

Abstract
Background
Malignant pleural mesothelioma (MPM) is an aggressive cancer that is refractory to current treatment modalities. Oncolytic herpes simplex viruses (HSV) used for gene therapy are genetically engineered, replication-competent viruses that selectively target tumor cells while sparing normal host tissue. The localized nature, the potential accessibility and the relative lack of distant metastasis, make MPM a particularly suitable disease for oncolytic viral therapy.
Methods
The infectivity, selective replication, vector spread and cytotoxic ability of three oncolytic HSV: G207, NV1020 and NV1066 were tested against eleven pathological types of MPM cell lines including those that are resistant to radiation therapy, gemcitabine or cisplatin. The therapeutic efficacy and the effect on survival of NV1066 were confirmed in a murine MPM model.
Results
All three oncolytic HSV were highly effective against all the MPM cell lines tested. Even at very low concentrations of MOI 0.01 (MOI: multiplicity of viral infection, ratio of viral particles per cancer cell), HSV were highly effective against MPM cells that are resistant to radiation, gemcitabine and cisplatin. NV1066, an oncolytic HSV that expresses green fluorescent protein (GFP) was able to delineate the extent of the disease in a murine model of MPM due to selective infection and expression of GFP in tumor cells. Furthermore, NV1066 was able to reduce the tumor burden and prolong survival even when treated at an advanced stage of the disease.
Conclusion
These findings support the continued investigation of oncolytic HSV as potential therapy for patients with therapy resistant malignant pleural mesothelioma.
Keywords: Gene therapy, Herpes simplex virus, NV1066
INTRODUCTION
Malignant pleural mesothelioma (MPM) is an aggressive cancer with a median survival of 4-12 months1, 2. There has been a steady rise in the incidence of this malignancy over the past 25 years. Because of patterns of occupational asbestos exposure and the long latency period (30 to 40 years), the annual incidence of 3000 new cases is expected to increase by more than 50% in the coming decade3, 4. Pleural mesothelioma is a diffuse disease. Therefore radiation therapy must include the entire hemithorax and the dose of radiation required to produce local control of the disease is high5. Mesothelioma is a chemoresistant malignancy. Even with combination chemotherapy (cisplatin and/or platinum-based combination chemotherapy), only a 13 to 18% response rate has been reported6. Surgery has been the mainstay of treatment for MPM as chemotherapy and radiation are relatively ineffective. Even with combined surgery, chemotherapy and radiation, only a minority of patients are rendered prolonged disease-free survivors6-8. There is a clear need for development of new treatment modalities.
Oncolytic Herpes Simplex type-1 (HSV-1) viruses have been efficacious in the treatment of number of human and animal cancers9-19 and are currently under evaluation in clinical trials20-23. HSV-1 viruses preferentially infect and lyse tumor cells because of their affinity for actively dividing cells 24-27. These viruses are replication-competent, allowing their progeny to infect other tumor cells and thus prolong antitumor activity. The localized nature, the potential accessibility, and the relative lack of distant metastasis make pleural malignant mesothelioma a particularly suitable disease for oncolytic viral therapy. Since the primary mechanism of oncolytic viral therapy is direct cell lysis, there is a good reason to believe that the bulk of the tumor may be reduced, thus allowing more patients to be eligible for surgical therapy.
One source of replication-competent HSV-1 vector selectivity for actively dividing cells, and hence cancer cells, is by virtue of mutations in viral enzymes involved in nucleotide metabolism (thymidine kinase [TK], ribonucleotide reductase [RR], and uracil deglycosylase [UNG]). There are functional similarities between the viral and cellular enzymes, which are upregulated in cancer cells and not expressed for the most part in postmitotic cells. Early attempts to utilize HSV-1 as an oncolytic vector focused on mutating/deleting one of these genes, leading to the development of first-generation attenuated vectors such as NV1020. This virus was originally developed as a herpes vaccine but was unsuccessful. However, building on the associated safety studies in rodents and primates, it has been used as an oncolytic agent against various non-CNS tumors. These first-generation HSV-1 vectors, thus, provided the foundation for examining the critical issues of safety, specificity, and efficacy for oncolytic virotherapy. In order to maximize safety, it was reasoned that HSV-1 vectors developed for clinical application contain multiple mutations, so that virulent strains would not arise from reversion or second site suppressor mutations. G207 was constructed as a second-generation vector from HSV-1 laboratory strain F, with both copies of 34.5 deleted and the ICP6 gene inactivated by insertion of the E. coli LacZ gene. Both NV1020 and G207 are currently in clinical trials20, 21, 28.
When administering HSV-1 mutants and other oncolytic viruses or viral vectors, attempts have been made to follow viral infection and spread in vivo by noninvasive imaging methods in several preclinical studies. Such imaging strategies have limitations similar to conventional radiological techniques and may only detect areas with large amounts of viral uptake. Genetically engineered herpes viruses may be useful in the treatment of cancer based upon their oncolytic properties alone, or as vectors to carry therapeutic or immunomodulatory transgenes to targeted tumors. NV1066 carries such a marker gene, a constitutively expressed transgene for EGFP, the protein product of which is identifiable 4–6 hr following viral entry into cells. In the current study, we sought to determine the efficacy of three oncolytic viral therapy; G207, NV1020 and NV1066 in the treatment of human MPM both in vitro and in vivo.
METHODS
Cell culture
Human malignant mesothelioma cell lines of various histological subtypes were studied including sarcomatoid (VAMT, H-28, H-2052, H-2373), epithelioid (H-2452, H-meso), biphasic (H-meso1A, MSTO-211H, JMN) and others (Meso-9, Meso-10). MSTO-211H, H28 and Vero cells (from the African green monkey kidney) were obtained from the American Type Culture Collection (ATCC®, Rockville, MD). H-meso and H-meso1A cell lines were obtained from National Cancer Institute (NCI, Bethesda, MD). JMN, VAMT, Meso-9 and Meso-10 cell lines were kind donation from Dr. Sirotnik from Memorial Sloan-Kettering Cancer Center, New York. H-2052, H-2452, H-2373 cell lines were kind donation from Dr. Pass from Karmanos Cancer Institute, Wayne State University, Detroit. All the cell lines were grown in appropriate media and were incubated in a humidified incubator supplied with 5% carbon dioxide.
Viruses
G207 is an engineered herpes simplex virus based on the wild-type HSV-1 strain-F, and was received as a gift from SD Rabkin and RL Martuza. As previously described, both γ134.5 genes have been deleted, and the Escherichia coli lacZ marker gene has been inserted into the UL39 gene, inactivating RR. NV1020 (gift of Medigene Inc, San Diego, CA) is an attenuated, replication-competent derivative of HSV-1. NV1020 is a non selected clonal derivative of R7020, an attenuated, replication-competent virus based on the HSV-1 strain-F, originally obtained from B Roizman29. It has a 15-kb deletion over the joint region of the HSV-1 genome. This deletion encompasses the region of the genome coding for the ICP0, ICP4, latency associated transcripts (LAT), and one copy of the neurovirulence gene (γ134.5). These deletions greatly attenuate virulence of the virus. Because it was originally designed as a vaccine against HSV-1 and HSV-2 infection, a fragment of HSV-2 DNA from the HindIIIL region encoding for several glycoprotein genes was inserted into the deleted joint region. It also has a 700-bp deletion of the endogenous TK locus that prevents expression of the overlapping transcripts belonging to the UL24 gene. An exogenous copy of the HSV-1 TK gene was inserted under control of the 4 promoter. NV1066 is a replication-competent, attenuated HSV-1 oncolytic virus with loss of single copies of the ICP-4, ICP-0, and γ134.5 genes have been deleted to increase tumor specificity and to decrease virulence30. NV1066 also contains the enhanced GFP (Green Florescent Protein) sequence under the control of a constitutive cytomegalovirus promoter. All virus preparations were formulated in D-phosphate-buffered saline solution (PBS)-10% glycerin and stored at −80°C. Viral stocks were propagated on Vero cells, harvested by freeze-thaw lysis and sonication, and titered by standard plaque assay.
