

Abstract
A deficiency in mitochondrial frataxin causes an increased generation of mitochondrial reactive oxygen species (ROS), which may contribute to the cell degenerative features of Friedreich’s ataxia. In this work the authors demonstrate mitochondrial iron–sulfur cluster (ISC) defects and mitochondrial heme defects, and suggest how both may contribute to increased mitochondrial ROS in lymphoblasts from human patients. Mutant cells are deficient in the ISC-requiring mitochondrial enzymes aconitase and succinate dehydrogenase, but not in the non-ISC mitochondrial enzyme citrate synthase; also, the mitochondrial iron–sulfur scaffold protein IscU2 co-immunoprecipitates with frataxin in vivo. Presumably as a consequence of the iron–sulfur cluster defect, cytochrome c heme is deficient in mutants, as well as heme-dependent Complex IV. Mitochondrial superoxide is elevated in mutants, which may be a consequence of cytochrome c deficiency. Hydrogen peroxide, glutathione peroxidase activity, and oxidized glutathione (GSSG) are each elevated in mutants, consistent with activation of the glutathione peroxidase pathway. Mutant status blunted the effects of Complex III and IV inhibitors, but not a Complex I inhibitor, on superoxide production. This suggests that heme defects late in the electron transport chain of mutants are responsible for increased mutant superoxide. The impact of ISC and heme defects on ROS production with age are discussed.
INTRODUCTION
Mitochondrial oxidative stress is thought to be an important contributor to age-related cell degeneration (1, 13, 20). Substantial experimental support exists in animal models for an increase in mitochondrial oxidative stress and damage with age (16, 25, 52). As a corollary, cell degeneration is triggered in animal models of human disease in which important mitochondrial antioxidant genes are mutated, including CuZnSOD, which diffuses to the mitochondrial inter-membrane space (40, 55), MnSOD which dismutates mitochondrial matrix superoxide (32), and glutathione peroxidase which catabolizes matrix peroxide (65).
Thus, increased mitochondrial reactive oxygen species (ROS) production is implicated as a potential causative agent in aging, and increased mitochondrial ROS production induces cell degenerative changes, particularly in weakly mitotic cells.
Friedreich’s ataxia (FRDA) is a human neuro- and car-diodegenerative disease which results from an inherited deficiency of the mitochondrial frataxin protein (8, 39, 45). The observation of clinical similarities between FRDA and AVED (Ataxia with Vitamin E Deficiency), which is caused by defects in the alpha-tocopherol (vitamin E) transfer protein, suggested that both FRDA and AVED were diseases of increased mitochondrial oxidative stress (26, 31).
The primary function of human frataxin is still disputed. Human frataxin protein or its yeast homologs, and their deficiency, have been demonstrated to have effects on iron metabolism (6, 9, 38); on iron–sulfur cluster (ISC) metabolism (15, 34, 54); on heme metabolism (21, 30); and on ROS metabolism (10, 23, 57, 62).
In this work we demonstrate (a) deficiency of ISC enzyme activity and a direct interaction of frataxin with the ISC scaffold in vivo; (b) a defect in heme, cytochrome c, and cytochrome oxidase activity in mutants; (c) increased mitochondrial superoxide, and blunting of the ROS response to Complex III and Complex IV inhibitors. We suggest that the increased ROS in FRDA cells are the consequence of defective expression of cytochrome c and heme present in Complex III and IV, and that the defective heme synthesis produces the Complex IV defect. Thus, a mitochondrial ISC defect appears to cause a heme defect that causes deficient cytochrome c and oxidase activity, and increased mitochondrial ROS in Friedreich’s ataxia. ROS damage to mitochondrial iron–sulfur clusters are thought to rise with aging. If so, then this ISC damage could cause heme defects, Complex IV defects, and increased ROS by the same mechanism we infer in Friedreich’s ataxia.
MATERIALS AND METHODS
Reagents
Biochemical reagents were purchased from Sigma (St. Louis, MO, USA) or Bio-Rad (Hercules, CA, USA).
Antibodies: a-cytochrome c was purchased by BD Biosciences (San Jose, CA, USA); a-frataxin polyclonal antibody was a kind gift of Dr. Franco Taroni (Instituto Neurologico Nazionale C. Besta, Milan, Italy); a-IscU2 was a kind gift from Dr. Tracy Rouault (National Institute of Child Health and Human Development, Bethesda, MD, USA). Protein G sepharose was purchased by Amersham (Piscataway, NJ, USA).
Cell culture
Whole cells and mitochondrial fraction from controls and FRDA lymphoblasts were used for this study. Immortalization of lymphocytes from three control lines and from three FRDA different patients was as previously described (56). All cells were maintained at 37°C in a humidified atmosphere containing 5% CO2 in RPMI 1640 supplemented with 500 mg/L glutamate, 1 mM sodium pyruvate, 50 μg/ml uridine, 100 μM nonessential amino acids (Invitrogen, Carlsbad, CA, USA), 20% fetal calf serum, and penicillin/streptomycin (100 U/ml each).
Frataxin levels were checked periodically in each cell line in order to evaluate the amount of protein expressed at different passage numbers. In fact, it is known that the GAA triplet repeat associated with Friedreich’s ataxia shows varying degrees of instability in lymphoblasts (4). The amount of frataxin has been checked in all the mitochondrial lysates, controls, and FRDA, used for our study (Fig. 1A). On average, frataxin expression in FRDA lymphoblasts was about 80% lower with respect to the control lines (Fig. 1B).
FIG. 1. Frataxin amount in controls and FRDA lymphoblasts.
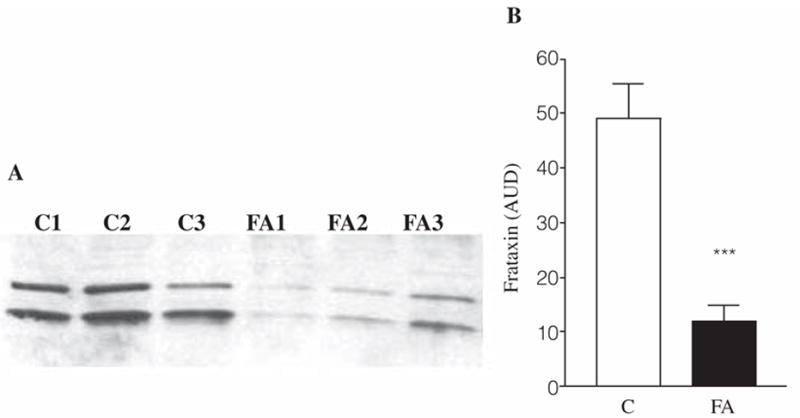
(A) Example of Western blot with anti-frataxin antibody (1:3000 dil) on three control and three FRDA mitochondrial lysates (30 μg). Both the intermediate (~18.8 KDa) and the mature (~17.2 KDa) form of frataxin are shown. The band intensities were analyzed by densitometry and the averages of six different controls and six different FRDA mitochondrial lysates were calculated (B). Results are expressed as means ± SEM of two experiments in duplicate for each cell line. AUD, arbitrary units of densitometry; C, controls; FA, Friedreich’s ataxia mutants. ***p < 0.001 calculated by Student’s t test.
Mitochondrial isolation
Mitochondria were isolated from three controls and three FRDA mutant lines of human immortalized lymphoblasts following the method of Trounce et al. (58) with slight modifications.
Approximately 1 × 108 cells were harvested by centrifugation and the pellet was resuspended with 4 ml of isolation buffer (210 mM mannitol, 70 mM sucrose, 1 mM EGTA, and 5 mM HEPES, pH 7.2) for each gram of packed cells, treated with a final concentration of 0.3 mg/ml digitonin for 1 min 30 sec and centrifuged at 3000 g. The pellet was then resuspended with 5 ml for each gram of initial packed cells and homogenized with a chilled class homogenator (20 passes). After several centrifugation steps at 625 g for 5 min, the final supernatant was centrifuged at 10,000 g and the final pellet was suspended with 0.1 ml of isolation buffer per gram of starting cells, giving a protein concentration of approximately 8–12 mg/ml. For determination of mitochondrial protein concentration, 5 μl of the mitochondrial suspension were diluted 1:20 in double distilled water and the protein concentration was estimated using the Bradford assay (Bio-Rad).
