

Abstract
Dengue virus is a mosquito-borne flavivirus that represents an important emerging infectious disease and is an international health concern. Currently, there is no vaccine or effective antiviral therapy to prevent or to treat dengue virus infection. The slow progress in developing antiviral agents might be alleviated by the availability of efficient high-throughput anti-dengue virus screening assays. In this study, we report an immunofluorescence image-based assay suitable for identification of small molecule inhibitors of dengue virus infection and replication. Using this assay, we have discovered that inhibitors of the c-Src protein kinase exhibit a potent inhibitory effect on dengue virus (serotypes 1–4) and murine flavivirus Modoc. Mechanism of action studies demonstrated that the c-Src protein kinase inhibitor dasatinib prevents the assembly of dengue virions within the virus-induced membranous replication complex. These results demonstrate that this cell-based screen may provide a powerful means to identify new potential targets for anti-dengue drug development while simultaneously providing pharmacological probes to investigate dengue virus–host cell interactions at the biochemical level. Given the simplicity and excellent reproducibility of the assay, it should be useful in high-throughput screens of both small molecule and RNAi libraries when implemented on a robotic image-based high-throughput screen (HTS) platform. Given the reasonable clinical safety of inhibitors such as dasatinib and AZD0530, inhibitors of c-Src protein kinase may have the potential to become a new class of anti-dengue viral therapeutic agents.
Keywords: antiviral, chemical biology, flavivirus, small molecule inhibitor, high-throughput screen
Dengue virus (DENV) is an emerging mosquito-borne pathogen that causes dengue fever (DF) and a severe life-threatening illness, dengue hemorrhagic fever/dengue shock syndrome (DHF/DSS) (1). DENV is a small, enveloped, positive-stranded RNA virus that belongs to the Flavivirus genus of the Flaviviridae family. Four distinct serotypes (DENV1 to -4) of dengue viruses are transmitted to humans through the bites of the mosquito species, Aedes aegypti and Aedes albopictus (2). It has been estimated that ≈50–100 million cases of DF, and ≈250,000–500,000 cases of DHF occur every year (3). Furthermore, 2.5 billion of people are at risk for infection in subtropical and tropical regions of the world (4) in the absence of effective intervention. The intracellular life cycle of DENV begins with receptor-mediated endocytosis of the virus into cells, followed by fusion of the viral envelope protein with the late endosomal membrane, which results in the release of the viral genome into the cytoplasm for replication. Replication of the viral RNA genome occurs within membrane-bound complexes formed from the endoplasmic reticulum membrane. Subsequently, virus particles are assembled and released via the host cell secretory machinery (5). Although replication of DENV involves complex interaction between viral proteins and cellular factors, many of these interactions remain unidentified and uncharacterized. Small molecules that specifically target different steps in the viral replication cycle could potentially be used as “tool compounds” to facilitate biochemical characterization of these host–virus interactions and might also be used to identify pharmacological intervention points for treatment of DENV infection. Although extensive studies have been carried out over the years to understand the pathogenicity of DENV infection, little progress has been made in the development of specific anti-DENV compounds. Currently, there are no specific treatments for DENV infection, and vaccines are unavailable.
In this article, we report the development of a microscopy-based immunofluorescence assay that allows screening for small molecules that inhibit any step(s) in the DENV replication cycle, including entry, viral RNA replication, and virion assembly and secretion. Phosphorylation of proteins by kinases is responsible for the transmission of biochemical signals in many signal transduction pathways, including those promoting cell survival (6, 7) and immune evasion (8, 9) during DENV infection as well as those regulating endocytosis of other viruses (10). In addition, phosphorylation of viral proteins such as DENV NS5 (11, 12) by cellular kinases is known to regulate their subcellular localization and, it is presumed, their functions. Hypothesizing that kinase inhibitors could be used to probe the impact of cellular kinases and their associated signaling pathways on DENV infection and replication, we screened a collection of 120 known inhibitors of mammalian Ser/Thr and Tyr kinases. A number of the protein kinase inhibitors were found to affect distinct steps in the DENV replication cycle and to cause multilog decreases in viral titer in the absence of cytotoxicity. These findings provide pharmacological evidence that host–cell kinase activity is essential for various stages of the DENV life cycle and may provide new insights for a possible anti-DENV therapy.