Cell proliferation assay
Each MPM cell line was plated at a concentration of 20,000 cells per well in 1 ml of respective media in 24-well plates (Becton Dickinson, Franklin Lake, NJ) and incubated. On days 3, 5, 6 and 7, viable cells from four individual wells were counted after trypsinization and staining with trypan blue. The average number of cells per well of each cell line were plotted logarithmically to demonstrate the growth properties.
Determination of therapy resistance
Exponentially growing cells were detached from the cell culture and were plated to achieve 15,000 cells per well for each of the eleven cell lines in 96-well plates. Plated cells were incubated for a 12-hr period before treatment. Cells were treated with chemotherapeutic agents at concentrations 1, 10, 100, 1000 and 10,000 ng / mL of gemcitabine alone, or 0.1, 1, 10, 100 and 1000 μM / mL of cisplatin alone. Each concentration of drug alone was completed in six wells. Also, controls (no drug) were included for each 96-well plate. At 72 hours, media was removed. Cells were washed with PBS, and lysed (1.35% Triton-X solution) to release intracellular lactate dehydrogenase (LDH). LDH was quantified using a Cytotox 96 nonradioactive cytotoxicity assay (Promega, Madison, WI) that measures the conversion of a tetrazolium salt into a red formazan product. The amount of color formed is directly proportional to the number of lysed cells. Absorbance was measured at 450 nm using a microplate reader (EL 312e: Bio-Tek Instruments, Winooski, VT). Results were expressed as surviving fraction, based on the measured absorbance of treated cellular lysates, compared to that of untreated, control cellular lysates. Experiments were repeated in triplicate to ensure reproducibility and the IC50 (dose required to achieve 50% growth inhibition) was determined from the mean percent growth inhibition. Similar experiments were repeated with 20,000 cells plated in 24-well plate with radiation alone (137Cs source irradiator, 224 cGy/min) at a dose of 1, 2, 3, 4 or 5 Gy or no radiation and IC50 for each cell line was derived. IC50 for each therapeutic agent for all the cell lines was calculated by using median-effect equation and plot of Chou and Talalay method.
In vitro cytotoxicity assay
Malignant mesothelioma cells were plated at a concentration of 20,000 in 24-well flat bottom plates in 1 ml of media. Cells were treated with media alone (control wells), or virus alone (G207, NV1020, NV1066). Viral infection was carried out at multiplicities of infection of 0.01, 0.1 or 1 in a total volume of 100 μl of medium. Typically cells were plated overnight and infected with virus in the morning. Percent survival for each group was determined on each day for 7 days after treatment using a standard LDH release bioassay. Results were expressed as surviving fraction, based on the measured absorbance of treated cellular lysates, compared to that of untreated, control cellular lysates. All samples were tested in six wells. Experiments were repeated at least three times to ensure reproducibility.
In vitro viral growth analysis
The ability of the oncolytic virus to replicate within malignant mesothelioma cells was evaluated by viral growth analysis. 50,000 cells / well were plated in 6-well plates. Cells were then infected with G207, NV1020 or NV1066 (MOI 0.01, 0.05, 0.1 or MOI 1). Cells and media were harvested at 48, 72, 96, 120 and 144 hours post-infection. After three cycles of freeze-thaw lysis, standard plaque assay was performed on Vero cells to evaluate viral titers. All samples were performed in triplicate.
Vector Spread Assay
Vector propagation as analyzed by GFP expression was determined by FACS analysis at a viral infective dose of MOI 0.01, 0.1 or 1. Percentage of GFP-positive live cells at 12, 24, 48, 72, 96,120, and 144 hours compared to control cells without viral infection was plotted to derive the GFP-expression trend. Cells were harvested with 0.25% trypsin in 0.02% EDTA, centrifuged, washed in PBS, and brought up in 100 μl of PBS. Five μl of 7-amino-actinomycin (7-AAD, BD Pharmingen, San Diego, CA) was added as an exclusion dye for cell viability. Data for GFP expression was acquired on a FACS Calibur machine equipped with Cell Quest software (Becton Dickinson, San Jose, CA). Results are reported as percent of live cells expressing GFP. All samples were performed in triplicate.
Establishment of malignant pleural mesothelioma
Athymic male mice were purchased from the National Cancer Institute (Bethesda, MD) and were provided food and water ad libitum. All animals received humane care in accordance with the “Guide for the care and use of laboratory animals (NIH)” and the animal protocols were approved by animal care committee of the institution. Anesthesia was induced by a mixture of Isoflurane (2 liters / minute) and oxygen (2 liters / minute) in an induction chamber and maintained by a nasal cone. Mice were placed in left lateral position. The right chest was prepared with 10% povidone-Iodine solution. A 3 to 5 mm incision was created over the fourth to fifth intercostal space. Sharp dissection was carried out exposing the parietal pleura without breaching it. The underlying expanding lung was thereby easily visualized through the thin membrane. One hundred microliters of MSTO-211H malignant mesothelioma cellular suspension (5 × 106 cells in mice for the treatment effect analysis and 1 × 107 cells in mice for survival analysis) was slowly injected through the pleura with a 27-gauge needle. Puncture of the lung was easily avoided because the needle tip was clearly observed to be superficial to the lung surface during injection through the transparent parietal pleura. After the injection the skin was closed with surgical staples. Recovery was observed for 15 minutes before the mice were returned to their cages.
Imaging pleural malignant mesothelioma
One × 107 MSTO-211H cells were injected into the pleural cavities of mice as described previously. Animals (n = 4) were treated with 1 × 107 plaque forming units (pfu) of intrapleural NV1066 as above at separate time points. For imaging of gross pleural disease, mice were treated 10 days after cell injection. Mice were examined 48 to 72 hours and one week later in both cases, using a fluorescent thoracoscopic system and by fluorescent stereomicroscopy. The thoracoscopic system was developed in concert with Olympus America, Inc. (Scientific Equipment Division, Melville, NY) to allow for the detection of GFP as well as routine white light. The light source was an Olympus Visera CLV-U40 model with an adaptable excitation filter set at 470 ± 20 nm. The camera processor was an Olympus Visera OTVS7V with an emission filter set at 510 nm. Imaging was performed in both bright-field and fluorescent modes. With the stereomicroscope (Olympus America Inc., Melville, NY), imaging was also performed in bright-field mode and after placement of both excitation and emission filters to detect GFP. The excitation filter was fixed passage 470 ± 40 nm wavelength light as GFP has a minor excitation peak at 475 nm. The emission filter was fixed at 500 nm, to accommodate the emission peak of GFP at 509 nm. The image-capture system consisted of a Retiga EX digital CCD camera (Qimaging, Burnaby, BC, Canada). For generation of topography maps, images were presented in Zeiss LSM510 software (v3.2) using the “pseudo 3D” tool. Individual pixel intensity values of the image are mapped upon a height grid to quantify fluorescent intensity. Pixels of high intensity are represented by the grid peaks, while pixels of low intensity are represented by grid valleys.
Treatment of malignant pleural mesothelioma
Intrapleural treatment with virus was performed in a similar fashion described above five days after tumor cell instillation (5 × 106 cells) into the pleural cavity. NV1066 (1 × 107 pfu) was administered in 100 μL of PBS. Control animals were treated with an equal volume of PBS. Mice were regularly assessed for weight loss and tachypnea throughout the experimental period. Mice were sacrificed on day 21, weighed and all visible tumor in the chest was graded (grade 1: disease limited to one side of the pleural cavity, grade 2: disease present on both sides of the pleural cavity, grade 3: disease present on both sides of the pleural cavity and mediastinum, grade 4: extensive disease on both sides of the pleural cavity, mediastinum, and diaphragm). Both lungs, heart along with mediastinal content was removed intact and weighed. In treated mice with gross disease, tissue from the chest cavity was harvested for immunohistochemistry.
