

Abstract
Progranulin (PGRN) is a pleiotropic protein that has gained the attention of the neuroscience community with recent discoveries of mutations in the gene for PGRN that cause frontotemporal lobar degeneration (FTLD). Pathogenic mutations in PGRN result in null alleles, and the disease is likely the result of haploinsufficiency. Little is known about the normal function of PGRN in the central nervous system apart from a role in brain development. It is expressed by microglia and neurons. In the periphery, PGRN is involved in wound repair and inflammation. High PGRN expression has been associated with more aggressive growth of various tumors. The properties of full length PGRN are distinct from those of proteolytically derived peptides, referred to as granulins (GRNs). While PGRN has trophic properties, GRNs are more akin to inflammatory mediators such as cytokines. Loss of the neurotrophic properties of PGRN may play a role in selective neuronal degeneration in FTLD, but neuroinflammation may also be important. Gene expression studies suggest that PGRN is up-regulated in a variety of neuroinflammatory conditions, and increased PGRN expression by microglia may play a pivotal role in the response to brain injury, neuroinflammation and neurodegeneration.
Background
Progranulin (PGRN) was discovered independently by several investigators and given several different names, including granulin-epithelin precursor, proepithelin, prostate cancer (PC) cell derived growth factor and acrogranin [1]. Encoded by a single gene on chromosome 17q21 (PGRN), PGRN is a 593-amino acid, cysteine-rich protein with an estimated molecular weight of 68.5 kDa that runs at 90 kDa on standard western blots due to heavy glycosylation [2]. It contains seven granulin-like domains, which consist of highly conserved tandem repeats of a rare 12 cysteinyl motif [3,4] (Figure 1). Proteolytic cleavage of the precursor protein by extracellular proteases, such as elastase, gives rise to smaller peptide fragments termed granulins (GRNs) or epithelins [1]. These fragments range in size from 6 to 25 kDa and have been implicated in a range of biological functions [1,5].
Figure 1.

The protein sequence of full length PGRN and its proteolytically cleaved GRNs. Amino acids shown in bold represent the granulin consensus sequence separated by variably long linker regions. Cysteine-rich parts of the sequence are denoted by CC.
Previous work on PGRN focused on its role in embryonic development and neoplasia (reviewed elsewhere [1]). The recent discovery that mutations in PGRN cause frontotemporal lobar degeneration with ubiquitin-immunoreactive neuronal inclusions (FTLD-U) has brought renewed interest in PGRN and its functions in the central nervous system (CNS). We review what is known about PGRN in peripheral tissues during injury, repair and inflammation and explore the relevance of these properties to CNS disorders, with a focus on FTLD-U.
PGRN in the periphery
Gene expression studies
Basal gene expression studies in mice and rats reveal widespread expression of PGRN in many different tissues, as well as in epithelial and hematopoietic cell lines [6,7]. Expression of PGRN mRNA is particularly high in epithelial cells that have a rapid turnover, such as those of the skin and gastrointestinal tract. Non-proliferating epithelia, such as lung alveolar cells, have relatively low levels of expression [6]. Epididymal cells have high PGRN expression, but are mitogenically stable, implying a pleiotropic role for PGRN. Mesenchymal tissues that lack PGRN mRNA are mitogenically responsive to PGRN in vitro. Both full length PGRN [1] and its proteolytic peptides [8] have mitogenic affects on epithelial cells in culture. Many transformed or immortalized epithelial cell lines express PGRN [3], while primary cells and cells in vivo have relatively low PGRN expression [6].
PGRN peptides were originally isolated and characterized in activated leukocytes [9]. In the periphery PGRN mRNA is abundant in lymphoid tissue of the lung, gut and spleen, and expression is also high in hematopoietic cell lines [6]. Hematopoietic myeloid cells, such as macrophages and tissue histiocytes in liver, spleen, lungs and brain, show no labeling by in situ hybridization even though a profile of human macrophages transcripts in vitro identified PGRN as one of the most highly expressed mRNAs [10]. All in vitro results suggests that there are high levels of expression in hematopoietic myeloid cells in the periphery, but low basal level of expression in vivo. In contrast to the mitogenic properties of PGRN on epithelial cells, there is little evidence to suggest that PGRN has mitogenic effects on hematopoietic cells [6,11].
PGRN in wound healing
PGRN is an important growth factor in the wound healing response [12], which can be separated into the distinct phases of inflammation, epithelialization, granulation, neovascularization and contraction (reviewed in [13]). In experimental skin wounds of mice, PGRN mRNA increases in fibroblasts, endothelial cells, macrophages and neutrophils. Fibroblasts and endothelial cells have no PGRN expression in normal skin, but there is constitutive expression of PGRN in keratinocytes. Addition of PGRN to the wound increases and prolongs infiltration of neutrophils and macrophages, and it enhances neovascularization [12], but it has no effect on the overall rate of healing [14]. The role of PGRN in the later stages of wound healing is minimal. Stimulation of fibroblasts and endothelial cells in vitro with PGRN causes proliferation and migration, suggesting that injury-induced expression of PGRN may have a paracrine effect. Supporting this hypothesis, PGRN was shown to have the same properties as known stimulators of neovascularization, such as vascular endothelial growth factor, in a cell culture model [12].
PGRN and inflammation
The wound repair response also sheds light on the roles of PGRN and GRNs during inflammation. Zhu and coworkers demonstrated an immunoregulatory role of PGRN and GRN peptides during wound healing and highlighted novel interactions between PGRN, secretory leukocyte protease inhibitor (SLPI) and elastase. SLPI is a 14-kDa protein encoded by a gene on chromosome 20 in a genomic region that has several genes with protease inhibitor domains [15] that has been implicated in regulating proteolysis. It is produced by macrophages and neutrophils and is known to inhibit the inflammatory response of these cells to various agents [16]. Elastase, a serine proteinase released in large quantities by neutrophils during inflammation, acts on PGRN to generate GRN peptides by cleaving short linker regions between the different GRN domains [14]. Interestingly, SLPI can inhibit this process by binding directly to the linker regions on PGRN as well as to elastase, thereby acting as a potent regulator of PGRN proteolytic processing.
The role of elastase and SLPI in regulating inflammation can be recognized by the contrasting effects of PGRN and GRN on epithelial cells and neutrophils. In cell culture, GRN(B) stimulates epithelial cells to secrete interleukin-8 (IL-8), a major chemoattractant for neutrophils and monocytes, whereas PGRN has no such effect. In response to proinflammatory cytokines, such as tumor necrosis factor alpha (TNFα), neutrophils adhere, spread, undergo degranulation and release a range of reactive species (including elastase) during respiratory burst. PGRN inhibits spreading, degranulation and respiratory burst of TNFα-activated neutrophils. In contrast, GRN (A) and GRN (B) peptides have no inhibitory effect. These observations implicate pro-inflammatory and anti-inflammatory roles for GRN and PGRN, respectively.
Although PGRN can inhibit TNFα-mediated neutrophil activation, it has no effect on neutrophils already undergoing a respiratory burst. This phenomena has also been seen when using reagents that disrupt signaling events that lead to the respiratory burst [17,18], suggesting PGRN somehow changes intracellular signaling events after TNFα binds to one of the TNF receptors [14]. In light of this, it is not surprising that wounds in SLPI deficient mice have increased leukocyte infiltration and elevated elastase activity, as well as impaired wound healing [19]. Addition of recombinant SLPI or PGRN to these animals normalizes wound response [14], suggesting that PGRN was being converted into GRNs as a result of unregulated proteolysis by elastase in wounds of SLPI-deficient mice. Deficiency in SLPI has the potential to disrupt the ratio of PGRN to GRNs resulting in excess GRNs and a net pro-inflammatory response. Collectively, these results suggest that activities of elastase (generating GRNs) and SLPI (inhibiting PGRN cleavage) are biological regulators of the innate immune response during wound healing.