Results
Screen Development.
In this study, a screen for small molecule inhibitors of DENV replication was developed to detect small molecules capable of interfering with the different step(s) of the DENV replication cycle through their direct effects on viral gene products or through their interactions with cellular factors that participate in viral processes. The image-based assay is based on the detection of DENV envelope protein and is outlined in supporting information (SI) Fig. 6. We first evaluated the ability of the assay to quantitatively detect inhibition of DENV infection by a small molecule, mycophenolic acid (MPA), which is known to inhibit the viral RNA synthesis of DENV (13). Vero cells cultured in a 384-well plate were first infected with DENV 2 at a multiplicity of infection (moi) of 1 and then incubated with different concentrations of MPA. Three days after infection, cells were fixed and stained for viral envelope protein (Env). Fig. 1A shows the reduction in the number of cells staining positively for DENV Env protein with increasing concentrations of MPA treatment when compared with the untreated, DENV-infected cells. By using an automated microscope, the cytoplasmic green fluorescence and nuclear blue fluorescence acquired from four randomly selected fields in each well were imaged and the average number of DENV-infected cells was then determined by automated data analysis. The percentage inhibition of DENV infection was measured as a function of MPA concentration (Fig. 1B) and found to be dose-dependent, with an EC50 of 5.7 μM. This result was consistent with previous measurements of the anti-DENV activity of MPA (13). To ensure that the assay has minimal signal variation and a consistently high signal-to-background ratio, we also determined the Z′ factor of the screening assay (14) based on data collected from 100 wells of DENV-infected cells treated with 18 μM of MPA (final concentration of 0.25% DMSO) and data collected from another 100 wells of the same 384-well plate that were infected with DENV and treated with 0.25% DMSO only in cell culture medium. A signal-to-background ratio of 8:1 and a Z′ factor of 0.75 were consistently observed, demonstrating the reliability and robustness of this assay. We then turned to use of this assay as a tool for the discovery of small molecule inhibitors of DENV.
Fig. 1.

Development of an image-based immunofluorescence assay for detection of DENV. (A) Detection of DENV infection of Vero cells using immunofluorescence staining for DENV envelope protein (Env) in the absence and presence of MPA. (B) Dosage-dependent inhibition of DENV 2 infection of cells treated with MPA.
Screening of a Protein Kinase Inhibitor Library.
With the hypothesis that kinase inhibitors can be used to unmask the cellular signal transduction pathways co-opted by DENV during its replication cycle, we used the immunofluorescence assay to screen a collection of over 120 inhibitors of mammalian Ser/Thr and Tyr kinases at concentrations predetermined to be noncytotoxic. The host cell target(s) reported to be affected by the members of the protein kinase panel are described in SI Table 2. This kinase inhibitor panel notably includes multiple distinct scaffold classes for many kinases, a feature intended to facilitate determination of which kinase is responsible for a particular anti-DENV effect.
Employing a 50% reduction in the number of fluorescently stained DENV-infected cells as the criterion for further evaluation, inhibitors of seventeen protein kinases were identified (Table 1). Of the 44 different cellular kinases known to be targeted by our inhibitor collection, inhibition of cyclin-dependent (CDKs), JAK, casein (CK), Src-family, and epidermal growth factor receptor tyrosine (EGFRs) kinases was associated with anti-DENV activity. Notably, multiple compounds with inhibitory activity against Src-family, Bcr-Abl/c-Abl, c-Kit, and platelet-derived growth factor receptor (PDGFR) kinases were associated with anti-DENV activity and had diverse chemical structures (SI Table 3).
Table 1.
