

Abstract
Antigen-dependent activation of T lymphocytes requires T cell receptor (TCR)-mediated recognition of specific peptides, together with the MHC molecules to which they are bound. To achieve this recognition in a reasonable time frame, the TCR must scan and discriminate rapidly between thousands of MHC molecules differing from each other only in their bound peptides. Kinetic analysis of the interaction between a TCR and its cognate peptide–MHC complex indicates that both association and dissociation depend heavily on the temperature, indicating the presence of large energy barriers in both phases. Thermodynamic analysis reveals changes in heat capacity and entropy that are characteristic of protein–ligand associations in which local folding is coupled to binding. Such an “induced-fit” mechanism is characteristic of sequence-specific DNA-binding proteins that must also recognize specific ligands in the presence of a high background of competing elements. Here, we propose that induced fit may endow the TCR with its requisite discriminatory capacity and suggest a model whereby the loosely structured antigen-binding loops of the TCR rapidly explore peptide–MHC complexes on the cell surface until some critical structural complementarity is achieved through localized folding transitions. We further suggest that conformational changes, implicit in this model, may also propagate beyond the TCR antigen-binding site and directly affect self-association of ligated TCRs or TCR–CD3 interactions required for signaling.
The immune system contains two distinct but structurally related antigen receptors, Igs and T cell receptors (TCRs). Unlike Igs, TCRs must recognize their antigens as peptide fragments that are complexed with cell-surface MHC molecules. Developing T cells are selected in the thymus for a minimal ability to recognize self-MHC molecules containing a collection of endogenous peptides (1). After thymic development, peripheral T cells must then distinguish self-MHC molecules containing peptide fragments of antigen from endogenous peptide complexes. Perhaps as a result of this necessity for dual recognition, T cells often have degenerate specificities (2–4), which are rarely seen in mature Ig. Another feature of TCR recognition that distinguishes it from antibodies is that interactions between TCRs and peptide–MHC (pMHC) are often of relatively low affinities and are characterized by unusually slow association and rapid dissociation rates (5).
The molecular basis for these interesting features of TCRs is now emerging. An explanation for crossreactivity was provided recently by Garcia et al. (6, 7), who showed poor surface complementarity in a crystal structure of a 2C TCR–pMHC complex. Poor surface complementarity may also explain the relatively low affinities of some TCR–pMHC interactions (7). By comparison to the unligated 2C TCR structure, it was evident that, on binding, large conformational changes occurred in some of the antigen-binding hypervariable (complementarity-determining region; CDR) loops (6, 7). Such “structural plasticity,” in turn, suggests that a single TCR may accommodate different pMHC. The conformational change occurring during binding would also seem to explain the unusually slow association rates, as was originally suggested as a possible explanation (8). Further support for this explanation comes from a thermodynamic study by Anton van der Merwe and colleagues (9), who described large energy barriers to association for two class I restricted TCRs. The rapid dissociation rate for TCR–pMHC complexes seems necessary for the coupling of kinetic discrimination and signal transduction (5, 10). For example, in the 2B4 TCR system at 25°C, TCR–pMHC complexes have half-lives of approximately 12, 2, and 0.2 seconds when the peptide was a strong agonist, a weak agonist, or an antagonist, respectively (5). The steep correspondence between complex stability and biological response suggests that an important event in signal transduction occurs on a time scale of seconds, with the caveat that half-lives were measured at 25°C. These facts, together with the observed crossreactivities, indicate that T cells are selected during thymic development to react with endogenous pMHC complexes at a threshold just below the requirements for peripheral biological response.
The requirement for dual recognition and the presence of structural plasticity in TCRs, however, present another paradox. If a given TCR possesses a minimal but significant affinity for endogenous pMHC complexes and can recognize more than one antigenic peptide, how does a T cell rapidly sort through the irrelevant pMHC complexes, which are present in vast excess, to find and react specifically to antigen? Here, we show that the binding of a particular TCR to its cognate pMHC complex becomes far more rapid as the temperature is increased to 37°C, thereby facilitating the ability of the TCR to sample pMHC complexes over a cell surface. Thermodynamic analysis indicates that binding is coupled to folding transitions, which are probably concentrated but not necessarily confined to the antigen-binding site of the TCR. Propagation of the structural rearrangements induced by binding to regions outside this site might be important for TCR–TCR or TCR–CD3 interactions implicated in signaling.
