

Abstract
Homeostasis of intravascular volume, Na+, Cl−, and K+ is interdependent and determined by the coordinated activities of structurally diverse mediators in the distal nephron and the distal colon. The behavior of these flux pathways is regulated by the renin–angiotensin–aldosterone system; however, the mechanisms that allow independent modulation of individual elements have been obscure. Previous work has shown that mutations in WNK4 cause pseudohypoaldosteronism type II (PHAII), a disease featuring hypertension with hyperkalemia, due to altered activity of specific Na-Cl cotransporters, K+ channels, and paracellular Cl− flux mediators of the distal nephron. By coexpression studies in Xenopus oocytes, we now demonstrate that WNK4 also inhibits the epithelial Na+ channel (ENaC), the major mediator of aldosterone-sensitive Na+ (re)absorption, via a mechanism that is independent of WNK4's kinase activity. This inhibition requires intact C termini in ENaC β- and γ-subunits, which contain PY motifs used to target ENaC for clearance from the plasma membrane. Importantly, PHAII-causing mutations eliminate WNK4's inhibition of ENaC, thereby paralleling other effects of PHAII to increase sodium balance. The relevance of these findings in vivo was studied in mice harboring PHAII-mutant WNK4. The colonic epithelium of these mice demonstrates markedly increased amiloride-sensitive Na+ flux compared with wild-type littermates. These studies identify ENaC as a previously unrecognized downstream target of WNK4 and demonstrate a functional role for WNK4 in the regulation of colonic Na+ absorption. These findings support a key role for WNK4 in coordinating the activities of diverse flux pathways to achieve integrated fluid and electrolyte homeostasis.
Keywords: aldosterone, hyperkalemia, hypertension, pseudohypoaldosteronism, renin–angiotensin system
The precise regulation of intravascular salt and water balance is essential for maintenance of normal tissue perfusion and survival. Under conditions of normal dietary intake and intestinal absorption of fluid and electrolytes, homeostasis is largely maintained by modulation of salt and water reabsorption in the kidney via alterations in the activities of renal ion channels and transporters. In the setting of intravascular volume depletion and/or dietary salt restriction, however, Na+ and Cl− absorption in the distal colon is augmented to help maintain salt and water balance (1).
The mechanisms underlying the handling of Na+ in the distal colon and distal nephron are similar. In both, the major regulated step for Na+ entry is electrogenic Na+ flux via the epithelial Na+ channel (ENaC); the lumen-negative potential resulting from ENaC activity provides the electrical driving force for paracellular Cl− flux across tight junctions and K+ secretion via luminal K+ channels (2, 3). The major regulator of colonic and renal ENaC activity is the steroid hormone aldosterone, which is secreted from the adrenal glomerulosa in response to reduced effective intravascular volume via activation of the renin–angiotensin system or by direct effects of high serum K+ levels. Aldosterone increases ENaC activity via an incompletely understood signaling pathway. The serine-threonine kinase SGK1, a direct transcriptional target of the activated mineralocorticoid receptor (4), is an upstream component of this signaling cascade. A downstream element is the ubiquitin ligase Nedd4-2, which specifically binds to C termini of ENaC subunits and targets ENaC for clearance from the plasma membrane (5). Nedd4-2 activity is inhibited by phosphorylation downstream of SGK1 signaling, thereby increasing ENaC surface expression and activity (6). Other intermediate components that couple aldosterone signaling to downstream targets have been obscure.
