

Abstract
Based on recent directed evolution of P450 2B1, six P450 2B11 mutants at three positions were created in an N-terminal modified construct termed P450 2B11dH and characterized for enzyme catalysis using five substrates. Mutant I209A demonstrated a 3.2-fold enhanced kcat/Km for 7-ethoxy-4-trifluoromethylcourmarin O-deethylation, largely due to a dramatic decrease in Km (0.72 vs. 18 μM). I209A also demonstrated enhanced selectivity for testosterone 16β-hydroxylation over 16α-hydroxylation. In contrast, V183L showed a 4-fold increased kcat for 7-benzyloxyresorufin debenzylation and a 4.7-fold increased kcat/Km for testosterone 16α-hydroxylation. V183L also displayed a 1.7-fold higher kcat/Km than P450 2B11dH with the anti-cancer prodrugs cyclophosphamide and ifosfamide, resulting from a ~4-fold decrease in Km. Introduction of the V183L mutation into full-length P450 2B11 did not enhance the kcat/Km. Overall, the re-engineered P450 2B11dH enzymes exhibited enhanced catalytic efficiency with several substrates including the anti-cancer prodrugs.
Keywords: cytochrome P450, P450 2B11, structure-function relationships, site-directed mutagenesis, enzyme catalysis
INTRODUCTION
Mammalian cytochromes P450 of the 2B subfamily metabolize many xenobiotics and endogenous compounds [1], and are among the best studied P450 enzymes from the standpoint of structure-function relationships [2, 3]. Dog P450 2B11 is a particularly efficient enzyme for activation of the anti-cancer prodrugs cyclophosphamide (CPA) and ifosafamide (IFA) and detoxification of environmental contaminants such as polychlorinated biphenyls [4, 5]. When expressed in tumor cells, P450 2B11 shows substantial activity with CPA and IFA, resulting in strong cytotoxicity and anti-tumor activity, suggesting the potential utility for P450 gene-directed enzyme prodrug therapy (GDEPT) applications for cancer treatment [6]. Engineering of P450 2B11 for enhanced catalytic efficiency would be helpful in enhancing the efficacy of this enzyme for GDEPT and studying structure-function relationships.
Comparison of the active site residues among P450 2B enzymes within and across species has led to several successful efforts to enhance and/or re-engineer their activities [2, 3]. For example, the regioselectivity of progesterone hydroxylation by P450 2B1 has been re-engineered to that of P450 2C5 by rational site-directed mutagenesis of the latter enzyme [7]. In addition, advances in x-ray crystallography, homology modeling, and site-directed mutagenesis of the P450 2B enzymes have yielded crucial insights into the structural basis of enzyme efficiency, differential substrate specificity, and stereo- and regioselectivity [2, 3, 9–15]. These studies have revealed that substrate binding and oxidation are influenced not only by the active site residues but also by residues outside of the active site that play a significant role in initial substrate recognition, substrate access, and redox partner binding.
Recently, directed evolution has been successfully applied to bacterial enzyme P450 BM3 for engineering new and more efficient P450s and identifying the role of non-active site residues [16–18]. Mammalian P450s have also been engineered by random mutagenesis, which in some cases has yielded 5- to 10-fold increases in catalytic efficiency [19–23]. More recently, we have employed this approach to create more active and stable forms of P450 2B1 and 3A4, and to identify functionally important residues outside of the active site [24–26]. To engineer P450 2B1, an N-terminal modified enzyme containing an L209A substitution, termed 2B1dH L209A, was used as the template in a directed evolution study that yielded additional non-active site mutants V183L, F202L, and S334P. These mutants, either individually or in combination, demonstrated higher catalytic efficiency with several model substrates and the anti-cancer prodrugs CPA and IFA. In particular, relative to L209A, the double mutant V183L/L209A showed enhanced catalytic efficiency (kcat/Km) for IFA activation, while L209A/S334P showed enhanced kcat/Km for CPA activation [24].
In this study, we investigated whether selected mutations that improve the activity of P450 2B1dH have similar effects in the context of P450 2B11dH. Six mutants at three positions (V183L, F202L, I209A, F202L/I209A, V183L/I209A, and V183L/F202L/I209A) were created in P450 2B11dH (N-terminal modified), and steady-state kinetic analysis was performed using the model substrates 7-ethoxy-4-trifluoromethylcourmarin (7-EFC), 7-benzyloxyresorufin (7-BR), testosterone and the anti-cancer prodrugs CPA and IFA. An enhanced catalytic efficiency was observed for P450 2B11dH V183L, suggesting the feasibility of re-engineering P450s from the same subfamily by replacing their respective non-active site residues identified by directed evolution.
Experimental Procedures
Materials
7-EFC and 7-BR were purchased from Invitrogen (Carlsbad, CA). CPA and NADPH were bought from Sigma Chemical Co. (St. Louis, MO). [4-14C]-testosterone was obtained from Amersham Biosciences (Piscataway, NJ). 4-hydroperoxy-CPA was obtained from ASTA Pharma (Beilefeld, Germany). IFA was obtained from the Drug Synthesis and Chemistry Branch of the National Cancer Institute (Bethesda, MD). Recombinant NADPH-cytochrome P450 reductase (CPR) and cytochrome b5 from rat liver were prepared as described previously [27]. Oligonucleotide primers for polymerase chain reaction (PCR) were obtained from Sigma Genosys (Woodlands, TX). QuikChange PCR and ligation kits were obtained from Stratagene (La Jolla, CA). All other chemicals were of the highest grade available and were obtained from standard commercial sources.