Cytokines and hormones act as regulators of PGRN expression. Proinflammatory cytokines of the innate immune system, interleukin1 beta (IL-1β) and TNFα, activate PGRN gene expression in murine embryo fibroblasts [20]. The promoter for both human [21] and murine [22]PGRN contain regulatory elements that are involved in cytokine and growth-factor – regulated gene expression, including IL-6 response factor [21]. There is also increased expression of PGRN in inflammatory and immune disorders, such as rheumatoid arthritis [23], a zebra fish model of chronic tuberculosis [24] and a murine model simulating chemokine-induced alveolar monocyte trafficking [25]. PGRN is preferentially associated with cells of the innate immune system, including macrophages and neutrophils. IL-4, which is an anti-inflammatory cytokine of the adaptive immune system, decreases PGRN expression in certain myeloid cells [11]. Together these studies suggest a pleiotropic role for PGRN during inflammation in peripheral tissues.
PGRN in the CNS
Gene expression in the CNS
Few studies have investigated the expression and function of PGRN in the CNS. Initial studies analyzing brain homogenates by northern blot analysis reported relatively low levels of expression of PGRN mRNA in the brain [7]. Using in situ hybridization techniques in adult rodent brains, PGRN mRNA was found to be abundant in specific neuronal subsets, including cortical pyramidal neurons, cerebellar Purkinje cells and pyramidal neurons of the hippocampus [6]. Immunohistochemical studies have also shown expression of PGRN in certain neuronal populations (Figure 2) [26,27]. The subcellular location of PGRN in neurons is currently unknown, but preliminary studies suggest that it may be associated with endosomal or lysosomal vacuoles.
Figure 2.

Immunohistochemical staining of human brain tissue using a PGRN-specific polyclonal antibody. In a neurologically normal individual, PGRN immunoreactivity is present in hippocampal pyramidal neurons, but particularly high in CA1 (A), dentate fascia (B) and endplate/CA4 (C). In AD, neurons and activated microglia (arrows) in the endplate are labeled (D), along with PGRN immunoreactivity associated with dystrophic neurites in senile plaques (inset).
During CNS development [2]PGRN expression is high in neuroepithelial cells in the embryo and then decreases in fetal development, where it is restricted to the forebrain, olfactory lobes, retinal ganglion and spinal cord. Later, PGRN is expressed throughout the neocortex, but not in regions where neurogenesis is known to occur, such as the subventricular zone [28]. The differential expression of PGRN in specific cell types during forebrain development suggests a role in the developing CNS.
Neurotrophic properties of PGRN
PGRN promotes growth of PC12 cells, a pheochromocytoma-derived neuronal cell line that responds poorly to most nerve growth factors [6]. The only other growth factors shown to have an effect on PC12 cells similar to PGRN are insulin growth factors-1 and 2 (IGF-1 and IGF-2). In embryonic fibroblasts PGRN activates similar signal transduction pathways as IGF-1 and IGF-2. [29]. Although these results suggest a neurotrophic role for PGRN, there are subtle, but potentially important, differences between PGRN and other growth factors. For example, in a blunt-force traumatic brain injury model in mice, most growth factors, such as neuregulin and brain derived neurotrophic factor, show robust increases in as few as three hours. In the same model, increases in PGRN mRNA do not occur until 24 hours, by which time the expression of the other growth factors have returned to normal [30]. The delayed induction of PGRN and its potential roles in normal and pathological conditions requires further investigation, but the available evidence suggests that PGRN may be important in long-term neuronal survival, but not a significant factor in response to acute neuronal injuries.
Microglial expression of PGRN
Non-neuronal cell types also show evidence of PGRN expression in the CNS. Although initial in situ hybridization studies of PGRN expression did not detect any signal in glial cells [6], more recent immunohistochemical studies have shown strong immunoreactivity in microglia [26,27,31] (Figure 2), especially when activated. Microglia are intrinsic CNS glial cells derived from the mononuclear phagocyte system [32,33], which fits with the fact that PGRN is a protein produced by hematopoietic cell types [6,7,11]. In contrast, astrocytes and oligodendroglia, which are derived from the neural tube, have no or very low levels of PGRN immunoreactivity [27].
Microglia represent about 5–20% of all CNS glia [32,33]. Current evidence suggests that microglia are derived from monocytes that enter the developing brain during embryogenesis, after which they differentiate into resident microglia [34]. There is little turnover of resident microglia, but more so for perivascular macrophages [35]. Under normal conditions microglia are quiescent and characterized morphologically by ramified processes and a small soma. In response to pathologic insults, such as traumatic injury, infection or neurodegeneration, microglia become activated. In the activated state microglia have proliferative potential, and they undergo migration and phagocytosis [36,37]. During injury-induced microglial activation, increases in microglia occurs through both mitosis of resident microglia [38] and migration of bone-marrow derived cells into the CNS [39-41].
Microglial activation is associated with changes in shape and functional properties, including increases in cell surface molecules (e.g., HLA-DR and β2-integrins) and production of proinflammatory cytokines (e.g., IL-1β and TNFα), chemokines, growth factors and inflammatory mediators (e.g., platelet activation factor) [42]. In most conditions, microglial activation is accompanied by reactive astrocytic gliosis [43]. Together these features are the face of neuroinflammation, the major response of the innate immune system in the CNS.
In the context of neurodegenerative diseases, sustained microglial activation has been linked to neuronal injury and loss, in part mediated by excessive production of proinflammatory cytokines and other toxic species [44-46]. According to this theory, neuronal injury occurs through a "by-stander" mechanism [47]. Considering the role of PGRN during inflammation in the periphery, along with the mechanism by which microglia respond, tight regulation of PGRN expression in the CNS would seem important. A model of ischemic stroke in rats [48] provides a theoretical means by which PGRN may be regulated during CNS inflammation (Figure 3). Microglia and astrocytes both respond to hypoxic-ischemic neuronal injury with abundant cross-talk between these cell populations. For example, IL-1β is produced by activated microglia and is a major activator of astrocytes [49]. Conversely, colony stimulating factor 1 (CSF-1) and granulocyte-macrophage colony stimulating factor are potent microglial growth factors that are produced by activated astrocytes [50,51]. Interestingly, astrocytes also express SLPI, the elastase- and PGRN-binding protein implicated as a regulator of PGRN proteolysis in the periphery [14], during ischemic stroke where it reduces ischemic-induced injury [48]. It remains to be shown whether SLPI regulates PGRN proteolysis in the CNS. As noted above, in the periphery the relative balance between the activities of elastase and SLPI influences the levels of the anti-inflammatory PGRN and the pro-inflammatory GRNs. It is worth noting that cultured microglia have been shown to produce elastase [52], which is also thought to be a key player in this inflammatory switch mechanism. Environmental or genetic factors that may affect the normal regulation of PGRN could have adverse consequences leading to neuroinflammation and neurodegeneration.
Figure 3.

Hypothetical interaction between PGRN, elastase and SLPI in the CNS. 1) PGRN expression in neurons in the absence of microglial-derived elastase has potentially growth factor and anti-inflammatory properties. 2) In response to CNS injury, microglia are activated and release inflammatory mediators, including proteases. 3) PGRN and elastase levels increase, resulting in the proteolytic cleavage of PGRN into GRNs, which may contribute to the inflammatory milieu. 4) Inflammatory signals such as IL-1β derived from activated microglia cause changes in nearby cells. 5) Astrocytes become reactive in response to inflammatory stimulus from activated microglia. 6) Reactive astrocytes produce SLPI, which along with its other anti-inflammatory properties, inhibits the proteolytic cleavage of PGRN into GRN as a means of feedback regulation of the inflammatory response.