Co-immunoprecipitation experiments
Lysis of mitochondria was performed in presence of 0 mM, 1 mM, and 10 mM EDTA. Two milligrams of mitochondrial lysate without EDTA were incubated with the antibody specific for frataxin (1:1000) for 1 h at 4°C. Protein A sepharose beads were then added and the lysates incubated 1 h at 4°C. Beads and supernatant were separated by centrifugation. Two aliquots of the immunocomplex obtained were separated from the beads, analyzed by SDS-PAGE, followed by immunostaining with anti-frataxin antibody and with anti-IscU antibody.
One milligram of mitochondrial extracts containing 1 mM EDTA or 10 mM EDTA were immunoprecipitated with a-frataxin antibody as described, analyzed by SDS-PAGE and immunoblotted with a-IscU antibody. 25 μg of total mitochondrial protein were loaded as positive control.
Western blot analysis
Mitochondria were lysed in 50 mM Tris, pH 8.0, 100 mM NaCl, 1 mM EDTA, 1 mM PMSF, 1% IGEPAL CA-630 (Sigma), at 4°C for 30 min and insoluble material removed by centrifugation. Protein concentration was estimated using the Bradford assay (Bio-Rad). Equal amounts of lysates (30 μg) were resolved on a 15% SDS–polyacrylamide gel and then transferred to a nitrocellulose membrane (Millipore, Bedford, MA) by electroblotting. After blocking with 4% nonfat dry milk, the blot was incubated with anti-frataxin polyclonal antibody (1:3000) or anti-cytochrome c (1:1500) and was developed with alkaline-phosphatase-conjugated secondary antibodies using a chemiluminescent substrate.
Superoxide ion production
The mitochondria-specific dye dihydroethidium (DHE) was used to detect intracellular superoxide. Superoxide oxidizes DHE to a red fluorescent signal.
Two control and two FRDA lymphoblast lines (4 × 106 cells) were collected, washed two times with PBS and incubated (0.5 × 106/200 μl) for 120 min at 37°C in the following buffer: PBS, 20 mM glucose, 2 μM DHE, with or without the following respiratory chain inhibitors: 10 μM rotenone (complex I inhibitor), 10 μM antimycin (complex III inhibitor), 0.5 mM KCN (complex IV inhibitor).
Fluorescence was recorded immediately after adding DHE (time zero) and after 120 min incubation (ex 530/25 nm, em 620/40 nm). Superoxide production was expressed as Δfluorescence/106 cells.
Mitochondrial enzyme activity assays
Succinate dehydrogenase, cytochrome c oxidase, and citrate synthase were evaluated in mitochondria isolated from two control and two FRDA patients’ lymphoblasts.
Succinate dehydrogenase activity was measured by following the reduction of 2,6-dichlorophenolindophenol (DCIP) at 600 nm as described (5).
Cytochrome c oxidase (COX) activity was evaluated by the single wavelength spectrophotometric assay previously described (33) with some modifications. Mitochondria (1.5 μg) were incubated with 2.5 mM of dodecylmaltoside in 0.17 M KH2PO4, pH 7.0 for 3 min at 30°C, and, after recording the baseline, the reaction was started with 10 mM of reduced cytochrome c. COX activity was calculated from the pseudo-linear rate of cytochrome c oxidation at 550 nm.
Citrate synthase was evaluated as described (53) following the disappearance of 5,5′-dithiobis(2-nitrobenzoic acid) at 412 nm.
Heme staining of cytochrome c
Mitochondrial extracts were separated on a 15% SDS/PAGE gel. Heme staining was performed as previously described (48). 30 μg of mitochondrial protein from three different control and FRDA lymphoblast lines were loaded onto a 15% polyacrylamide gel and the electrophoresis was performed at 130 V for 60 min.
After the run, the gel was fixed in 10% TCA for 10 min, washed four times for 5 min in double distilled water. The heme staining is based on the oxidation of o-dianisidine, a probe which, in presence of hydrogen peroxide (H2O2), can be oxidized by the peroxidase activity of some heme-proteins (such as cytochrome c), changing color. The gel was soaked in a solution of 50 mM trisodium citrate pH 4.4, 0.7% H2O2, and 1 mg/ml o-dianisidine for 40–60 min at room temperature. 0.5 μg of cytochrome c purified from horse heart was used as positive controls.
Glutathione peroxidase activity assay
Control and FRDA lymphoblasts (20 × 106) were washed three times with PBS before treatment with a solution consisting of 0.25 M sucrose,10 mM Tris-HCl (pH 7.5), 1 mM EDTA, 0.5 mM phenylmethanesulfonyl fluoride (PMSF), 0.5 mM 1,4-dithio-dl-threitol, and 0.1% (v/v) Igepal CA-630, for 30 min at 0°C to obtain complete lysis of intracellular organelles. Samples were then centrifuged for 30 min at 105,000 g. Protein measurements and enzyme assays were carried out on the clear supernatant fractions.
Total glutathione peroxidase (GPx) activity was measured by the coupled enzyme procedure with glutathione reductase, as described (42), using cumene hydroperoxide as substrate. Enzyme activity was monitored by following the disappearance of NADPH at 340 nm for 3 min at 25°C. The incubation medium (final volume 1 ml) had the following composition: 50 mM KH2PO4, pH 7.0, 3 mM EDTA, 1 mM GSH, 0.1 mM NADPH, 2 U glutathione reductase, and ~150 μg protein. After a 3-min equilibration period at 25°C, the reaction was started by the addition of 0.1 mM cumene hydroperoxide. The specific activity was calculated by using a molar absorption coefficient obtained from a standard curve of NADPH (0.02 to 0.1 μmol·ml−1), and GPx activity was expressed in nmol NADPH consumed per mg protein min−1.
RESULTS
Frataxin physically interacts with the mitochondrial iron–sulfur cluster scaffold protein IscU2 in vivo
Frataxin deficiency causes partial deficiency of ISC biogenesis in yeast (15, 18, 34), and microarray analysis of human cells indicate alteration of iron–sulfur and sulfur amino acid pathways specifically (57). Furthermore, purified human holofrataxin has been demonstrated to interact in vitro with purified human mitochondrial IscU (binding constant, KD of 0.15 μM) (63) and studies in yeast (44) and most recently in cardiac cells (35) have shown physical interaction between frataxin and the mitochondrial isoforms of IscU (Isu1 in yeast and IscU2 in human).
In order to investigate whether frataxin binds mitochondrial IscU2 in human lymphoblasts in vivo, we carried out immunoprecipitation experiments in mitochondria isolated from human lymphoblasts (Fig. 2). After mitochondrial lysis, frataxin was immuno-purified with a polyclonal anti-frataxin antibody and the immunocomplexes, separated from the beads, were then analyzed by SDS-PAGE and immunostaining with anti-frataxin (left side) and anti-IscU2 (right side) antibodies. We observed that anti-frataxin antibody co-immunoprecipitated IscU2 (lane 5) and that this binding was dependent on an EDTA-chelatable factor, which should be iron, as demonstrated by Yoon and Cowan (63). The amount of IscU2 associated with the immunobeads is significantly lower in mitochondria lysed with a buffer containing 1 mM EDTA (lane 6).
FIG. 2. Co-immunoprecipitation of mitochondrial lysate with anti-frataxin antibody.
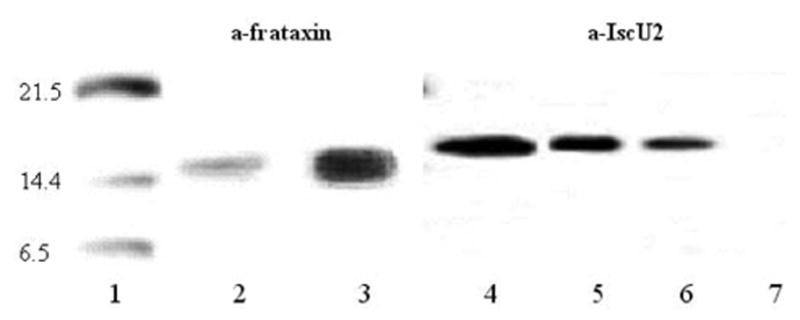
Two aliquots of the immunocomplex obtained in absence of EDTA were separated from the beads, analyzed by SDS-PAGE and subsequent immunostaining with anti-frataxin antibody (lane 3) and with anti-ISCU2 antibody (lane 5). 1 mg of mitochondrial extracts containing 1 mM EDTA (lane 6) or 10 mM EDTA (lane 7) were immunoprecipitated with anti-frataxin antibody, analyzed by SDS-PAGE, and immunoblotted with anti-IscU2 antibody. 25 μg of total mitochondrial protein (lines 2 and 4) were loaded as positive control. Lane 1: molecular weight standard.