Survival analysis
Efficacy of NV1066 in prolonging the survival was assessed in two groups of animals with early and advanced disease. In the first experiment, the ability of NV1066 to treat microscopic disease and prolong survival was assessed. Twenty mice (n = 10 / group) were injected with MSTO-211H cells (1 × 107 cells) into the pleural space, and chest incisions were closed. Five days later, mice were treated with either intrapleural PBS or intrapleural NV1066 (1 × 107 pfu). In a second experiment, the ability of NV1066 to treat macroscopic disease and prolong the survival was assessed. Two groups of mice (n = 10 / group) were implanted with MSTO-211H (1 × 107 cells) and treated 11 days later with NV1066 (1 × 107 pfu). Three additional mice were injected with tumor and sacrificed at 10 days to verify the presence of macroscopic disease. Similar survival experiments were repeated with treatment groups receiving three doses of either PBS or NV1066 (1 × 107 pfu each) either injected intrapleurally or given by systemic tail vein injection.
Viral specificity for tumor
Samples of tissue emitting GFP under stereomicroscopy from the chest cavity were frozen in Tissue-Tek embedding medium (Sakura Finetek, Torrance, CA) and sectioned by cryotome for histological examination. Similar sections were taken from other organs (heart, lungs, liver, spleen, kidney, intestine and bone). Following paraformaldehyde fixation, slides were first examined under fluorescent microscopy for GFP expression, then stained with hematoxylin and eosin (H & E) to determine whether GFP expression localized to foci of cancer. To confirm that GFP expression was localizing virus, serial sections that expressed GFP were stained with rabbit polyclonal HSV-1 antibody using a Histomouse-SP Bulk Staining Kit (Zymed Laboratories Inc., San Francisco, CA) and compared for GFP expression and viral antibody binding. An institutional animal pathologist confirmed all results.
Evaluation of viral dissemination and toxicity
Following the establishment of intrathoracic disease as described above, 1 × 107 pfu NV1066 was injected into the pleural cavity. Tissue from normal organs (lung, heart, liver, spleen, kidney, brain, urinary bladder, bone and serum) (n = 3) were harvested at serial time points, 24, 48, and 72 hours, and snap frozen in liquid nitrogen. Genomic DNA was isolated per standard protocols (Wizard Genomic DNA Isolation Kit, Promega, Madison, WI). Real-time quantitative PCR was performed using a Bio-Rad iCycler thermal cycler (Bio-Rad Laboratories, Hercules, CA). Forward (5′-ATGTTTCCCGTCTGG TCCAC-3′) and reverse (5′-CCCTGTCGCCTTACGTGAA-3′) primers and a dual-labeled fluorescent TaqMan probe (5′-6FAM-CCCCGTCTCCATGTCCAGGATGG-TAMRA-3′) were designed for the 111-bp fragment of the HSV-1 ICP0 immediate-early gene. Forward (5′-CGCCTACCACATCCAAGGAA-3′) and reverse (5′-GCTGGAATTACCGCGGCT-3′) primers and a dual-labeled fluorescent TaqMan probe (5′-VICTGCTGGCACCAGCTTGCCCTC-TAMRA-3′) were designed for the 87-bp coding sequence for 18s rRNA to normalize the amount of DNA. The PCR consisted of 50 cycles (step 1: 94°C for 30 sec.; step 2: 55°C for 30 sec.; and step 3: 72°C for 30 sec.). Results are reported as fold increase in ICP0, using the 6 hour time point as the reference. Normal mice without disease were injected with either saline or NV1066 into the pleural cavity and organs were collected to serve as controls.
Statistical analysis
All data were expressed as mean ± standard error of the mean. Comparisons between groups were made using the two-tailed Student's t-test. ANOVA test, where appropriate, was used to identify statistical significance for multiple comparisons. Log rank analysis was performed to analyze the mean survival difference between treated and untreated group.
RESULTS
Mesothelioma cells differ in cell proliferation rates
There was significant variability in the proliferation rates of the 11 malignant mesothelioma cells. Compared to the initial cell number, there is a 7 (Meso-10 cells) to 56 (MSTO-211H cells) –fold increase in cell number by day 7. The most rapidly dividing cells were MSTO-211H, H-Meso, and HMeso1A. JMN, H-2373, VAMT, H-2452 and Meso-9 demonstrated intermediate growth rates. The most slowly dividing cell line was Meso-10.
Oncolytic HSV are highly cytotoxic to malignant mesothelioma cells
All three HSV: G207, NV1020 and NV1066, progressively killed all the mesothelioma cells tested in vitro at all MOI's (MOI: ratio of viral plaque forming units per tumor cell). Higher dose of virus was able to achieve an early high cell kill (MOI of 1, Figure 1A). At lower infective doses (MOI 0.1 and 0.01, Figures 1B and 1C), virus progressively replicated with minimal cell kill at earlier time points, but higher cell kill at later time points. In the most sensitive cell lines (JMN, Meso-1A, Meso-9 and H-2373), 95-100% cell kill is achieved with NV1066 even at a very low dose of virus (MOI of 0.01: one viral pfu per 100 cancer cells). Even in the most resistant cell lines (Meso-10, H-28 and H-2052), > 50% cell kill is achieved at a low MOI of only 0.1 by day 7. In the biphasic cell line, MSTO-211H, after one week in culture, NV1066 was able to achieve a cell kill of 89%, 98% and 99% of cells, at an MOI of 0.01, 0.1 and 1 respectively (p < 0.01). Similar cytotoxicity was observed with other viruses NV1020 and G207 (data not shown). Infected MM cells support viral replication: Significant in vitro viral replication occurred in all malignant mesothelioma cells after infection at all MOI's as determined by viral plaque assay. After in vitro treatment with oncolytic HSV-1 mutants, higher peak viral titers often occur after lower initial MOIs, because early cell death in the populations that are treated with the higher MOI limits the time and cell number for productive cellular replication of virus. In MSTO-211H cell line, infection at an MOI of 0.05 and 0.01 resulted in progressive increase in viral titers until all the cells are lysed (Figure 2A). At an MOI of 0.01, 0.1 and 1.0; NV1066 is able to replicate over 20,000, 1500 and 70 -fold as compared to the initial dose (Figure 2B). In all the cell lines tested, NV1066 is able to replicate between 5-fold (most resistant cell line) to 20,000-fold (most sensitive cell line). Both G207 and NV1020 showed similar replicative pattern (data not shown).
Figure 1.

Cytotoxicity of NV1066 on mesothelioma cells. Eleven mesothelioma cells, epithelioid (H-2452, H-Meso), sarcomatoid (VAMT, H-28, H-2052, H-2373), biphasic (H-Meso1A, MSTO-211H, JMN) and miscellaneous (Meso-9 and Meso-10) were infected in vitro with NV1066. In all these cell lines, NV1066 was cytotoxic at all MOIs of 1 (A), 0.1 (B) and 0.01 (C). Cell kill was represented as percentages of live cells compared to control untreated cells grown under identical conditions. MOI = Multiplicity of infection, ratio of viral particles to tumor cells.
Figure 2.

NV1066 replicates in MSTO-211H cell line at all MOIs. MSTO-211H cells were infected with NV1066 at an MOI of 0.05 or 0.1. Cells and supernatant was collected on days 2, 3, 4, 5 and 7. Viral titers were measured by viral plaque assay on Vero cells. Average numbers of viral plaque forming units in individual well were represented (A). When infected at MOI's 0.01, 0.1 and 1, at all MOIs, infected MSTO-211H cells supported viral replication (B). Initial infective dose was represented as white bars and viral plaque forming units on the day of maximal burst as crossed bars (B). MOI = Multiplicity of infection, ratio of viral particles to tumor cells.
