitochondria found in GFP control neurons. This suggested that the striking change in mitochondrial distribution following disruption of the STB–KHC interaction by CBD or KBD expression is specific.
Syntabulin mediates anterograde, but not retrograde, movement of mitochondria along axonal processes
One explanation for the observed reduction in mitochondrial density in neuronal processes is a defect in the microtubule (MT)-based trafficking machinery that normally delivers mitochondria by a complex interplay of anterograde and retrograde movements over long distances (Jellali et al., 1994; Nangaku et al., 1994; Tanaka et al., 1998). To evaluate the possible involvement of syntabulin in mitochondrial motility, we monitored and quantified the movement of DsRed-mitotracker–labeled mitochondria by time-lapsed confocal microscopy of live hippocampal neurons at DIV9–10 after transfection with stb-siRNA or control siRNA. In neurons that were transfected with a control siRNA, individual mitochondria moved in a saltatory manner. Despite the tendency of mitochondria to reverse their direction, most mitochondria showed sustained net movement in a single direction (Video 2; available at http://www.jcb.org/cgi/content/full/jcb.200506042/DC1).
We selected axonal processes for quantitative analysis of mitochondrial mobility. The net direction (anterograde or retrograde) of movement for each mitochondrial organelle during the entire recording time (15 min; 100 frames taken in 9-s intervals) within axonal processes was determined, and the relative percentages of stationary and net plus and minus end-directed events were calculated. In axonal processes that expressed the control siRNA (n = 259 from 11 cells), approximately one third (29.5 ± 3.0%) of the mitochondria were in motion at least once, of which most (18.4 ± 2.9%) moved in the net anterograde direction; the remainder (11.2 ± 1.6%) moved in the net retrograde direction (Fig. 6 A, Video 2). In contrast, transfection with stb-siRNA (n = 383 from 17 cells) resulted in reduced mitochondrial density within the axonal processes accompanied by a marked reduction in anterograde movements (3.1 ± 0.8%; P < 0.001), and no significant change in the mitochondria moving retrogradely (14.8 ± 3.2%; P = 0.4) (Fig. 6 A; Video 3, available at http://www.jcb.org/cgi/content/full/jcb.200506042/DC1). This observation in living neurons suggests that the accumulation of mitochondria in cell bodies is due to a failure of MT-based mitochondrial transport out of somata, whereas dynein-mediated retrograde trafficking into cell bodies dominates mitochondrial mobility following knockdown of syntabulin. These results suggest that syntabulin is required for maintaining anterograde, but not retrograde, transport of mitochondria within axons of developing hippocampal neurons.
Figure 6.
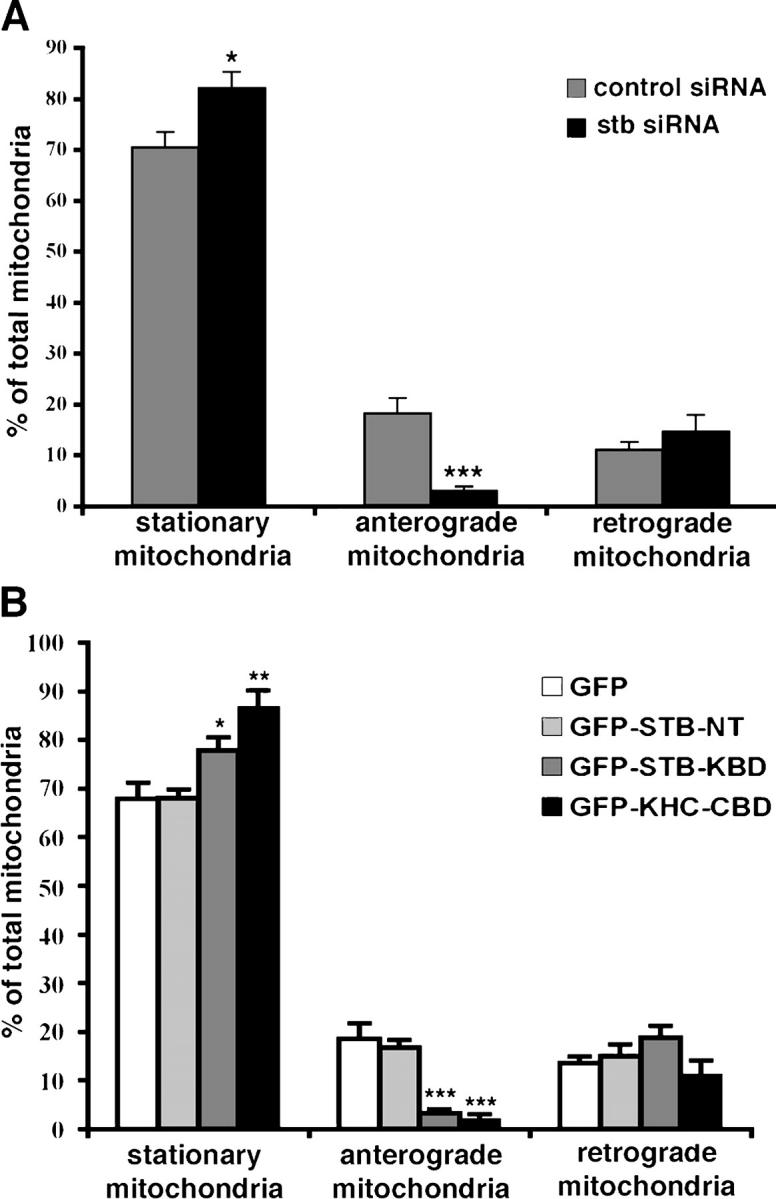
Syntabulin loss-of-function impairs anterograde movement of mitochondria along axonal processes. Quantification of relative mitochondrial movement in the neurons that were transfected with constructs expressing siRNAs or GFP-fusion proteins. The motility of DsRed-mitotracker–labeled mitochondria was examined in live hippocampal neurons at DIV9–10 after transfection with siRNAs or 48–72 h after transfection at DIV5 with GFP-fusion proteins. The direction of net movement for each mitochondrial organelle along axonal processes was determined during the same time window (15 min) of time-lapse imaging. The relative percentages of stationary, net anterograde, or net retrograde events were calculated. (A) Knockdown of syntabulin inhibits anterograde, but not retrograde, movement of mitochondria along axonal processes. Of a total of 259 mitochondrial clusters from 11 cells that were transfected with the control siRNA, one third of the mitochondrial organelles in the axons are mobile (29.6 ± 3.0%), and moved anterogradely (18.4 ± 2.9%) or retrogradely (11.2 ± 1.6%). Knockdown of syntabulin (total 383 mitochondria from 17 cells) exhibited a marked reduction in anterograde transport (***P < 0.001), a slight but significant increase in stationary mitochondria (*P < 0.02), and no significant change in retrograde movement of mitochondria (P = 0.4). (B) Interfering with the STB–KHC interaction selectively inhibits anterograde trafficking of mitochondria along axonal processes. Note that in the axonal processes of GFP-transfected control neurons, 67.9 ± 3.4%, 18.6 ± 3.1%, or 13.6 ± 1.4% of the mitochondria were immobile, moved in the anterograde direction, or