Although the neurotoxic potential of chronically activated microglia has been the focus of most studies, a growing body of research suggests that microglia may play a neuroprotective role, as well [53]. This has prompted some researchers to suggest that the loss of normal physiologic functions of microglia may contribute to neurodegeneration. Since PGRN has been shown to have trophic functions in the periphery, and it is expressed by activated microglia, it is intriguing to speculate that microglia-derived PGRN may support neuronal viability or possibly perform a role equivalent to wound healing in the periphery mediated by its neurotrophic activity. There are several lines of evidence that lend support to this hypothesis. First, microglia have the ability to produce neurotrophic factors, such as thrombospondin [53,54]. Second, microglia produce other growth factors, such as TGF-β, during injury and repair [55,56]. Third, as mentioned previously, PGRN has growth factor properties on neuronal cells in culture [6]. Obviously, much remains to be learned about potential neurotrophic properties of microglia-derived PGRN as it relates to neurodegeneration.
PGRN in CNS disorders and animal models of CNS disease
Much of the available information on PGRN expression in various disorders comes from unbiased expression array studies in which PGRN was shown to be one of the responsive genes increased compared to controls. Interestingly, almost all the mRNA expression studies that have shown differential expression of PGRN share the common property of microglial activation and inflammation, leading some researchers to speculate that the increase in PGRN expression is closely related to microglial activation and neuroinflammation [1].
PGRN in models of CNS viral infection
PGRN has been shown to be increased in young mice during the host response to two different strains of Sindbis virus with varying neurovirulence [57]. Virus replication, viral burden and evidence of apoptosis were greater with the more virulent strain, even though neuronal cell tropism was the same. Histologic evidence of inflammation was mild, but the gene expression profile highlighted differences between virulent and non-virulent strains. In particular, a number of chemokine genes, as well as PGRN, that are up-regulated in microglia during inflammation were increased during infection by the virulent strain. Given that both microglia and neurons expressed PGRN, it is not possible to know the cellular origin of the PGRN in this and other models.
PGRN in models of Creutzfeldt-Jakob disease (CJD)
Studies of the gene expression profile of microglial cell cultures taken from mice infected with CJD (M-CJD) have provided some information on the microglial expression of PGRN in response to different activating stimuli [58]. The pathogenic prion protein has been shown to accumulate in activated microglia, although the mechanism for this remains to be determined [59]. Proinflammatory transcripts, such as IL-1β and complement factors, as well as PGRN were increased in M-CJD. When normal microglia were challenged with endotoxin (LPS) and interferon-gamma (IFNγ), to mimic the expression profile of activated microglia, PGRN expression was substantially suppressed compared to that seen in M-CJD. In addition, both IL1β and TNFα were highly expressed in M-CJD and LPS treated mice, but PGRN was only increased in MCJD. This paradoxical response of PGRN may relate to the pleiotropic functional properties of PGRN not expected with more traditional growth factors; PGRN may function as a growth factor or anti-inflammatory agent as an intact molecule, but as a source of diverse inflammatory mediators, when it undergoes proteolysis to GRNs. The only other transcripts that had an expression pattern similar to PGRN were LY5, leukocytes common antigen and CD84, which are involved in inflammation and intracellular communication [58]. These particular studies suggest that microglial expression of PGRN increases during neuroinflammation in neurodegenerative disease, but not during simple microglial induction through LPS treatment. As a result, fundamental questions need to be addressed concerning the role of PGRN in microglial function. In particular, it is not known if PGRN is differentially expressed by intrinsic microglia or by bone-marrow derived macrophages.
PGRN in models of lysosomal storage disease
Mucopolysaccharidoses (MPS) type I and type IIIB are lysosomal storage diseases that affect the CNS. Mouse models have suggested that activated microglia accelerate the neuronal degeneration caused by lysosomal storage [60]. Microarray gene analysis of cortical brain tissue from these mouse models showed a prominent inflammatory pattern of gene expression, which was accompanied by increases in PGRN [61]. They also demonstrated that microglia in these mice contain massive lysosomal vacuoles. It remains to be determined if the increase in PGRN in these models is due to neuronal degeneration or to microglial activation in response to abnormal lysosomal storage.
PGRN in ALS
PGRN mRNA expression in ALS spinal cord has been found to be increased by 400% compared with controls [62]. This is likely related to microglial activation, which has been implicated in the pathogenesis of ALS [63]. As discussed later, the neuronal cytoplasmic inclusions in motor neurons of ALS are composed of the same protein (TDP-43) as those in FTLD-U [64,65], which suggests a fundamental linkage between ALS and FTLD.
PGRN in Alzheimer's disease (AD)
Several recent studies have shown PGRN immunoreactivity in AD is associated with amyloid plaques [31] (Figure 2), including labeling of microglia and dystrophic neurites [27]. Plaque-related dystrophic neurites are large axonal varicosities that are associated with reduced dendritic spine density and shaft diameter [66]. Considering that PGRN expression is increased during injury and repair in the periphery, the presence of PGRN immunoreactivity in dystrophic neurites could reflect a reparative response in damaged axons. Complementing this hypothesis is the observation that dystrophic neurites are constantly being formed and resolved; however, the rate of dystrophic neurite formation exceeds their resolution [66]. Considering the growth factor properties of PGRN in the periphery, it is tempting to speculate that PGRN may be involved in neuritic remodeling.
Most studies have shown PGRN to be present in the perikarya of neurons, but it is unknown whether PGRN is also normally present in axons or dendrites. If PGRN is normally present in axons, then the accumulation of PGRN in dystrophic neurites may also reflect disruption of axonal transport, similar to what is found for other axonally transported proteins such as amyloid precursor protein [67].
PGRN in FTLD-U
Mutations in PGRN cause FTLD-U
Recent interest in PGRN has been fueled by the discovery of mutations in PGRN in some families with autosomal dominant FTLD-U [27,31,68-70]. Prior to this discovery, no mutations in PGRN had been associated with any human disorder [1]. FTLD-U is a member of a diverse group of neurodegenerative disorders that produce frontotemporal dementia, which accounts for 5–15% of all dementia disorders [71]. The clinical phenotype in patients with PGRN mutations is similar to those with sporadic frontotemporal dementia. They have a variable age of onset, and the dementia tends to be characterized by prominent behavioral and language dysfunction, usually a progressive non-fluent aphasia [72]. Mild parkinsonism is common, but motor neuron disease is usually absent [26,73].
Mutations in the gene for the microtubule associated protein tau (MAPT) on chromosome 17q21, which incidentally is in close proximity to the PGRN gene, are known to be responsible for 10–20% of familial frontotemporal dementia [74]. These cases are pathologically characterized by the abnormal accumulation of hyperphosphorylated tau protein in neurons and glia, which is distinct from the FTLD-U pathology in PGRN-related frontotemporal dementia. To date no MAPT mutations have been detected in FTLD-U [75].
A recent study reported the frequency of PGRN mutations in FTLD to be 5% in a patient referral series, which is a similar frequency to that of MAPT mutations in the same series of patients [69]. At least 35 different pathogenic PGRN mutations have now been identified, all of which are predicted to create functional null alleles with the majority causing premature termination of the coding sequence [69]. The introduction of a premature termination codon results in degradation of the mutant mRNA species by nonsense mediated decay [31]. As a result, there is no production of mutant protein. It therefore appears that PGRN mutations cause FTLD-U due to a partial loss of functional PGRN, (haploinsufficiency), rather than accumulation of mutant protein characteristic of frontotemporal dementia due to MAPT mutations.
Several other genes or chromosomal loci have been identified for FTLD-U, including mutations in the gene for valosin-containing protein (VCP) [76] and CHMP2b (charged multivesicular body protein 2b) [77]. VCP is an endoplasmic reticulum-associated protein that is involved in ER-stress related protein degradation [78,79]. While little is known about CHMP2b, it appears to play a role in endosomal trafficking through the ESCRT (endosomal secretory complex required for trafficking) III complex, which may be involved in degradation of growth factors [80]. Given the preliminary evidence that PGRN is a neurotrophic factor that may be associated with endosomal or lysosomal vacuoles, it raises the intriguing possibility that the three major genes for FTLD-U are associated with defects in protein degradation linked to membranous cytoplasmic organelles.