In vitro imaging and vector spread
The GFP transgene carried by NV1066 was used as a marker of viral infection in mesothelioma cells. Under microscopy, cells were visualized under bright-field, cell nucleus stained with Hoechst stain (blue) was visualized with DAPI filter and GFP expression in the cytoplasm was visualized with GFP filter. Representative pictures were taken and overlapped to identify the infected mesothelioma cells (Figures 3A-D). In vitro, with progressive viral replication and vector spread percentage of GFP expressing cells increased over time (Figures 3A-D). In MSTO-211H cells, after the treatment with NV1066 at MOI 0.01, 0.1 and 1.0, nearly 79%, 94% and 98% of the cells expressed GFP by day 6 (p < 0.001 versus untreated control cells) as measured by flow cytometry.
Figure 3.
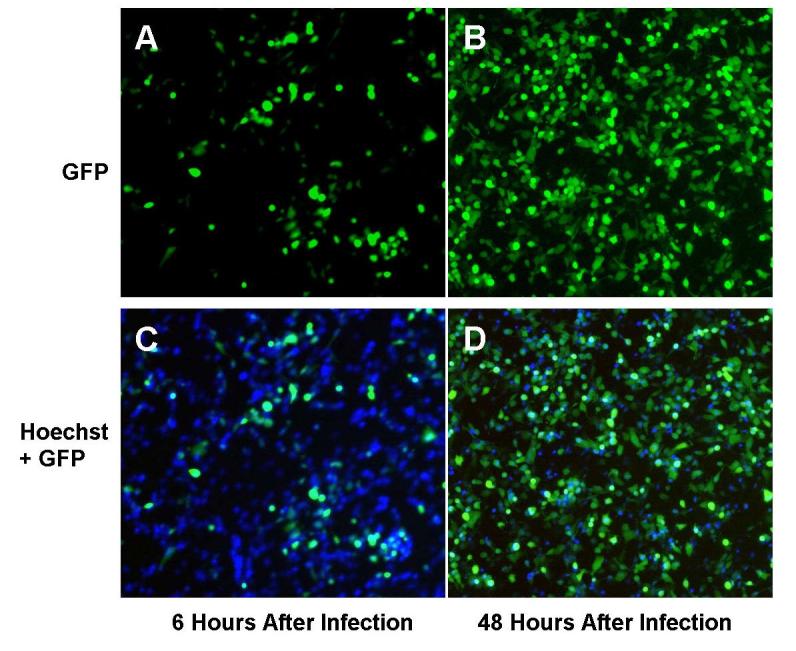
In vitro vector spread as identified by GFP expression. Following treatment with NV1066, infected MSTO-211H cells express GFP, as determined by fluorescent microscopy. GFP expressing cells increased over time, to nearly 100% at all MOIs, indicating that all the cells were infected by NV1066. Malignant mesothelioma cells were infected by NV1066 and their nucleus was stained with Hoechst staining (blue). Images were taken under DAPI filter (to identify individual cells by blue nuclear staining) and GFP filter (to identify GFP expressing infected cells) at 6 and 48 hours. Images were overlapped using metamorph software. Over time, NV1066 was able to infect, replicate, propagate, and express GFP in all malignant mesothelioma cells. MOI = Multiplicity of infection, ratio of viral particles to tumor cells, GFP = Green fluorescent protein.
Cytotoxicity in relation to cell growth
Oncolytic herpes simplex viruses are effective in all malignant mesothelioma cells irrespective of their growth properties (Figures 1B-D). By day 7, Meso-10, JMN and MSTO-211H cells have grown 7-, 27- and 61-fold compared to initial plating concentration of 20,000 cells. In all the cell lines, oncolytic herpes simplex viruses were effective in killing the cancer. By day 7, 49% (Meso-10), 93% (JMN) and 97% (MSTO-211H) of the cells were killed compared to control untreated cells at an infective dose of MOI 0.1. Viral replication and cell kill is more efficacious in rapidly dividing cells as viral replication is dependent upon the cellular replicative machinery.
Oncolytic viral therapy is effective against therapy resistant mesothelioma cells
Malignant mesothelioma cells were relatively resistant to radiation therapy. IC50 was calculated for each cell line to calculate dose required to cause a growth inhibition of 50%. Mesothelioma cells were characterized as ‘radiation therapy resistant’ when the dosage required to achieve IC50 is > 5Gy (JMN, Meso, MSTO-211H, H-28, H-2052 and Meso-10). Gemcitabine and cisplatin demonstrated a dose- and time- dependent decrease in cell viability. Cell lines with gemcitabine IC50 > 100 nM / ml were characterized as ‘gemcitabine resistant’ (VAMT, H28, MSTO-211H, Meso9, Meso10, Meso1A, H2052, H2452 and H-Meso) and those with cisplatin IC50 > 100 μM / ml were characterized as ‘cisplatin resistant’ (Meso9, H2052, H28, and Meso10). These concentrations were chosen basing upon the published peak plasma concentrations achieved with these therapeutic agents when used to treat malignant mesothelioma. HSV were effective in all the mesothelioma cell lines that are resistant to other therapeutic modalities. The IC50 values for three oncolytic viruses in all mesothelioma cell lines were documented in Table 1. In highly gemcitabine resistant cell line, VAMT (IC50 > 1000 nM / ml), at an MOI of 0.1, all three HSV killed 90 ± 5% cells by day 7. In highly cisplatin resistant cell line, Meso-10 (IC50 > 1000 μM / ml), at an MOI of 0.1, all three HSV killed 77 ± 24% cells by day 7. In highly radiation resistant cell line, H-28 (IC50 > 10 Gy), at an MOI of 0.1, all three HSV killed 72 ± 22% cells by day 7.
TABLE 1.
Sensitivity of malignant mesothelioma cells to oncolytic viruses G207, NV1020 and NV1066 Listed are the required MOI to achieve 50% cell kill (IC50) by day 7. ▲ - Radiation resistant cell lines (IC50 > 5Gy), ● -Gemcitabine resistant cell line (IC50 > 100 nM / ml), ◙ - Cisplatin resistant cell line (IC50 > 100 μM / ml). (MOI: Multiplicity of infection, ratio of viral particles per cancer cell).
| Cell Line | Pathological Type |
G207 (MOI) |
NV1020 (MOI) |
NV1066 (MOI) |
|
|---|---|---|---|---|---|
| VAMT | ● | Sarcomatoid | 0.01 | < 0.01 | < 0.01 |
| H-2052 | ▲ ● ◙ | Sarcomatoid | 0.02 | 0.01 | < 0.01 |
| H-2373 | Sarcomatoid | 0.01 | < 0.01 | < 0.01 | |
| H-28 | ▲ ● ◙ | Sarcomatoid | 0.1 | 0.01 | 0.02 |
| H-2452 | ● | Epithelioid | < 0.01 | < 0.01 | < 0.01 |
| H-Meso | ▲ ● | Epithelioid | < 0.01 | < 0.01 | < 0.01 |
| H-Meso1A | ● | Biphasic | < 0.01 | < 0.01 | < 0.01 |
| MSTO-211H | ▲ ● | Biphasic | 0.01 | < 0.01 | < 0.01 |
| JMN | ▲ | Mixed | 0.01 | < 0.01 | < 0.01 |
| Meso-9 | ● ◙ | - | < 0.01 | < 0.01 | < 0.01 |
| Meso-10 | ▲ ● ◙ | - | 0.1 | 0.5 | 0.5 |
A single intrapleural instillation of NV1066 is effective in reducing pleural mesothelioma tumor burden
In these experiments, treated animals continued to demonstrate normal activity with normal feeding and grooming habits. Mice treated with NV1066 retained their body weights. The average body weight of normal, PBS treated and NV1066 treated mice were 30.3 ± 5.9 gm, 26.4 ± 2.6 gm and 30.9 ± 2 gm respectively (p < 0.01). By day 21, only 2/10 NV1066 treated animals (grade 2 and 3) had macroscopic pleural disease compared to 10/10 PBS treated control mice (grade 3 - 4). Organ and mediastinal weights of PBS treated, NV1066 treated and non-tumor bearing animals were 669.3 ± 97.1 mg, 394.9 ± 47.9 mg, and 359.5 ± 80.2 mg, p < 0.01).