moved in the retrograde direction, respectively. In contrast, the expression of GFP-STB-KBD (n = 376 from 14 cells) or GFP-KHC-CBD (n = 200 from 10 cells) resulted in a remarkable increase in the percentage of immobile mitochondrial during the 15-min observation time (GFP-KHC-CBD: P < 0.005; GFP-STB-KBD: P = 0.05), and a significant reduction in mitochondria moving anterogradely (GFP-STB-KBD: P<0.001; GFP-KHC-CBD: P<0.001). However, retrograde trafficking of mitochondria along the axonal processes exhibited no significant changes in the neurons that were transfected with GFP-STB-KBD (18.8 ± 2.5%, P = 0.23), GFP-KHC-CBD (11.2 ± 2.9%, P = 0.59), or GFP-STB-NT (15.1 ± 2.5%, P = 0.64) relative to those found in GFP-transfected neurons (13.6 ± 1.3%). (Error bars = SEM; *P = 0.05; **P < 0.005; ***P < 0.001 relative to GFP controls by t test).
To confirm further whether the interaction of syntabulin and KHC is involved in the anterograde transport of mitochondria, we visualized and quantified the motility of mitochondria in live hippocampal neurons 48–72 h after cotransfection of DsRed-mitotracker with GFP, GFP-STB-NT, GFP-STB-KBD, or GFP-KHC-CBD at DIV4–5. Our time-lapse imaging showed that in the control cells that expressed GFP (n = 73 from 4 cells) or GFP-STB-NT (n = 199 from 7 cells), 67.9 ± 3.4% or 68.1 ± 1.7% of mitochondria in axonal processes were immobile, whereas 18.6 ± 3.1% or 16.9 ± 1.5% moved in the anterograde direction, and 13.6 ± 1.4% or 15.1 ± 2.4% moved in the retrograde direction, respectively (Fig. 6 B; Video 4, available at http://www.jcb.org/cgi/content/full/jcb.200506042/DC1). This is similar to results observed from control siRNA-transfected cells (Fig. 6 A), and consistent with the reported ratio for axonal mitochondrial mobility (Ligon and Steward, 2000). In contrast, the expression of GFP-STB-KBD (n = 376 from 14 cells) or GFP-KHC-CBD (n = 200 from 10 cells) resulted in a remarkable decrease in mitochondrial density and disruption of anterograde movement along axonal processes. Most mitochondrial organelles became immobile during the 15-min observation (CBD: 86.8 ± 3.5%, P < 0.01; KBD: 77.9 ± 2.6%, P = 0.05), and a significantly lower percentage of mitochondria moved anterogradely (KBD: 3.3 ± 0.8%, P < 0.001; CBD: 2.0 ± 1.0%, P < 0.001) relative to those found in GFP-transfected neurons. Thus, retrograde trafficking in axonal processes dominates mitochondrial mobility events when the STB-KHC interaction is disrupted (CBD: 11.2 ± 2.9%; KBD: 18.8 ± 2.5%) (Fig. 6 B; Video 5, available at http://www.jcb.org/cgi/content/full/jcb.200506042/DC1). The results from loss-of-function studies that used dominant negative mutants of syntabulin and KHC are consistent with observations taken from syntabulin knockdown studies and further support our conclusion that the STB–KHC interaction is required for maintaining anterograde, but not retrograde, transport of mitochondria within axons.
Although the possibility that axonal blockage and resultant transport failures might result from other halted organelles or protein aggregates could not be excluded, log jams in the axon generally have an effect on total flux of transporting vesicles in both directions. This possible mechanism likely is not involved in the defective transport of mitochondria that we observed, because syntabulin loss-of-function did not demonstrate any significant effect on retrograde movement of mitochondria along axonal processes. To exclude the possibility that other organelles are halted after syntabulin loss-of-function, we extended our observations to include ER and Golgi. Because the distribution of ER and Golgi and the Golgi-to-ER traffic seemed to be normal in the KIF5B null mutant cells (Tanaka et al., 1998), KIF5B is unlikely to be a primary motor protein for the transport of both organelles. Thus, these organelles are good controls to test whether syntabulin loss-of-function causes a general traffic jam. Expression of either syntabulin-targeted siRNA or dominant negative transgene did not disturb the proper distribution of these organelles in neurons (Figs. S3 and S4; available at http://www.jcb.org/cgi/content/full/jcb.200506042/DC1), which suggests that a general transport blockage is unlikely. Thus, the effects of stb-siRNA and dominant negative mutant seem to be relatively specific for mitochondrial organelles; the results further support our conclusion that the reduced density of mitochondria in processes results from a defect in syntabulin-mediated anterograde transport.