Mutagenesis, Expression, and Purification
The mutants were constructed by site-directed mutagenesis using the QuikChange XL site-directed mutagenesis kit. Appropriate template and forward and reverse primers used for the mutagenesis are presented in Figure 1. All constructs were sequenced at the University of Texas Medical Branch Protein Chemistry Laboratory (Galveston, TX) to confirm the desired mutation and verify the absence of unintended mutations. P450 2B11dH and its mutants were expressed in E. coli TOPP3 cells (Stratagene, La Jolla, CA) and purified with a Ni-NTA affinity column as described previously [28]. Because full-length P450 2B11 and P450 2B11 V183L lack the COOH-terminal His-tag, the proteins were not purified, and membrane preparations were used for enzymatic studies as described [29]. Protein concentrations were determined using the Bradford protein assay kit (Biorad, Hercules, CA). The specific contents of the purified samples were between 6 and 15 nmol P450/mg protein. Upon purification, the yield, purity, and stability of the mutant proteins were similar or slightly lower than the respective wild-type enzymes.
Figure 1. Primers for the construction of P450 2B11 site-directed mutants.
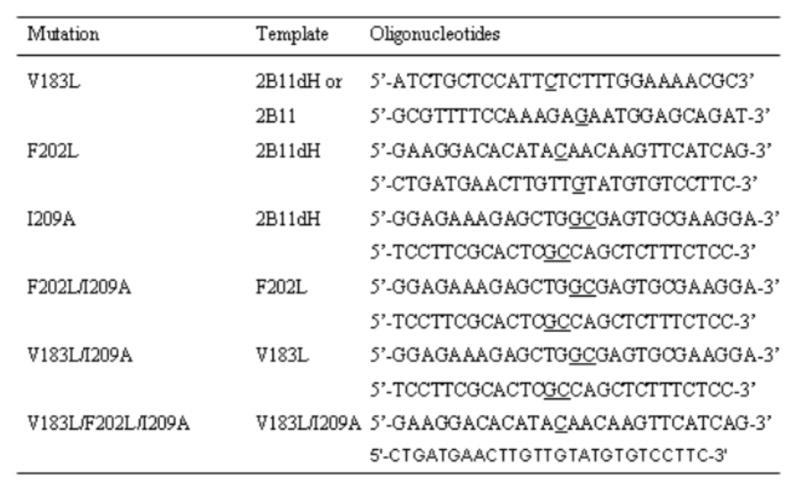
The nucleotide(s) that were changed to make the desired mutation are underlined. The V183L mutant was created using the P450 2B11 and 2B11dH templates, whereas the other single mutants were created using P450 2B11dH template only. The multiple mutants were created using the template as indicated in the Figure.
Enzyme assays
Enzyme assays were carried out with five structurally distinct substrates: 7-EFC, 7-BR, testosterone, CPA, and IFA (Fig. 2). 7-EFC, 7-BR, and testosterone are model substrates for P450 2B enzymes, whereas CPA and IFA are anti-cancer prodrugs that are activated efficiently by P450 2B11. 7-EFC and 7-BR oxidation reactions were assayed using a fluorescence method as described [10]. Testosterone hydroxylation was assayed by TLC using radiolabeled substrate as described [7]. Reconstitution of P450 2B11 and 2B11dH with CPR and cytochrome b5 for NADPH-supported monoxygenation was carried out at a 1:4:2 molar ratio as described earlier [28, 29] and included 10 μg DLPC/100 μl reaction volume with full-length P450 2B11. The reconstitution system for assaying CPA and IFA metabolism included P450 and purified rat liver CPR in a 1:4 molar ratio (5 pmol P450/0.1 ml of 0.1 M KPi buffer, pH 7.4 containing 0.1 mM EDTA) and was devoid of lipid and cytochrome b5. CPA and IFA metabolism were assayed by HLPC as described [4].
Figure 2. Structure of the substrates used for enzyme assays.

These structures were built using Chemdraw Ultra Version 6.0 (Cambridge, MA). The site of oxidation is shown by arrows.
Data analysis
Steady state kinetic parameters for metabolism of 7-EFC, 7-BR, and testosterone were determined by regression analysis using Sigma Plot (Jandel, San Rafel, CA). The Km value was determined using the Michaelis-Menten equation, whereas the S50 and n values were determined using the Hill equation. Kinetic analysis of CPA and IFA metabolism was carried out using Kinetics v0.44 software (Jacek Stanislawski, Novato, CA) based on graphical analysis using the Eadie-Hofstee method. Wild-type and mutant enzymes (presented in Tables 1–3) were assayed in parallel for more accurate comparison of the data.
Table 1.