Although aldosterone secretion increases in response to both intravascular volume depletion and hyperkalemia (increased plasma K+ levels), the mechanisms that permit maximal NaCl reabsorption in response to volume depletion versus K+ secretion in response to hyperkalemia have been poorly understood. It seems likely that branches downstream of aldosterone signaling operate to determine the balance between these alternative responses. Recent studies have identified the serine–threonine kinase WNK4 as a regulator of diverse electrolyte flux pathways and as a candidate element in aldosterone signaling that might serve to determine the balance between salt reabsorption and K+ secretion (7). WNK4 was first identified by positional cloning as a gene responsible for pseudohypoaldosteronism type II (PHAII) [Online Mendelian Inheritance in Man (OMIM) no. 145260], an autosomal-dominant disease featuring hypertension (high blood pressure) due to increased renal NaCl reabsorption and hyperkalemia due to impaired renal K+ excretion (8). Intriguingly, the mutations in WNK4 that cause PHAII are all missense and cluster within a highly conserved, negatively charged 10-aa domain (Fig. 1a) (8). Wild-type WNK4 has been shown to inhibit the activity of the thiazide-sensitive Na-Cl cotransporter (NCC) (9, 10) and the renal outer medullary K+ channel (ROMK) (11) by increasing the clearance of each from the cell surface. PHAII-causing mutations in WNK4 dramatically alter its regulation of its downstream targets, abrogating inhibition of NCC, increasing inhibition of ROMK, and augmenting the paracellular permeability of epithelia for Cl− (9–13). These in vitro observations can explain the hypertension and hyperkalemia seen among PHAII patients and suggest that WNK4 is a molecular switch that can be locked into different regulatory states, one of which promotes NaCl reabsorption while inhibiting K+ secretion. Moreover, these findings predicted that an increased dosage of wild-type WNK4 should lower blood pressure and serum K+, whereas PHAII-mutant WNK4 should have the opposite effects in vivo. This hypothesis was recently confirmed by the development of BAC transgenic mice that harbor additional copies of either wild-type or PHAII-mutant WNK4 (14). As predicted, these two different transgenes impart opposite effects on blood pressure and K+ homeostasis (14).
Fig. 1.

Wild-type WNK4, but not PHAII-mutant WNK4, inhibits ENaC by way of a kinase-independent mechanism. cRNA encoding the indicated proteins was injected into Xenopus oocytes, and amiloride-sensitive, whole-cell Na+ currents were recorded as describe in Methods. (a) A diagram showing the clustering of PHAII-causing mutations in WNK4. (b) Representative current tracings from oocytes expressing ENaC alone, ENaC plus wild-type WNK4, or ENaC plus PHAII–WNK4. (c) Current–voltage (I–V) relationships of oocytes expressing indicated constructs from a representative experiment plotted as mean ± SE of the Na+ currents for each experimental group. (d Upper) Cumulative results of amiloride-sensitive currents measured at −100 mV are shown as a proportion of the ENaC control. Each group expresses ENaC plus the indicated construct; a minimum of 29 oocytes were studied in each group. The significance of differences between groups was calculated with two-tailed Student's t test. (d Lower) Results of Western blotting of oocyte lysates shows the expression of WNK4-HA in the corresponding experimental groups.
These findings together suggest that WNK4 coordinates hormonal signaling at structurally diverse but functionally related ion transport pathways involved in Na+, Cl−, K+, and volume homeostasis. If correct, one might speculate that WNK4 should also regulate ENaC activity and that this effect should be consistent with the other effects of WNK4 on integrated physiology. We now present evidence that WNK4 regulates ENaC activity both in vitro and in vivo.
Results
Wild-Type WNK4, but Not PHAII-WNK4, Inhibits ENaC.
We coinjected Xenopus laevis oocytes with cRNA encoding HA-tagged wild-type or PHAII mutant WNK4 and the α-, β-, and γ-subunits of ENaC. We then measured amiloride-sensitive, whole-cell currents by using a two-electrode voltage clamp. Wild-type WNK4 inhibited ENaC activity >50% compared with oocytes expressing ENaC alone (Fig. 1 b–d). This result was reproduced in all experiments and was highly significant (P = 1.7 × 10−7). WNK4's kinase activity is required for its inhibition of some (e.g., NCC) (9, 10), but not all (e.g., ROMK; see ref. 11) of its targets. To determine whether WNK4's inhibition of ENaC depends on its catalytic activity, we coexpressed kinase-dead WNK4 (WNK4-D318A) and ENaC subunits in oocytes. Kinase-dead WNK4 continues to inhibit ENaC like its wild-type counterpart (Fig. 1d), demonstrating that WNK4's inhibition of ENaC is independent of its kinase activity. We next expressed ENaC with WNK4 containing a single, PHAII-causing missense mutation (Q562E); PHAII–WNK4 had no significant inhibitory effect on ENaC activity, a result that was significantly different from the effect of wild-type WNK4 (P = 10−5) (Fig. 1d). Coexpression of wild-type or mutant WNK4 in oocytes did not change ENaC reversal potentials, and the observed WNK4-mediated inhibition of ENaC activity was consistent across a wide range of different holding potentials (Fig. 1c). Western blots of injected oocytes with anti-HA antibody revealed that the expression levels of HA-tagged wild type WNK4, kinase-dead WNK4-D318A, and PHAII–WNK4–Q562E were similar, indicating that the observed effects are not attributable to varying levels of WNK4 expression (Fig. 1d).