Discussion
We reported previously that syntabulin mediates transport of syntaxin-containing vesicles to neuronal processes by linking these vesicles to KHC (Su et al., 2004). In the present study, we discovered a novel role for syntabulin in anterograde trafficking of mitochondria along neuronal processes. First, we showed that syntabulin is a peripheral membrane-associated protein, relatively enriched in the mitochondrial fraction, and associates with mitochondria by way of its carboxyl-terminal sequence. Second, immunocytochemical and time-lapse imaging studies demonstrated that syntabulin colocalized and co-migrated with mitochondria within neuronal processes. Finally, loss-of-function studies showed that knockdown of syntabulin and competitive blocking of the STB–KHC interaction reduced mitochondrial density remarkably within neuronal processes by impairing anterograde trafficking of mitochondria. Thus, in addition to serving as a linker for syntaxin cargoes, syntabulin plays a critical role in mitochondrial transport, and consequently, contributes to the proper distribution of mitochondria within the neuronal processes.
To accommodate the specific delivery of mitochondria to axonal and dendritic domains, neurons must use mechanisms that attach the organelles to various molecular motors and transport them by the MT-based trafficking machinery (Hollenbeck, 1996). Several members of the kinesin superfamily (KIF1B, KIF5B, and KLP67A) were shown to localize preferentially to mitochondria (Nangaku et al., 1994; Pereira et al., 1997; Tanaka et al., 1998). KIF1B (kinesin-3) colocalized and copurified with mitochondria and moved mitochondria along MTs in an in vitro assay (Leopold et al., 1992; Jellali et al., 1994; Nangaku et al., 1994). Analysis of KIF5B provided direct evidence of a role for kinesin in mitochondrial distribution. Targeted disruption of kif5b resulted in abnormal perinuclear clustering of mitochondria instead of spreading throughout the cytoplasm and toward the cell periphery in undifferentiated extraembryonic cells from mice (Tanaka et al., 1998). Presenilin-1 mutations, found in a type of familial Alzheimer's disease, caused a defect in kinesin-1–based axonal transport and reduced mitochondrial density in processes of hippocampal neurons (Pigino et al., 2003).
To preserve organelle identity and the proper flow of transport organelles within cells, linking of mitochondria with appropriate transport motors must occur with a high degree of specificity (Goldstein and Yang, 2000; Manning and Snyder, 2000). Biochemical and genetic analysis suggest that there are at least two mechanisms through which motors connect with their cargoes: a direct linkage through organelle membrane receptors, and an indirect linkage by way of linker molecules. Recent studies indicate that indirect motor–cargo matchmaking through a linker molecule is a particularly important mechanism in neurons (Kamal et al., 2000; Takeda et al., 2000; Setou et al., 2000; Bowman et al., 2000; Verhey et al., 2001). However, little is known about how mitochondrial organelles physically associate with their MT-based transport motors. Thus, identification of these linking molecules will be crucial in elucidating the cellular mechanisms underlying the trafficking of mitochondria in neurons.
The regulation of mitochondrial mobility along MTs may involve dynamics of sequential binding and release between mitochondrial outer membrane proteins and cytoskeletal scaffolding components or molecular motors. However, no adaptor–receptor complex associating with the mitochondrial outer membrane has been identified. The Drosophila protein Milton was proposed as a candidate “adaptor” for attaching the kinesin motor to mitochondria, because Milton associated with mitochondria and KHC, and the milton mutation in Drosophila resulted in a loss of mitochondria at synaptic terminals and axons (Stowers et al., 2002; Gorska-Andrzejak et al., 2003). However, it remains unclear whether Milton directly attaches mitochondria to MT-based motor proteins, or requires additional linker(s) or an adaptor complex to guide the motor to associate with the mitochondrial outer membrane. Recently, AMPA receptor interacting factor (GRIF-1) was reported as a rat ortholog of Milton, because both share ∼44% amino acid homology (Brickley et al., 2005). GRIF-1 associated with motors KIF5 in vitro, and exogenously expressed GRIF-1 colocalized with mitochondria in HEK293 cells, which implicated it as a candidate in mitochondrial transport in a manner similar to the motor-dependent transport of AMPA receptors to dendrites. However, the relative distribution of endogenous GRIF-1 in neurons has not been examined.
Although syntabulin associates with syntaxin cargoes through its coiled-coil domain (Su et al., 2004), our current findings indicate that syntabulin targets to mitochondria by way of its carboxyl-terminal tail, which suggests a general role for syntabulin in mediating the transport of proteins and organelles with its specific targeting sequences. syntabulin loss-of-function caused a dramatic change in mitochondrial distribution in cultured hippocampal neurons: mitochondria were clustered in the soma but were distributed sparsely in processes. This distribution profile was consistent with the observation from our mobility analysis; syntabulin loss-of-function impaired anterograde, but not retrograde, transport of mitochondria along axonal processes. The loss-of-function phenotype suggests a failure of mitochondrial loading onto the kinesin motor for anterograde transport, or a defect in the regulation of the motor activity. This provides direct evidence that syntabulin is an important component of the mitochondrial trafficking machinery.
Although Milton and syntabulin are crucial for the proper distribution of mitochondria in neurons, it is unlikely that both proteins regulate mitochondrial trafficking by way of the same mechanism. First, syntabulin directly binds to the CBD of KHC (Su et al., 2004), whereas a direct interaction between KHC and Milton was not observed (Stowers et al., 2002). Second, syntabulin is a peripheral membrane-associated protein, because it was present in the aqueous phase after Triton X-114 phase partitioning. syntabulin associates with mitochondria by way of its carboxyl-terminal tail, which has the predicted hydrophobic anchor and three positively charged residues flanking the extreme COOH terminus that are characteristics of other outer mitochondrial membrane proteins (Wattenberg and Lithgow, 2001). Thus, it is possible that syntabulin serves as a linker molecule which associates loosely with the phospholipid of the mitochondrial outer membrane by way of its hydrophobic tail, or which associates with mitochondria indirectly by binding to the hydrophobic domain of an unknown receptor in the outer membrane. Alternatively, syntabulin could be a component of an adaptor complex that is required for the assembly of the mitochondrial transport machinery.