We also looked for the existence of interaction between frataxin and the human homologs of other proteins known to be involved in ISC synthesis/repair in bacteria, such as ISC-S, which is a cysteine desulfurase (61), whose expression has already been proved lower in FRDA mutants (57) and Hsc20, which is encoded by a gene which has shown an identical phylogenetic distribution with frataxin, involved in ISC assembly (22) but we observed no evidence for interaction (data not shown).
Frataxin deficiency causes deficiency in iron–sulfur cluster enzymes
Frataxin deficiency causes defects specifically ISC enzymes in yeast (17, 54), in mice (43), and human cells (56). Intimate contact of frataxin with ISCU suggests that frataxin deficiency should cause deficiency in ISC production, and hence enzyme activity. We previously demonstrated deficient aconitase activity in human FRDA lymphoblasts (56). The activity of mitochondrial ISC enzyme succinate dehydrogenase was also deficient in mutant cells (Fig. 3A). By contrast, the mean activity of mitochondrial citrate synthase, which does not require iron–sulfur clusters, was not significantly altered (Fig. 3B), means ± SEM reported as % of controls average. Statistical analysis was performed by Student’s t test. *p < 0.05.
FIG. 3. Succinate dehydrogenase (SDH) activity, but not citrate synthase (CS) activity, is lower in FRDA mitochondria.
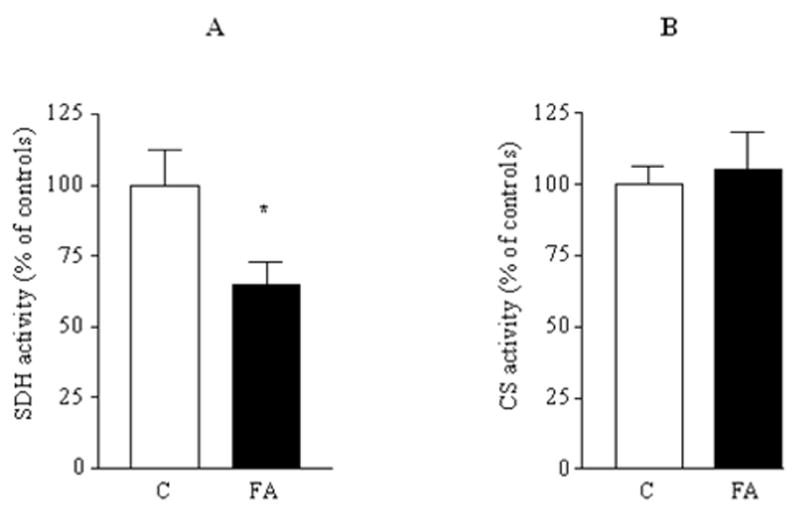
(A) SDH activity was measured in mitochondrial lysates from two controls and two FRDA mutant lymphoblasts. (B) Citrate synthase activity was measured in isolated mitochondria from three control and three FRDA cell lines following the reduction of DTNB at 412 nm. Both the activities were calculated as nmol/mg/min and the results (expressed as means ± SEM) reported as % of controls average. Statistical analysis was performed by Student’s t test. *p < 0.05.
Higher superoxide production in FRDA lymphoblasts
The amount of mitochondrial O2•− was evaluated in FRDA lymphoblasts (Fig. 4A), using the dihydroethidium (DHE) method. A significant (p = 0.01) overproduction of mitochondrial superoxide was observed in FRDA lymphoblasts relative to the controls, a near doubling of the fluorescence observed (e.g., 60 f.u. vs. 34 f.u.).
FIG. 4. Superoxide production and glutathione peroxidase activity in control and FRDA patient lymphoblasts.
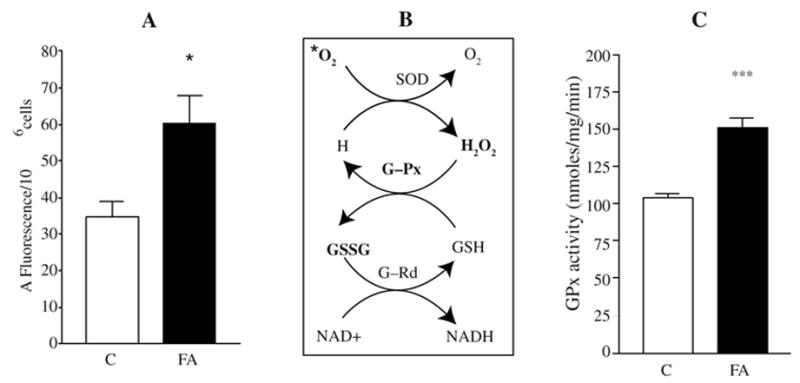
(A) Lymphoblasts from two controls and two FRDA mutants were incubated with 2 μM DHE (dihydroethidium), a fluorescent probe specific for superoxide anion detection. Superoxide production was recorded after 120 min at 37°C and expressed as ΔFluorescence/106 cells. (B) GSH-dependent detoxifying mechanism of superoxide; significantly increased reactants or products are in bold. (C) Total GPx activity was measured by the coupled enzyme procedure with glutathione reductase. The results are expressed as means ± SEM of at least two experiments in duplicate for two controls and two FRDA cell lines. C, controls; FA, Friedreich’s ataxia mutants. Statistical analysis was done by Student’s t test using the Prism Graph Pad software. *p < 0.05, ***p < 0.0001.
Previously, we demonstrated an increase in mitochondrial peroxide production in mutant lymphoblast mitochondria that is rescued by frataxin transfection (57). Since the product of mitochondrial superoxide is hydrogen peroxide, these data are consistent with the idea of a near doubling of mitochondrial superoxide in mutants, which is then converted to hydrogen peroxide.
Activation of the glutathione peroxidase pathway in FRDA lymphoblasts
The observed higher levels steady-state mitochondrial superoxide and hydrogen peroxide might be expected to stimulate the activity of glutathione peroxidases (GPx), which remove H2O2 by coupling its reduction to H2O with oxidation of reduced glutathione (GSH) (Fig. 4B).
GPx activity is significantly increased in FRDA lymphoblasts, to about 150% of control activity (Fig. 4C). The higher GPx activity is also consistent with our previous observations that oxidized glutathione (GSSG) levels are significantly elevated by about 100% in frataxin-deficient cells, and GSSG/GSH levels are also higher, an indication of increased oxidative stress (57). Thus, superoxide, hydrogen peroxide, gluthathione peroxidase, GSSG, and the GSSG/GSH ratio, are each elevated in mutant cells, consistent with the idea of increased oxidative stress as a consequence of frataxin deficiency. However, the oxidative stress was not so intensive that the levels of GSH, were significantly decreased in mutants (57).
Attribution of site of increased ROS using electron transport chain inhibitors
Mitochondrial superoxide is thought to be generated at two main sites in the electron transport chain (i.e., at complex I, by reverse electron flow, and complex III, by reduction of Q to ubisemiquinone radical) (14, 60). In an attempt to clarify the source of the increased mitochondrial superoxide in FRDA cells, we treated cells with inhibitors of these respiratory sites.
Rotenone is a specific inhibitor of mitochondrial complex I. Complex I contains multiple iron–sulfur clusters. Previous studies have demonstrated a rotenone-dependent increase in ROS, which is the result of reverse electron flow from succinate dehydrogenase, and is thought to involve an ISC donor in complex I (11, 19, 27). The mean increase in superoxide over untreated cells was not significantly different between the control cells (235%) versus mutant cells (209%). Thus although superoxide production is elevated in both mutants and controls by rotenone, there is no significant difference in the fold-change of superoxide increase versus untreated cells. If complex I ISCs were being depleted as a consequence of frataxin deficiency to the extent that complex I was blocked, then we should observe no increase in ROS in rotenone-inhibited mutants (Fig. 5A). Because we observe no significant decrease in rotenone-dependent ROS in mutants relative to our expectation derived from controls, complex I defects appear to be a minor or insignificant contributor to the increased mitochondrial superoxide in FRDA lymphoblasts.
FIG. 5. Superoxide production in controls and FRDA patient lymphoblasts.