Protein kinase inhibitors exhibiting inhibitory effects on DENV infection
| Inhibitor | Src | Abl | c-Kit, PDGFR, VEGFR | Other kinase targets | Pre | Post |
|---|---|---|---|---|---|---|
| K002 | CDKs | X | ||||
| K014 (Imatinib) | X | X | X | |||
| K039 | c-Raf | X | ||||
| K040 | JAK1, -2, -3 | X | ||||
| K003 | X | X | X | X | X | |
| K013 (GNF2) | X | X | X | |||
| K030 | X | Kdr | X | X | ||
| K032 | CK II | X | X | |||
| K117 | X | X | X | |||
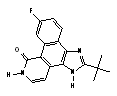
| K045 (AZD0530) | X | X | X | |||
| K005 (Dasatinib) | X | X | X | X | ||
| K025 (SU11652) | X | FGFR | X | |||
| K028 (Lavendustin A) | X | EGFR | X | |||
| K026 (SU5271) | EGFR | X | ||||
| K144 (Kenpaullone) | X | CDKs, GSK3-b | X | |||
| K115 (Lavendustin C) | X | CaMK II | X | |||
| K116 (MC7) | MLCK | X | ||||
| K118 (Tyrphostin46) | X | X | X | Multi-targeted | X |
Because kinase inhibitors (including the ones used in this study) often target multiple intracellular enzymes, we used the partially overlapping selectivities of these inhibitors to determine which cellular kinases might actually be responsible for the observed anti-DENV effects. An additional inhibitor, AZD0530 (K045), was added to the secondary screen to assist in deconvolution of the kinase inhibitory activities associated with compounds discovered in the primary screen. The eighteen compounds in Table 1 were evaluated in a set of secondary assays to quantify changes in viral titer when cells were treated with inhibitors either pre- or post-inoculation (SI Fig. 6). These experiments enabled us to measure the effect of each inhibitor on the output of DENV infection and to distinguish between the effects of individual kinase inhibitors on processes occurring early in the DENV replication cycle (e.g., viral entry, genome release) and that can be blocked by treatment of host cells before inoculation with DENV from effects on processes occurring later in the DENV replication cycle (e.g., viral RNA replication, virion assembly and egress) and that can only be targeted by continuous kinase inhibition. All compounds were assayed at two noncytotoxic concentrations and were considered to have anti-DENV activity if associated with at least a five-fold (0.7 log unit) decrease in viral titer.
All compounds were confirmed to have anti-DENV activity when added pre- or postinoculation with some compounds exhibiting anti-DENV activity under both sets of conditions (Table 1). Inhibitors of cyclin-dependent kinases (K002), c-Raf (K039), the JAK (K040), and the multitargeted inhibitor imatinib (K014), seemed to affect events early in viral infection (e.g., viral entry, genome release) because incubation of host cells with these compounds for a three-hour period before inoculation was sufficient to cause significant reductions in DENV titer. Five compounds collectively targeting Src-family (K003, K030, K117), Abl (K003, K013), c-Kit (K003), PDGFR (K003), VEGFR (K003, K030) kinases and casein kinase II (CK II, K032) were found to have anti-DENV activity in both pre- and postinoculation assays. These data suggested that these compounds may inhibit kinases that are important for multiple viral processes that occur during or after viral entry.
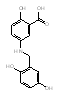
The remaining nine compounds, all of which are known to target two or more kinases, exhibited anti-DENV activity exclusively in the postinoculation assay. Of these nine inhibitors, six are known to have activity against Src-family kinases, making this the strongest association between inhibition of a cellular kinase and anti-DENV activity. Notably, all compounds in our library known to have potent activity against Src-family kinases (nine of 120 compounds tested in the primary screen and nine of seventeen compounds tested in the secondary screen) were also potent inhibitors of DENV (Table 1). This result included Src family inhibitors from the pyrimidopyrimidine compound class (K003), the quinazoline class (K030), the indoyl sulfonamide compound K117, as well as Kenpaullone (K144), and Lavendustins A (K028) and C (K115) (SI Table 3). This group of six compounds included two clinical stage candidates: dasatinib (SPRYCEL, K005), a thiazolylamino-pyrimidine compound currently approved for the treatment of imatinib-resistant chronic myelogenous leukemia (CML) (15, 16) and AZD0530 (K045), a quinazoline which is currently being tested in solid tumors and leukemia (17). Because of their respective stages of clinical development, we were particularly interested in understanding the mechanism responsible for the anti-DENV activity of these compounds.