Mitochondrial transport in axons and dendrites is similar; at any given time approximately one third of the mitochondria are in motion; ∼70% move in the anterograde direction and the remainder move in the retrograde direction (Ligon and Steward, 2000). Mitochondria are capable of anterograde and retrograde movements; however, the regulation of anterograde motor activity most often is responsible for the proper distribution of mitochondria (Morris and Hollenbeck, 1993; Hollenbeck, 1996). Our mobility analysis in live neurons after syntabulin loss-of-function demonstrates that, although kinesin-mediated anterograde movement along axonal processes was impaired significantly, the dynein-mediated retrograde trafficking remained normal, and consequently, dominated mitochondrial mobility. This indicates that syntabulin is involved in anterograde trafficking by regulating mitochondria–kinesin transport process. Although we consistently found a dramatic reduction in mitochondrial distribution in neuronal processes and a significantly lower percentage of mitochondria moving anterogradely along axonal processes, the loss-of-function approaches by siRNA knockdown and by dominant negative transgenes might have some limitation on the effects of the disruption of syntabulin's function. The limitation could be dependent on the expression levels of targeted siRNA and dominant negative transgenes, the timing of observations following loss-of-function, the life-time of endogenous syntabulin in neurons, and the stability of the STB-KHC complex. Alternatively, the limited effect could be explained by the contribution of other mitochondrial adaptor proteins, such as the rat homologue of Drosophila protein Milton (Stowers et al., 2002), to the trafficking of mitochondria in our neuronal cultures.
Based on these and our previous findings, we propose that syntabulin functions as a linker molecule or a component of the adaptor complex involved in the KIF5-mediated trafficking of syntaxin cargoes and mitochondrial organelles. Thus, our findings provide new information on the nature of the specific KHC–mitochondria interaction, and reveal the cellular mechanisms that are likely to underlie the anterograde transport of mitochondria in neurons. However, several important questions remain to be addressed. How is the function of syntabulin as a linker molecule regulated? Is syntabulin-mediated mobility and distribution of mitochondria modulated in response to synapse formation and synaptic activity? Does syntabulin participate in more general intracellular membrane trafficking pathways? Identification of a mitochondrial receptor or adaptor complex that binds to syntabulin, and characterization of the nature of syntabulin's association with the mitochondrial outer membrane will contribute to understanding the molecular and cellular details of how this protein modulates intracellular membrane trafficking pathways in neurons.
Materials and methods
Preparation of mitochondria-enriched fractions and immunoisolation of mitochondria
Brain homogenates were prepared from the forebrains of adult rats as reported previously (Su et al., 2004). Following homogenization, proteins were solubilized with Tris buffered saline supplemented with 1% Triton. Protein concentrations were determined by BCA protein assay (Pierce Chemical Co.) using BSA standards. Mitochondria-enriched fraction was prepared by Percoll gradient centrifugation as described previously (Sims, 1990). In brief, rat forebrains were homogenized in ice-cold mitochondrial isolation buffer (IB, 0.32 M sucrose, 1 mM EDTA, 10 mM Tris-HCl, pH 7.4). Homogenates were centrifuged at 1,300 g for 3 min, the supernatant was collected, and the pellet was resuspended with IB and recentrifuged at 1,300 g for 3 min. The combined first and second supernatant was centrifuged at 21,000 g for 10 min and the pellet was resuspended in 12 ml 15% Percoll. 2 ml of the 15% Percoll suspension was overlaid on freshly prepared Percoll gradient containing 3.5 ml of 23% Percoll layered above 3.5 ml of 40% Percoll. The gradient was separated by centrifugation at 30,000 g for 5 min. The mitochondrial fraction was harvested from the interface of the 23% and 40% Percoll layers, supplemented with 0.5 ml of 10 mg/ml BSA in 3 ml IB, centrifuged at 16,000 g for 10 min, and then resuspended in IB. Mitochondria-enriched membrane fraction was subjected to phase partitioning with Triton X-114 as described (Bordier, 1981), and equal proportions of the aqueous and detergent phases were immunoblotted sequentially with affinity purified antibodies against syntabulin, cytochrome c (BD Biosciences), and TOM20 (Santa Cruz Biotechnology, Inc.). The immunoisolation of mitochondria was performed with antibody-coated magnetic beads. In brief, goat anti–mouse IgG (Fc fragment specific) (linker, Jackson ImmunoResearch Laboratories) was incubated for 24 h at 37°C on a rotator with M-500 Dynabeads (Dynal) at a ratio of 7 μg of linker per 107 beads in 0.1 M borate buffer (pH 9.5) at a final concentration of 4 × 108 beads/ml. The coated beads were washed twice, 5 min each, in PBS pH 7.4 with 0.1% BSA at 4°C on a rotator, and incubated for 20 h in 0.2 M Tris pH 8.5 with 0.1% BSA at RT. After washing once for 5 min in PBS pH 7.4 with 0.1% BSA at 4°C, the linker-coated beads (1.4 mg) were incubated with 5 μg anti-TOM20 mAb or control mouse IgG overnight at 4°C on a rotator. After incubation, the beads were washed four times (5 min each) in PBS pH 7.4 with 0.1% BSA at 4°C, and then resuspended in buffer A containing PBS, pH 7.4, 2 mM EDTA, and 5% BSA. Mitochondrial fraction was mixed with buffer A containing beads (final reaction volume 1 ml) and incubated overnight at 4°C on a rotator. After incubation, the beads were collected with a magnetic device (Dynal) and washed five times with the incubation buffer and three times with PBS at 10 min each. After the final wash, the beads were saved as bound fractions, and were analyzed by immunoblotting for detection of syntabulin, cytochrome c, and TOM20.