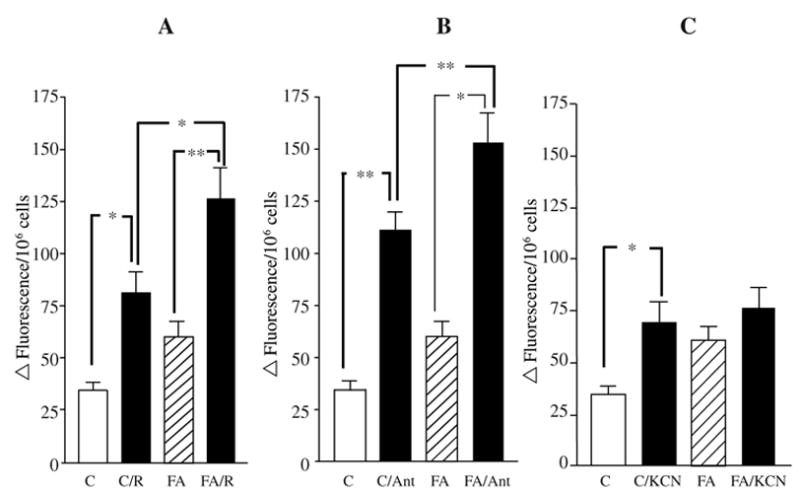
Lymphoblasts from two controls and two FRDA mutants were incubated with 2 μM DHE. Preincubation with 10 μM of rotenone (A), which specifically inhibits complex I activity, increased the superoxide levels in both controls and FRDAs. Treatment of mutants with 10 μM antimycin a (B), complex III inhibitor, produces a blunted superoxide response. Treatment of mutants with 0.5 mM of KCN (C), complex IV inhibitor, does not significantly increase superoxide production in FRDA lymphoblasts. Data are reported as means ± SEM of at least 10 experiments in duplicate for controls and 12 for FRDA mutants. Ant, antimycin a; C, controls; FA, Friedreich’s ataxia mutants; R, rotenone. Statistical analysis was done by one-way ANOVA followed by Bonferroni’s multiple comparison post test. *p < 0.05, **p < 0.001.
Effects of antimycin a on superoxide production are blunted in mutant cells
Antimycin a is a specific inhibitor of complex III. Inhibition of complex III by antimycin a is thought to generate increased superoxide by the Q-cycle, in which Q is reduced by a single electron to Q radical, which then reduces O2 to O2 (59, 60).
We observed that, although antimycin stimulated a significant increase in mitochondrial superoxide in both mutants and controls, the mean ROS stimulation was 321% in controls versus 254% in mutants (Fig. 5B). This suggested that some feature of the mutant cells blunted the induction of ROS by antimycin a (i.e., that either the Q-cycle was already partially inhibited, or that cytochrome c was decreased). Two factors have been demonstrated to protect from antimycin a-induced ROS production (i.e., a decrease in cytochrome c, or in the activity of the Rieske iron–sulfur protein) (59, 60). Since a specific deficiency in ISCs has already been demonstrated above and in other work on frataxin deficiency, we also investigated cytochrome c levels, two paragraphs below.
Effects of cyanide on superoxide production are blunted in mutant cells
Cyanide (KCN) is a volatile inhibitor of cytochrome oxidase. Mutant lymphoblasts were preincubated with 0.5 mM of the complex IV inhibitor KCN (Fig. 5C). Cyanide treatment produced a statistically significant increase in superoxide in the controls (p < 0.05), but no significant increase in FRDA cells. Cyanide is a specific inhibitor of cytochrome oxidase, which binds at heme a3. The lack of a significant increase in superoxide in the mutants could be explained as a consequence of frataxin-dependent defects at Complex III and IV, for example, defects in cytochrome c and cytochrome oxidase activity, which are described below.
Heme c and cytochrome c levels are decreased in FRDA mitochondria
Studies in yeast and humans have suggested that frataxin deficiency may alter the heme biosynthetic pathway (30, 66). One of the products of the heme pathway is heme c (the heme that associates with apocytochrome c). Thus a defect in the mitochondrial steps of the heme pathway might be expected to cause a defect in the level of heme c. Furthermore, a defect in cytochrome c would be expected to cause increased superoxide (because cytochrome c oxidizes superoxide), and a blunting of the antimycin a-dependent stimulation of superoxide production.
Cytochrome c levels were evaluated by immunoblotting followed by densitometry (Fig. 6A). Cytochrome c expression was found to be decreased by 18% in mutants with respect to controls (p < 0.05, Student’s t test).
FIG. 6. Depletion of cytochrome c and heme c in FRDA mitochondria.
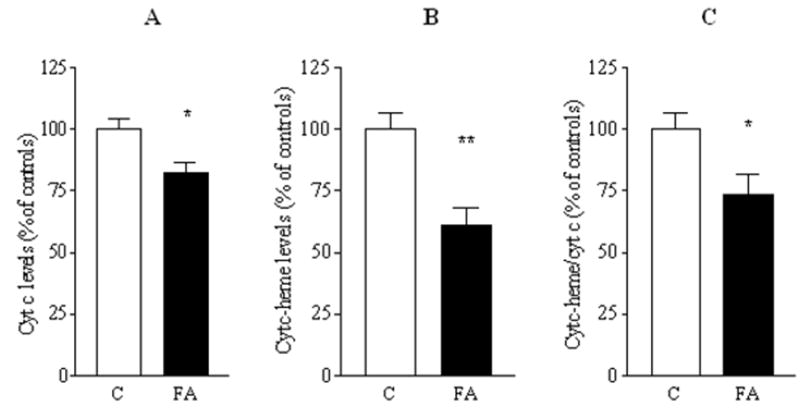
(A) Cytochrome c levels were analyzed by Western blot followed by densitometry. (B) Cytochrome c specific heme content was analyzed by the o-dianisidine staining as described in Materials and Methods. (C) Heme c/cytochrome c ratio. The results are reported as means ± SEM of eight experiments in duplicate for controls and FRDA and expressed as band intensities normalized as a percent of controls. C, controls; FA, Friedreich’s ataxia mutants. Statistical analysis by Student’s t test. *p < 0.05, **p < 0.005.
One possible cause of the decreased cytochrome c is deficient heme production. The amount of heme c was evaluated by gel staining with o-dianisidine, an artificial substrate which, in presence of hydrogen peroxide (H2O2) is oxidized by heme-containing proteins to give a colored product. Densitometric analysis of total heme staining showed a significant decrease in mutants (Fig. 6B). The densitometric analysis demonstrated that there was a further decrease in heme than was explainable by decrease in cytochrome c molecules alone, that is, that heme per cytochrome c molecule significantly decreased in mutants, by a mean 27% (Fig. 6C). Thus the residual functional cytochrome c in mutants should be on the order of the product of the residual cytochrome c molecules (82%) multiplied by the amount to which they are heme-replete (73%), or 82% × 73% = 60% residual cytochrome c activity in mutants versus controls.
Cytochrome oxidase activity is decreased in mutants
Protoheme that is produced by the heme pathway is also converted to heme a, which only is used in cytochrome oxidase (i.e., as hemes a + a3). The fact that ROS stimulation by the volatile inhibitor cyanide (which binds the a3 heme) was blunted in mutants, suggested that complex IV was at least partially inhibited in mutants. Complex IV activity was measured (Fig. 7), and was observed to be significantly decreased in frataxin-deficient cells (p < 0.005), on average by about 25%.
FIG. 7. Complex IV activity is impaired in FRDA lymphoblasts.