NV1066 is effective in achieving cures and improving survival in mice with pleural malignant mesothelioma
In mice with established pleural mesothelioma (MSTO-211H), the median survival for the early NV1066 treated group (single dose) was 95 days versus 19 days for control PBS treated animals (logrank p value < 0.001, n = 10 each, Figure 4A). For the late NV1066 treated group (single dose), the median survival was 48 days versus 20 days for control animals (logrank p = 0.002, n = 10 each, Figure 4B). Treatment with three doses of NV1066 intrapleurally improved survival (logrank p < 0.001, n = 10 each, Figure 4B). The median survival for this group was 86 (47, not reached) and the PBS treated group was 21 (17, 27). In mice treated with NV1066 systemically (three doses by tail vein injection), survival was prolonged (logrank p value < 0.001, n = 10 each, Figure 4C). The median survival for the systemic treated group was 89 (49, 103) and for the PBS treated group was 23 (15, 27). None of the NV1066-treated animals suffered from any clinically apparent side effects attributable to viral administration. A post mortem examination was performed after sacrificing mice in both groups. In control PBS treated group, gross disease was confirmed in all mice. In the NV1066 treated group, in mice that died or sacrificed after prolonged survival, no intrathoracic disease was noted. In these mice, metastatic disease was seen in the retroperitoneal lymph nodes, and in the liver, and adrenal glands. NV1066 was able to spread systemically and selectively infect metastatic disease as identified by GFP fluorescence under fluorescent microscope. But the metastatic tumor burden was high and only part of the metastatic disease expressed GFP.
Figure 4.

NV1066 prolongs survival in murine malignant pleural mesothelioma model. MPM was established in mice after intrapleural instillation of MSTO-211H cells (1 × 107). Mice were treated with either PBS or NV1066 (1 × 107 pfu) injected intrapleurally. NV1066 significantly improved the median survival in the early treatment (day 5) group (A), (p < 0.001); late treatment (day 11) group with one dose (B), (p = 0.002) or three doses (B), (p = 0.002); and with systemic treatment with three doses (C), (p < 0.001). Each dose was administered as 1 × 107 pfu either intrapleurally or systemically by tail vein injection. pfu = plaque forming unit.
In vivo imaging
After the administration of NV1066 in vivo, GFP expression was visualized easily by fluorescent microscopy, in both flank and pleural tumor models. GFP expression was localized to tumor deposits, sparing normal tissues following intrapleural administration of virus. GFP expression could be visualized to localize intrapleural tumor deposits, identifying foci of tumor <1 mm in diameter. Because both pleural cavities are connected in mice, even a single dose of NV1066 was able to spread, infect, replicate and propagate to adjacent pleural disease. This phenomenon was noted in mice that were sacrificed on consecutive days. Mice that were examined a week following NV1066 demonstrated robust GFP expression in all the tumor tissue. The virus was able to spread and infect pleural (Figure 5A-B), and mediastinal mesothelioma (Figure 5C-D) and spare normal tissue. These findings were reproduced when pleural cavity is examined with thoracoscope fitted with fluorescent filter to identify the GFP expression. Specific infection of malignant mesothelioma was clearly seen with thoracoscope in diaphragmatic (Figure 6A-C) and pleural (Figure 6D-E) position. Similar results were obtained after NV1066 treatment of mice with malignant pleural Mesothelioma established by VAMT (sarcomatoid) and JMN (mixed) pathological type.
Figure 5.

Thoracoscopic identification of diaphragmatic and pleural mesothelioma. In vivo, viral uptake and GFP expression in pleural cavities and diaphragmatic mesothelioma can be easily identified by using a thoracoscope with GFP filter. All the mice were injected with NV1066 intrapleurally. GFP expression can be visualized selectively only in malignant mesothelioma tissue as early as 48 - 72 hours. Normal mouse diaphragm (A), diaphragmatic mesothelioma (B) were examined under bright-field. Examination under GFP mode identifies no GFP expression in normal diaphragm (C) and selective infection of diaphragmatic mesothelioma by NV1066 (D). Normal parietal pleura (E), pleural malignant mesothelioma (F) was identified under bright-field. No GFP expression in normal parietal pleura (G) and selective infection of pleural mesothelioma (H) was identified by thoracoscope fitted with fluorescent filters for GFP. GFP = Green fluorescent protein.
Figure 6.
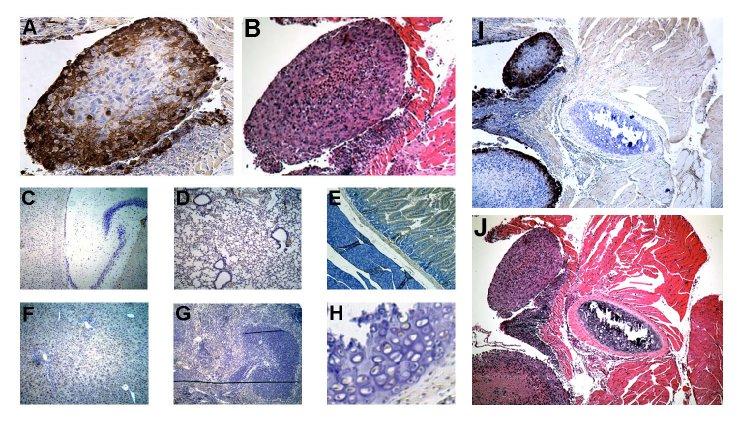
NV1066 selectively infects malignant mesothelioma and spares normal tissue. Tissue specimens from thoracic cavity were selected by their GFP expression. Serial sectioning was performed and specimens were stained with polyclonal HSV-1 antibody. Representative section shown here includes mesothelioma nodule (A, HSV immunohistochemistry and B, H&E), HSV immunohistochemistry of normal tissue from brain (C), lung (D), intestine (E), liver (F), spleen (G) and bone (H). Viral infection was localized to mesothelioma nodules. No GFP expression or viral staining was evident in normal tissue surrounding mesothelioma nodules as shown in figures I and J. GFP = Green fluorescent protein
Viral specificity for tumor
After the intrapleural administration of NV1066, within 48 hours, strong GFP expression was noted on the periphery of the tumor nodules after serial sections. All sections that expressed GFP were found to have tumor cell infiltrations that correspond to the areas of expression. When a cross section was taken from the frozen tissue and stained with HSV-1 specific polyclonal antibody, NV1066 infection and GFP expression was localized clearly to mesothelioma nodule and no evidence of infection was noted in normal tissue. The specificity of tumor infection was also demonstrated by HSV-1 specific immunohistochemistry after intrapleural delivery to detect the presence of herpes virus in vivo. Immunohistochemistry of other organs (lung, heart, liver, spleen, kidney and bone) by HSV-1 specific antibody confirmed absence of viral infection in other organs. To assess the risk of the virus spreading systemically following intrapleural administration, serum and other normal organs were analyzed for viral infective gene, ICP0 and no evidence was noted (data not shown).
DISCUSSION
For MPM, therapeutic success with current therapies is a rarity31, 32. In the majority of patients, a palliative treatment approach remains the only choice. Combined systemic chemotherapy and intrapleural chemotherapy33 results in partial responses of between 15% and 20%; with only rare complete responses34, 35. It is not feasible to administer radiotherapy as a single treatment modality because of the large target volume and the toxicity incurred by the adjacent organs5. Because of the diffuse nature of this malignancy, complete resection even by extrapleural pneumonectomy or decortication can only be achieved in those rare cases discovered at an earlier stage7, 8, 36. It is also difficult to achieve a microscopically complete resection because of the anatomy of the pleura and the characteristic infiltrative nature of MPM. Therefore, surgery alone is associated with a high recurrence rate. Recently, most efforts have been directed towards multimodality systemic treatment options to supplement surgical resection37.