Steady-state kinetics of 2B11dH and its mutants: NADPH-dependent oxidation of 7-BR and 7-EFC
| 7-BR | 7-EFC | |||||
|---|---|---|---|---|---|---|
| P450s | kcat(min−1) | S50 (μM) | n | kcat(min−1) | Km (μM) | kcat/Km |
| 2B11dH | 0.44 (0.03)a | 0.41 (0.08) | 1.5 | 13 (0.35) | 18 (3.8) | 0.68 |
| V183L | 1.8 (0.05) | 0.55 (0.07) | 1.3 | 15 (0.71) | 56 (4.7) | 0.25 |
| F202L | 0.14 (0.06) | 0.44 (0.08) | 1.2 | 15 (0.71) | 45 (5.8) | 0.29 |
| I209A | 0.25 (0.07) | 0.31 (0.04) | 1.3 | 1.6 (0.03) | 0.72 (0.11) | 2.2 |
| F202L/I209A | 0.04 (0.00) | 0.15 (0.03) | 2.0 | 1.3 (0.04) | 1.4 (0.34) | 0.93 |
| V183L/I209A | 0.96 (0.05) | 0.31 (0.07) | 1.7 | 2.2 (0.05) | 2.0 (0.27) | 1.1 |
| V183L/F202L/I209A | 0.38 (0.01) | 0.25 (0.04) | 1.5 | 2.9 (0.09) | 4.4 (0.62) | 0.66 |
Results are the representative of 2–3 independent determinations. The variation between experiments was ≤ 15%.
Standard errors for fit to Michaelis-Menten or Hill equations are shown in parenthesis. Kinetics with 7-BR fit well with the Hill equations with n values >1.0, as shown.
Table 3.
Steady-state kinetics of 2B11 and 2B11 V183L: NADPH-dependent oxidation of various substrates
| P450 2B11 | P450 2B11 V183L | |||||
|---|---|---|---|---|---|---|
| Substrate | kcat(min−1) | Km (μM) | kcat/Km | kcat(min−1) | Km (μM) | kcat/Km |
| 7-BR | 2.7b (0.1)a | 5.2 (0.1) | ||||
| 7-EFC | 19 (0.8) | 19 (2.6) | 1.0 | 15 (0.6) | 19 (2.7) | 0.79 |
| Testosterone (16α-OH) | 29 (1) | 42 (6.0) | 0.69 | 44 (2.2) | 57 (4.0) | 0.76 |
| Testosterone (16β-OH) | 2.2 (0.2) | 57 (10) | 0.04 | 2.3 (0.2) | 50 (8.0) | 0.05 |
| CPAc | 28 | 160 | 0.17 | 24 | 140 | 0.18 |
| IFAc | 5.4 | 100 | 0.05 | 6.4 | 150 | 0.04 |
Results are representative of 2–3 independent determinations. The variation between the experiments is ≤ 20%.
Standard errors for fit to Michaelis-Menten are shown in parenthesis.
Activity with 7-BR follows the Hill equation with an n value of 1.6 and S50 of 1.2 μM in 2B11 and n value of 1.3 and S50 of 2.5 μM in 2B11 V183L.
Km and kcat values for each individual experiment, typically based on n = 7 substrate concentrations, were determined by the Eadie-Hofstee method. 4-hydroxylation was assayed.
Molecular Modeling
The P450 2B11dH model was made essentially as described earlier for P450 2B1dH [7]. In brief, the initial molecular model was constructed using the InsightII software package (Molecular Simulations, Inc., San Diego, CA). The models were constructed based on the crystal structure of a ligand-bound P450 2B4dH/H226Y complex (PDB entry 1SUO) [11]. The sequence of P450 2B11dH was obtained from SwissProt (accession number P00176). The coordinates of the conserved residues were assigned based on the corresponding residues of the P450 2B4dH complex. The heme group was copied from P450 2B4dH into the P450 2B11dH model. For the P450 2B11dH mutants, the coordinates of the corresponding residues were changed in the respective 3D models, and the resulting mutants were energy minimized.
7-EFC was automatically docked into the 3D models of P450 2B11dH, I209A, F202L, and F202L/I209A in an orientation leading to O-deethylation using InsightII as described [7, 25]. Similarly, testosterone was docked into the 3D models of P450 2B11dH and I209A in a reactive binding orientation leading to 16α- or 16β-hydroxylation. Since the initial oxidation step involves hydrogen abstraction, the reactive carbon atom was placed 3.7 Å from ferryl oxygen with C-H-ferryl O angle of 180° to promote hydrogen bond formation. During the energy minimization process, the substrate molecule and side chains of protein residues within 10 Å of the substrate were allowed to move. The non-bond interaction energies were evaluated with the Docking module of Insight II, and the lowest energy orientation obtained after molecular mechanics minimization of P450 2B11dH and mutants are shown in Figure 4.
Figure 4. Docking of 7-EFC into the active site of P450 2B11 models.

7-EFC was docked in the P450 models in an orientation leading to formation of 7-hydroxy-4-trifluorcoumarin. The heme (red sticks), 7-EFC (cyan sticks), and mutant residues (blue sticks) are shown. The distance between the closest atom of 7-EFC and amino acid side chain is as follows. 1) P450 2B11dH: Ile209-7-EFC = 2.3 Å, Phe202 = 7.7 Å; 2) I209A: Ala209-7-EFC = 2.7 Å, Phe202-7-EFC = 8.4 Å; 3) F202L: Ile209-7-EFC = 2.2 Å, Leu202-7-EFC = 8.9 Å; 4) F202L/L209A; Ala209-7-EFC = 2.6 Å, Leu202-7-EFC = 9.5 Å.