Fusion protein preparation and in vitro binding
Truncated mutants of syntabulin were constructed into the GST-fusion vectors, pGEX-2 or pGEX-4T (GE Healthcare); the His-tagged fusion protein vectors, pET28a (Novagen); and pEGFP-C1 vectors (CLONTECH Laboratories, Inc.). GST- and His-tagged fusion proteins were prepared as crude bacterial lysates by mild sonication in PBS (50 mM sodium phosphate at pH 8.0, 300 mM NaCl, 1% Triton X-100, plus protease inhibitors of 1 mM phenylmethylsulfonyl fluoride, 10 μg ml−1 leupeptin, 2 μg ml−1 aprotinin). In vitro binding studies were performed as described previously (Su et al., 2004). In brief, GST fusion proteins were bound to glutathione-Sepharose beads (GE Healthcare) in TBS buffer (50 mM Tris-HCl at pH 7.5, 140 mM NaCl) with 0.1% Triton X-100 and protease inhibitors, incubated at 4°C for 1 h with constant agitation, and washed with TBS buffer to remove unbound protein. Glutathione-Sepharose beads coupled with ∼1 μg GST fusion protein were added to His-fusion proteins, and then incubated with gentle mixing for 3 h at 4°C. The beads were washed three times with TBS buffer, and bound proteins were eluted and processed by electrophoresis on a 10–20% SDS-tricine gradient gel followed by immunoblotting. Bound His-tagged proteins and GST fusion proteins were detected with antibodies against T7-His (Novagen) and GST (GE Healthcare). HRP-conjugated secondary antibodies and enhanced chemiluminescence reagents (GE Healthcare) were used to visualize detected proteins. For multiple detection with different antibodies, blots were stripped in a solution of 62.5 mM Tris-HCl, pH 7.5, 20 mM dithiothreitol, and 1% SDS for 20 min at 50°C with agitation, and washed twice with TBS/0.1% Tween-20 for 15 min each time.
Transfection and immunocytochemistry of cultured hippocampal neurons
Rat hippocampal neuron cultures were prepared at a density of 25,000 cells per cm2 on polyornithine- and fibronectin-coated coverslips from hippocampi that were dissected from embryonic day (E) 18 or E19 rat embryos as described (Goslin et al., 1998). Cultures were grown for 2–3 d in a 40:60 mix of conditioned glial feed (10% FBS in Neurobasal, Invitrogen) and neuronal feed (1 × B27 supplemented with 100 × L-glutamine, Invitrogen). Starting at DIV2–3, cultures were maintained in conditioned medium with half-feed changes of neuronal feed lacking glutamine every 3–4 d. Transfection was performed by using Lipofectamine 2000 (Life Technologies) in two-chambered tissue culture chambers. Opti-MEM (100 μl; Invitrogen) and 2–3 μl Lipofectamine per chamber were preincubated at RT for 5 min. During the time of incubation, 100 μl Opti-MEM was combined with the indicated DNA constructs (1–2 μg per chamber), and the mixture was incubated for another 20 min at RT to allow complex formation. The entire mixture was added directly to cultured hippocampal neurons at DIV4–5. Following transfection, cells were cultured for an additional 48–120 h before imaging or immunocytochemistry. For immunostaining, cultured cells were fixed in a physiologic solution containing 4% paraformaldehyde (119 mM NaCl, 2.5 mM KCl, 2 mM CaCl2, 2 mM MgCl2, 30 mM glucose, pH 7.4) at RT for 15 min as described in Su et al. (2004), washed with PBS three times for 5 min, permeabilized in 0.2% Triton X-100 or 0.2% saponin for 15 min, and incubated in 5% normal goat serum in PBS for 1 h. Fixed cultures were incubated with primary antibodies against syntabulin (1:100) and cytochrome c (1:100, BD Biosciences) in PBS/1% BSA and 0.05% saponin overnight at 4°C or for 4 h at RT. The cells were washed four times with PBS at RT for 5 min each, incubated with secondary fluorescent antibodies (Jackson ImmunoResearch Laboratories) at 1:100 dilution in PBS for 1 h, washed again with PBS, and mounted with antifade mounting medium for imaging.
Image acquisition and quantification
Confocal images were obtained using a Zeiss LSM 510 oil immersion 40× or 63× objective with sequential-acquisition setting. For quantification of the fluorescence signal, images of labeled cells were acquired by using all of the same settings below saturation at a resolution of 1,024 × 1,024 pixels (12 bit). 8 to 10 sections were taken from top to bottom of the specimen, and brightest point projections were made. Transfected neurons were chosen for quantification from more than six chambers of Lab-Tek from at least three independent experiments for each DNA construct. The number of neurons used for quantification is indicated in the figure legends. Morphometric measurements were performed using Image-Pro Plus analysis software (Media Cybernetics); the values were imported into Excel software for data analysis.
Because the density of mitochondria throughout neuronal development is more stable in main processes (axons) than in minor processes (Ruthel and Hollenbeck, 2003), we chose to examine the main neuronal processes—the longest process relative to the minor processes in each neuron. We manually traced the main neuronal processes >200 μm length starting from the cell body; regions of interest were measured. Fluorescence intensity of DsRed-mitotracker was measured as mean intensity. The average background intensity from neighboring regions of untransfected cells was subtracted. Relative mitochondria distribution in main neuronal processes was expressed as the mitochondrial mean intensity, number of mitochondrial clusters per μm length of process, and relative mitochondrial area per μm length of process as described previously (De Vos et al., 2003; Pigino et al., 2003; Ruthel and Hollenbeck, 2003; Miller and Sheetz, 2004). These data were estimated automatically and scored by the Image-Pro Plus analysis software based on the fluorescence intensity profile. All statistical analyses were performed using the t test and all measurements are presented as mean ± SEM.