Oncolytic HSV-1 viruses have been shown to be efficacious in the treatment of a number of human cancers in the laboratory9-19, 38. Two such viruses are currently in clinical trials for recurrent malignant gliomas22, 23 and for unresectable colorectal metastases to the liver20. Oncolytic HSV maintain several biologic properties that make them promising vectors for the treatment of cancer, in particular as therapy for diffusely spread malignancy such as malignant pleural mesothelioma. The ability of these viruses to selectively replicate within tumor cells means that they do not have to be directly injected into a mass to produce an antitumor response. Furthermore, smaller initial doses can be delivered with the expectation that viral progeny will kill neighboring, uninfected cells. These viruses are engineered to be safe through strategic deletions in nonessential herpes genes that attenuate virulence, yet still allow recombinants to replicate in tumor cells. Previous studies have shown the efficacy of Oncolytic HSV, HSV-1716, a mutant lacking both copies of the gene coding ICP-34.5, in the treatment of localized intraperitoneal malignant mesothelioma39. Human malignant mesothelioma cells supported the growth of HSV-1716 and were efficiently lysed in vitro. Intraperitoneal injection of HSV-1716 into animals with established tumor nodules reduced tumor burden and significantly prolonged survival without dissemination or persistence. These findings suggest that HSV virus may be efficacious and safe for use in localized malignant mesothelioma.
The viruses used in the current studies are viruses that are multi-mutated to avoid a wild-type reversion. In the current study, the HSV-1 viruses, G207, NV1020 and NV1066, effectively killed a wide variety of malignant mesothelioma cells. In vitro, initial doses of the viruses as low as MOI 0.01, one viral pfu per one hundred cancer cells killed more than 90% of cell populations after one week in culture. This is due to the ability of the malignant mesothelioma cells to support viral replication, with increase in viral titers from 5 to 20,000- fold. The efficacy of oncolytic viruses was also proven in vivo. In addition to efficacy in flank tumor models established with JMN and VAMT cells (data not shown), a model of pleural malignant mesothelioma was chosen for this study because of its clinical relevance. NV1066 replicated in vivo, highly specific to tumor cells and not only decreased the tumor burden, but increased the survival with a single dose of the viral administration without any evidence of toxicity. When used as an adjunct to prior to surgical procedure, these viruses may reduce the tumor burden and help increase the percentage of patient population that is eligible for surgical resection. Furthermore, oncolytic viral therapy can be combined with postoperative adjuvant modalities to eliminate the residual disease. The current data demonstrate promise for the three HSV oncolytic viruses as therapy for diffuse malignant pleural mesothelioma.
Previous studies have suggested that rapidly dividing cells may be most susceptible to cytotoxicity by oncolytic HSV. Cytotoxicity by the ribonucleotide reductase (RR) deficient G207 HSV has been correlated to the proliferation rates of colorectal carcinomas17. But unlike G207, NV1020 and NV1066 carry an intact RR gene and might therefore be less dependent on host cell RR to sustain viral replication. Because pleural malignant mesotheliomas proliferate at varying rates, we assessed whether this factor might influence sensitivity to oncolytic HSV therapy. All MPM cell lines were susceptible to G207, NV1020 and NV1066. In rapidly dividing cell lines, all three viruses were very highly effective in killing tumor cells even at a very low MOI of 0.01 and no differences can be discerned. Both viral replication and cytotoxicity of NV1020 and NV1066 were greater than G207 for all cell lines. Even in the slowly proliferating MPM cell lines (Meso-10, H-28 and H-2052); NV1066 was able to kill > 70% cells at a low MOI of 0.1 when compared to control untreated cells after one week in culture. Therefore, even slowly proliferating malignant mesothelioma cell lines remain viable targets for herpes oncolytic therapy.
In published studies, the epithelioid variant has the most favorable histology for malignant mesothelioma. In our study, in addition to epithelioid histology, oncolytic HSV are effective in all other pathological types of MPM cell lines. In addition, even tumors that were resistant to radio- or chemotherapy were sensitive to oncolytic herpes viruses. MPM cells tested in this study are uniformly radiation therapy resistant. Oncolytic viruses are highly effective in the radiation therapy resistant MPM cells even at a low dose of one viral pfu per ten cancer cells. Such efficacy is also demonstrated against gemcitabine and cisplatin resistant MPM cells. Furthermore, the efficacy of oncolytic HSV is not isolated to one particular type of virus. All three viruses G207, NV1020 and NV1066 are highly effective in killing therapy-resistant MPM cells.
The safety of these multi mutant viruses has been well established in previous studies40, 41. G207 has been directly injected into the brains of Aotus monkeys with no virally-related toxic side effects42. When delivered similarly in human trials showed no major toxicity at titers above 1 × 109 pfu. NV1020 was also shown to be safe in Aotus monkeys when delivered systemically in doses ten-thousand fold greater than the known lethal dose of the wild-type virus43, 44. The basis for attenuation of NV1020 may be secondary to deletion of the internal repeated region, and deletions of the UL5/6 gene and the L/S junction. The NV1020 vector is currently being studied in a trial for patients with hepatic colorectal metastases. Doses of up to 1 × 108 pfu were administered by hepatic arterial infusion without dose-limiting toxicity20. NV1020 therefore has a favorable safety profile, a finding encouraging our study of the related NV1066 vector for clinical application. In the current study, intrapleural NV1066 was well tolerated in all animals. The specificity of tumor infection was also demonstrated by immunohistochemistry after intrapleural delivery to detect the presence of herpes virus in vivo. Histologic assessment of peripheral organs revealed no signs of inflammation or injury, whereas tumor tissue was characterized by viral presence and necrosis.
NV1066 carries a marker gene, a constitutively expressed transgene for GFP, GFP, the protein product of which is identifiable 4-6 hours following viral entry into cells. While GFP and its derivative proteins have been used extensively as reporters and/or markers in numerous biologic studies, their use to determine infection and spread of replication-competent viral vectors in vivo is an emerging technology45. In this study, we demonstrated the feasibility of using GFP expression to localize the virus, and secondarily to localize tumor deposits in vivo due to the inherent specificity of NV1066 for cancer cells. Samples of tissue expressing GFP were harvested, sectioned serially, and examined for the presence of virus. All tissue that expressed GFP was noted to stain positively for HSV-1 polyclonal antibody, while no viral staining was evident in tissues not expressing GFP. In vitro, GFP expression increases in infected cell populations with viral replication. Furthermore, our laboratory has shown that following intrapleural administration of NV1066, in situ metastatic tumor deposits could be distinguished by green fluorescence using fluorescent stereomicroscopy or a thoracoscopic system with appropriate fluorescent filters45. This suggests that fluorescent protein expression can be used as a surrogate to identify viral distribution in vivo without the need for extensive tissue sampling. The specificity of viral replication and of GFP expression for tumor cells serves the further purpose of identification of tumor deposits in vivo.
In conclusion, this study demonstrates that an oncolytic HSV may efficiently infect, replicate within, and lyse a variety of human malignant mesothelioma cells. Patients with pleural malignant mesothelioma failing standard therapies are in need of alternate treatment options. Our finding that a wide variety of human malignant mesothelioma cells are highly sensitive to HSV oncolysis therefore might have important clinical applicability. These findings support the continued investigation of replication-competent, attenuated, oncolytic HSV as potential therapy for patients with therapy resistant malignant mesothelioma.
ACKNOWLEDGMENTS
The authors thank Brian Horsburgh, Ph.D. and Medigene, Inc. for constructing and providing us with the NV1066 virus. We thank Liza Marsh and Scott Tuorto of the Department of Surgery at Memorial Sloan-Kettering Cancer Center for their editorial assistance. Special thanks to Kan Matsumoto from Olympus America Inc., for design and construction of the fluorescent thoracoscopic system.