FTLD-U is the most common pathology associated with frontotemporal dementia and is characterized by focal cortical atrophy with spongiosis, gliosis and ubiquitin-immunoreactive neuronal cytoplasmic inclusions (NCI) and neurites in layer II of affected cortices and in the hippocampal dentate fascia [81] (Figure 4). Most cases also have neuronal loss in the hippocampus consistent with hippocampal sclerosis [82]. In postmortem series, pathology was more severe and associated with marked neuronal loss and gliosis in cases with PGRN mutations compared to those without mutations [73]. The presence of lentiform shaped neuronal intranuclear inclusions (NII) is a consistent feature of cases with PGRN mutations, but is not entirely specific [83,84] (Figure 4).
Figure 4.

Neuropathology of FTLD-U with PGRN mutations. Gross cortical atrophy is visible in frontal and superior temporal lobes (A). In coronal sections (B), the lateral ventricle is dilated and the caudate nucleus is flat (arrow). Laminar spongiosis in the layer II of the cortical ribbon (C) is associated with TDP-43-positive neuronal cytoplasmic (D, arrows) and "lentiform" intranuclear inclusions (inset). Severe neuronal loss (D) in these regions is associated with microgliosis (E) and astrogliosis (F), shown by a microglial marker [ionized calcium-binding adapter molecule 1 (Iba-1)) and glial fibrillary acidic protein (GFAP) specific immunohistochemistry, respectively.
As in any neurodegenerative disorder neuronal loss in FTLD-U is accompanied by reactive astrogliosis and microglial activation (Figure 4). There are no studies of microglial functional properties in FTLD-U cases with and without PGRN mutations. A closer look at the inflammatory response in cases with and without the PGRN mutation would also be helpful in understanding the biological role of PGRN in FTLD-U.
Neuronal inclusions in FTLD-U contain TDP-43
Consistent with the haploinsufficiency mechanism, NCI and NII in familial FTLD-U with PGRN mutations do not contain mutant PGRN [26]. A major component of the inclusion bodies was recently shown to be TAR DNA binding protein-43 (TDP-43) [64,65] (Figure 4). The same protein was also found to be present in the neuronal inclusions in ALS. Very little is known about the biological function of TDP-43. Originally identified as a ubiquitously expressed 43-kDa protein, TDP-43 binds to the TAR DNA in the long terminal repeat region of human immunodeficiency virus (HIV)-1, resulting in repression of promoter activity [85]. It was also independently identified as a regulator of alternative splicing of exon 9 of the cystic fibrosis transmembrane conductance regulator transcript, through its ability to bind (UG)n-repeated RNA sequences [86]. It is thought that these DNA and RNA binding properties implicate TDP-43 as a transcription regulator through one of its two RNA-recognition motifs [85,87]. TDP-43 is widely expressed in many tissues, including the brain [85], where immunohistochemistry highlights diffuse, but grainy expression of nuclei of neurons and other CNS cells. In FTLD-U, the normal nuclear staining pattern is absent in neurons that contain NCI and NII, leading investigators to suggest that TDP-43 is translocated from the nucleus to the cytoplasm [64] or that TDP-43 is prevented from crossing the nuclear membrane possibly, as a result of hyperphosphorylation of TD-43 [88]. This abnormal metabolism of TDP-43 in the FTLD-U seems central to the disease pathogenesis given that the gene encoding TDP-43 is highly conserved among different species [89] consistent with an essential, yet currently unknown biological function.
TDP-43 and PGRN
The relationship between PGRN and TDP-43 and their respective roles in neurodegeneration is currently unknown. Although most of the biological considerations in this review have focused on PGRN as a secreted protein; there is data suggesting an intracellular and possibly even an intra-nuclear role for PGRN. While most growth factors function through binding to cell surface receptors with subsequent intracellular signaling, there is recent evidence to suggest that some growth factors or inflammatory mediators may cross the cell membrane through currently poorly defined means to gain access to the cytosol and even the nucleus (reviewed in [90]). It is intriguing to speculate that PGRN may be involved in intracellular trafficking. There is little experimental evidence to support this hypothesis at present, but studies of transcriptional regulation hint at a possible link between PGRN and TDP-43. In these studies intracellular trafficking of certain proteins has been shown to be modulated differentially by PGRN and GRN. PGRN and one of the GRN peptides, CDE, have the ability to bind cyclin T1 [91], an essential protein component of the positive transcription elongation factor needed to phosphorylate the largest subunit of RNA polymerase II, resulting in transcriptional elongation (reviewed by [92]). When PGRN and cyclin T1 are co-expressed in the same cell, both are restricted to cytoplasm, thereby inhibiting transcription elongation [91]. In contrast, when co-expressed with GRN (CDE), both proteins are localized mainly in the nucleus. Given these results and the abnormal location of TDP-43 in the cytoplasm rather than the nucleus in FTLDU, it raises the possibility that PGRN might be involved in the trafficking of TDP-43. Interestingly, PGRN is known to bind HIV-1 and HIV-2 tat proteins [91,93]. Tat proteins associate with TAR DNA, a property that is also shared by TDP-43 [85]. If TDP-43 has PGRN-binding properties, coupled with what is known about the effects of PGRN and GRN on nuclear-cytoplasmic trafficking of certain proteins, one can envisage how dysfunction or dysregulation of PGRN might contribute to abnormal compartmentalization of TDP-43 (Figure 5).
Figure 5.
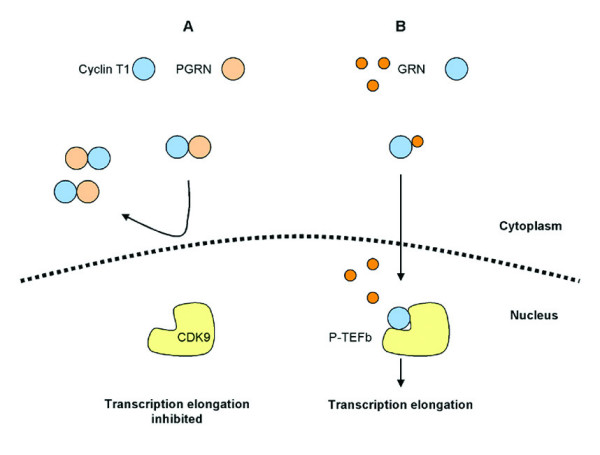
Schematic model of nuclear trafficking of cyclin T1 and it alterations by PGRN and GRN. Cyclin T1 binds PGRN and some of its GRN derivatives when co-expressed in COS7 cells [91]. A) When cyclin T1 is expressed with full length PGRN, both proteins are localized in the cytoplasm. B) In contrast, when expressed with the C-terminal GRN, CDE, cyclin T1 and GRN are found in the nucleus, enabling down-stream transcription elongation. Given the evidence that PGRN and GRN are associated with translocation of proteins such as cyclin T1 from cytoplasm to nucleus, it is tempting to hypothesize that PGRN might be involved in similar regulation of TDP-43 nuclear-cytoplasmic translocation. With decreased functional PGRN in FTLD-U associated with PGRN mutations, TDP-43 translocation might be perturbed, leading to accumulation in the cytoplasm and formation of NCI.
Conclusion
PGRN is a complex protein that has distinct functional properties as an intact precursor protein compared to GRN peptides derived from its proteolytic cleavage. In peripheral tissues, it has been implicated in development, maintenance, repair, inflammation and neoplasia. The interactions of elastase, SLPI and PGRN leads to speculation that PGRN may have pro- or anti-inflammatory properties depending upon the extent of regulated proteolysis of PGRN and generation of pro-inflammatory GRN peptides.
PGRN is expressed in the developing CNS, where its growth promoting function has been suggested. In the mature CNS immunohistochemical and in situ hybridization studies have shown that certain populations of neurons express PGRN. There is evidence that full-length PGRN may function as a neuronal growth factor. In this light, pathogenic PGRN mutations that lead to decreases in functional PGRN may produce neurodegeneration in FTLDU as a result of decreases in neurotrophic activity.