Image acquisition from living cells
Transfected hippocampal neurons were transferred to the Tyrode's solution (10 mM Hepes, 10 mM glucose, 3 mM KCl, and 145 mM NaCl, pH 7.4). The temperature was maintained at 37°C with an air stream incubator. Cells were visualized with an oil immersion 40/63× NA 1.4 Planacromat objective on an LSM 510 Zeiss confocal microscope, using 488-nm excitation for GFP and 543-nm excitation for DsRed. For imaging live neurons, 1% of the intensity of the argon laser was used to minimize bleaching and damage to neurons; maximum pinhole opening was set; at least three sections were taken from top to bottom of the specimen for brightest point projections to cover most of the thickness of the processes; and 1.4–2.0× digital zoom was used for better viewing. Images of 640 × 512-pixel resolution were taken at 5-s intervals (6 s of scanning and 3-s cycle delay) for a total of 100 frames. The short interval was used to minimize laser-induced neuronal damages. All recordings started 6 min after the coverslip was placed in the chamber.
Measurement of mitochondrial mobility
The polarity orientation of MTs differs in different process regions of neurons. In axons, MTs are oriented uniformly with their plus-ends directed distally (Burton and Paige, 1981; Heidemann et al., 1981; Baas et al., 1988). In contrast, in the proximal region of dendrites, MTs are of mixed orientation and the plus-end–directed motor kinesins can function in both directions (Baas et al., 1988, 1989). Thus, to simplify interpretation for our results, we selected axonal processes for quantitative analysis of mitochondrial mobility. Axonal processes were chosen for recordings based on known morphologic characteristics (Banker and Cowan, 1979). A process was considered an axon if it was at least twice the length of any of the other processes. The other processes were considered dendrites. Only those that appeared to be single axons and isolated from other processes in the field were chosen. Regions where crossings or fasciculation occurred were excluded from analysis. To ensure that we were observing true mitochondrial motility, rather than the results of possible axonal retraction or bulk axoplasmic flow, we used two selection criteria: (1) only axons that did not move relative to the substratum were considered and (2) only sections of axons that were thin and of uniform caliber were studied. Furthermore, to avoid bias in selecting moving or stationary mitochondria for study, only mitochondria that met the above criteria were traced for the period of time during which they were in focus. An organelle was considered to be stopped if it remained stationary for the entire recording period; a movement was counted only if the displacement was at least 5 μm. The net direction of resumed movement was recorded as either the same or changed, which is determined by comparing the net displacement between its initial and final positions relative to the cell body. For quantitative analysis, two motility characteristics were recorded for each mitochondrion observed: (1) whether it moved and (2) its net direction of movement (anterograde or retrograde) as described previously (Overly et al., 1996; Ligon and Steward, 2000; De Vos et al., 2003; Miller and Sheetz, 2004). Recordings were analyzed with the Image-Pro Plus image analysis software. Subsequently, the relative percentage of stationary, net plus- and minus-end–directed events was calculated (Mandelkow et al., 2004). Counts often were repeated two to five times and then averaged to ensure accuracy. All statistical analyses were performed using the t test and all measurements are presented as mean ± SEM.
Online supplemental material
Fig. S1 shows that the carboxyl-terminal tail of syntabulin (600–663) is required for its association with mitochondria. Fig. S2 shows that syntabulin (145–230) is required for binding to the kinesin CBD. Figs. S3 and S4 show that syntabulin loss-of-function has no effect on the proper distribution of ER and Golgi in neurons. Video 1 shows that the carboxyl-terminal tail of syntabulin is sufficient to target to mobile mitochondria along processes of living neurons. Videos 2–5 show relative motility of DsRed-mitotracker–labeled mitochondria in live hippocampal neurons expressing control-siRNA, stb-siRNA, GFP control, or GFP-STB-KBD. Online supplemental material available at http://www.jcb.org/cgi/content/full/jcb.200506042/DC1.
Acknowledgments
We gratefully acknowledge R.J. Youle, C. Blackstone, and B. McNeil for their critical reading and thoughtful comments on the manuscript; and J.-S. Kang, M. Leenders, P. Zald, J.-H. Tian, J. Stadler, and other members of Sheng's lab for discussion on imaging quantification and mitochondrial purification. We also acknowledge numerous contributions from many laboratories whose references we were unable to cite because of space limitations.
Q. Cai is a graduate student of the National Institutes of Health-Shanghai Second Medical University (NIH-SSMU) Joint Ph.D. Program in Neuroscience. We acknowledge P.-H. Lu for her support to the joint NIH-SSMU training program. This work was supported by the intramural research program of National Institute of Neurological Disorders and Stroke, NIH (Z.-H. Sheng).
Abbreviations used in this paper: CBD, cargo-binding domain; DIV, day in vitro; IB, isolation buffer; KBD, kinesin-binding domain; KHC, kinesin-1 heavy chain or KIF5; MT, microtubule; stb-siRNA, syntabulin-targeted small interfering RNA.