RESULTS
Oxidation of 7-EFC and 7-BR by P450 2B11dH and mutants
Initially, oxidation of two model fluorogenic substrates, 7-BR and 7-EFC, was assayed in reactions catalyzed by P450 2B11dH and six mutants encompassing three amino acids: V183L, F202L, I209A, F202L/I209A, V183L/I209A, and V183L/F202L/I209A (Table 1). Sigmoidal kinetics were observed for 7-BR oxidation by 2B11dH and all of the mutants. Compared with P450 2B11dH, the V183L mutant showed a 4-fold enhanced kcat with 7-BR with a slight increase in S50 and decrease in n values. However, F202L and I209A both showed 2- to 3-fold decreased kcat values, and combination of the mutants (F202L/I209A) further decreased the kcat to < 10% of 2B11dH (Table 1). Introduction of V183L into either I209A or F202L/I209A increased the kcat compared with I209A and F202L/I209A, respectively, but resulted in a lower kcat than V183L alone. In the case of 7-EFC oxidation, Michaelis-Menten kinetics were observed for all samples. I209A exhibited a markedly lower kcat than P450 2B11dH with 7-EFC (1.6 min−1 vs. 13 min−1), but showed a 3.2-fold overall increase in kcat/Km due to a 25-fold decrease in Km (0.72 vs. 18 μM). V183L and F202L showed > 2-fold lower kcat/Km than P450 2B11dH. As expected, incorporation of I209A into F202L or V183L increased the kcat/Km but yielded a lower kcat/Km than I209A (Table 1). Overall, compared with P450 2B11dH, V183L showed enhanced kcat with 7-BR, whereas I209A showed enhanced kcat/Km with 7-EFC, suggesting distinct roles of these residues in substrate oxidation.
Testosterone hydroxylation by P450 2B11dH and mutants
Another model reaction of the P450 2B enzymes, testosterone hydroxylation, was initially studied with P450 2B11dH and mutants at 200 μM. V183L showed enhanced activity, whereas I209A showed increased selectivity for 16β- over 16α-hydroxylation compared with P450 2B11dH (data not shown). Therefore, steady-state kinetic analysis of testosterone 16α- and 16β-hydroxylation by P450 2B11dH and its mutants was performed (Table 2). While V183L showed a 4.7-fold increase in kcat/Km for testosterone 16α-hydroxylation, F202L and I209A showed a 4- and 1.5-fold decreased kcat/Km, respectively. Incorporation of I209A, or F202L/I209A into V183L yielded a lower kcat/Km than V183L and a higher kcat/Km than F202L, I209A, or F202L/I209A (Table 2). Interestingly, I209A showed an altered stereoselectivity of testosterone hydroxylation, leading to preferred catalysis at the 16β-site (kcat/Km of 1.1 at 16β vs. 0.23 at 16α), whereas P450 2B11dH and the other mutants showed no detectable testosterone 16β-hydroxylation activity. As expected, incorporation of I209A or F202L/I209A into V183L resulted in a decreased kcat/Km for 16α-hydroxylation and an increased kcat/Km for 16β-hydroxylation compared with V183L (Table 2). Overall, V183L, I209A, V183L/I209A, and V183L/F202L/I209A demonstrated ~5-fold higher kcat/Km for total testosterone hydroxylation than P450 2B11dH. These results suggested that both V183 and I209 in P450 2B11dH have important roles in testosterone metabolism.
Table 2.
Steady-state kinetics of 2B11dH and its mutants: NADPH-dependent hydroxylation of testosterone
| Testosterone 16α-OH | Testosterone 16β-OH | |||||
|---|---|---|---|---|---|---|
| P450s | kcat(min−1) | Km (μM) | kcat/Km | kcat(min−1) | Km (μM) | kcat/Km |
| 2B11dH | 5.6 (0.20)a | 16 (1.6) | 0.34 | ND | ND | ND |
| V183L | 9.7 (0.44) | 6.1 (1.0) | 1.6 | ND | ND | ND |
| F202L | 1.2 (0.04) | 14 (1.3) | 0.08 | ND | ND | ND |
| I209A | 3.2 (0.06) | 14 (0.85) | 0.23 | 15 (0.3) | 13 (0.9) | 1.1 |
| F202L/I209A | 2.0 (0.12) | 34 (2.1) | 0.06 | 2.7 (0.15) | 37 (3.2) | 0.07 |
| V183L/I209A | 3.1 (0.16) | 8.8 (1.6) | 0.35 | 13 (0.4) | 9.7 (1.0) | 1.4 |
| V183L/F202L/I209A | 6.7 (0.27) | 8.5 (1.2) | 0.79 | 7.3 (0.2) | 9.5 (1.0) | 0.77 |
Results are the representative of at 2–3 independent determinations. The variation between the experiments is ≤ 15%.
Standard errors for fit to Michaelis-Menten are shown in parenthesis. ND - Not determined due to very low activity.