MATERIALS AND METHODS
Proteins.
The recombinant class II MHC molecule Ek containing a single C-terminal 13-aa biotinylation site was produced as described (11, 12). Briefly, inclusion bodies containing Ek chains were prepared from the BL21(DE3)pLysS strain of Escherichia coli by repeated freeze/thaw cycles in 50 mM Tris, pH 8.0/1 mM EDTA/25% (vol/vol) sucrose/1% Triton X-100/1 mg/ml lysozyme. Chains were dissolved individually in 5.8 M guanidine-HCl/50 mM Tris, pH 8.8/2 mM EDTA at less than 1 mg/ml and allowed to oxidize to form disulfide bonds for 5–7 days. After concentration, folding was initiated by combining and diluting subunits to 2 μM in 50 mM sodium phosphate, pH 7.5/25% (vol/vol) glycerol/5 mM reduced glutathione/0.5 mM oxidized glutathione/0.5 mM EDTA/0.1 mM PMSF/1 μg/ml pepstatin/1 μg/ml leupeptin/10 μM moth cytochrome c (MCC) peptide. After 4–5 days of incubation at 4°C, correctly folded MCC–Ek complexes were isolated by immunoaffinity chromatography on a 14.4.4 mAb column. Complexes then were biotinylated enzymatically with the birA enzyme in the presence of protease inhibitors and were size purified to remove aggregates and free biotin. Biotinylation was confirmed by gel-shift assays by using excess streptavidin and was typically greater than 75%. 2B4 TCR was prepared as described (13) as a glycosylphosphatidylinositol-anchored chimera in Chinese hamster ovary cells. Molecules were recovered from cells grown to high density on hollow-fiber bioreactors by phosphatidyl inositol-specific phospholipase C digestion. Heterodimers were purified by tandem immunoaffinity chromatography on an anti-α-chain column (A2B4) followed by an anti-β-chain column (H57). Protein was then size purified on a Superdex-200 FPLC column immediately before BIAcore analyses. Quantification was made by using an extinction coefficient (280 nm) of 1.3 ml⋅mg−1⋅cm−1.
BIAcore Analysis.
The BIAcore streptavidin chip was prewashed with two short pulses of 0.05% SDS, and biotinylated MCC–Ek was immobilized to an approximate level of 3,500 resonance units. Soluble 2B4 TCR binding was monitored at a flow rate of 15 μl/min in PBS containing 0.005% P-20 detergent (BIAcore, Uppsala). PBS was filtered and degassed before use. Binding curves were fit by using the Marquardt–Levenberg nonlinear least squares algorithm provided in the biaevaluation 2.1 package. A BIAcore1000 was used to collect data from 20°C to 37°C. Because of its stability at lower temperatures, a BIAcore2000 was used to collect data from 10°C to 20°C. At 20°C, kinetic parameters on the two devices were identical. BIAcore-binding data generally do not seem to be limited by mass transport in this system (8). Potential mass-transport regions of BIAcore curves, when present, were not included in the analysis. Great care was taken to remove existing aggregates (above) before analysis, and the purified individual molecules (2B4 TCR and MCC–Ek) do not self-associate at the concentrations tested (14). Calculated dissociation-rate constants showed little or no dependence on analyte concentration. Calculated association-rate constants showed some analyte concentration dependence (as much as 2- to 3-fold across a 10-fold range in analyte concentration). Calculated rate constants at each analyte concentration and for each experiment were averaged to give the combined data set (see Fig. 2). χ2 values for individual fits were typically less than 1.0.
Figure 2.

Temperature dependence of 2B4–MCC–Ek association-rate (A) and dissociation-rate (B) constants. (Inset) Variation of the reaction half-life with temperature. Data points shown represent the mean value of three to seven independent measurements with at least three different analyte concentrations. Error bars represent SEM; uncertainty involved in temperature setting was ±0.1°C. Data were fit to a rearranged form of Eq. 1 (detailed in Materials and Methods) with a reference temperature T0 of 298.15 K. Note, we chose to illustrate the data by using temperature on the x axis, as opposed to the reciprocal of temperature that is common to Van’t Hoff analysis.