References
- Allen, R.D., J. Metuzals, I. Tasaki, S.T. Brady, and S.P. Gilbert. 1982. Fast axonal transport in squid giant axon. Science. 218:1127–1129. [DOI] [PubMed] [Google Scholar]
- Baas, P.W., J.S. Deitch, M.M. Black, and G.A. Banker. 1988. Polarity orientation of microtubules in hippocampal neurons: uniformity in the axon and nonuniformity in the dendrite. Proc. Natl. Acad. Sci. USA. 85:8335–8339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baas, P.W., M.M. Black, and G.A. Banker. 1989. Changes in microtubule polarity orientation during the development of hippocampal neurons in culture. J. Cell Biol. 109:3085–3094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banker, G.A., and W.M. Cowan. 1979. Further observations on hippocampal neurons in dispersed cell culture. J. Comp. Neurol. 187:469–493. [DOI] [PubMed] [Google Scholar]
- Bordier, C. 1981. Phase separation of integral membrane proteins in Triton X-114 solution. J. Biol. Chem. 256:1604–1607. [PubMed] [Google Scholar]
- Bowman, A.B., A. Kamal, B.W. Ritchings, A.V. Philp, M. McGrail, J.G. Gindhart, and L.S. Goldstein. 2000. Kinesin-dependent axonal transport is mediated by the sunday driver SYD protein. Cell. 103:583–594. [DOI] [PubMed] [Google Scholar]
- Brickley, K., M.J. Smith, M. Beck, and F.A. Stephenson. 2005. Grif-1 and OGT interacting protein, OIP106, members of a novel gene family of coiled-coil domain proteins: association in vivo and in vitro with kinesin. J. Biol. Chem. 280:14723–14732. [DOI] [PubMed] [Google Scholar]
- Burton, P.R., and J.L. Paige. 1981. Polarity of axoplasmic microtubules in the olfactory nerve of the frog. Proc. Natl. Acad. Sci. USA. 78:3269–3273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Vos, K.J., J. Sable, K.E. Miller, and M.P. Sheetz. 2003. Expression of phosphatidylinositol 4,5 bisphosphate–specific pleckstrin homology domains alters direction but not the level of axonal transport of mitochondria. Mol. Biol. Cell. 14:3636–3649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diefenbach, R.J., J.P. Mackay, P.J. Armati, and A.L. Cunnimgham. 1998. The C-terminal region of the Stalk domain of ubiquitous human kinesin heavy chain contains the binding site for kinesin light chain. Biochemistry. 37:16663–16670. [DOI] [PubMed] [Google Scholar]
- Goldstein, L.S., and Z. Yang. 2000. Microtubule-based transport systems in neurons: the roles of kinesins and dyneins. Annu. Rev. Neurosci. 23:39–71. [DOI] [PubMed] [Google Scholar]
- Gorska-Andrzejak, J., R.S. Stowers, J. Borycz, R. Kostyleva, T.L. Schwarz, and I.A. Meinertzhagen. 2003. Mitochondria are redistributed in Drosophila photoreceptors lacking milton, a kinesin-associated protein. J. Comp. Neurol. 463:372–388. [DOI] [PubMed] [Google Scholar]
- Goslin, K., H. Asmussen, and G. Banker. 1998. Rat hippocampal neurons in low density. Culturing Nerve Cells. 2nd ed. G. Banker and K. Goslin, editors. M.I.T. Press, Cambridge, MA. 339–370.
- Gunawardena, S., and L.S. Goldstein. 2001. Disruption of axonal transport and neuronal viability by amyloid precursor protein mutations in Drosophila. Neuron. 32:389–401. [DOI] [PubMed] [Google Scholar]
- Gunawardena, S., L.S. Her, R.G. Brusch, R.A. Laymon, I.R. Niesman, B. Gordesky-Gold, L. Sintasath, N.M. Bonini, and L.S. Goldstein. 2003. Disruption of axonal transport by loss of huntingtin or expression of pathogenic polyQ proteins in Drosophila. Neuron. 40:25–40. [DOI] [PubMed] [Google Scholar]
- Gunawardena, S., and L.S. Goldstein. 2004. Cargo-carrying motor vehicles on the neuronal highway: transport pathways and neurodegenerative disease. J. Neurobiol. 58:258–271. [DOI] [PubMed] [Google Scholar]
- Heidemann, S.R., J.M. Landers, and M.A. Hamborg. 1981. Polarity orientation of axonal microtubules. J. Cell Biol. 91:661–665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollenbeck, P.J. 1996. The pattern and mechanism of mitochondrial transport in axons. Front. Biosci. 1:91–102. [DOI] [PubMed] [Google Scholar]
- Hurd, D.D., and W.M. Saxton. 1996. Kinesin mutations cause motor neuron disease phenotypes by disrupting fast axonal transport in Drosophila. Genetics. 144:1075–1085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jellali, A., M.H. Metz-Boutigue, I. Surgucheva, V. Jancsik, C. Schwartz, D. Filliol, V.I. Gelfand, and A. Rendon. 1994. Structural and biochemical properties of kinesin heavy chain associated with rat brain mitochondria. Cell Motil. Cytoskeleton. 28:79–93. [DOI] [PubMed] [Google Scholar]
- Kamal, A., G.B. Stokin, Z. Yang, C.H. Xia, and L.S. Goldstein. 2000. Axonal transport of amyloid precursor protein is mediated by direct binding to the kinesin light chain subunit of kinesin-I. Neuron. 28:449–459. [DOI] [PubMed] [Google Scholar]
- Leopold, P.L., A.W. McDowall, K.K. Pfister, G.S. Bloom, and S.T. Brady. 1992. Association of kinesin with characterized membrane-bounded organelles. Cell Motil. Cytoskeleton. 23:19–33. [DOI] [PubMed] [Google Scholar]
- Li, Z., K. Okamoto, Y. Hayashi, and M. Sheng. 2004. The importance of dendritic mitochondria in the morphogenesis and plasticity of spines and synapses. Cell. 119:873–887. [DOI] [PubMed] [Google Scholar]
- Ligon, L.A., and O. Steward. 2000. Movement of mitochondria in the axons and dendrites of cultured hippocampal neurons. J. Comp. Neurol. 427:340–350. [DOI] [PubMed] [Google Scholar]