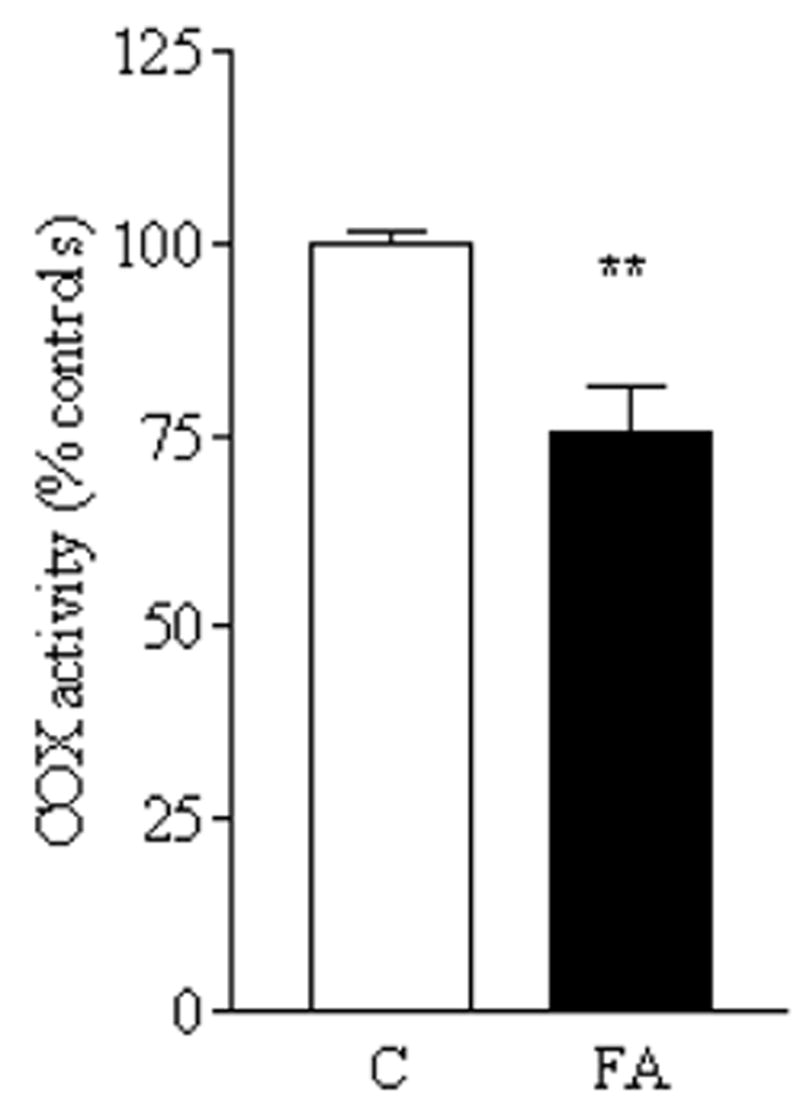
Cytochrome c oxidase activity was measured as described in Materials and Methods and expressed as nmol/mg/min. The bar graph shows the means ± SEM of four experiments in duplicate for controls (C) and FRDA (FA) reported as percentage of controls. Statistical analysis done by Student’s t test. **p < 0.005.
DISCUSSION
Frataxin-deficiency affects iron–sulfur clusters, heme, and ROS
Three sets of data have been presented here. First, we demonstrate that there is a defect specifically in ISC-dependent enzyme activity in FRDA lymphoblasts, and that the frataxin protein interacts directly with mitochondrial IscU2 in vivo, but not with ISC-S, rhodanese, HSC-20, or ferrochelatase.
Second, we demonstrate that there is a deficiency of cytochrome c, heme, and cytochrome oxidase activity in mutant cells, presumably the consequence of the iron–sulfur cluster defect.
Third, we demonstrate that there is increased superoxide production from FRDA mitochondria, a stimulation of the glutathione pathway, and a blunted stimulation of ROS by inhibitors of complex III and complex IV, which could be explained by the heme defects described above.
Frataxin deficiency and ISC
There is now substantial evidence that frataxin is intimately involved in ISC biogenesis and repair. The pattern of complex I, II, and III deficiency in frataxin-deficient yeast first prompted the suggestion of an ISC defect (17). In yeast, multiple groups have reported that frataxin is a facilitator, but is not essential, for ISC biogenesis (15, 34). In conditionally frataxin-deficient mice, defects in iron–sulfur cluster enzymes precede iron accumulation and cell death (43).
Previous work from our laboratory has shown a preferential effect of frataxin deficiency on transcripts related to ISC metabolism, including the mitochondrial rhodanese transcript whose product repairs ISCs, and transcripts in the sulfur amino acid pathway (57). In this work we demonstrate an in vivo affinity of frataxin with mitochondrial IscU2. Previously, we have demonstrated decreased expression of IscU2 in lymphoblasts of FRDA patients. Thus, frataxin is clearly attached to ISC metabolism, presumably as a consequence of its interaction with (and possibly, stabilization of) IscU2.
Frataxin, heme, and cytochrome c
Frataxin also has been shown to affect heme metabolism in yeast (21, 28, 30). A late step in the heme pathway is catalyzed by the ISC enzyme ferrochelatase. Thus it is possible that frataxin’s function in iron or ISC metabolism primarily affects the heme pathway secondarily (Schoenfeld and Napoli et al., unpublished observations). We observe that cytochrome c levels are decreased in FRDA cells, and that in addition, the ratio of heme per cytochrome c molecule is decreased in FRDA cells. Cytochrome oxidase activity, which requires two hemes produced by the heme pathway, is also significantly reduced in mutant cells.
Frataxin deficiency causes increased ROS and alters antioxidant enzyme activity
Cells from FRDA patients are sensitive to increased ROS (10, 23, 62). We show here that mitochondrial superoxide is elevated in mutant cells, consistent with our earlier demonstration that mitochondrial hydrogen peroxide is elevated in mutant mitochondria, and rescued by the frataxin gene (57). Presumably it is the increased burden of mitochondrial superoxide and hydrogen peroxide that stimulates the higher activity of glutathione peroxidase, and the higher GSSG concentration, and higher GSSG/GSH ratio. Thus FRDA cells are under increased oxidative stress.
General mechanisms for ROS formation in Friedreich’s ataxia
Given three frataxin-dependent parameters, ISCs, heme, and ROS, there are at least three possibilities for a primary mechanism, that is, ROS→ISCs + heme, ISCs→heme + ROS, heme→ISCs + ROS.
It was recently demonstrated that the cardiomyopathy in knockout mice was not rescuable by the MnSOD mimetic mntbap, and that markers of ROS occurred late in pathology (50, 51), suggesting that ROS are not the primary cause of FeS cluster destruction (or a heme defect), which had been a mechanistic proposal for FRDA (10, 36, 37, 46).
So the second and third explanations, that either ISC or heme are the primary consequences of frataxin-deficiency, are both viable.
Superoxide production by inhibition at complex I in FRDA mitochondria
The two main sites of mitochondrial superoxide production are thought to be complex I (by reverse electron flow) and complex III (by Q formation). It was previously proposed that an ISC defect might cause a more reduced electron transport chain and higher ROS production (36, 47). However, with respect to complex I, we observe similar amounts of rotenone-stimulated excess superoxide in both mutants (209%) and controls (235%). If ISC deficiency in mutants caused a complex I defect to the extent that reverse electron flow occurred, then complete inhibition of complex I by rotenone should produce a much smaller increase in superoxide in mutants with respect to controls, which is not what we observe. Thus we doubt that complex I is a major source of mitochondrial superoxide in FRDA mitochondria. This could be resolved by future studies of superoxide production in purified mitochondria fed different respiratory substrates.
Superoxide production by inhibition at complex III and IV
By contrast, we propose that it is inhibition at the latter end of the electron transport chain in mutant cells that produces both the increased mitochondrial superoxide, and the unique mutant responses to inhibition of complex III and complex IV.
First, we demonstrate that both cytochrome c and heme c/cytochrome c are deficient in mutant cells, so that the effective concentration of functional cytochrome c is about 60% of normal. Cytochrome c can act as an antioxidant, that is, it can oxidize O2•− to O2 (41, 60). Therefore, decreased cytochrome c in vivo should cause both a partial defect in complex III/IV function in vivo, because cytochrome c carries electrons between these complexes, and also increased ROS, because less cytochrome c molecules are available to oxidize superoxide to molecular oxygen. Furthermore, depletion of cytochrome c is known to blunt the increased ROS produced by antimycin a (60), which is exactly what we observe in the mutant cells. Thus, cytochrome c deficiency is likely to explain at least part of the increased ROS in FRDA cells (Fig. 8).
FIG. 8.
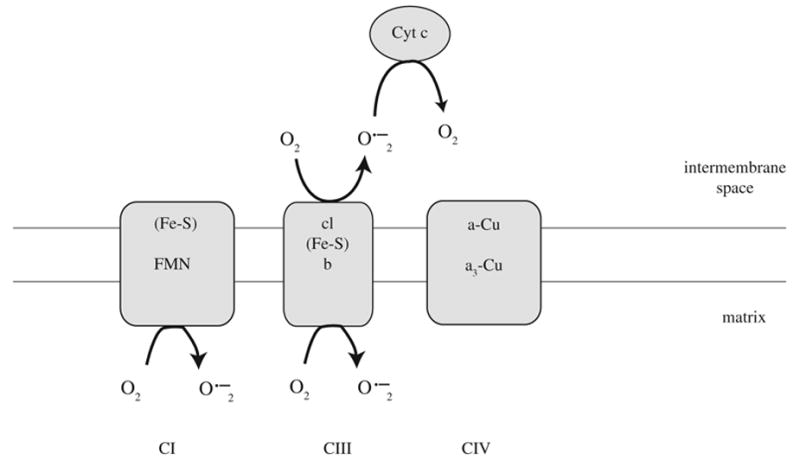
Mechanism of cytochrome c-dependent superoxide detoxification.