Microglia are the other major cell type that expresses PGRN in the CNS. Trauma, infection and neurodegeneration are all accompanied by increases in PGRN mRNA expression. These results are consistent with the notion that PGRN expression is directly or indirectly related to microglial proliferation and activation, implicating PGRN in neuroinflammation and potentially brain repair. The role of altered PGRN in microglia in this regard needs further investigation since some studies have suggested trophic rather than toxic functions of microglia in specific circumstances. Studies of microglia and the inflammatory responses in PGRN mutation carriers and controls, as well as in PGRN knock out mice will be useful in determining not only the normal function of PGRN in the CNS, but also its role in the pathogenesis of FTLDU.
Competing interests
The author(s) declare that they have no competing interests.
Authors' contributions
ZA wrote the initial draft and produced the hypothetical mechanisms and pathological images. Modification suggested by IM, MH and DD were applied by ZA and DD to the final draft. MH and IM made particular contributions to the sections on the genetics and pathology of FTLD-U, respectively. All authors read and approved the final version.
Acknowledgments
Acknowledgements
Z.A. is a Doctoral Candidate for the Department of Neuroscience, King's College London, Institute of Psychiatry, De Crespigny Park, London, SE5 8AF, UK. The authors acknowledge the valuable histologic support of Monica Casey-Castanedes, Virginia Phillips and Linda Rousseau. Supported by NIH R01-AG20216; P50-AG16574, P50-AG25711, P50-NS40256, P01-AG17216, P01-AG03949.
Contributor Information
Zeshan Ahmed, Email: ahmed.zeshan@mayo.edu.
Ian RA Mackenzie, Email: ian.mackenzie@vch.ca.
Michael L Hutton, Email: hutton.michael@mayo.edu.
Dennis W Dickson, Email: dickson.dennis@mayo.edu.
References
- He Z, Bateman A. Progranulin (granulin-epithelin precursor, PC-cell-derived growth factor, acrogranin) mediates tissue repair and tumorigenesis. J Mol Med. 2003;81:600–612. doi: 10.1007/s00109-003-0474-3. [DOI] [PubMed] [Google Scholar]
- Daniel R, Daniels E, He Z, Bateman A. Progranulin (acrogranin/PC cell-derived growth factor/granulin-epithelin precursor) is expressed in the placenta, epidermis, microvasculature, and brain during murine development. Dev Dyn. 2003;227:593–599. doi: 10.1002/dvdy.10341. [DOI] [PubMed] [Google Scholar]
- Bhandari V, Palfree RG, Bateman A. Isolation and sequence of the granulin precursor cDNA from human bone marrow reveals tandem cysteine-rich granulin domains. Proc Natl Acad Sci USA. 1992;89:1715–1719. doi: 10.1073/pnas.89.5.1715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avrova AO, Stewart HE, De Jong WD, Heilbronn J, Lyon GD, Birch PR. A cysteine protease gene is expressed early in resistant potato interactions with Phytophthora infestans. Mol Plant Microbe Interact. 1999;12:1114–1119. doi: 10.1094/MPMI.1999.12.12.1114. [DOI] [PubMed] [Google Scholar]
- Parnell PG, Wunderlich J, Carter B, Halper J. Transforming growth factor e: amino acid analysis and partial amino acid sequence. Growth Factors. 1992;7:65–72. doi: 10.3109/08977199209023938. [DOI] [PubMed] [Google Scholar]
- Daniel R, He Z, Carmichael KP, Halper J, Bateman A. Cellular localization of gene expression for progranulin. J Histochem Cytochem. 2000;48:999–1009. doi: 10.1177/002215540004800713. [DOI] [PubMed] [Google Scholar]
- Bhandari V, Giaid A, Bateman A. The complementary deoxyribonucleic acid sequence, tissue distribution, and cellular localization of the rat granulin precursor. Endocrinology. 1993;133:2682–2689. doi: 10.1210/en.133.6.2682. [DOI] [PubMed] [Google Scholar]
- Shoyab M, McDonald VL, Byles C, Todaro GJ, Plowman GD. Epithelins 1 and 2: isolation and characterization of two cysteine-rich growth-modulating proteins. Proc Natl Acad Sci USA. 1990;87:7912–7916. doi: 10.1073/pnas.87.20.7912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bateman A, Belcourt D, Bennett H, Lazure C, Solomon S. Granulins, a novel class of peptide from leukocytes. Biochem Biophys Res Commun. 1990;173:1161–1168. doi: 10.1016/S0006-291X(05)80908-8. [DOI] [PubMed] [Google Scholar]
- Chantry D, DeMaggio AJ, Brammer H, Raport CJ, Wood CL, Schweickart VL, Epp A, Smith A, Stine JT, Walton K, Tjoelker L, Godiska R, Gray PW. Profile of human macrophage transcripts: insights into macrophage biology and identification of novel chemokines. J Leukoc Biol. 1998;64:49–54. doi: 10.1002/jlb.64.1.49. [DOI] [PubMed] [Google Scholar]
- Ong CH, He Z, Kriazhev L, Shan X, Palfree RG, Bateman A. Regulation of progranulin expression in myeloid cells. Am J Physiol Regul Integr Comp Physiol. 2006;291:R1602–1612. doi: 10.1152/ajpregu.00616.2005. [DOI] [PubMed] [Google Scholar]
- He Z, Ong CH, Halper J, Bateman A. Progranulin is a mediator of the wound response. Nat Med. 2003;9:225–229. doi: 10.1038/nm816. [DOI] [PubMed] [Google Scholar]
- Singer AJ, Clark RA. Cutaneous wound healing. N Engl J Med. 1999;341:738–746. doi: 10.1056/NEJM199909023411006. [DOI] [PubMed] [Google Scholar]
- Zhu J, Nathan C, Jin W, Sim D, Ashcroft GS, Wahl SM, Lacomis L, Erdjument-Bromage H, Tempst P, Wright CD, Ding A. Conversion of proepithelin to epithelins: roles of SLPI and elastase in host defense and wound repair. Cell. 2002;111:867–878. doi: 10.1016/S0092-8674(02)01141-8. [DOI] [PubMed] [Google Scholar]
- Clauss A, Lilja H, Lundwall A. A locus on human chromosome 20 contains several genes expressing protease inhibitor domains with homology to whey acidic protein. Biochem J. 2002;368:233–242. doi: 10.1042/BJ20020869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin FY, Nathan C, Radzioch D, Ding A. Secretory leukocyte protease inhibitor: a macrophage product induced by and antagonistic to bacterial lipopolysaccharide. Cell. 1997;88:417–426. doi: 10.1016/S0092-8674(00)81880-2. [DOI] [PubMed] [Google Scholar]
- Nathan CF. Respiratory burst in adherent human neutrophils: triggering by colony-stimulating factors CSF-GM and CSF-G. Blood. 1989;73:301–306. [PubMed] [Google Scholar]
- Nathan CF. Neutrophil activation on biological surfaces. Massive secretion of hydrogen peroxide in response to products of macrophages and lymphocytes. J Clin Invest. 1987;80:1550–1560. doi: 10.1172/JCI113241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashcroft GS, Lei K, Jin W, Longenecker G, Kulkarni AB, Greenwell-Wild T, Hale-Donze H, McGrady G, Song XY, Wahl SM. Secretory leukocyte protease inhibitor mediates non-redundant functions necessary for normal wound healing. Nat Med. 2000;6:1147–1153. doi: 10.1038/80489. [DOI] [PubMed] [Google Scholar]