Activation of the anti-cancer prodrugs IFA and CPA by P450 2B11dH and mutants
P450 2B11 is the most efficient enzyme for conversion of the anti-cancer prodrugs CPA and IFA into therapeutically active metabolites. Therefore, P450 2B11dH and its mutants were further characterized with CPA and IFA. Compared with P450 2B11dH, V183L showed a 1.7-fold higher kcat/Km for CPA and IFA 4-hydroxylation (Fig. 3). The increase in kcat/Km was primarily due to a ~4-fold decrease in Km (0.06 mM vs. 0.21 mM for CPA and 0.03 mM vs. 0.12 mM for IFA). However, F202L and I209A showed a > 12- and > 20-fold decrease in kcat/Km with CPA and IFA, respectively (Fig. 3), reflecting both a decrease in kcat and an increase in Km. Overall, our results suggested that the Val183→Leu substitution is the only non-active site mutation that enhances the activation of CPA and IFA.
Figure 3. Steady state kinetics of CPA and IFA 4-hydroxylation.

4-hydroxylase activities were determined for CPA and for IFA as described in Material and Methods. The Y-axis displays Km, kcat, and kcat/Km, values. Km and kcat values for each experiment, typically based on n = 7 substrate concentrations, were determined by the Eadie-Hofstee method. Data points fit the Eadie-Hofstee plot with r values of 0.93–0.97.
Substrate metabolism by full-length P450 2B11 and P450 2B11 V183L
Next, we investigated whether the enhanced activity of V183L in the P450 2B11dH background is also manifested in full-length P450 2B11. Results of the kinetic analysis of P450 2B11 and V183L with 7-EFC, 7-BR, testosterone, CPA, and IFA are shown in Table 3. Compared with P450 2B11dH, full-length P450 2B11 showed a > 6-fold higher kcat with 7-BR, a > 1.5-fold increased kcat/Km with 7-EFC, and a 2-fold increased kcat/Km with testosterone (Tables 1 and 2 vs. Table 3). In contrast, compared with P450 2B11dH, P450 2B11 showed > 20% decreased kcat/Km with CPA and IFA (Fig 4 vs. Table 3). Although V183L showed a 2-fold increase in kcat with 7-BR, it did not show increases in kcat/Km with 7-EFC or testosterone; rather, kcat/Km was decreased with 7-EFC compared with P450 2B11. In addition, V183L showed a further decrease in kcat/Km with CPA and IFA in the P450 2B11 background (Table 3), indicating that the Val183→Leu substitution is not beneficial when incorporated into full-length P450 2B11.
Molecular docking of 7-EFC and testosterone
7-EFC was docked in the active site of P450 2B11dH, F202L, I209A, and F202L/I209A (Fig. 4) to help explain the observed changes in substrate metabolism, including: 1) the very low Km with 7-EFC of I209A (Table 1) and 2) the decreased kcat/Km with 7-EFC caused by the Phe202→Leu substitution in P450 2B11dH and I209A backgrounds (Table 1). Ile-209 was close to the active site of P450 2B11dH (2.3 Å from the closest atom of 7-EFC) compared with the 5.8 Å between Leu-209 and 7-EFC in P450 2B1 [25]. In 2B11dH I209A, the distance between Ala-202 and 7-EFC was increased (2.7 Å). Docking of 7-EFC in the active site of F202L oriented Leu-202 away from the substrate and increased its distance from 7-EFC (8.9 Å; Fig. 4), compared with the distance between Phe-202 and 7-EFC in P450 2B11dH (7.2 Å). When 7-EFC was docked in the active site of I209A/F202L, the distance between Leu-202 and 7-EFC was increased further (9.5 Å).
Testosterone was docked in the active site of P450 2B11dH and I209A to better understand the enhanced selectivity of I209A for 16β-hydroxylation over 16α-hydroxylation compared with P450 2B11dH (Table 2). The results showed that testosterone fits well in the P450 2B11dH active site, with no van der Waals overlaps when docked in an orientation that leads to formation of 16α-OH testosterone, while it fits well in the 16β orientation when docked in the active site of I209A (data not shown). However, the distance (C16-ferryl oxygen) and angle (C-H-ferryl O) were unfavorable when testosterone was docked in the 16β orientation in P450 2B11dH or in the 16α orientation in I209A.
Discussion
Mammalian P450s have been engineered for enhanced activity, substrate specificity, and stereo- and regioselectivity within and across subfamilies by site-directed mutagenesis of active site residues [reviewed in 2, 3]. Because residues outside of the active site may also be critical for substrate metabolism, we recently developed a directed evolution approach to engineer P450 2B1 for enhanced activity with several substrates including the anti-cancer prodrugs CPA and IFA [24]. In the present study we engineered P450 2B11, the most efficient P450 enzyme for the activation of the anti-cancer prodrugs CPA and IFA [4, 6], by replacing residues V183, F202 and I209 with the corresponding residues identified by directed evolution of P450 2B1 [24]. When introduced into P450 2B11dH, V183L showed enhanced metabolism of the majority of the substrates tested, including CPA and IFA. To the best of our knowledge this is the first report on rational re-engineering of a mammalian P450 based on non-active site mutations created by directed evolution of a P450 from the same subfamily.