Thermodynamic Analysis.
The equilibrium-association constants (K) were calculated from the ratio of the rate constants (ka/kd). Thermodynamic parameters were determined by calculating ΔG° values (free energy change) by using ΔG° = −RTlnKT and fitting them as a function of temperature (T) according to
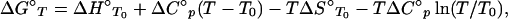 |
1 |
where ΔH°T0 (enthalpy change), ΔS°T0 (entropy change), and ΔC°p (heat-capacity change) are used as fitting parameters (15). ΔC°p was assumed to be independent of temperature; inclusion of a ΔC°p/dT term in the analysis did not improve the quality of the fits and gave larger standard errors for the returned parameters. Such insensitivity of ΔC°p to the temperature in the physiological range is consistent with processes dominated by the hydrophobic effect, such as an induced fit (16). Eq. 1, rearranged to include kinetic terms, was used to fit some of the data (see Fig. 2). ΔH° and TΔS° plots (see Fig. 3B) were made by using
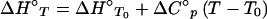 |
2 |
and
 |
3 |
Figure 3.

The thermodynamics of the 2B4–MCC–Ek association. (A) ΔG°assoc data were fit to Eq. 1 (T0 = 298.15 K) to yield the parameters and the best-fit line shown. Errors for the indicated parameters represent the estimated SEM obtained from data fitting. (B) ΔH° and TΔS° values were derived from Eqs. 2 and 3 detailed in Materials and Methods.
Model Building of the 2B4–MCC–Ek Complex.
The 2B4 TCR α- and β-chains were modeled separately by using 2C TCR as a template (coordinates kindly provided by K. C. Garcia, Stanford Univ., Stanford, CA, and I. A. Wilson, The Scripps Research Institute, La Jolla, CA). Sequence identity between target and template was 60.6% (α) and 65.6% (β). Sequence alignment, model building, energy minimization, and three-dimensional profile analysis were all made by using the swiss-model automated comparative protein modeling server (refs. 17–19; www.expasy.ch/swissmod/SWISS-MODEL.html). After this step, the two chains were combined, and the structure was energy minimized further by using gromos96 (W. F. van Gunsteren; BIOMOS b.v, Laboratory of Physical Chemistry, Eidgenössische Technische Hochschule Zentrum, Zurich). Computations were made in vacuo by using the 43B1 parameters set. The energy minimized 2B4 heterodimer, along with its ligand, MCC–Ek (Protein Data Bank ID code 1IEA; with MCC residues 88–103 substituted for the original peptide), was then superimposed on 2C–Kb template, and the complex was energy minimized by gromos96.
Accessible Surface Areas.
Accessible surface areas were calculated with the program naccess (20) by using a 1.4-Å probe sphere.
RESULTS AND DISCUSSION
The class II MHC molecule Ek complexed to an antigenic peptide fragment of MCC (residues 88–103) was attached to a streptavidin sensor chip (BIAcore) via a single biotin positioned at the C terminus of the MHC α-chain. Binding of the αβ TCR, 2B4, which recognizes and specifically interacts with this complex (5), was then measured at various temperatures by surface plasmon resonance. Fig. 1 illustrates the large temperature dependence of the overall reaction. The kinetics in both directions becomes more rapid as the temperature is increased, leading to a faster approach to equilibrium.
Figure 1.

Binding of 2B4 to MCC–Ek monitored at different temperatures on a BIAcore device. Injection of TCR (0.45 mg/ml) was from ≈70 to ≈160 sec, after which dissociation was initiated. Each curve in A (20–37°C) can be distinguished by the faster approach to equilibrium at higher temperatures. Binding curves in A are not directly comparable to those in B (10–20°C), because they were obtained in separate experiments, on different instruments, and at slightly different densities of immobilized ligand. RU, resonance units.