WNK4 Regulation Requires ENaC C-Terminal Domains.
The plasma membrane expression of ENaC is regulated by the E3 ubiquitin ligase Nedd4-2 through a direct interaction between its three WW domains and C-terminal PY motifs in each ENaC subunit (5). After ubiquitinylation by Nedd4-2, ENaC is cleared from the cell surface via endocytosis by clathrin-coated pits (15). The importance of this mechanism in vivo is demonstrated by the finding that mutations in ENaC that eliminate PY motifs result in increased ENaC activity and the Mendelian form of hypertension known as Liddle syndrome (16–21). SGK1 activity increases ENaC activity, in part, by inhibiting Nedd4-2 (6). We tested whether WNK4's inhibition of ENaC requires the short C termini of ENaC subunits, which contain the PY motifs by expressing ENaC containing C-terminal truncations in the β- and γ-subunits like those seen in Liddle syndrome (Fig. 2a; see Methods). In the presence of these mutations, there was no significant inhibition of ENaC by WNK4 (Fig. 2 b and c), demonstrating that inhibition of ENaC by WNK4 requires the ENaC segments that are used by Nedd4-2 to target ENaC for degradation.
Fig. 2.

Inhibition of ENaC by WNK4 requires C-terminal domains in the channel. (a) A diagram of wild-type ENaC subunit and Liddle's syndrome mutations that truncate the C terminus of either β- or γ-subunits and delete the PY motif (in green) is shown (β and γLT represent Liddle's truncations in the respective ENaC subunits). (b and c) Activity of ENaC with Liddle's mutations in the presence or absence of WNK4. cRNA encoding ENaC harboring a Liddle's mutation in either the β-subunit (at least 11 oocytes per group) (b) or the γ-subunit (at least 22 oocytes per group) (c) with or without wild-type WNK4 was injected into Xenopus oocytes, and amiloride-sensitive, whole-cell Na+ currents were recorded and analyzed as in Fig. 1. Western blotting of oocyte lysates confirms the presence of WNK4-HA in the corresponding experimental groups. WNK4 does not inhibit ENaC activity in the presence of Liddle's mutations.
PHAII-Mutant WNK4 Stimulates Amiloride-Sensitive Na+ Absorption in the Aldosterone-Sensitive Distal Colon.
Our findings in oocytes suggest that WNK4 is a previously unrecognized regulator of ENaC. To examine the physiological relevance of these observations in vivo, we investigated ENaC activity in the distal colonic epithelium, an aldosterone-sensitive tissue. We first localized WNK4 in the mouse gastrointestinal (GI) tract by using specific anti-WNK4 antibodies. Immunostaining reveals that WNK4 is expressed in epithelia of the GI tract. As reported in ref. 22, expression is high in the distal colon; WNK4 prominently labels the lateral membrane of cells in the surface epithelium and upper part of crypts (Fig. 3); this localization is similar to the localization to the tight junction and lateral membrane seen in the renal epithelium (8). The α-, β-, and γ-subunits of ENaC also localize to the surface epithelium and upper part of crypts in these colon segments (23). Thus, WNK4 and ENaC are present in the same cells of the distal colon and show similar quantitative distributions, being more highly expressed in the surface epithelium than in the crypts.
Fig. 3.