Heme and ISC deficiency effects on ROS production at complex III and IV
The heme pathway, in which there is a defect in FRDA cells, produces not only heme c of cytochrome c, but also cytochromes b566 and b540 of complex III, and heme a and aa3 of complex IV. So heme deficiency should preferentially affect complex III and IV. A partial defect in cytochromes b566 and b540 should produce the identical consequence as antimycin treatment, which inhibits at this point, to increase superoxide radical in mutant cells, and would explain the blunted antimycin superoxide response. Other contributory factors to the blunted increase in antimycin-stimulated super-oxide production could be decreased ISP function, decreased cytochrome c, and decreased cytochrome oxidase activity, which are required at maximal activity for maximal an-timycin a-dependent ROS production (60).
Cytochrome oxidase, heme, and ROS production
Although complex IV is not thought to be a major site of ROS production, its inhibition does increase ROS production in some cellular models (7, 24, 64); in our cells, inhibition of complex IV with KCN produced a stimulation of mitochondrial superoxide. It was demonstrated previously that inhibition of the heme pathway produces a preferential defect in complex IV and cytochrome c (2), and an increase in ROS (7), which is just what we observe in the mutant cells.
Presumably, partial or transient inhibition of cytochrome oxidase produces superoxide through reduction of complex III and the Q cycle. However, there was no cyanide–dependent significant increase in superoxide in mutants (i.e., mutant cells completely resisted the cyanide-stimulated increase in ROS). This result is consistent with our observations in mutants of 1) decreased cytochrome oxidase activity, 2) decreased cytochrome c level, and decreased FeS availability (e.g., Rieske ISP). The simplest interpretation is that as a result of frataxin deficiency and the consequent heme deficiency, complex IV, cytochrome c, Rieske ISP, and cytochromes b566 and b540 are already deficient in mutants, that is, there is limited electron flow from Q to O2. Thus a further inhibition of complex IV by cyanide does not have a major additional effect on superoxide production.
SUMMARY
Thus, the data presented here support a role for frataxin in iron–sulfur cluster metabolism, heme metabolism, and mitochondrial ROS metabolism. We observe intimate contact of frataxin with IscU2, and thus a frataxin deficiency could explain defects in IscU2 function for the biosynthesis of new mitochondrial iron–sulfur clusters, and the deficient activity of ISC enzymes observed. We believe that it is this iron–sulfur function which produces the defect in heme, that produces the selective defect on cytochrome c and cytochrome oxidase activity, that produces excess mitochondrial ROS. The elevated superoxide levels in FRDA mitochondria, and the blunting of the effects of antimycin a and cyanide on them, can be understood as a consequence of heme deficiency which should preferentially affect complex III and IV. These data explain how a mitochondrial iron–sulfur defect could cause a mitochondrial heme defect and a cytochrome defect, leading to the increased ROS observed.
Relevance to cell degenerative disease and aging
Multiple genetic defects, biochemical defects, bioenergetic defects, and ROS-related defects have been demonstrated with aging in the mitochondria (1, 12, 25, 29, 52). In the context of these age-related mitochondrial defects, which may involve ROS and iron–sulfur clusters, it is not surprising that defects in the mitochondrial heme pathway occur with aging (3, 49). In the degenerative disease Friedreich’s ataxia, a defect in mitochondrial frataxin expression causes a defect in iron–sulfur clusters, and apparently a consequent defect in heme metabolism, and we present here a mechanism by which the heme defect may trigger increased ROS. These suggest an alternative pathway for a ‘deleterious cycle’ in which ROS damage to mitochondrial iron–sulfur clusters triggers heme deficiency, and further generation of ROS.
Acknowledgments
The authors wish to thank Dr. Robert Schoenfeld and Dr. Alice Wong for their helpful comments and advice, and the NIH for support via USPHS AG11967, AG16719, and EY12245.
Abbreviations
- AVED
ataxia with vitamin E deficiency
- COX
cytochrome c oxidase
- DCIP
2,6-dichlorophenolindophenol
- DHE
dihydroethidium
- FRDA
Friedrich’s ataxia
- GPx
glutathione peroxidase
- GSH
reduced glutathione
- GSSG
oxidized glutathione
- ISC
iron-sulfur cluster
- PMSF
phenylmethanesulfonyl fluoride
- ROS
reactive oxygen species
References
- 1.Ames BN, Shigenaga MK, Hagen TM. Oxidants, antioxidants, and the degenerative diseases of aging. Proc Natl Acad Sci USA. 1993;90:7915–7922. doi: 10.1073/pnas.90.17.7915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Atamna H, Liu J, Ames BN. Heme deficiency selectively interrupts assembly of mitochondrial complex IV in human fibroblasts: revelance to aging. J Biol Chem. 2001;276:48410–48416. doi: 10.1074/jbc.M108362200. [DOI] [PubMed] [Google Scholar]
- 3.Atamna H, Killilea DW, Killilea AN, Ames BN. Heme deficiency may be a factor in the mitochondrial and neuronal decay of aging. Proc Natl Acad Sci USA. 2002;99:14807–14812. doi: 10.1073/pnas.192585799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bidichandani SI, Purandare SM, Taylor EE, Gumin G, Machkhas H, Harati Y, Gibbs RA, Ashizawa T, Patel PI. Somatic sequence variation at the Friedreich ataxia locus includes complete contraction of the expanded GAA triplet repeat, significant length variation in serially passaged lymphoblasts and enhanced mutagenesis in the flanking sequence. Hum Mol Genet. 1999;8:2425–2436. doi: 10.1093/hmg/8.13.2425. [DOI] [PubMed] [Google Scholar]
- 5.Birch-Machin MA, Turnbull DM. Assaying mitochondrial respiratory complex activity in mitochondria isolated from human cells and tissues. Methods Cell Biol. 2001;65:97–117. doi: 10.1016/s0091-679x(01)65006-4. [DOI] [PubMed] [Google Scholar]
- 6.Branda SS, Yang ZY, Chew A, Isaya G. Mitochondrial intermediate peptidase and the yeast frataxin homolog together maintain mitochondrial iron homeostasis in Saccharomyces cerevisiae. Hum Mol Genet. 1999;8:1099–1110. doi: 10.1093/hmg/8.6.1099. [DOI] [PubMed] [Google Scholar]
- 7.Campian JL, Qian M, Gao X, Eaton JW. Oxygen tolerance and coupling of mitochondrial electron transport. J Biol Chem. 2004;279:46580–46587. doi: 10.1074/jbc.M406685200. [DOI] [PubMed] [Google Scholar]
- 8.Campuzano V, Montermini L, Lutz Y, Cova L, Hindelang C, Jiralerspong S, Trottier Y, Kish SJ, Faucheux B, Trouillas P, Authier FJ, Durr A, Mandel JL, Vescovi A, Pandolfo M, Koenig M. Frataxin is reduced in Friedreich ataxia patients and is associated with mitochondrial membranes. Hum Mol Genet. 1997;6:1771–1780. doi: 10.1093/hmg/6.11.1771. [DOI] [PubMed] [Google Scholar]
- 9.Cavadini P, Gellera C, Patel PI, Isaya G. Human frataxin maintains mitochondrial iron homeostasis in Saccharomyces cerevisiae. Hum Mol Genet. 2000;9:2523–2530. doi: 10.1093/hmg/9.17.2523. [DOI] [PubMed] [Google Scholar]