Fig. 2 summarizes the temperature dependence of the association-rate (Fig. 2A) and dissociation-rate (Fig. 2B) constants. Slow association kinetics (owing to a large association activation energy) and steep temperature dependence, for both phases, are evident. As also shown, the plots are not linear but have a gentle, but clear, concave up (kd) and down (ka) curvature, indicating, in addition to a deviation from a simple reaction mechanism, a significant heat-capacity change (ΔC°p) in both phases (463 ± 53 and −200 ± 48 cal⋅mol−1⋅K−1, respectively; 1 cal = 4.18 J). These data cannot be explained by aggregation, because great care was taken to eliminate aggregates before analysis (see Materials and Methods) and because neither molecule alone aggregates at these concentrations (14). Owing to the marked variation of kd with temperature, the reaction half-life (t1/2) also has a steep temperature dependence, decreasing from 240 sec at 10°C to almost 3 sec at 37°C (Fig. 2B Inset). This last value (3 sec) is much smaller than those reported for TCR–pMHC interactions (reviewed in ref. 5), most of which have been measured at 25°C. It is also much faster than the t1/2 determined by Sykulev et al. (21) for the 2C TCR and one of its ligands at 37°C (46 sec). However, the 2C TCR–pMHC interaction is of unusually high affinity, and the same interaction measured at 25°C is five times slower (21), consistent with the ratio observed here. As it is more typical of TCRs, the t1/2 value for the 2B4 TCR may be useful in models concerning ligand discrimination (10, 22) and self-association (14). We note, however, that the t1/2 values reported here are likely to be shorter than those prevailing in vivo because of a reduction in the TCR’s translational and rotational freedom on the cell surface and the presence of coreceptors that interact and stabilize the ternary complex. The increase in the reaction on-and-off rates with temperature is also compatible with the proposed model of serial engagement, whereby a single pMHC complex could interact transiently with and trigger a large number of TCRs (23).
The thermodynamics of the 2B4–MCC–Ek association is summarized in Fig. 3. Of the parameters shown (Fig. 3A), of particular interest is a large negative ΔC°p assoc, which is indicative of the removal of substantial amounts of nonpolar surface area from solvent on binding. It also gives rise to thermodynamics that typify an entropy–enthalpy compensation process (i.e., with |ΔC°p assoc| ≫ |ΔS°assoc|) in which enthalpy contributes favorably to the interaction throughout the entire temperature range studied (Fig. 3B). Entropy, on the other hand, mostly opposes association and makes a favorable contribution only at low temperatures (below 290 K).
For rigid-body associations, observed changes in heat capacity often can be predicted very accurately based solely on the amount of surface area buried on complex formation (see ref. 24 and references therein). We have calculated ΔC°p assoc for two known TCR–pMHC complex structures and for a model of 2B4–MCC–Ek. As indicated (Table 1), all three complexes bury roughly the same amount of nonpolar and polar surfaces, yielding similar ΔC°p assoc of approximately −250 cal⋅mol−1⋅K−1. Predicted values are thus only one-half to one-third as large as the experimentally determined value of −663 cal⋅mol−1⋅K−1 for the 2B4 TCR and its ligand. The large discrepancy between calculated and observed |ΔC°p| argues against a rigid-body association and indicates that binding involves an induced fit. In this case, the excess |ΔC°p| results from the burial of additional nonpolar surfaces through the folding transitions concomitant to binding.
Table 1.
Predicted and observed heat-capacity changes for TCR–pMHC complexes
| TCR–pMHC | Water-accessible surface area, Å2
|
Heat-capacity change, cal·mol−1·K−1
|
||
|---|---|---|---|---|
| −ΔAnp | −ΔAp | ΔC°p calc | ΔC°p obs | |
| A6–HLA-A2* | 1,153 | 837 | −252 | n/a |
| 2C–Kb† | 1,066 | 819 | −227 | n/a |
| 2B4–Ek‡ | 1,226 | 762 | −286 | −663 |
Calculated ΔCp° values were derived according to ΔCp°= (0.32·ΔAnp) − (0.14·ΔAp) (24), in which Anp and Ap are the respective changes in water-accessible nonpolar (np) and polar (p) surface area in Å2.
*Brookhaven Protein Data Bank ID code 1A07.
Coordinates kindly provided by K. C. Garcia and I. A. Wilson.
Model structure based on 2C–Kb template and published Ek coordinates (see Materials and Methods).