- Li X, Massa PE, Hanidu A, Peet GW, Aro P, Savitt A, Mische S, Li J, Marcu KB. IKKalpha, IKKbeta, and NEMO/IKKgamma are each required for the NF-kappa B-mediated inflammatory response program. J Biol Chem. 2002;277:45129–45140. doi: 10.1074/jbc.M205165200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhandari V, Daniel R, Lim PS, Bateman A. Structural and functional analysis of a promoter of the human granulin/epithelin gene. Biochem J. 1996;319:441–447. doi: 10.1042/bj3190441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baba T, Nemoto H, Watanabe K, Arai Y, Gerton GL. Exon/intron organization of the gene encoding the mouse epithelin/granulin precursor (acrogranin) FEBS Lett. 1993;322:89–94. doi: 10.1016/0014-5793(93)81544-A. [DOI] [PubMed] [Google Scholar]
- Justen HP, Grunewald E, Totzke G, Gouni-Berthold I, Sachinidis A, Wessinghage D, Vetter H, Schulze-Osthoff K, Ko Y. Differential gene expression in synovium of rheumatoid arthritis and osteoarthritis. Mol Cell Biol Res Commun. 2000;3:165–172. doi: 10.1006/mcbr.2000.0211. [DOI] [PubMed] [Google Scholar]
- Meijer AH, Verbeek FJ, Salas-Vidal E, Corredor-Adamez M, Bussman J, van der Sar AM, Otto GW, Geisler R, Spaink HP. Transcriptome profiling of adult zebrafish at the late stage of chronic tuberculosis due to Mycobacterium marinum infection. Mol Immunol. 2005;42:1185–1203. doi: 10.1016/j.molimm.2004.11.014. [DOI] [PubMed] [Google Scholar]
- Srivastava M, Jung S, Wilhelm J, Fink L, Buhling F, Welte T, Bohle RM, Seeger W, Lohmeyer J, Maus UA. The inflammatory versus constitutive trafficking of mononuclear phagocytes into the alveolar space of mice is associated with drastic changes in their gene expression profiles. J Immunol. 2005;175:1884–1893. doi: 10.4049/jimmunol.175.3.1884. [DOI] [PubMed] [Google Scholar]
- Mackenzie IR, Baker M, Pickering-Brown S, Hsiung GY, Lindholm C, Dwosh E, Gass J, Cannon A, Rademakers R, Hutton M, Feldman HH. The neuropathology of frontotemporal lobar degeneration caused by mutations in the progranulin gene. Brain. 2006;129:3081–3090. doi: 10.1093/brain/awl271. [DOI] [PubMed] [Google Scholar]
- Mukherjee O, Pastor P, Cairns NJ, Chakraverty S, Kauwe JS, Shears S, Behrens MI, Budde J, Hinrichs AL, Norton J, Levitch D, Taylor-Reinwald L, Gitcho M, Tu PH, Tenenholz Grinberg L, Liscic RM, Armendariz J, Morris JC, Goate AM. HDDD2 is a familial frontotemporal lobar degeneration with ubiquitin-positive, tau-negative inclusions caused by a missense mutation in the signal peptide of progranulin. Ann Neurol. 2006;60:314–322. doi: 10.1002/ana.20963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Austin CP, Cepko CL. Cellular migration patterns in the developing mouse cerebral cortex. Development. 1990;110:713–732. doi: 10.1242/dev.110.3.713. [DOI] [PubMed] [Google Scholar]
- Nagtegaal ID, Lakke EA, Marani E. Trophic and tropic factors in the development of the central nervous system. Arch Physiol Biochem. 1998;106:161–202. doi: 10.1076/apab.106.3.161.4380. [DOI] [PubMed] [Google Scholar]
- Matzilevich DA, Rall JM, Moore AN, Grill RJ, Dash PK. High-density microarray analysis of hippocampal gene expression following experimental brain injury. J Neurosci Res. 2002;67:646–663. doi: 10.1002/jnr.10157. [DOI] [PubMed] [Google Scholar]
- Baker M, Mackenzie IR, Pickering-Brown SM, Gass J, Rademakers R, Lindholm C, Snowden J, Adamson J, Sadovnick AD, Rollinson S, Cannon A, Dwosh E, Neary D, Melquist S, Richardson A, Dickson D, Berger Z, Eriksen J, Robinson T, Zehr C, Dickey CA, Crook R, McGowan E, Mann D, Boeve B, Feldman H, Hutton M. Mutations in progranulin cause tau-negative frontotemporal dementia linked to chromosome 17. Nature. 2006;442:916–919. doi: 10.1038/nature05016. [DOI] [PubMed] [Google Scholar]
- Jordan FL, Thomas WE. Brain macrophages: questions of origin and interrelationship. Brain Res. 1988;472:165–178. doi: 10.1016/0165-0173(88)90019-7. [DOI] [PubMed] [Google Scholar]
- Cuadros MA, Navascues J. The origin and differentiation of microglial cells during development. Prog Neurobiol. 1998;56:173–189. doi: 10.1016/S0301-0082(98)00035-5. [DOI] [PubMed] [Google Scholar]
- Alliot F, Godin I, Pessac B. Microglia derive from progenitors, originating from the yolk sac, and which proliferate in the brain. Brain Res Dev Brain Res. 1999;117:145–152. doi: 10.1016/S0165-3806(99)00113-3. [DOI] [PubMed] [Google Scholar]
- Hickey WF, Kimura H. Perivascular microglial cells of the CNS are bone marrow-derived and present antigen in vivo. Science. 1988;239:290–292. doi: 10.1126/science.3276004. [DOI] [PubMed] [Google Scholar]
- Kreutzberg GW. Microglia: a sensor for pathological events in the CNS. Trends Neurosci. 1996;19:312–318. doi: 10.1016/0166-2236(96)10049-7. [DOI] [PubMed] [Google Scholar]
- Nakajima K, Kohsaka S. Functional roles of microglia in the brain. Neurosci Res. 1993;17:187–203. doi: 10.1016/0168-0102(93)90047-T. [DOI] [PubMed] [Google Scholar]
- Ladeby R, Wirenfeldt M, Garcia-Ovejero D, Fenger C, Dissing-Olesen L, Dalmau I, Finsen B. Microglial cell population dynamics in the injured adult central nervous system. Brain Res Brain Res Rev. 2005;48:196–206. doi: 10.1016/j.brainresrev.2004.12.009. [DOI] [PubMed] [Google Scholar]
- Simard AR, Rivest S. Bone marrow stem cells have the ability to populate the entire central nervous system into fully differentiated parenchymal microglia. Faseb J. 2004;18:998–1000. doi: 10.1096/fj.04-1517fje. [DOI] [PubMed] [Google Scholar]
- Flugel A, Bradl M, Kreutzberg GW, Graeber MB. Transformation of donor-derived bone marrow precursors into host microglia during autoimmune CNS inflammation and during the retrograde response to axotomy. J Neurosci Res. 2001;66:74–82. doi: 10.1002/jnr.1198. [DOI] [PubMed] [Google Scholar]
- Lassmann H, Schmied M, Vass K, Hickey WF. Bone marrow derived elements and resident microglia in brain inflammation. Glia. 1993;7:19–24. doi: 10.1002/glia.440070106. [DOI] [PubMed] [Google Scholar]
- Lee SC, Dickson DW. Common immune pathway of neural injury in neurodegenerative disorders. In: Gendelman HE, Grant I, Everall IP, Lipton SA, Swindells S, editor. The Neurology of AIDS. Second. London: Oxford University Press; 2005. pp. 85–93. [Google Scholar]
- Town T, Nikolic V, Tan J. The microglial "activation" continuum: from innate to adaptive responses. J Neuroinflammation. 2005;2:24. doi: 10.1186/1742-2094-2-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Streit WJ, Walter SA, Pennell NA. Reactive microgliosis. Prog Neurobiol. 1999;57:563–581. doi: 10.1016/S0301-0082(98)00069-0. [DOI] [PubMed] [Google Scholar]
- Hanisch UK. Microglia as a source and target of cytokines. Glia. 2002;40:140–155. doi: 10.1002/glia.10161. [DOI] [PubMed] [Google Scholar]