Footnotes
Meeting presentation: Presented at Surgical Forum, American College of Surgeons 90th Annual Clinical Congress, New Orleans, Louisiana, October, 2004
Supported in part by AACR-AstraZeneca Cancer Research and Prevention fellowship (P.S.A.), grants RO1 CA 75416 and RO1 CA/DK80982 (Y.F.) from the National Institutes of Health, grant MBC-99366 (Y.F.) from the American Cancer Society, grant BC024118 from the US Army (Y.F.), grant IMG0402501 from the Susan G. Komen Foundation (Y.F. and P.S.A.) and grant 032047 from Flight Attendant Medical Research Institute (Y.F. and P.S.A.)
References
- 1.Antman KH. Natural history and epidemiology of malignant mesothelioma. Chest. 1993;103(4 Suppl):373S. doi: 10.1378/chest.103.4_supplement.373s. [DOI] [PubMed] [Google Scholar]
- 2.Price B. Analysis of current trends in United States mesothelioma incidence. Am.J.Epidemiol. 1997;145(3):211. doi: 10.1093/oxfordjournals.aje.a009093. [DOI] [PubMed] [Google Scholar]
- 3.Connelly RR, Spirtas R, Myers MH, Percy CL, Fraumeni JF., Jr Demographic patterns for mesothelioma in the United States. J.Natl.Cancer Inst. 1987;78(6):1053. [PubMed] [Google Scholar]
- 4.Walker AM, Loughlin JE, Friedlander ER, Rothman KJ, Dreyer NA. Projections of asbestos-related disease 1980-2009. J.Occup.Med. 1983;25(5):409. [PubMed] [Google Scholar]
- 5.Rusch VW, Rosenzweig K, Venkatraman E, Leon L, Raben A, Harrison L, et al. A phase II trial of surgical resection and adjuvant high-dose hemithoracic radiation for malignant pleural mesothelioma. J.Thorac.Cardiovasc.Surg. 2001;122(4):788. doi: 10.1067/mtc.2001.116560. [DOI] [PubMed] [Google Scholar]
- 6.Jaklitsch MT, Grondin SC, Sugarbaker DJ. Treatment of malignant mesothelioma. World Journal of Surgery. 2001;25(2):210. doi: 10.1007/s002680020021. [DOI] [PubMed] [Google Scholar]
- 7.Rusch VW. Indications for pneumonectomy. Extrapleural pneumonectomy. Chest Surg.Clin.N.Am. 1999;9(2):327. [PubMed] [Google Scholar]
- 8.Rusch VW, Venkatraman ES. Important prognostic factors in patients with malignant pleural mesothelioma, managed surgically. Ann.Thorac.Surg. 1999;68(5):1799. doi: 10.1016/s0003-4975(99)01038-3. [DOI] [PubMed] [Google Scholar]
- 9.Advani SJ, Sibley GS, Song PY, Hallahan DE, Kataoka Y, Roizman B, et al. Enhancement of replication of genetically engineered herpes simplex viruses by ionizing radiation: a new paradigm for destruction of therapeutically intractable tumors. Gene Ther. 1998;5(2):160. doi: 10.1038/sj.gt.3300546. [DOI] [PubMed] [Google Scholar]
- 10.Bennett JJ, Delman KA, Burt BM, Mariotti A, Malhotra S, Zager J, et al. Comparison of safety, delivery, and efficacy of two oncolytic herpes viruses (G207 and NV1020) for peritoneal cancer. Cancer Gene Therapy. 2002;9(11):935. doi: 10.1038/sj.cgt.7700510. [DOI] [PubMed] [Google Scholar]
- 11.Bennett JJ, Kooby DA, Delman K, McAuliffe P, Halterman MW, Federoff H, et al. Antitumor efficacy of regional oncolytic viral therapy for peritoneally disseminated cancer. Journal of Molecular Medicine. 2000;78(3):166. doi: 10.1007/s001090000092. [DOI] [PubMed] [Google Scholar]
- 12.Bennett JJ, Malhotra S, Wong RJ, Delman K, Zager J, St Louis M, et al. Interleukin 12 secretion enhances antitumor efficacy of oncolytic herpes simplex viral therapy for colorectal cancer. Annals Of Surgery. 2001;233(6):819. doi: 10.1097/00000658-200106000-00012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chung SM, Advani SJ, Bradley JD, Kataoka Y, Vashistha K, Yan SY, et al. The use of a genetically engineered herpes simplex virus (R7020) with ionizing radiation for experimental hepatoma. Gene Ther. 2002;9(1):75. doi: 10.1038/sj.gt.3301620. [DOI] [PubMed] [Google Scholar]
- 14.Cozzi PJ, Burke PB, Bhargav A, Heston WD, Huryk B, Scardino PT, et al. Oncolytic viral gene therapy for prostate cancer using two attenuated, replication-competent, genetically engineered herpes simplex viruses. Prostate. 2002;53(2):95. doi: 10.1002/pros.10138. [DOI] [PubMed] [Google Scholar]
- 15.Cozzi PJ, Malhotra S, McAuliffe P, Kooby DA, Federoff HJ, Huryk B, et al. Intravesical oncolytic viral therapy using attenuated, replication-competent herpes simplex viruses G207 and Nv1020 is effective in the treatment of bladder cancer in an orthotopic syngeneic model. FASEB Journal. 2001;15(7):1306. doi: 10.1096/fj.00-0533fje. [DOI] [PubMed] [Google Scholar]
- 16.Delman KA, Zager JS, Bennett JJ, Malhotra S, Ebright MI, McAuliffe PF, et al. Efficacy of multiagent herpes simplex virus amplicon-mediated immunotherapy as adjuvant treatment for experimental hepatic cancer. Annals Of Surgery. 2002;236(3):337. doi: 10.1097/00000658-200209000-00010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kooby DA, Carew JF, Halterman MW, Mack JE, Bertino JR, Blumgart LH, et al. Oncolytic viral therapy for human colorectal cancer and liver metastases using a multimutated herpes simplex virus type-1 (G207) FASEB.J. 1999;6:499. doi: 10.1096/fasebj.13.11.1325. [DOI] [PubMed] [Google Scholar]
- 18.Stanziale SF, Petrowsky H, Joe JK, Roberts GD, Zager JS, Gusani NJ, et al. Ionizing radiation potentiates the antitumor efficacy of oncolytic herpes simplex virus G207 by upregulating ribonucleotide reductase. Surgery. 2002;132(2):353. doi: 10.1067/msy.2002.125715. [DOI] [PubMed] [Google Scholar]
- 19.Zager JS, Delman KA, Malhotra S, Ebright MI, Bennett JJ, Kates T, et al. Combination vascular delivery of herpes simplex oncolytic viruses and amplicon mediated cytokine gene transfer is effective therapy for experimental liver cancer. Molecular Medicine. 2001;7(8):561. [PMC free article] [PubMed] [Google Scholar]
- 20.Fong Y, Kemeny N, Jarnagin W, Stanziale S, Guilfoyle B, Gusani N, et al. Phase 1 study of a replication-competent herpes simplex oncolytic virus for treatment of hepatic colorectal metastases. Proceedings of the American Society of Clinical Oncology. 2002;21:8a. [Google Scholar]
- 21.Markert JM, Medlock MD, Rabkin SD, Gillespie GY, Todo T, Hunter WD, et al. Conditionally replicating herpes simplex virus mutant, G207 for the treatment of malignant glioma: results of a phase I trial. Gene Ther. 2000;7(10):867. doi: 10.1038/sj.gt.3301205. [DOI] [PubMed] [Google Scholar]
- 22.Rampling R, Cruickshank G, Papanastassiou V, Nicoll J, Hadley D, Brennan D, et al. Toxicity evaluation of replication-competent herpes simplex virus (ICP 34.5 null mutant 1716) in patients with recurrent malignant glioma. Gene Ther. 2000;7(10):859. doi: 10.1038/sj.gt.3301184. [DOI] [PubMed] [Google Scholar]