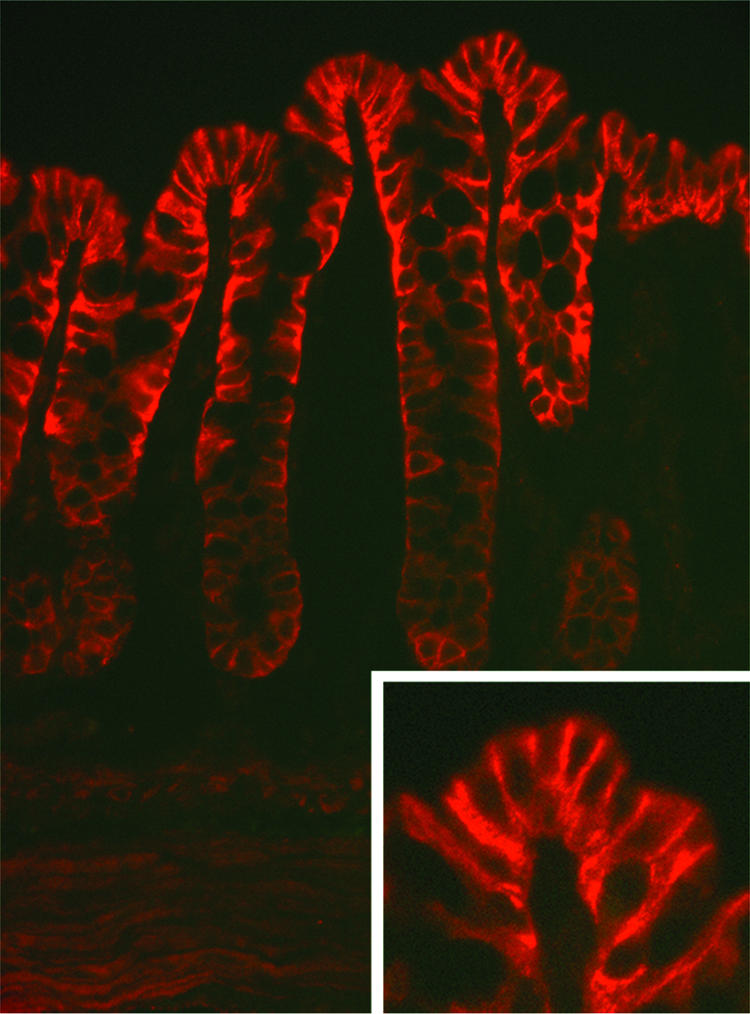
Immunolocalization of WNK4 in the mouse colon. Distal colons of mice were dissected, prepared, and stained with antibodies specific for WNK4 as described in Methods. A transverse section is shown. The results show strong staining of the surface epithelium and upper part of crypts. (Magnification: ×400.) (Inset) A higher magnification, demonstrating the strongest staining of the basolateral membrane. (Magnification: ×800.)
We used a modified Ussing chamber apparatus to compare amiloride-sensitive short-circuit currents (Isc) across epithelia of the distal colon from mice with two copies of a PHAII-mutant WNK4 BAC transgene (denoted Tg-WNK4PHAII) and their wild-type littermates. The expression of this mutant WNK4 transgene is qualitatively and quantitatively similar to expression of endogenous WNK4, and the quantitative effect of this transgene on total WNK4 expression is additive to the expression of the endogenous gene (14). As a result, this model allows us to determine the impact of increased expression of PHAII mutant WNK4 on ENaC in vivo.
Colonic epithelia from PHAII-mutant mice show a significant increase in amiloride-inhibitable basal Isc, with mean currents 6.0-fold higher in the PHAII epithelia than in epithelia from their wild-type littermates. (Fig. 4; P = 0.01; n = 11 wild-type and 11 Tg-WNK4PHAII mice). In contrast, forskolin-induced, bumetanide-sensitive Isc was not significantly different between Tg-WNK4PHAII mice and their wild-type littermates (data not shown). These results reveal that PHAII-mutant WNK4 specifically stimulates ENaC-mediated Na+ absorption, but not NKCC1-dependent Cl− secretion, in vivo. This increase in ENaC activity is not attributable to increased levels of aldosterone in the PHAII mice, because plasma aldosterone levels are not significantly different among wild-type mice (368 ± 135 pg/ml; n = 27), heterozygous PHAII-mutant WNK4 mice (485 ± 185 pg/ml; n = 21), and homozygous PHAII-mutant WNK4 mice (433 ± 225 pg/ml; n = 29).
Fig. 4.

Increased amiloride-sensitive Na+ transport in the distal colon of PHAII mice. Segments of distal colonic epithelia were dissected and mounted in modified Ussing chambers, and amiloride-sensitive Na+ flux was measured as described in Methods. (a) A representative current tracing of amiloride-sensitive Isc from wild-type and PHAII mice is shown, demonstrating a larger amiloride-sensitive current in PHAII versus wild-type mice. (b) Mean and SE of amiloride-sensitive Na+ flux across colonic epithelia from wild-type mice (n = 11) and PHAII mice (n = 11) is shown, demonstrating a significant increase in PHAII mice.