- 10.Chantrel-Groussard K, Geromel V, Puccio H, Koenig M, Munnich A, Rotig A, Rustin P. Disabled early recruitment of antioxidant defenses in Friedreich’s ataxia. Hum Mol Genet. 2001;10:2061–2067. doi: 10.1093/hmg/10.19.2061. [DOI] [PubMed] [Google Scholar]
- 11.Cino M, Del Maestro RF. Generation of hydrogen peroxide by brain mitochondria: the effect of reoxygenation following postdecapitative ischemia. Arch Biochem Bio-phys. 1989;269:623–638. doi: 10.1016/0003-9861(89)90148-3. [DOI] [PubMed] [Google Scholar]
- 12.Cortopassi GA, Arnheim N. Detection of a specific mitochondrial DNA deletion in tissues of older humans. Nucleic Acids Res. 1990;18:6927–6933. doi: 10.1093/nar/18.23.6927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cortopassi GA, Wong A. Mitochondria in organismal aging and degeneration. Biochim Biophys Acta. 1999;1410:183–193. doi: 10.1016/s0005-2728(98)00166-2. [DOI] [PubMed] [Google Scholar]
- 14.Demin OV, Kholodenko BN, Skulachev VP. A model of O2•− eneration in the complex III of the electron transport chain. Mol Cell Biochem. 1998;184:21–33. [PubMed] [Google Scholar]
- 15.Duby G, Foury F, Ramazzotti A, Herrmann J, Lutz T. A non-essential function for yeast frataxin in iron–sulfur cluster assembly. Hum Mol Genet. 2002;11:2635–2643. doi: 10.1093/hmg/11.21.2635. [DOI] [PubMed] [Google Scholar]
- 16.Edwards MG, Sarkar D, Klopp R, Morrow JD, Weindruch R, Prolla TA. Age-related impairment of the transcriptional responses to oxidative stress in the mouse heart. Physiol Genomics. 2003;13:119–127. doi: 10.1152/physiolgenomics.00172.2002. [DOI] [PubMed] [Google Scholar]
- 17.Foury F, Cazzalini O. Deletion of the yeast homologue of the human gene associated with Friedreich’s ataxia elicits iron accumulation in mitochondria. FEBS Lett. 1997;411:373–377. doi: 10.1016/s0014-5793(97)00734-5. [DOI] [PubMed] [Google Scholar]
- 18.Foury F. Low iron concentration and aconitase deficiency in a yeast frataxin homologue deficient strain. FEBS Lett. 1999;456:281–284. doi: 10.1016/s0014-5793(99)00961-8. [DOI] [PubMed] [Google Scholar]
- 19.Handsford RG, Hogue BA, Mildaziene V. Dependence of H2O2 formation by rat heart mitochondria on substrate availability and donor age. J Bioenerg Biomembr. 1997;29:89–95. doi: 10.1023/a:1022420007908. [DOI] [PubMed] [Google Scholar]
- 20.Harman D. The biologic clock: the mitochondria? J Am Geriatr Soc. 1972;20:145–147. doi: 10.1111/j.1532-5415.1972.tb00787.x. [DOI] [PubMed] [Google Scholar]
- 21.He Y, Aalam SL, Proteasa SV, Zhang Y, Lesuisse E, Dancis A, Stemmler TL. Yeast frataxin solution structure, iron binding, and ferrochelatase interaction. Biochemistry. 2004;43:16254–16262. doi: 10.1021/bi0488193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Huynen MA, Snel B, Bork P, Gibson TJ. The phylogenetic distribution of frataxin indicates a role in iron–sulfur cluster protein assembly. Hum Mol Genet. 2001;10:2463–2468. doi: 10.1093/hmg/10.21.2463. [DOI] [PubMed] [Google Scholar]
- 23.Jauslin ML, Meier T, Smith RA, Murphy MP. Mitochondria-targeted antioxidants protect Friedreich ataxia fibroblasts from endogenous oxidative stress more effectively than untargeted antioxidants. FASEB J. 2003;17:1972–1974. doi: 10.1096/fj.03-0240fje. [DOI] [PubMed] [Google Scholar]
- 24.Johnson WT, Thomas AC. Copper deprivation potentiates oxidative stress in HL-60 cell mitochondria. Proc Soc Exp Biol Med. 1999;222:147–152. doi: 10.1046/j.1525-1373.1999.d01-68.x. [DOI] [PubMed] [Google Scholar]
- 25.Judge S, Jang YM, Smith A, Hagen T, Leeuwenburgh C. Age-associated increases in oxidative stress and antioxidant enzyme activities in cardiac interfibrillar mitochondria: implications for the mitochondrial theory of aging. FASEB J. 2005;19:419–421. doi: 10.1096/fj.04-2622fje. [DOI] [PubMed] [Google Scholar]
- 26.Koenig M. Friedreich ataxia and AVED. In: Scriver C, Beaudet A, Sly W, Valle D, editors. The Metabolic and Molecular Basis of Inherited Disease. New York: McGraw Hill; 2001. pp. 5845–5855. [Google Scholar]
- 27.Korshunov SS, Skulachev VP, Starkov AA. High protonic potential actuates a mechanism of production of reactive oxygen species in mitochondria. FEBS Lett. 1997;416:15–18. doi: 10.1016/s0014-5793(97)01159-9. [DOI] [PubMed] [Google Scholar]
- 28.Lange H, Muhlenhoff U, Denzel M, Kispal G, Lill R. The heme synthesis defect of mutants impaired in mitochondrial iron–sulfur protein biogenesis is caused by reversible inhibition of ferrochelatase. J Biol Chem. 2004;279:29101–29108. doi: 10.1074/jbc.M403721200. [DOI] [PubMed] [Google Scholar]
- 29.Lee CK, Klopp RG, Weindruch R, Prolla TA. Gene expression profile of aging and its retardation by caloric restriction. Science. 1999;285:1390–1393. doi: 10.1126/science.285.5432.1390. [DOI] [PubMed] [Google Scholar]
- 30.Lesuisse E, Santos R, Matzanke BF, Knight SA, Camadro JM, Dancis A. Iron use for haeme synthesis is under control of the yeast frataxin homologue (Yfh1) Hum Mol Genet. 2003;15:879–889. doi: 10.1093/hmg/ddg096. [DOI] [PubMed] [Google Scholar]
- 31.Mariotti C, Di Donato S. Cerebellar/spinocerebellar syndromes. Neurol Sci. 2001;22:S88–S92. doi: 10.1007/s100720100042. [DOI] [PubMed] [Google Scholar]
- 32.Melov S, Ravenscroft J, Malik S, Gill MS, Walker DW, Clayton PE, Wallace DC, Malfroy B, Doctrow SR, Lithgow GJ. Extension of life-span with superoxide dismutase/catalase mimetics. Science. 2000;289:1567–1569. doi: 10.1126/science.289.5484.1567. [DOI] [PubMed] [Google Scholar]
- 33.Miro O, Cardellach F, Barrientos A, Casademont J, Rotig A, Rustin P. Cytochrome c oxidase assay in minute amounts of human skeletal muscle using single wavelength spectrophotometers. J Neurosci Methods. 1998;80:107– 111. doi: 10.1016/s0165-0270(97)00204-5. [DOI] [PubMed] [Google Scholar]
- 34.Muhlenhoff U, Richhardt N, Ristow M, Kispal G, Lill R. The yeast frataxin homolog Yfh1p plays a specific role in the maturation of cellular Fe/S proteins. Hum Mol Genet. 2002;11:2025–2036. doi: 10.1093/hmg/11.17.2025. [DOI] [PubMed] [Google Scholar]
- 35.O’Neill HA, Gakh O, Isaya G. Supramolecular assemblies of human frataxin are formed via subunit-subunit interactions mediated by a non-conserved amino-terminal region. J Mol Biol. 2005;345:433–439. doi: 10.1016/j.jmb.2004.10.074. [DOI] [PubMed] [Google Scholar]
- 36.Pandolfo M. Molecular pathogenesis of Friedreich ataxia. Arch Neurol. 1999;56:1201–1208. doi: 10.1001/archneur.56.10.1201. [DOI] [PubMed] [Google Scholar]
- 37.Pandolfo M. Iron metabolism and mitochondrial abnormalities in Friedreich ataxia. Blood Cells Mol Dis. 2002;29:536–547. doi: 10.1006/bcmd.2002.0591. [DOI] [PubMed] [Google Scholar]
- 38.Park S, Gakh O, O’Neill HA, Mangravita A, Nichol H, Ferreira GC, Isaya G. Yeast frataxin sequentially chaperones and stores iron by coupling protein assembly with iron oxidation. J Biol Chem. 2003;278:31340–31351. doi: 10.1074/jbc.M303158200. [DOI] [PubMed] [Google Scholar]