An induced-fit mechanism is supported further and more definitively by a dissection of the total entropy change ΔS°assoc of the 2B4–MCC–Ek association. ΔS°assoc includes several contributions and, for protein–ligand interactions, may be expressed as
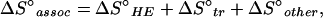 |
4 |
where ΔS°HE and ΔS°tr correspond to entropy changes arising from the hydrophobic effect and changes in the molecules’ rotational and translational entropy, respectively, and ΔS°other includes any other entropic effects (e.g., changes in vibrational or conformational entropy) and is assumed to be temperature independent (24). For rigid-body associations, contributions arising from ΔS°other are usually very small, and ΔS°assoc is determined by only ΔS°HE and ΔS°tr (24). For an induced fit, however, a significant negative ΔS°other term, amounting to the loss in conformational entropy on structure formation, should be present. For the 2B4–MCC–Ek association at Ts (290 K; where ΔS°assoc = 0; Fig. 3), we calculate (by using equation 3 in ref. 24) ΔS°HE to be 256 entropy units. Because ΔS°tr is relatively constant and, for bimolecular protein–protein associations, averages to −50 ± 10 entropy units (24–26), summation of these two contributions leaves a substantial unfavorable ΔS°other term of −206 entropy units. This value of ΔS°other falls in the midrange of values reported by Spolar and Record (24) for processes known to couple local or long-range folding to site-specific binding. The low Ts (290 K) determined here is likewise a value typical of such processes (24). Finally, the number of residues that change their conformation on binding can be estimated from ΔS°other (24) and, for the reaction investigated, is calculated to be 36.
The binding of 2B4 to MCC–Ek therefore fits a thermodynamic signature characteristic of reactions involving an induced fit. Although folding transitions, implicit in this model, may occur anywhere in the MHC or the TCR, they are most likely to be dominated by the TCR hypervariable loops (CDRs), which seem likely to be inherently flexible binding interfaces. Some of these loops have indeed been shown by crystallographic studies (6, 7) to undergo substantial rearrangements on binding. In contrast, the structure of pMHC molecules changes very little when bound to the TCR (7, 27). Thus, we propose that the TCR CDR loops are particularly flexible in the free state and become ordered only on binding as a result of localized folding transitions. Results consistent with the formation of structure during TCR–pMHC binding also were obtained recently by thermodynamic analyses for two different class I restricted TCRs (9). Folding, as determined here, is substantial and is estimated to involve about 36 residues. This number of residues undergoing conformational changes on binding is significantly larger than indicated by x-ray analyses of the bound and free 2C TCR (7). Thus, in general, the values derived here suggest that TCRs may be significantly less ordered in solution than in the crystalline state. The extensive change in conformational entropy measured here could also arise, in part, from subtle rearrangements that propagate beyond the antigen-binding loops. Of note in this regard is the fact that small shifts in VαVβ pairing were observed in the 2C TCR on binding (6, 7). Such structural shifts or other subtle conformational rearrangements could potentially enhance the interface complementarity required for the self-association of ligated TCRs (14) or TCR–CD3 interactions required for signaling.
In trying to understand TCR-mediated recognition in the context of an induced-fit model, appealing parallels can be drawn to the recognition of DNA by site-specific binding proteins. Like the TCR, DNA-binding proteins must rapidly discriminate between numerous competing and chemically similar elements, and the presence of an induced fit has been shown in several such proteins (24). In these cases, recognition does not follow a rigid alignment of preexisting complementary surfaces. Instead, regions in the protein (and, in some cases, in the DNA), which are unstructured in the free state, fold on binding to create key parts of the contact interface. This “on-site” construction of the protein–DNA interface indicates that the final conformation will depend strongly on the DNA sequence bound, as indeed has been observed (e.g., refs. 28–30). As expected, engagement of the binding protein with the DNA during scanning is mediated by nonspecific electrostatic interactions.