- 23.Mineta T, Rabkin SD, Yazaki T, Hunter WD, Martuza RL. Attenuated multi-mutated herpes simplex virus-1 for the treatment of malignant gliomas. Nature Medicine. 1995;1(9):938. doi: 10.1038/nm0995-938. [DOI] [PubMed] [Google Scholar]
- 24.Carew JF, Kooby D, Halterman MW, Federoff HJ, Fong Y. Selective infection and cytolysis of human head and neck squamous cell carcinoma with sparing of normal mucosa by a cytolytic herpes simplex virus type 1 (G207) Proceedings of the American Head and Neck Society. 1999 doi: 10.1089/10430349950017608. [DOI] [PubMed] [Google Scholar]
- 25.Detta A, Harland J, Hanif I, Brown SM, Cruickshank G. Proliferative activity and in vitro replication of HSV1716 in human metastatic brain tumours. Journal of Gene Medicine. 2003;5(8):681. doi: 10.1002/jgm.396. [DOI] [PubMed] [Google Scholar]
- 26.Jia WW, McDermott M, Goldie J, Cynader M, Tan J, Tufaro F. Selective destruction of gliomas in immunocompetent rats by thymidine kinase-defective herpes simplex virus type 1. Journal of the National Cancer Institute. 1994;86(16):1209. doi: 10.1093/jnci/86.16.1209. [DOI] [PubMed] [Google Scholar]
- 27.McKie EA, Brown SM, MacLean AR, Graham DI. Histopathological responses in the CNS following inoculation with a non-neurovirulent mutant (1716) of herpes simplex virus type 1 (HSV 1): relevance for gene and cancer therapy. Neuropathology & Applied Neurobiology. 1998;24(5):367. doi: 10.1046/j.1365-2990.1998.00133.x. [DOI] [PubMed] [Google Scholar]
- 28.Varghese S, Rabkin SD. Oncolytic herpes simplex virus vectors for cancer virotherapy. Cancer Gene Ther. 2002;9(12):967. doi: 10.1038/sj.cgt.7700537. [DOI] [PubMed] [Google Scholar]
- 29.Pass H, Dwyer A, Makuch R, Roth J. Detection of pulmonary metastases in patients with osteogenic and soft-tissue sarcomas: the superiority of CT-scans compared with conventional linear tomograms using dynamic anlysis. Journal of Clinical Oncology. 1985;3:1261. doi: 10.1200/JCO.1985.3.9.1261. [DOI] [PubMed] [Google Scholar]
- 30.Meignier B, Martin B, Whitley RJ, Roizman B. In vivo behavior of genetically engineered herpes simplex viruses R7017 and R7020. II. Studies in immunocompetent and immunosuppressed owl monkeys (Aotus trivirgatus) Journal Of Infectious Diseases. 1990;162(2):313. doi: 10.1093/infdis/162.2.313. [DOI] [PubMed] [Google Scholar]
- 31.Rusch VW. Diagnosis and treatment of pleural mesothelioma. Semin.Surg.Oncol. 1990;6(5):279. doi: 10.1002/ssu.2980060510. [DOI] [PubMed] [Google Scholar]
- 32.Wong RJ, Joe JK, Kim SH, Shah JP, Horsburgh B, Fong YM. Oncolytic herpesvirus effectively treats murine squamous cell carcinoma and spreads by natural lymphatics to treat sites of lymphatic Metastases. Human Gene Therapy. 2002;13(10):1213. doi: 10.1089/104303402320138998. [DOI] [PubMed] [Google Scholar]
- 33.Giaccone G. Pleural mesothelioma: combined modality treatments. Annals of Oncology. 2002;13(Suppl 4):217. doi: 10.1093/annonc/mdf663. [DOI] [PubMed] [Google Scholar]
- 34.Rusch VW, Niedzwiecki D, Tao Y, Menendez-Botet C, Dnistrian A, Kelsen D, et al. Intrapleural cisplatin and mitomycin for malignant mesothelioma following pleurectomy: pharmacokinetic studies. Journal of Clinical Oncology. 1992;10(6):1001. doi: 10.1200/JCO.1992.10.6.1001. [DOI] [PubMed] [Google Scholar]
- 35.Tomek S, Emri S, Krejcy K, Manegold C. Chemotherapy for malignant pleural mesothelioma: past results and recent developments. British Journal of Cancer. 2003;88(2):167. doi: 10.1038/sj.bjc.6600673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Vogelzang NJ, Rusthoven JJ, Symanowski J, Denham C, Kaukel E, Ruffie P, et al. Phase III study of pemetrexed in combination with cisplatin versus cisplatin alone in patients with malignant pleural mesothelioma. Journal of Clinical Oncology. 2003;21(14):2636. doi: 10.1200/JCO.2003.11.136. [DOI] [PubMed] [Google Scholar]
- 37.Rusch VW. Pleurectomy/decortication in the setting of multimodality treatment for diffuse malignant pleural mesothelioma. Semin.Thorac.Cardiovasc.Surg. 1997;9(4):367. [PubMed] [Google Scholar]
- 38.Rusch VW. Pemetrexed and cisplatin for malignant pleural mesothelioma: a new standard of care? Journal of Clinical Oncology. 2003;21(14):2629. doi: 10.1200/JCO.2003.02.043. [DOI] [PubMed] [Google Scholar]
- 39.Kucharczuk JC, Randazzo B, Chang MY, Amin KM, Elshami AA, Sterman DH, et al. Use of a “replication-restricted” herpes virus to treat experimental human malignant mesothelioma. Cancer Research. 1997;57(3):466. [PubMed] [Google Scholar]
- 40.Stiles BM, Bhargava A, Adusumilli PS, Stanziale SF, Kim TH, Rusch VW, et al. The replication-competent oncolytic herpes simplex mutant virus NV1066 is effective in the treatment of esophageal cancer. Surgery. 2003;134(2):357. doi: 10.1067/msy.2003.244. [DOI] [PubMed] [Google Scholar]
- 41.Todo T, Feigenbaum F, Rabkin SD, Lakeman F, Newsome JT, Johnson PA, et al. Viral shedding and biodistribution of G207, a multimutated, conditionally replicating herpes simplex virus type 1, after intracerebral inoculation in aotus. Mol.Ther. 2000;2(6):588. doi: 10.1006/mthe.2000.0200. [DOI] [PubMed] [Google Scholar]
- 42.Varghese S, Newsome JT, Rabkin SD, McGeagh K, Mahoney D, Nielsen P, et al. Preclinical safety evaluation of G207, a replication-competent herpes simplex virus type 1, inoculated intraprostatically in mice and nonhuman primates. Hum.Gene Ther. 2001;12(8):999. doi: 10.1089/104303401750195944. [DOI] [PubMed] [Google Scholar]
- 43.Hunter WD, Martuza RL, Feigenbaum F, Todo T, Mineta T, Yazaki T, et al. Attenuated, replication-competent herpes simplex virus type 1 mutant G207: safety evaluation of intracerebral injection in nonhuman primates. J Virol. 1999;73(8):6319. doi: 10.1128/jvi.73.8.6319-6326.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pass HI, Stevens EJ, Oie H, Tsokos MG, Abati AD, Fetsch PA, et al. Characteristics of nine newly derived mesothelioma cell lines. Ann.Thorac.Surg. 1995;59(4):835. doi: 10.1016/0003-4975(95)00045-m. [DOI] [PubMed] [Google Scholar]
- 45.Meignier B, Longnecker R, Roizman B. In vivo behavior of genetically engineered herpes simplex viruses R7017 and R7020: Construction and evaluation in rodents. Journal Of Infectious Diseases. 1999;158:602. doi: 10.1093/infdis/158.3.602. [DOI] [PubMed] [Google Scholar]