Discussion
WNK kinases have emerged in recent years as important regulators of integrated electrolyte flux; they achieve these effects by modulating the activities of diverse electrolyte flux pathways. The present findings add several important elements to this developing story. First, WNK4 inhibits the Na+ channel ENaC, adding a structurally diverse member to the flux pathways that WNK4 regulates. This inhibition is kinase-independent and is alleviated by mutations that cause PHAII. These findings extend and underscore the observation that PHAII–WNK4's effects on diverse electrolyte flux pathways all serve the same physiologic end: increasing net salt balance while preserving serum K+. We presume these effects mimic the normal integrated physiological response to intravascular volume depletion. Similar to WNK4's localization and targets in kidney, WNK4 localizes predominantly to the lateral membrane of colonic epithelia, whereas its target acts at the apical membrane. This finding implies either signaling intermediates or trafficking of WNK4 or its target between cellular compartments.
It is noteworthy that WNK4's effects on ENaC are distinguishable from its effects on other targets. Thus, WNK4 inhibits ENaC in a kinase-independent fashion, and PHAII mutations abrogate this inhibition; WNK4 inhibits NCC in a kinase-dependent fashion, and PHAII mutation abrogates inhibition; and WNK4 inhibits ROMK activity in a kinase-independent fashion, but PHAII mutation increases inhibition. These differential effects underscore the potential for WNK4 to independently regulate the activities of specific downstream flux mediators, potentially allowing distinct responses to different physiologic perturbations.
Second, we demonstrate that the short cytoplasmic C termini of ENaC, which are used by Nedd4-2 to target the channel for degradation, are also essential for WNK4's inhibition of ENaC. This finding raises the question of whether WNK4 is acting through Nedd4-2 or an independent pathway; moreover, they pose the question of whether WNK4's effects at ENaC are downstream of SGK1 signaling, because SGK1 is a known aldosterone-responsive regulator of ENaC activity (see ref. 24 in this issue of PNAS). Further investigation will be required to elucidate the details of this mechanism.
Third, the physiological significance of these effects of WNK4 on ENaC in vivo is demonstrated by the transport properties of aldosterone-sensitive colonic epithelia of mice harboring PHAII-mutant WNK4. These mice show increased ENaC activity compared with their wild-type littermates. This observation contributes importantly to the understanding of WNK4 as a regulator of distinct but functionally related ion transport pathways. Increased intestinal Na+ absorption via ENaC promotes increased intravascular volume (25), an effect that closely parallels the action of PHAII-mutant WNK4 to maximize salt reabsorption in the kidney. These net effects are adaptive in restoring intravascular volume in the setting of volume depletion and lend further support to WNK4 being a downstream mediator of aldosterone–angiotensin II signaling. Moreover, this result demonstrates the physiologic importance of WNK4 activity outside the kidney and has potential implications for its activity in other tissues in which ENaC plays an important physiological role, such as the lung.
Last, the physiological importance of WNK4's regulation of ENaC in the kidney is not known. PHAII-mutant WNK4 causes hyperplasia of the distal convoluted tubule and increased levels of NCC in mice, and both the hypertensive and hyperkalemic phenotypes of these mice can be corrected by genetic deficiency for NCC (14). These large effects of WNK4 on NCC, however, do not exclude significant effects of WNK4 on other determinants of renal Na+, Cl−, and K+ homeostasis (e.g., ENaC, ROMK, and paracellular Cl− flux) that have been observed in vitro. Further studies will be required to directly measure the activities of these and other distal nephron transport pathways in PHAII.
Methods
Molecular Cloning.
The complete coding sequence of mouse WNK4 with an appended 3′ hemagglutinin A (HA) tag was subcloned from pcDNA3.1− into pGH19 through a blunt-end ligation. Full-length and truncated clones of the α- (pSD5), β- (pSPORT), and γ- (pSPORT) subunits of the epithelial Na+ channel (ENaC) were as described in ref. 18. All point mutations were introduced using the QuikChange site-directed mutagenesis system (Stratagene, La Jolla, CA).