As N-terminal truncated mammalian P450 enzymes have been used for all x-ray crystal structures determined to date [3], we investigated the effect of the N-terminal modification on the steady state kinetics of P450 2B11 with five substrates. Compared with P450 2B11, the modified P450, P450 2B11dH, showed decreased kcat/Km with 7-BR, 7-EFC, and testosterone, mainly as the result of a decreased kcat (Fig. 5). However, P450 2B11dH showed higher kcat/Km than P450 2B11 with CPA and IFA, as the result of a decrease in Km. The basis for such substrate-specific differences in kcat or Km is not clear, primarily because of a lack of x-ray crystal structure of a full-length mammalian P450 enzyme. These findings are similar to our earlier observations with P450 2B1 and 2B1dH [4, 9, 24, 28]. Relative activity of the full-length and truncated P450 2B enzymes is dependent on the substrate and on the availability of DLPC [28]. However, in this case the only difference between the assays encompassing the two sets of substrates (7-EFC, 7-BR, and testosterone vs. CPA and IFA) is the availability of cytochrome b5. Cytochrome b5 can either activate or inhibit P450 activity [30–31], with the effect dependent on the types of P450, lipid, substrate, and oxidant used [26, 30–32].
Figure 5. Relative kcat/Km.
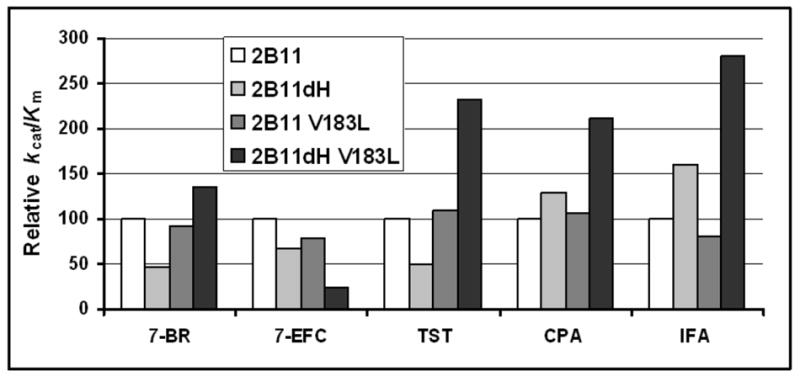
The kcat/Km with five distinct substrates was re-plotted to compare the effect of the N-terminal truncation and Val183→Leu substitution. kcat/S50 was used in the case of 7-BR for better comparison. TST: testosterone. In each case, the kcat/Km of full-length P450 2B11 is set to 100%.
Interestingly, the Val183→Leu substitution had distinct effects on P450s 2B11 and 2B11dH (Fig. 5). Val183→Leu substitution in the full-length P450 2B11 had no significant effect on catalytic efficiency with any of the substrates. However, Val183→Leu substitution in P450 2B11dH increased catalytic efficiency with 7-BR, testosterone, CPA, and IFA. Thus, it appears that the reduced activity of P450 2B11dH with 7-BR and testosterone compared with P450 2B11 is fully reversed by the Val183→Leu substitution. This is not due to the absence of DLPC in the standard assay with the dH proteins. Accordingly, although there was a slight increase in the 7-BR debenzylation activity by P450 2B11dH and V183L in the presence of DLPC, the Val183→Leu substitution still caused a > 2-fold increase in activity (data not shown). Val183→Leu appears to be an optimal substitution based on analysis of several additional substitutions at residue 183, including Ala, Ile, Trp, Tyr, and Thr. While mostly showing higher kcat/Km than P450 2B11dH, none of these mutants exhibited a higher kcat/Km than V183L with 7-EFC, 7-BR, or testosterone (data not shown).
Val183→Leu substitution in P450 2B1dH and 2B11dH yielded similarities as well as differences in substrate metabolism with a panel of substrates [24]. For example, with both P450s, V183L showed increased kcat/Km for CPA and IFA 4-hydroxylation and decreased kcat/Km for 7-EFC O-deethylation [24], whereas with 7-BR and testosterone, V183L showed decreased and increased activity in P450 2B1dH and 2B11dH, respectively. The ligand-free and 4-(4-chlorophenyl)imidazole (4-CPI)- and bifonazole (BIF)-bound P450 2B4 x-ray crystal structures as well as homology models of the P450 2B enzymes based on the CPI-bound 2B4 crystal structure do not predict any functional role of Val-183 (2, 3, 10–12). However, comparison of the ligand-free, and the 4-CPI-, and BIF-bound x-ray crystal structures identified five plastic regions (PR) in P450 2B4, where there are > 1 Å variations in the Fe-Cα distances [12]. Region PR3 spans residues 177 and 188, which might explain a possible role of the Val183→Leu substitution. The only other evidence for a role of this residue is that an Ile187→Thr substitution in a P450 3A26/3A12 (182/183 in 2B11) hybrid enzyme increased progesterone hydroxylase activity [33].