Analogously, the TCR must discriminate amongst thousands of chemically similar pMHCs on the cell surface through transient interactions that are relatively insensitive to the peptide antigen. These interactions are most likely made with epitopes located over the MHC helices. Mutagenesis and structural data indicating a common, peptide-independent orientation of all TCRs over the pMHC ligand (5, 7) are consistent with such a model. Once a cognate complex is found, the free energy of binding could drive local rearrangements in the CDRs, a process by which surface complementarity is maximized and complex stability is enhanced. As noted above, this structural adaptation may, in principle, be ligand dependent; different structures may form on different pMHC ligands. That this model may well be the case was suggested earlier by Ehrich et al. (31), who found that the pattern of MHC mutants that disrupted recognition by a particular TCR varied markedly when different peptides were recognized. More recently, Mason (32) has suggested that each TCR must be inherently able to interact with many peptides to account for thymic selection and crossreactivities (e.g., refs. 2–4). Such plasticity, as noted by Garcia et al. (6, 7) would give rise to a wide spectrum of complexes of various stabilities and lifetimes and, hence, may lead to markedly different signaling events, as has also been observed in this system (10, 33, 34). We further suggest that the energy necessary to form encounter pairs during the search mode is provided predominantly by the CDR loops directly contacting the MHC moiety. Stable complex formation, on the other hand, most likely involves CDRs engaged in peptide recognition, such as the CDR3 loops and, hence, is ligand dependent.
An interesting feature of αβ TCRs is that their most hypervariable loops (CDR3) are very constrained in size when compared with those of Ig heavy chains. Thus, the CDR3 of human TCR α- or β-chains have a median length of 9 aa and a range of 6–12 aa (35). This median length is compared with a one of 12 aa and a range of 3–25 aa observed for Ig heavy-chain CDR3s (35). This selection for certain CDR3 lengths in αβ TCRs may reflect a compromise. A minimum number of residues are clearly needed to afford specificity and conformational flexibility. On the other hand, the energetic costs associated with structure formation set a limit to this number, above which the expenditure of free energy would become too costly to sustain the low-affinity-based scanning of the MHC surfaces. In contrast, the length diversity seen in Ig heavy-chain CDR3s may be the result of the fact that there is no need for a scanning function, because there is no commonality between most ligands and no preferred binding orientation. It is also interesting to note that Ig CDR loops are optimized in a preferred high-affinity conformation through somatic hypermutation as an immune response matures. Therefore, the necessity for an induced fit in the already selected, matured forms becomes significantly reduced. Consequently, in many cases in which mature antibody–antigen interactions have been studied, evidence for an induced fit is lacking (36–41), and, in cases where it has been described (42–44), structural changes are significantly smaller than those seen in the 2C TCR (7) and those predicted here. That an induced-fit mechanism may be more prevalent in nonsomatically mutated antibodies has been elegantly established by Wedemayer et al. (45), who have shown that a nonmutated antibody to a hapten binds by an induced-fit mechanism, whereas a mutated form does not.
Recently, Willcox et al. (9) described temperature effects similar to those reported here and, in particular, a striking loss of entropy on binding for two TCRs that recognize class I MHC ligands. These data, together with existing structural data and the similarity in thermodynamics of binding in TCRs and DNA-binding proteins, suggest that an induced-fit mechanism may be general to systems that require the rapid scanning of many similar molecular entities.
Acknowledgments
We thank R. Ghirlando, E. J. Wachtel, S. Demer, J. Samayao, and J. Goldberg for advice and helpful discussions; K. C. Garcia, J. Samayao, and L. Wu for insightful comments on the manuscript; and A. Watta for assistance with BIAcore2000 measurements. This work was supported by the National Institutes of Health and the Howard Hughes Medical Institute. D.S.L. was funded by a Predoctoral Fellowship from the Howard Hughes Medical Institute.