- Mandelkow, E.M., E. Thies, B. Trinczek, J. Biernat, and E. Mandelkow. 2004. MARK/PAR1 kinase is a regulator of microtubule-dependent transport in axons. J. Cell Biol. 167:99–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manning, B.D., and M. Snyder. 2000. Drivers and passengers wanted! The role of kinesin-associated proteins. Trends Cell Biol. 10:281–289. [DOI] [PubMed] [Google Scholar]
- Miller, K.E., and M.P. Sheetz. 2004. Axonal mitochondrial transport and potential are correlated. J. Cell Sci. 117:2791–2804. [DOI] [PubMed] [Google Scholar]
- Morris, R.L., and P.J. Hollenbeck. 1993. The regulation of bidirectional mitochondrial transport is coordinated with axonal outgrowth. J. Cell Sci. 104:917–927. [DOI] [PubMed] [Google Scholar]
- Nangaku, M., R. Sato-Yoshitake, Y. Okada, Y. Noda, R. Takemura, H. Yamazaki, and N. Hirokawa. 1994. KIF1B, a novel microtubule plus end-directed monomeric motor protein for transport of mitochondria. Cell. 79:1209–1220. [DOI] [PubMed] [Google Scholar]
- Overly, C.C., H.I. Rieff, and P.J. Hollenbeck. 1996. Organelle motility and metabolism in axons vs dendrites of cultured hippocampal neurons. J. Cell Sci. 109:971–980. [DOI] [PubMed] [Google Scholar]
- Pereira, A.J., B. Dalby, R.J. Stewart, S.J. Doxsey, and L.S. Goldstein. 1997. Mitochondrial association of a plus end-directed microtubule motor expressed during mitosis in Drosophila. J. Cell Biol. 136:1081–1090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pigino, G., G. Morfini, A. Pelsman, M.P. Mattson, S.T. Brady, and J. Busciglio. 2003. Alzheimer's presenilin 1 mutations impair kinesin-based axonal transport. J. Neurosci. 23:4499–4508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raha, S., and B.H. Robinson. 2000. Mitochondria, oxygen free radicals, disease and ageing. Trends Biochem. Sci. 25:502–508. [DOI] [PubMed] [Google Scholar]
- Rowland, K.C., N.K. Irby, and G.A. Spirou. 2000. Specialized synapse-associated structures within the calyx of Held. J. Neurosci. 20:9135–9144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruthel, G., and P.J. Hollenbeck. 2003. Response of mitochondrial traffic to axon determination and differential branch growth. J. Neurosci. 23:8618–8624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawa, A. 2001. Mechanisms for neuronal cell death and dysfunction in Huntington's disease: pathological cross-talk between the nucleus and the mitochondria? J. Mol. Med. 79:375–381. [DOI] [PubMed] [Google Scholar]
- Setou, M., D.H. Seog, Y. Tanaka, Y. Kanai, Y. Takei, M. Kawagishi, and N. Hirokawa. 2002. Glutamate-receptor-interacting protein GRIP1 directly steers kinesin to dendrites. Nature. 417:83–87. [DOI] [PubMed] [Google Scholar]
- Setou, M., T. Nakagawa, D.H. Seog, and N. Hirokawa. 2000. Kinesin superfamily motor protein KIF17 and mLin-10 in NMDA receptor-containing vesicle transport. Science. 288:1796–1802. [DOI] [PubMed] [Google Scholar]
- Shepherd, G.M., and K.M. Harris. 1998. Three-dimensional structure and composition of CA3–CA1 axons in rat hippocampal slices: implications for presynaptic connectivity and compartmentalization. J. Neurosci. 18:8300–8310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sims, N.R. 1990. Rapid isolation of metabolically active mitochondria from rat brain and subregions using Percoll density gradient centrifugation. J. Neurochem. 55:698–707. [DOI] [PubMed] [Google Scholar]
- Stowers, R.S., L.J. Megeath, J. Gorska-Andrzejak, I.A. Meinertzhagen, and T.L. Schwarz. 2002. Axonal transport of mitochondria to synapses depends on Milton, a novel Drosophila protein. Neuron. 36:1063–1077. [DOI] [PubMed] [Google Scholar]
- Su, Q., Q. Cai, C. Gerwin, C.L. Smith, and Z.H. Sheng. 2004. Syntabulin is a microtubule-associated protein implicated in syntaxin transport in neurons. Nat. Cell Biol. 6:941–953. [DOI] [PubMed] [Google Scholar]
- Swerdlow, R.H., and S.J. Kish. 2002. Mitochondria in Alzheimer's disease. Int. Rev. Neurobiol. 53:341–385. [DOI] [PubMed] [Google Scholar]
- Takeda, S., H. Yamazaki, D.H. Seog, Y. Kanai, S. Terada, and N. Hirokawa. 2000. Kinesin superfamily protein 3 KIF3 motor transports fodrin-associating vesicles important for neurite building. J. Cell Biol. 148:1255–1265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka, Y., Y. Kanai, Y. Okada, S. Nonaka, S. Takeda, A. Harada, and N. Hirokawa. 1998. Targeted disruption of mouse conventional kinesin heavy chain, Kif5B, results in abnormal perinuclear clustering of mitochondria. Cell. 93:1147–1158. [DOI] [PubMed] [Google Scholar]
- Verhey, K.J., D. Meyer, R. Deehan, J. Blenis, B.J. Schnapp, T.A. Rapoport, and B. Margolis. 2001. Cargo of kinesin identified as JIP scaffolding proteins and associated signaling molecules. J. Cell Biol. 152:959–970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wattenberg, B., and T. Lithgow. 2001. Targeting of C-terminal tail-anchored proteins: understanding how cytoplasmic activities are anchored to intracellular membranes. Traffic. 2:66–71. [DOI] [PubMed] [Google Scholar]
- Werth, J.L., and S.A. Thayer. 1994. Mitochondria buffer physiological calcium loads in cultured rat dorsal root ganglion neurons. J. Neurosci. 14:348–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zucker, R.S. 1999. Calcium- and activity-dependent synaptic plasticity. Curr. Opin. Neurobiol. 9:305–313. [DOI] [PubMed] [Google Scholar]