I209A showed a > 3-fold higher kcat/Km than P450 2B11dH, primarily as the result of a dramatic decrease in the Km value (> 25-fold). This is the lowest Km ever reported for a P450 2B enzymes with 7-EFC. In our earlier studies, L209A showed a > 5-fold higher kcat or kcat/Km than P450 2B1dH with 7-EFC and testosterone [9]. Unlike P450 2B11dH I209A, 2B1dH L209A showed an increased kcat without a significant change in Km or S50. In modeling simulations Ile-209 was 2.3 Å from 7-EFC compared with the 5.8 Å between Leu-209 and 7-EFC in P450 2B1 [25], which may introduce an unfavorable interaction and steric constraint in the 7-EFC structure (Fig. 4). The Ile209→Ala substitution in P450 2B11dH appears to slightly relieve this constraint by increasing the distance of Ala-209 from the 7-EFC (2.7 Å), which may lead to a decreased Km.
F202L is deleterious when introduced into 2B11dH I209A, whereas it is beneficial in the context of 2B1dH L209A [25]. Interestingly, docking of 7-EFC in the active site of P450 2B11dH showed that the Phe202→Leu substitution in the presence of I209A orients Leu-202 away from the substrate and increases its distance from 7-EFC (Fig. 4). In contrast, Leu-202 faces towards 7-EFC in P450 2B1dH L209A and V183L/F202L/L209A/S334P mutants and is present in the active site [25]. The opposite orientations of Leu-202 may explain the contrasting effects of F202L in the background of P450 2B1dH L209A and 2B11dH I209A.
In summary, the key findings of the current study are: 1) P450 2B11dH V183L is the most efficient P450 2B11 enzyme for the metabolism of 7-BR and testosterone; 2) P450 2B11dH V183L is the most efficient P450 enzyme for the activation of CPA and IFA; 3) P450 2B11dH I209A exhibits the lowest Km for 7-EFC among the P450s studied; 4) compared with P450 2B11dH, I209A shows enhanced stereoselectivity and activity for testosterone 16β-hydroxylation. The current study serves as an example of re-engineering P450 from the same subfamily by replacing non-active site residues identified by directed evolution. This strategy may be applicable to the engineering of mammalian P450s across P450 subfamilies for a variety of applications in industry, such as biotechnology and environmental bioremediation, as well as novel applications in drug metabolism and gene therapy. P450 2B11 has shown strong promise in prodrug activation-based gene therapy for cancer using the anti-cancer prodrugs CPA and IFA [6, 34], and mutants with improved catalytic efficiency, such as those characterized here, may ultimately find applications in the clinic.
Acknowledgments
The Authors acknowledge Mr. Tori Strong (Pharmacology and Toxicology, UTMB) for generating the substrate structures, presented in Figure 2. Financial support was provided by NIH Grant ES03619 and Center grant ES06676 (to J.R.H.), NIH Grant CA49248 (to D.J.W.) and the Superfund Basic Research Center at Boston University, NIH grant 5 P42 ES07381.
Abbreviations used
- CPA
cyclophosphamide
- IFA
ifosafamide
- GDEPT
gene-directed enzyme prodrug therapy
- 7-EFC
7-ethoxy-4-trifluoromethylcourmarin
- 7-BR
7-benzyloxyresorufin
- CPR
NADPH-cytochrome P450 reductase
- PCR
polymerase chain reaction
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Anzenbacher P, Anzenbacherova E. Cell Mol Life Sci. 2001;58:737–747. doi: 10.1007/PL00000897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Domanski TL, Halpert JR. Curr Drug Metab. 2001;2:117–137. doi: 10.2174/1389200013338612. [DOI] [PubMed] [Google Scholar]
- 3.Zhao YH, Halpert JR. Arch Biochem Biophys. 2006 E. Pub. July 22. [Google Scholar]
- 4.Chen CS, Lin JT, Goss KA, He YA, Halpert JR, Waxman DJ. Mol Pharmacol. 2004;65:1278–1285. doi: 10.1124/mol.65.5.1278. [DOI] [PubMed] [Google Scholar]
- 5.Waller SC, He YA, Harlow GR, He YQ, Mash EA, Halpert JR. Chem Res Toxicol. 1999;12:690–699. doi: 10.1021/tx990030j. [DOI] [PubMed] [Google Scholar]
- 6.Jounaidi Y, Chen CS, Veal GJ, Waxman DJ. Mol Cancer Therap. 2006;5:541–555. doi: 10.1158/1535-7163.MCT-05-0321. [DOI] [PubMed] [Google Scholar]