Functional Studies in X. laevis Oocytes.
Plasmids used for functional studies encoded HA-tagged mouse WNK4 (pGH19-WNK4-HA) and rat α, β, and γ ENaC (pSD5-α rENaC, pSPORT1-β rENaC, and pSPORT1-γ rENaC). Plasmids were linearized by restriction digestion, and cRNA was transcribed by using T7 or SP6 RNA polymerase (mMessage mMachine kit; Ambion, Austin, TX). cRNA quality was assessed with UV spectrometry and agarose gel electrophoresis. Stage V–VI X. laevis oocytes were harvested and defolliculated in compliance with institutional regulations. Oocytes were injected with 2.5 ng of cRNA encoding the α-, β-, and γ-subunits of rat ENaC and with 10.0 ng of cRNA encoding mouse WNK4-HA. These quantities correspond to a 1:1 molar ratio of ENaC/WNK4. Injected oocytes were incubated in ND96 solution [96 mM NaCl/2.0 mM KCl/2.0 mM CaCl2/1.0 mM MgCl2/5.0 mM Hepes buffer (pH 7.4)] and 10 μM amiloride at 18°C for 36–48 h. Microelectrodes were pulled to a tip resistance of 0.9–1.5 MΩ and backfilled with 3 M KCl. Whole-cell, amiloride-sensitive Na+ currents were then determined by a two-electrode voltage clamp in a buffer containing 96 mM NaCl, 5 mM KCl, 1 mM MgCl2, 1 mM CaCl2, and 10 mM Hepes buffer (pH 7.4), in the presence and absence of amiloride. All Na+ currents reported refer to Na+ currents measured in the absence of amiloride. Oocyte membrane potentials were held at −40 mV, and currents were measured at 20-mV steps from −160 to +80 mV. Results of consecutive experiments were pooled by normalizing the currents to the average of the ENaC control group. Each experimental group contains measurements from at least three different frogs.
Western Blotting.
For Western blots with lysates from oocytes, five oocytes from each experimental group were lysed in 100 μl of RIPA buffer [150 mM NaCl/10 mM Tris (pH 7.2)/0.1% SDS/1.0% Triton X-100/1% deoxycholate/5 mM EDTA]. Lysates were cleared by centrifugation at 30,000 × g and 4°C for 15 min, and protein concentrations were quantified. Twenty micrograms of protein was fractionated by SDS/PAGE gradient gel electrophoresis. Proteins were transferred to a 0.2-μm PVDF membrane (Bio-Rad, Hercules, CA), blocked, and probed with rabbit anti-HA antibodies (Santa Cruz Biotechnology, Santa Cruz, CA) (1:5,000 dilution). Membranes were washed and incubated with secondary antibody (HRP-conjugated goat anti-rabbit IgG; Sigma, St. Louis, MO), and chemiluminescence was performed (ECL chemiluminescence system; Amersham, Piscataway, NJ) following the manufacturer's protocol.
Immunolocalization of WNK4 in the Mouse Colon.
Mice were killed by cervical dislocation at age 10 to 12 weeks in accordance with approved procedures of the Yale Animal Care and Use Committee. Tissue was embedded in OCT compound and snap-frozen in isopentane at −140°C. Sections of 5 μm were used for immunolocalization. Tissue sections were processed and incubated with rabbit anti-WNK4 (1:400) and secondary antibodies as described in ref. 14. Stained sections were visualized by immunofluorescence light microscopy. Results displayed are representative of results from at least three different mice. All immunostaining with anti-WNK4 was competed with a three-fold molar excess of the immunizing peptide, and staining with secondary antibody alone revealed no signal.
Ussing Chamber Isc Measurements from Distal Colons of WNK4 Transgenic Mice.
Mice heterozygous for a single copy BAC transgene containing WNK4 with a PHAII-causing mutation (Q562E) were crossed, and wild-type and homozygous transgenic mice were identified in their offspring by genotyping tail DNA as described in ref. 14. Mice were euthanized, and segments of distal colon were isolated as described in ref. 26 in accordance with the humane practices of animal care established by the Yale Animal Care and Use Committee. Segments of 0.3-cm2 surface area were mounted in modified Ussing chambers (Physiologic Instruments, San Diego, CA), bathed in Hepes-buffered saline solution containing 110 mM NaCl, 1.25 mM CaCl2, 1.25 mM MgCl2, 5 mM KCl, 10 mM glucose, and 22 mM Hepes. Tetrodotoxin (2 μM) (Sigma) was added to the serosal solution to inhibit the neuronal activity.