- Simard AR, Rivest S. Neuroprotective properties of the innate immune system and bone marrow stem cells in Alzheimer's disease. Mol Psychiatry. 2006;11:327–335. doi: 10.1038/sj.mp.4001809. [DOI] [PubMed] [Google Scholar]
- Streit WJ, Mrak RE, Griffin WS. Microglia and neuroinflammation: a pathological perspective. J Neuroinflammation. 2004;1:14. doi: 10.1186/1742-2094-1-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Li X, Xu L, Zhan Y, Yaish-Ohad S, Erhardt JA, Barone FC, Feuerstein GZ. Up-regulation of secretory leukocyte protease inhibitor (SLPI) in the brain after ischemic stroke: adenoviral expression of SLPI protects brain from ischemic injury. Mol Pharmacol. 2003;64:833–840. doi: 10.1124/mol.64.4.833. [DOI] [PubMed] [Google Scholar]
- Giulian D, Lachman LB. Interleukin-1 stimulation of astroglial proliferation after brain injury. Science. 1985;228:497–499. doi: 10.1126/science.3872478. [DOI] [PubMed] [Google Scholar]
- Thery C, Mallat M. Influence of interleukin-1 and tumor necrosis factor alpha on the growth of microglial cells in primary cultures of mouse cerebral cortex: involvement of colony-stimulating factor 1. Neurosci Lett. 1993;150:195–199. doi: 10.1016/0304-3940(93)90534-R. [DOI] [PubMed] [Google Scholar]
- Lee SC, Liu W, Brosnan CF, Dickson DW. GM-CSF promotes proliferation of human fetal and adult microglia in primary cultures. Glia. 1994;12:309–318. doi: 10.1002/glia.440120407. [DOI] [PubMed] [Google Scholar]
- Nakajima K, Shimojo M, Hamanoue M, Ishiura S, Sugita H, Kohsaka S. Identification of elastase as a secretory protease from cultured rat microglia. J Neurochem. 1992;58:1401–1408. doi: 10.1111/j.1471-4159.1992.tb11356.x. [DOI] [PubMed] [Google Scholar]
- Streit WJ. Microglia as neuroprotective, immunocompetent cells of the CNS. Glia. 2002;40:133–139. doi: 10.1002/glia.10154. [DOI] [PubMed] [Google Scholar]
- Chamak B, Morandi V, Mallat M. Brain macrophages stimulate neurite growth and regeneration by secreting thrombospondin. J Neurosci Res. 1994;38:221–233. doi: 10.1002/jnr.490380213. [DOI] [PubMed] [Google Scholar]
- Lehrmann E, Kiefer R, Christensen T, Toyka KV, Zimmer J, Diemer NH, Hartung HP, Finsen B. Microglia and macrophages are major sources of locally produced transforming growth factor-beta1 after transient middle cerebral artery occlusion in rats. Glia. 1998;24:437–448. doi: 10.1002/(SICI)1098-1136(199812)24:4<437::AID-GLIA9>3.0.CO;2-X. [DOI] [PubMed] [Google Scholar]
- Streit WJ, Semple-Rowland SL, Hurley SD, Miller RC, Popovich PG, Stokes BT. Cytokine mRNA profiles in contused spinal cord and axotomized facial nucleus suggest a beneficial role for inflammation and gliosis. Exp Neurol. 1998;152:74–87. doi: 10.1006/exnr.1998.6835. [DOI] [PubMed] [Google Scholar]
- Johnston C, Jiang W, Chu T, Levine B. Identification of genes involved in the host response to neurovirulent alphavirus infection. J Virol. 2001;75:10431–10445. doi: 10.1128/JVI.75.21.10431-10445.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker CA, Manuelidis L. Unique inflammatory RNA profiles of microglia in Creutzfeldt-Jakob disease. Proc Natl Acad Sci USA. 2003;100:675–679. doi: 10.1073/pnas.0237313100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker CA, Martin D, Manuelidis L. Microglia from Creutzfeldt-Jakob disease-infected brains are infectious and show specific mRNA activation profiles. J Virol. 2002;76:10905–10913. doi: 10.1128/JVI.76.21.10905-10913.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wada R, Tifft CJ, Proia RL. Microglial activation precedes acute neurodegeneration in Sandhoff disease and is suppressed by bone marrow transplantation. Proc Natl Acad Sci USA. 2000;97:10954–10959. doi: 10.1073/pnas.97.20.10954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohmi K, Greenberg DS, Rajavel KS, Ryazantsev S, Li HH, Neufeld EF. Activated microglia in cortex of mouse models of mucopolysaccharidoses I and IIIB. Proc Natl Acad Sci USA. 2003;100:1902–1907. doi: 10.1073/pnas.252784899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malaspina A, Kaushik N, de Belleroche J. Differential expression of 14 genes in amyotrophic lateral sclerosis spinal cord detected using gridded cDNA arrays. J Neurochem. 2001;77:132–145. doi: 10.1046/j.1471-4159.2001.t01-1-00231.x. [DOI] [PubMed] [Google Scholar]
- Hall ED, Oostveen JA, Gurney ME. Relationship of microglial and astrocytic activation to disease onset and progression in a transgenic model of familial ALS. Glia. 1998;23:249–256. doi: 10.1002/(SICI)1098-1136(199807)23:3<249::AID-GLIA7>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- Neumann M, Sampathu DM, Kwong LK, Truax AC, Micsenyi MC, Chou TT, Bruce J, Schuck T, Grossman M, Clark CM, McCluskey LF, Miller BL, Masliah E, Mackenzie IR, Feldman H, Feiden W, Kretzschmar HA, Trojanowski JQ, Lee VM. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science. 2006;314:130–133. doi: 10.1126/science.1134108. [DOI] [PubMed] [Google Scholar]
- Arai T, Hasegawa M, Akiyama H, Ikeda K, Nonaka T, Mori H, Mann D, Tsuchiya K, Yoshida M, Hashizume Y, Oda T. TDP-43 is a component of ubiquitin-positive tau-negative inclusions in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Biochem Biophys Res Commun. 2006;351:602–611. doi: 10.1016/j.bbrc.2006.10.093. [DOI] [PubMed] [Google Scholar]
- Tsai J, Grutzendler J, Duff K, Gan WB. Fibrillar amyloid deposition leads to local synaptic abnormalities and breakage of neuronal branches. Nat Neurosci. 2004;7:1181–1183. doi: 10.1038/nn1335. [DOI] [PubMed] [Google Scholar]
- Suzuki T, Araki Y, Yamamoto T, Nakaya T. Trafficking of Alzheimer's disease-related membrane proteins and its participation in disease pathogenesis. J Biochem (Tokyo) 2006;139:949–955. doi: 10.1093/jb/mvj121. [DOI] [PubMed] [Google Scholar]
- Cruts M, Gijselinck I, van der Zee J, Engelborghs S, Wils H, Pirici D, Rademakers R, Vandenberghe R, Dermaut B, Martin JJ, van Duijn C, Peeters K, Sciot R, Santens P, De Pooter T, Mattheijssens M, Van den Broeck M, Cuijt I, Vennekens K, De Deyn PP, Kumar-Singh S, Van Broeckhoven C. Null mutations in progranulin cause ubiquitin-positive frontotemporal dementia linked to chromosome 17q21. Nature. 2006;442:920–924. doi: 10.1038/nature05017. [DOI] [PubMed] [Google Scholar]
- Gass J, Cannon A, Mackenzie IR, Boeve B, Baker M, Adamson J, Crook R, Melquist S, Kuntz K, Petersen R, Josephs K, Pickering-Brown SM, Graff-Radford N, Uitti R, Dickson D, Wszolek Z, Gonzalez J, Beach TG, Bigio E, Johnson N, Weintraub S, Mesulam M, White CL, 3rd, Woodruff B, Caselli R, Hsiung GY, Feldman H, Knopman D, Hutton M, Rademakers R. Mutations in progranulin are a major cause of ubiquitin-positive frontotemporal lobar degeneration. Hum Mol Genet. 2006;15:2988–3001. doi: 10.1093/hmg/ddl241. [DOI] [PubMed] [Google Scholar]