ABBREVIATIONS
- TCR
T cell receptor
- CDR
complementarity-determining region
- pMHC
peptide–MHC
- MCC
moth cytochrome c
References
- 1.Saito T, Watanabe N. Crit Rev Immunol. 1998;18:359–370. doi: 10.1615/critrevimmunol.v18.i4.40. [DOI] [PubMed] [Google Scholar]
- 2.Bhardwaj V, Kumar V, Geysen H M, Sercarz E E. J Immunol. 1993;151:5000–5010. [PubMed] [Google Scholar]
- 3.Evavold B D, Sloan-Lanchaster J, Wilson K J, Rothbard J B, Allen P M. Immunity. 1995;2:655–663. doi: 10.1016/1074-7613(95)90010-1. [DOI] [PubMed] [Google Scholar]
- 4.Wucherpfennig K W, Strominger J L. Cell. 1995;80:695–705. doi: 10.1016/0092-8674(95)90348-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Davis M M, Boniface J J, Reich Z, Lyons D, Hampl J, Arden B, Chien Y. Annu Rev Immunol. 1998;16:523–544. doi: 10.1146/annurev.immunol.16.1.523. [DOI] [PubMed] [Google Scholar]
- 6.Garcia K C, Teyton L. Curr Opin Biotechnol. 1998;4:338–343. doi: 10.1016/s0958-1669(98)80004-9. [DOI] [PubMed] [Google Scholar]
- 7.Garcia K C, Degano M, Pease L R, Huang M, Peterson P A, Teyton L, Wilson I A. Science. 1998;279:1166–1172. doi: 10.1126/science.279.5354.1166. [DOI] [PubMed] [Google Scholar]
- 8.Matsui K, Boniface J J, Steffner P, Reay P A, Davis M M. Proc Natl Acad Sci USA. 1994;91:2862–2866. doi: 10.1073/pnas.91.26.12862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Willcox B E, Gao G F, Wyer J R, Ladbury J E, Bell J I, Jakobsen B K, Anton van der Merwe P. Immunity. 1999;10:357–365. doi: 10.1016/s1074-7613(00)80035-7. [DOI] [PubMed] [Google Scholar]
- 10.Rabinowitz J D, Beeson C, Lyons D S, Davis M M, McConnell H M. Proc Natl Acad Sci USA. 1996;93:1401–1405. doi: 10.1073/pnas.93.4.1401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Altman J D, Reay P A, Davis M M. Proc Natl Acad Sci USA. 1993;90:10330–10334. doi: 10.1073/pnas.90.21.10330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Boniface J J, Rabinowitz J D, Wulfing C, Hampl J, Reich Z, Altman J D, Kantor R M, Beeson C, McConnell H M, Davis M M. Immunity. 1998;9:459–466. doi: 10.1016/s1074-7613(00)80629-9. [DOI] [PubMed] [Google Scholar]
- 13.Lin A Y, Devaux B, Green A, Sagerstrom C, Elliott J F, Davis M M. Science. 1990;249:677–679. doi: 10.1126/science.1696397. [DOI] [PubMed] [Google Scholar]
- 14.Reich Z, Boniface J J, Lyons D S, Borochov N, Wachtel E J, Davis M M. Nature (London) 1997;387:617–620. doi: 10.1038/42500. [DOI] [PubMed] [Google Scholar]
- 15.Yoo S H, Lewis M S. Biochemistry. 1995;34:632–638. doi: 10.1021/bi00002a030. [DOI] [PubMed] [Google Scholar]
- 16.Ha J-H, Spolar R S, Record M T., Jr J Mol Biol. 1989;209:801–816. doi: 10.1016/0022-2836(89)90608-6. [DOI] [PubMed] [Google Scholar]
- 17.Peitsch M C. Bio/Technology. 1995;13:658–660. [Google Scholar]
- 18.Peitsch M C. Biochem Soc Trans. 1996;24:274–279. doi: 10.1042/bst0240274. [DOI] [PubMed] [Google Scholar]
- 19.Guex N, Peitsch M C. Electrophoresis. 1997;18:2714–2723. doi: 10.1002/elps.1150181505. [DOI] [PubMed] [Google Scholar]
- 20.Hubbard S J, Thornton J M. naccess. London: Univ. Coll.; 1993. [Google Scholar]
- 21.Sykulev Y, Brunmark A, Tsomides T J, Kageyama S, Jackson M, Peterson P A, Eisen H N. Proc Natl Acad Sci USA. 1994;91:11487–11491. doi: 10.1073/pnas.91.24.11487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.McKeithan T W. Proc Natl Acad Sci USA. 1995;92:5042–5046. doi: 10.1073/pnas.92.11.5042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Valitutti S, Muller S, Cella M, Padovan E, Lanzavecchia A. Nature (London) 1995;375:148–151. doi: 10.1038/375148a0. [DOI] [PubMed] [Google Scholar]