Short circuit current, Isc (μA/cm2), was monitored and recorded by computer every 10–20 s. For measurements of amiloride-sensitive Isc, 10 μM amiloride was applied to the luminal surface segments after 400 s, and the difference between stable Isc after and before amiloride treatment was calculated. For measurements of forskolin-stimulated, butmetanide-sensitive Isc, 10 μM forksolin was applied to both surfaces of the segments, and the resulting Isc was monitored for 20 min, followed by the addition of 100 μM bumetanide to the serosal surface. Forskolin-stimulated, butmetanide-sensitive Isc was calculated by determining the difference between stable Isc after forskolin treatment and stable Isc after bumetanide treatment.
Statistical Analysis.
For all statistical comparisons, results were compared by two-tailed unpaired t tests, or by nonparametric Mann–Whitney U tests if the data violated a normal distribution.
Acknowledgments
We thank Peter Aronson, Gerhard Giebisch, Cecelia Canessa, and Carol Nelson-Williams for advice and discussions. This work was supported in part by a National Institutes of Health Specialized Center of Research in Hypertension grant (to R.P.L.) and a Beckman Scholar Award (to A.M.R.).
Abbreviations
- ENaC
epithelial Na+ channel
- Isc
short-circuit current
- NCC
Na-Cl contransporter
- PHAII
pseudohypoaldosteronism type II
- ROMK
renal outer medullary K+ channel.
Footnotes
The authors declare no conflict of interest.
References
- 1.Schultheis PJ, Clarke LL, Meneton P, Miller ML, Soleimani M, Gawenis LR, Riddle TM, Duffy JJ, Doetschman T, Wang T, et al. Nat Genet. 1998;19:282–285. doi: 10.1038/969. [DOI] [PubMed] [Google Scholar]
- 2.Kunzelmann K, Mall M. Physiol Rev. 2002;82:245–289. doi: 10.1152/physrev.00026.2001. [DOI] [PubMed] [Google Scholar]
- 3.Lifton RP, Gharavi AG, Geller DS. Cell. 2001;104:545–556. doi: 10.1016/s0092-8674(01)00241-0. [DOI] [PubMed] [Google Scholar]
- 4.Pearce D. Trends Endocrinol Metab. 2001;12:341–347. doi: 10.1016/s1043-2760(01)00439-8. [DOI] [PubMed] [Google Scholar]
- 5.Kamynina E, Debonneville C, Bens M, Vandewalle A, Staub O. FASEB J. 2001;15:204–214. doi: 10.1096/fj.00-0191com. [DOI] [PubMed] [Google Scholar]
- 6.Debonneville C, Flores SY, Kamynina E, Plant PJ, Tauxe C, Thomas MA, Munster C, Chraibi A, Pratt JH, Horisberger JD, et al. EMBO J. 2001;20:7052–7059. doi: 10.1093/emboj/20.24.7052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kahle KT, Wilson FH, Lifton RP. Trends Endocrinol Metab. 2005;16:98–103. doi: 10.1016/j.tem.2005.02.012. [DOI] [PubMed] [Google Scholar]
- 8.Wilson FH, Disse-Nicodeme S, Choate KA, Ishikawa K, Nelson-Williams C, Desitter I, Gunel M, Milford DV, Lipkin GW, Achard JM, et al. Science. 2001;293:1107–1112. doi: 10.1126/science.1062844. [DOI] [PubMed] [Google Scholar]
- 9.Wilson FH, Kahle KT, Sabath E, Lalioti MD, Rapson AK, Hoover RS, Hebert SC, Gamba G, Lifton RP. Proc Natl Acad Sci USA. 2003;100:680–684. doi: 10.1073/pnas.242735399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yang CL, Angell J, Mitchell R, Ellison DH. J Clin Invest. 2003;111:1039–1045. doi: 10.1172/JCI17443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kahle KT, Wilson FH, Leng Q, Lalioti MD, O'Connell AD, Dong K, Rapson AK, MacGregor GG, Giebisch G, Hebert SC, et al. Nat Genet. 2003;35:372–376. doi: 10.1038/ng1271. [DOI] [PubMed] [Google Scholar]