- Pickering-Brown SM, Baker M, Gass J, Boeve BF, Loy CT, Brooks WS, Mackenzie IR, Martins RN, Kwok JB, Halliday GM, Kril J, Schofield PR, Mann DM, Hutton M. Mutations in progranulin explain atypical phenotypes with variants in MAPT. Brain. 2006;129:3124–3126. doi: 10.1093/brain/awl289. [DOI] [PubMed] [Google Scholar]
- Ikeda M, Ishikawa T, Tanabe H. Epidemiology of frontotemporal lobar degeneration. Dement Geriatr Cogn Disord. 2004;17:265–268. doi: 10.1159/000077151. [DOI] [PubMed] [Google Scholar]
- Snowden JS, Pickering-Brown SM, Mackenzie IR, Richardson AM, Varma A, Neary D, Mann DM. Progranulin gene mutations associated with frontotemporal dementia and progressive non-fluent aphasia. Brain. 2006;129:3091–3102. doi: 10.1093/brain/awl267. [DOI] [PubMed] [Google Scholar]
- Josephs KA, Ahmed Z, Katsuse O, Parisi JF, Boeve BF, Knopman DS, Petersen RC, Davies P, Duara R, Graff-Radford NR, Uitti RJ, Rademakers R, Adamson J, Baker M, Hutton ML, Dickson DW. Neuropathologic features of frontotemporal lobar degeneration with ubiquitin-positive inclusions with progranulin gene (PGRN) mutations. J Neuropathol Exp Neurol. 2007 doi: 10.1097/nen.0b013e31803020cf. [DOI] [PubMed] [Google Scholar]
- Rademakers R, Cruts M, van Broeckhoven C. The role of tau (MAPT) in frontotemporal dementia and related tauopathies. Hum Mutat. 2004;24:277–295. doi: 10.1002/humu.20086. [DOI] [PubMed] [Google Scholar]
- Mackenzie IR, Baker M, West G, Woulfe J, Qadi N, Gass J, Cannon A, Adamson J, Feldman H, Lindholm C, Melquist S, Pettman R, Sadovnick AD, Dwosh E, Whiteheart SW, Hutton M, Pickering-Brown SM. A family with tau-negative frontotemporal dementia and neuronal intranuclear inclusions linked to chromosome 17. Brain. 2006;129:853–867. doi: 10.1093/brain/awh724. [DOI] [PubMed] [Google Scholar]
- Guyant-Marechal L, Laquerriere A, Duyckaerts C, Dumanchin C, Bou J, Dugny F, Le Ber I, Frebourg T, Hannequin D, Campion D. Valosin-containing protein gene mutations: clinical and neuropathologic features. Neurology. 2006;67:644–651. doi: 10.1212/01.wnl.0000225184.14578.d3. [DOI] [PubMed] [Google Scholar]
- Skibinski G, Parkinson NJ, Brown JM, Chakrabarti L, Lloyd SL, Hummerich H, Nielsen JE, Hodges JR, Spillantini MG, Thusgaard T, Brandner S, Brun A, Rossor MN, Gade A, Johannsen P, Sorensen SA, Gydesen S, Fisher EM, Collinge J. Mutations in the endosomal ESCRTIII-complex subunit CHMP2B in frontotemporal dementia. Nat Genet. 2005;37:806–808. doi: 10.1038/ng1609. [DOI] [PubMed] [Google Scholar]
- Waugh MG, Minogue S, Anderson JS, Balinger A, Blumenkrantz D, Calnan DP, Cramer R, Hsuan JJ. Localization of a highly active pool of type II phosphatidylinositol 4-kinase in a p97/valosin-containing-protein-rich fraction of the endoplasmic reticulum. Biochem J. 2003;373:57–63. doi: 10.1042/BJ20030089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wojcik C. VCP – the missing link in protein degradation? Trends Cell Biol. 2002;12:212. doi: 10.1016/S0962-8924(02)02286-9. [DOI] [PubMed] [Google Scholar]
- Bowers K, Piper SC, Edeling MA, Gray SR, Owen DJ, Lehner PJ, Luzio JP. Degradation of endocytosed epidermal growth factor and virally ubiquitinated major histocompatibility complex class I is independent of mammalian ESCRTII. J Biol Chem. 2006;281:5094–5105. doi: 10.1074/jbc.M508632200. [DOI] [PubMed] [Google Scholar]
- Munoz DG, Dickson DW, Bergeron C, Mackenzie IR, Delacourte A, Zhukareva V. The neuropathology and biochemistry of frontotemporal dementia. Ann Neurol. 2003;54:S24–28. doi: 10.1002/ana.10571. [DOI] [PubMed] [Google Scholar]
- Josephs KA, Jones AG, Dickson DW. Hippocampal sclerosis and ubiquitin-positive inclusions in dementia lacking distinctive histopathology. Dement Geriatr Cogn Disord. 2004;17:342–345. doi: 10.1159/000077168. [DOI] [PubMed] [Google Scholar]
- Mackenzie IR, Feldman H. Neuronal intranuclear inclusions distinguish familial FTD-MND type from sporadic cases. Dement Geriatr Cogn Disord. 2004;17:333–336. doi: 10.1159/000077166. [DOI] [PubMed] [Google Scholar]
- Woulfe J, Kertesz A, Munoz DG. Frontotemporal dementia with ubiquitinated cytoplasmic and intranuclear inclusions. Acta Neuropathol (Berl) 2001;102:94–102. doi: 10.1007/s004010000346. [DOI] [PubMed] [Google Scholar]
- Ou SH, Wu F, Harrich D, Garcia-Martinez LF, Gaynor RB. Cloning and characterization of a novel cellular protein, TDP-43, that binds to human immunodeficiency virus type 1 TAR DNA sequence motifs. J Virol. 1995;69:3584–3596. doi: 10.1128/jvi.69.6.3584-3596.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buratti E, Dork T, Zuccato E, Pagani F, Romano M, Baralle FE. Nuclear factor TDP-43 and SR proteins promote in vitro and in vivo CFTR exon 9 skipping. Embo J. 2001;20:1774–1784. doi: 10.1093/emboj/20.7.1774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buratti E, Baralle FE. Characterization and functional implications of the RNA binding properties of nuclear factor TDP-43, a novel splicing regulator of CFTR exon 9. J Biol Chem. 2001;276:36337–36343. doi: 10.1074/jbc.M104236200. [DOI] [PubMed] [Google Scholar]
- Davidson Y, Kelley T, Mackenzie IR, Pickering-Brown S, Du Plessis D, Neary D, Snowden JS, Mann DM. Ubiquitinated pathological lesions in frontotemporal lobar degeneration contain the TAR DNA-binding protein, TDP-43. Acta Neuropathol (Berl) 2007 doi: 10.1007/s00401-006-0189-y. [DOI] [PubMed] [Google Scholar]
- Wang HY, Wang IF, Bose J, Shen CK. Structural diversity and functional implications of the eukaryotic TDP gene family. Genomics. 2004;83:130–139. doi: 10.1016/S0888-7543(03)00214-3. [DOI] [PubMed] [Google Scholar]
- Olsnes S, Klingenberg O, Wiedlocha A. Transport of exogenous growth factors and cytokines to the cytosol and to the nucleus. Physiol Rev. 2003;83:163–182. doi: 10.1152/physrev.00021.2002. [DOI] [PubMed] [Google Scholar]
- Hoque M, Young TM, Lee CG, Serrero G, Mathews MB, Pe'ery T. The growth factor granulin interacts with cyclin T1 and modulates P-TEFb-dependent transcription. Mol Cell Biol. 2003;23:1688–1702. doi: 10.1128/MCB.23.5.1688-1702.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Luca A, De Falco M, Baldi A, Paggi MG. Cyclin T: three forms for different roles in physiological and pathological functions. J Cell Physiol. 2003;194:101–107. doi: 10.1002/jcp.10196. [DOI] [PubMed] [Google Scholar]
- Trinh DP, Brown KM, Jeang KT. Epithelin/granulin growth factors: extracellular cofactors for HIV-1 and HIV-2 Tat proteins. Biochem Biophys Res Commun. 1999;256:299–306. doi: 10.1006/bbrc.1999.0317. [DOI] [PubMed] [Google Scholar]