- 7.Kumar S, Scott EE, Liu H, Halpert JR. J Biol Chem. 2003;278:17178–17184. doi: 10.1074/jbc.M212515200. [DOI] [PubMed] [Google Scholar]
- 8.Strobel SM, Halpert JR. Biochemistry. 1997;36:11697–11706. doi: 10.1021/bi9710176. [DOI] [PubMed] [Google Scholar]
- 9.Scott EE, He YQ, Halpert JR. Chem Res Toxicol. 2002;15:1407–1413. doi: 10.1021/tx020057u. [DOI] [PubMed] [Google Scholar]
- 10.Scott EE, He YA, Wester MR, White MR, Chin C, Halpert JR, Johnson EF, Stout CD. Proc Nat Acad Sci. 2003;100:13196–13201. doi: 10.1073/pnas.2133986100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Scott EE, White MA, He YA, Johnson EF, Halpert JR. J Biol Chem. 2004;279:27294–27301. doi: 10.1074/jbc.M403349200. [DOI] [PubMed] [Google Scholar]
- 12.Zhao Y, White MA, Muralidhara BK, Sun L, Halpert JR, Stout CD. J Biol Chem. 2006;281:5973–5981. doi: 10.1074/jbc.M511464200. [DOI] [PubMed] [Google Scholar]
- 13.Scott EE, Liu H, He YQ, Li W, Halpert JR. Arch Biochem Biophys. 2004;423:266–276. doi: 10.1016/j.abb.2003.12.035. [DOI] [PubMed] [Google Scholar]
- 14.Honma W, Li W, Liu H, Scott EE, Halpert JR. Arch Biochem Biophys. 2005;435:157–165. doi: 10.1016/j.abb.2004.12.014. [DOI] [PubMed] [Google Scholar]
- 15.Hernandez CE, Kumar S, Liu H, Halpert JR. Arch Biochem Biophys. 2006;455:61–67. doi: 10.1016/j.abb.2006.08.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Farinas ET, Schwaneberg U, Glieder A, Arnold FH. Adv Synth Catl. 2001;343:6–7. [Google Scholar]
- 17.Glieder A, Farinas ET, Arnold FH. Nature Biotechnology. 2002;20:1135–1139. doi: 10.1038/nbt744. [DOI] [PubMed] [Google Scholar]
- 18.Joo H, Lin Z, Arnold FH. Nature. 1999;399:670–673. doi: 10.1038/21395. [DOI] [PubMed] [Google Scholar]
- 19.Parikh A, Josephy PD, Guengerich FP. Biochemistry. 1999;38:5283–5289. doi: 10.1021/bi990142+. [DOI] [PubMed] [Google Scholar]
- 20.Gillam EM, Aguinaldo JAMA, Notley LM, Kim D, Mundkowski RG, Volkov AA, Arnold FH, Soucek P, DeVoss JJ, Guengerich FP. Biochem Biophy Res Comm. 1999;265:468–472. doi: 10.1006/bbrc.1999.1702. [DOI] [PubMed] [Google Scholar]
- 21.Nakamura K, Martin MV, Guengerich FP. Arch Biochem Biophy. 2001;395:25–31. doi: 10.1006/abbi.2001.2569. [DOI] [PubMed] [Google Scholar]
- 22.Kim D, Guengerich FP. Biochemistry. 2004;43:981–988. doi: 10.1021/bi035593f. [DOI] [PubMed] [Google Scholar]
- 23.Kim D, Guengerich FP. Arch Biochem Biophys. 2004;432:102–108. doi: 10.1016/j.abb.2004.09.001. [DOI] [PubMed] [Google Scholar]
- 24.Kumar S, Chen CS, Waxman DJ, Halpert JR. J Biol Chem. 2005;280:19569–19575. doi: 10.1074/jbc.M500158200. [DOI] [PubMed] [Google Scholar]
- 25.Kumar S, Sun L, Liu H, Muralidhara BK, Halpert JR. Protein Eng Des Select. 2006;19:547–554. doi: 10.1093/protein/gzl042. [DOI] [PubMed] [Google Scholar]
- 26.Kumar S, Liu H, Halpert JR. Drug Metab Dispos. 2006;34:1958–1965. doi: 10.1124/dmd.106.012054. [DOI] [PubMed] [Google Scholar]
- 27.Harlow GR, He YA, Halpert JR. Biochem Biophys Acta. 1997;1338:259–266. doi: 10.1016/s0167-4838(96)00209-9. [DOI] [PubMed] [Google Scholar]
- 28.Scott EE, Spatzenegger M, Halpert JR. Arch Biochem Biophys. 2001;395:57–68. doi: 10.1006/abbi.2001.2574. [DOI] [PubMed] [Google Scholar]
- 29.John GH, Hasler JA, He YA, Halpert JR. Arch Biochem Biophys. 1994;314:67–375. doi: 10.1006/abbi.1994.1455. [DOI] [PubMed] [Google Scholar]
- 30.Porter TD. J Biochem Mol Pharmacol. 2002;16:311–316. doi: 10.1002/jbt.10052. [DOI] [PubMed] [Google Scholar]
- 31.Schenkman JB, Jansson I. Pharmacol Therap. 2003;97:139–152. doi: 10.1016/s0163-7258(02)00327-3. [DOI] [PubMed] [Google Scholar]
- 32.Kumar S, Davydov DR, Halpert JR. Drug Metab Dispos. 2005;33:1131–1136. doi: 10.1124/dmd.105.004606. [DOI] [PubMed] [Google Scholar]
- 33.Fraser DF, Feyereisen R, Harlow GR, Halpert JR. Mol Pharmacol. 1999;55:241–247. doi: 10.1124/mol.55.2.241. [DOI] [PubMed] [Google Scholar]
- 34.Roy P, Waxman DJ. Toxicology in Vitro. 2006;20:176–186. doi: 10.1016/j.tiv.2005.06.046. [DOI] [PubMed] [Google Scholar]