- 24.Spolar R S, Record T R., Jr Science. 1994;263:777–784. doi: 10.1126/science.8303294. [DOI] [PubMed] [Google Scholar]
- 25.Finkelstein A V, Janin J. Protein Eng. 1989;3:1–3. doi: 10.1093/protein/3.1.1. [DOI] [PubMed] [Google Scholar]
- 26.Janin J, Chothia C. Biochemistry. 1978;17:2943–2948. doi: 10.1021/bi00608a001. [DOI] [PubMed] [Google Scholar]
- 27.Ding Y H, Smith K J, Garboczi D N, Utz U, Biddison W E, Wiley D C. Immunity. 1998;8:403–411. doi: 10.1016/s1074-7613(00)80546-4. [DOI] [PubMed] [Google Scholar]
- 28.Winkler F K. EMBO J. 1993;12:1781–1795. doi: 10.2210/pdb4rve/pdb. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Clarke N D, Beamer L J, Goldberg H R, Berkower C, Pabo C O. Science. 1991;254:267–270. doi: 10.1126/science.254.5029.267. [DOI] [PubMed] [Google Scholar]
- 30.O’Neil K T, Hoess R H, DeGrado W F. Science. 1990;249:774–778. doi: 10.1126/science.2389143. [DOI] [PubMed] [Google Scholar]
- 31.Ehrich E W, Devaux B, Rock E P, Jorgensen J L, Davis M M, Chien Y. J Exp Med. 1993;178:713–722. doi: 10.1084/jem.178.2.713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mason D. Immunol Today. 1998;9:395–404. doi: 10.1016/s0167-5699(98)01299-7. [DOI] [PubMed] [Google Scholar]
- 33.Lyons D S, Lieberman S A, Hampl J, Boniface J J, Chien Y, Berg L J, Davis M M. Immunity. 1996;5:53–61. doi: 10.1016/s1074-7613(00)80309-x. [DOI] [PubMed] [Google Scholar]
- 34.Kersh E N, Shaw A S, Allen P M. Science. 1998;281:572–575. doi: 10.1126/science.281.5376.572. [DOI] [PubMed] [Google Scholar]
- 35.Rock E P, Sibbald P R, Davis M M, Chien Y-h. J Exp Med. 1994;179:323–328. doi: 10.1084/jem.179.1.323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Murphy K P, Xie D, Garcia K C, Amzel L M, Freire E. Proteins. 1993;15:113–120. doi: 10.1002/prot.340150203. [DOI] [PubMed] [Google Scholar]
- 37.Bhat T N, Bentley G A, Boulot G, Greene M I, Tello D, Dall’Acqua W, Souchon H, Schwarz F P, Mariuzza R A, Poljak R J. Proc Natl Acad Sci USA. 1994;91:1089–1093. doi: 10.1073/pnas.91.3.1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hibbits K A, Gill D S, Willson R C. Biochemistry. 1994;33:3584–3590. doi: 10.1021/bi00178a015. [DOI] [PubMed] [Google Scholar]
- 39.Murphy K P, Freire E, Patterson Y. Proteins. 1995;21:83–90. doi: 10.1002/prot.340210202. [DOI] [PubMed] [Google Scholar]
- 40.Fields B A, Goldbaum F A, Dall’Acqua W, Malchiodi E L, Cauerhff A, Schwarz F P, Ysern X, Poljak R J, Mariuzza R A. Biochemistry. 1996;35:15494–15503. doi: 10.1021/bi961709e. [DOI] [PubMed] [Google Scholar]
- 41.Tsumoto K, Ogasahara K, Ueda Y, Watanabe K, Yutani K, Kumagai I. J Biol Chem. 1996;271:32612–32616. doi: 10.1074/jbc.271.51.32612. [DOI] [PubMed] [Google Scholar]
- 42.Schulze-Gahmen U, Rini J M, Wilson I A. J Mol Biol. 1993;234:1098–1118. doi: 10.1006/jmbi.1993.1663. [DOI] [PubMed] [Google Scholar]
- 43.Stanfield R L, Takimoto-Kamimura M, Rini J M, Profy A T, Wilson I A. Structure. 1993;1:83–93. doi: 10.1016/0969-2126(93)90024-b. [DOI] [PubMed] [Google Scholar]
- 44.Rini J M, Schulze-Gahmen U, Wilson I A. Science. 1992;255:959–965. doi: 10.1126/science.1546293. [DOI] [PubMed] [Google Scholar]
- 45.Wedemayer G J, Patten P A, Wang L H, Schultz P G, Stevens R C. Science. 1997;276:1665–1669. doi: 10.1126/science.276.5319.1665. [DOI] [PubMed] [Google Scholar]