- 12.Yamauchi K, Rai T, Kobayashi K, Sohara E, Suzuki T, Itoh T, Suda S, Hayama A, Sasaki S, Uchida S. Proc Natl Acad Sci USA. 2004;101:4690–4694. doi: 10.1073/pnas.0306924101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kahle KT, Macgregor GG, Wilson FH, Van Hoek AN, Brown D, Ardito T, Kashgarian M, Giebisch G, Hebert SC, Boulpaep EL, et al. Proc Natl Acad Sci USA. 2004;101:14877–14882. doi: 10.1073/pnas.0406172101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lalioti MD, Zhang J, Volkman HM, Kahle KT, Hoffmann KE, Toka HR, Nelson-Williams C, Ellison DH, Flavell R, Booth CJ, et al. Nat Genet. 2006;38:1124–1132. doi: 10.1038/ng1877. [DOI] [PubMed] [Google Scholar]
- 15.Shimkets RA, Lifton RP, Canessa CM. J Biol Chem. 1997;272:25537–25541. doi: 10.1074/jbc.272.41.25537. [DOI] [PubMed] [Google Scholar]
- 16.Shimkets RA, Warnock DG, Bositis CM, Nelson-Williams C, Hansson JH, Schambelan M, Gill JR, Jr, Ulick S, Milora RV, Findling JW, et al. Cell. 1994;79:407–414. doi: 10.1016/0092-8674(94)90250-x. [DOI] [PubMed] [Google Scholar]
- 17.Hansson JH, Nelson-Williams C, Suzuki H, Schild L, Shimkets R, Lu Y, Canessa C, Iwasaki T, Rossier B, Lifton RP. Nat Genet. 1995;11:76–82. doi: 10.1038/ng0995-76. [DOI] [PubMed] [Google Scholar]
- 18.Tamura H, Schild L, Enomoto N, Matsui N, Marumo F, Rossier BC. J Clin Invest. 1996;97:1780–1784. doi: 10.1172/JCI118606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schild L, Lu Y, Gautschi I, Schneeberger E, Lifton RP, Rossier BC. EMBO J. 1996;15:2381–2387. [PMC free article] [PubMed] [Google Scholar]
- 20.Schild L, Canessa CM, Shimkets RA, Gautschi I, Lifton RP, Rossier BC. Proc Natl Acad Sci USA. 1995;92:5699–5703. doi: 10.1073/pnas.92.12.5699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hansson JH, Schild L, Lu Y, Wilson TA, Gautschi I, Shimkets R, Nelson-Williams C, Rossier BC, Lifton RP. Proc Natl Acad Sci USA. 1995;92:11495–11499. doi: 10.1073/pnas.92.25.11495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kahle KT, Gimenez I, Hassan H, Wilson FH, Wong RD, Forbush B, Aronson PS, Lifton RP. Proc Natl Acad Sci USA. 2004;101:2064–2069. doi: 10.1073/pnas.0308434100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Coric T, Hernandez N, Alvarez de la Rosa D, Shao D, Wang T, Canessa CM. Am J Physiol Gastrointest Liver Physiol. 2004;286:G663–G670. doi: 10.1152/ajpgi.00364.2003. [DOI] [PubMed] [Google Scholar]
- 24.Ring AM, Leng Q, Rinehart J, Wilson FH, Kahle KT, Hebert SC, Lifton RP. Proc Natl Acad Sci USA. 2007;104:4025–4029. doi: 10.1073/pnas.0611728104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Will PC, Cortright RN, Groseclose RG, Hopfer U. Am J Physiol. 1985;248:G133–G141. doi: 10.1152/ajpgi.1985.248.1.G133. [DOI] [PubMed] [Google Scholar]
- 26.Geibel J, Sritharan K, Geibel R, Geibel P, Persing JS, Seeger A, Roepke TK, Deichstetter M, Prinz C, Cheng SX, et al. Proc Natl Acad Sci USA. 2006;103:9390–9397. doi: 10.1073/pnas.0602996103. [DOI] [PMC free article] [PubMed] [Google Scholar]