- 39.Patel PI, Isaya G. Friedreich ataxia: from GAA triplet-repeat expansion to frataxin deficiency. Am J Hum Genet. 2001;69:15–24. doi: 10.1086/321283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Peled-Kamar M, Lotem J, Wirguin I, Weiner L, Hermalin A, Groner Y. Oxidative stress mediates impairment of muscle function in transgenic mice with elevated level of wild-type Cu/Zn superoxide dismutase. Proc Natl Acad Sci USA. 1997;94:3883–3887. doi: 10.1073/pnas.94.8.3883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pereverzev MO, Vygodina TV, Konstantinov AA, Skulachev VP. Cytochrome c, an ideal antioxidant. Biochem Soc Trans. 2003;31:1312–1315. doi: 10.1042/bst0311312. [DOI] [PubMed] [Google Scholar]
- 42.Prohaska JR, Ganther HE. Glutathione peroxidase activity of glutathione-S-transferases purified from rat liver. Biochem Biophys Res Commun. 1976;76:437–445. doi: 10.1016/0006-291x(77)90744-6. [DOI] [PubMed] [Google Scholar]
- 43.Puccio H, Simon D, Cossee M, Criqui-Filipe P, Tiziano F, Melki J, Hindelang C, Matyas R, Rustin P, Keonig M. Mouse models for Friedreich ataxia exhibit cardiomyopathy, sensory nerve defect and Fe-S enzyme deficiency followed by intramitochondrial iron deposits. Nat Genet. 2001;27:181–186. doi: 10.1038/84818. [DOI] [PubMed] [Google Scholar]
- 44.Ramazzotti A, Vanmansart V, Foury F. Mitochondrial functional interactions between frataxin and Isu1p, the iron–sulfur cluster scaffold protein, in Saccharomyces cerevisiae. FEBS Lett. 2004;557:215–220. doi: 10.1016/s0014-5793(03)01498-4. [DOI] [PubMed] [Google Scholar]
- 45.Rotig A, De Lonlay P, Chretien D, Foury F, Koenig M, Sidi D, Munnich A, Rustin P. Aconitase and mitochondrial iron-sulphur protein deficiency in Friedreich ataxia. Nat Genet. 1997;17:215–217. doi: 10.1038/ng1097-215. [DOI] [PubMed] [Google Scholar]
- 46.Rotig A, Sidi D, Munnich A, Rustin P. Molecular insights into Friedreich’s ataxia and antioxidant-based therapies. Trends Mol Med. 2002;8:221–224. doi: 10.1016/s1471-4914(02)02330-4. [DOI] [PubMed] [Google Scholar]
- 47.Rustin P. The use of antioxidants in Friedreich’s ataxia treatment. Expert Opin Investig Drugs. 2003;12:569–575. doi: 10.1517/13543784.12.4.569. [DOI] [PubMed] [Google Scholar]
- 48.Schulz H, Pellicioli EC, Thony-Meyer L. New insights into the role of CcmC, CcmD and CcmE in the haem delivery pathway during cytochrome c maturation by a complete mutational analysis of the conserved tryptophan-rich motif of CcmC. Mol Microbiol. 2000;37:1379–1388. doi: 10.1046/j.1365-2958.2000.02083.x. [DOI] [PubMed] [Google Scholar]
- 49.Scotto AW, Rinehart RW, Beattie DS. Aging-related decreases in hepatic mitochondrial and cytosolic delta-aminolevulinic acid synthase during experimental porphyria. Arch Biochem Biophys. 1983;222:150–157. doi: 10.1016/0003-9861(83)90512-x. [DOI] [PubMed] [Google Scholar]
- 50.Seznech H, Simon D, Monassier L, Criqui-Filipe P, Gansmuller A, Rustin P, Koenig M, Puccio H. Idebenone delays the onset of cardiac functional alteration without correction of Fe-S enzymes deficit in a mouse model for Friedreich ataxia. Hum Mol Genet. 2004;13:1017–1024. doi: 10.1093/hmg/ddh114. [DOI] [PubMed] [Google Scholar]
- 51.Seznec H, Simon D, Bouton C, Reutenauer L, Hertzog A, Golik P, Procaccio V, Patel M, Drapier JC, Koenig M, Puccio H. Friedreich ataxia: the oxidative stress paradox. Hum Mol Genet. 2005;14:463–474. doi: 10.1093/hmg/ddi042. [DOI] [PubMed] [Google Scholar]
- 52.Sohal RS. Role of oxidative stress and protein oxidation in the aging process. Free Radic Biol Med. 2002;33:37–44. doi: 10.1016/s0891-5849(02)00856-0. [DOI] [PubMed] [Google Scholar]
- 53.Srere PA. Citrate synthase. In: Lowenstein J, editor. Methods in Enzymology: Oxidation and Phosphorylation. New York, NY: Academic Press; 1969. pp. 3–11. [Google Scholar]
- 54.Stehling O, Elsasser HP, Bruckel B, Muhlenhoff U, Lill R. Iron–sulfur protein maturation in human cells: evidence for a function of frataxin. Hum Mol Genet. 2004;13:3007–3015. doi: 10.1093/hmg/ddh324. [DOI] [PubMed] [Google Scholar]
- 55.Sturtz LA, Diekert K, Jensen LT, Lill R, Culotta VC. A fraction of yeast Cu,Zn-superoxide dismutase and its metallochaperone, CCS, localize to the intermembrane space of mitochondria. A physiological role for SOD1 in guarding against mitochondrial oxidative damage. J Biol Chem. 2001;276:38084–38089. doi: 10.1074/jbc.M105296200. [DOI] [PubMed] [Google Scholar]
- 56.Tan G, Chen LS, Lonnerdal B, Gellera C, Taroni FA, Cortopassi GA. Frataxin expression rescues mitochondrial dysfunctions in FRDA cells. Hum Mol Genet. 2001;10:2099–2107. doi: 10.1093/hmg/10.19.2099. [DOI] [PubMed] [Google Scholar]
- 57.Tan G, Napoli E, Taroni F, Cortopassi G. Decreased expression of genes involved in sulfur amino acid metabolism in frataxin-deficient cells. Hum Mol Genet. 2003;12:1699–1711. doi: 10.1093/hmg/ddg187. [DOI] [PubMed] [Google Scholar]
- 58.Trounce IA, Kim YL, Jun AS, Wallace DC. Assessment of mitochondrial oxidative phosphorylation in patient muscle biopsies, lymphoblasts, and transmitochondrial cell lines. Methods Enzymol. 1996;264:484–509. doi: 10.1016/s0076-6879(96)64044-0. [DOI] [PubMed] [Google Scholar]
- 59.Turrens JF, Alexandre A, Lehninger AL. Ubisemiquinone is the electron donor for superoxide formation by complex III of heart mitochondria. Arch Biochem Biophys. 1985;237:408–414. doi: 10.1016/0003-9861(85)90293-0. [DOI] [PubMed] [Google Scholar]
- 60.Turrens JF. Mitochondrial formation of reactive oxygen species. J Physiol. 2003;552:335–344. doi: 10.1113/jphysiol.2003.049478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Urbina HD, Silberg JJ, Hoff KG, Vickery LE. Transfer of sulfur from IscS to IscU during Fe/S cluster assembly. J Biol Chem. 2001;276:44521–44526. doi: 10.1074/jbc.M106907200. [DOI] [PubMed] [Google Scholar]
- 62.Wong A, Yang J, Cavadini P, Gellera C, Lonnerdal B, Taroni F, Cortopassi G. The Friedreich’s ataxia mutation confers cellular sensitivity to oxidant stress which is rescued by chelators of iron and calcium and inhibitors of apoptosis. Hum Mol Genet. 1999;8:425–430. doi: 10.1093/hmg/8.3.425. [DOI] [PubMed] [Google Scholar]
- 63.Yoon T, Cowan JA. Iron–sulfur cluster biosynthesis. Characterization of frataxin as an iron donor for assembly of [2Fe-2S] clusters in ISU-type proteins. J Am Chem Soc. 2003;125:6078–6084. doi: 10.1021/ja027967i. [DOI] [PubMed] [Google Scholar]
- 64.Yuyama K, Yamamoto H, Nishizaki I, Kato T, Sora I, Yamamoto T. Caspase-independent cell death by low concentrations of nitric oxide in PC12 cells: Involvement of cytochrome C oxidase inhibition and the production of reactive oxygen species in mitochondria. J Neurosci Res. 2003;73:351–363. doi: 10.1002/jnr.10669. [DOI] [PubMed] [Google Scholar]
- 65.Zhang J, Graham DG, Montine TJ, Ho YS. Enhanced N-methyl-4-phenyl-1,2,3,6-tetrahydropyridine toxicity in mice deficient in CuZn-superoxide dismutase or glutathione per-oxidase. J Neuropathol Exp Neurol. 2000;59:53–61. doi: 10.1093/jnen/59.1.53. [DOI] [PubMed] [Google Scholar]
- 66.Schoenfeld RA, Napoli E, Wong A, Zhan S, Reutenauer L, Morer D, Buckpitt AR, Taroni F, Lonnerdal B, Ristow M, Puccio H, Cortopassi GA. Frataxin deficiency alters heme pathway transcripts and decreases heme metabolites in mammalian cells. Hum Mol Genet. 2006;14:3787–3799. doi: 10.1093/hmg/ddi393. [DOI] [PubMed] [Google Scholar]