Dasatinib is an ATP-competitive inhibitor that exhibits subnanomolar cellular activity against BCR-Abl (the primary target in CML patients) and Src-family kinases, and can also inhibit platelet derived growth factor receptor (PDGFR, IC50 28 nM), stem cell factor (c-kit, IC50 13 nM), ephrin A2 (EPHA2, IC50 137 nM) receptor tyrosine kinases and p38 MAP kinase (IC50 100 nM) (reviewed in (18)). AZD0530 is a considerably more selective kinase inhibitor, exhibiting potent activity across the Src-family of kinases and has even been shown to be ≈10-fold more active against Src family kinases (IC50 1–5 nM) than against Abl kinase (17). Although dasatinib and AZD0530 target other cellular kinases, the association of Src kinase activity with DENV infection prompted us to focus our follow-up efforts on the role of Src family kinases in the postentry steps of DENV infection and replication.
c-Src Protein Kinase Inhibitors Reduce DENV Infection.
To confirm that c-Src protein is indeed necessary for DENV2 replication, we examined the effect of dasatinib (K005) and AZD0530 (K045) on DENV2 infection in Vero, Huh-7, and C6/36 cells. Cells were infected with DENV2 (moi of 1) and treated with noncytotoxic concentrations (50 nM–5 μM) of dasatinib or AZD0530 for three days. Quantitation of viral titers in culture supernatants by plaque assay revealed a dose-dependent inhibition of DENV2 infection in all three cell lines treated with dasatinib (Fig. 2A) and AZD0530 (SI Fig. 7A). These results suggested that DENV replication requires a cellular kinase(s). Given the structurally diverse classes of Src kinase inhibitors uncovered in our screen, it likely that host cell Src kinase is the primary target rather than a viral protein.
Fig. 2.

Antiviral activity of dasatinib, an inhibitor of c-Src and Abl kinases, on DENV infection. (A) Dosage dependent inhibition of DENV2 infection was observed in C6/36, Vero, and Huh-7 cells. (B) Viral reduction immunofluorescence assays were performed for the indicated viruses to determine the dose-dependent anti-DENV activity of dasatinib.
The viral selectivity of dasatinib was evaluated by measuring its inhibitory effect under similar treatment conditions against viral clones representative of the other serotypes of DENV, as well as against Modoc virus (a murine flavivirus) and poliovirus type 1 (an RNA virus of the picornavirus family). Inhibition of DENV1, DENV3, DENV4, and Modoc virus infections of Vero cells by dasatinib occurred in a dosage-dependent manner similar to that observed for inhibition of DENV2 (Fig. 2B). In contrast, dasatinib exhibited no inhibitory effect against poliovirus type 1 (Fig. 2B). These results may suggest that cellular kinases targeted by dasatinib and AZD0530 are specific for the DENV sero-complex and may also extend to related flaviviruses but not to more distantly related RNA viruses. Although Src kinases are the most likely candidate for this cellular kinase, we could not rule out the possibility that dasatinib and AZD0530 share additional kinase targets that may be required for DENV replication.
siRNA Mediated Silencing of c-Src Protein Inhibits DENV Replication.
To determine whether c-Src is indeed the cellular kinase mediating the anti-DENV effect of dasatinib and AZD0530, we determined the effect of siRNA-mediated “knockdown” of c-Src protein expression on DENV replication. Reverse transfection of Huh-7 cells with a pool of four siRNAs specific for c-Src protein at final siRNA concentrations of 200 nM and 300 nM resulted in reduction of c-Src protein levels by 63% and 74%, respectively, when compared with cells transfected with the transfection control (Fig. 3A). Minimal cytotoxicity was observed with Huh-7 cells transfected with 200 nM and 300 nM of c-Src siRNA (data not shown). Huh-7 cells treated with the c-Src-specific siRNA pools were then infected with DENV2 (moi of 1), and production of virus by these cells was evaluated by viral plaque assay of culture supernatants collected on day 3 postinfection. Treatment of cells with 200 nM of an siRNA pool directed against c-Src resulted in a greater than 1.5 log unit inhibition of DENV titers whereas treatment with 200 nM of negative control siRNA pools had no effect on DENV titers (Fig. 3B). These data strongly suggest that Src family kinases are indeed responsible for the anti-DENV activity of dasatinib and AZD0530.
Fig. 3.

siRNA Knockdown of c-Src protein levels inhibits DENV infection. Huh-7 cells were transfected with different concentrations of a c-Src-specific siRNA pool or control siRNA. (A) Knockdown of c-Src protein expression was confirmed by Western blot analysis by using a c-Src specific antibody. Lane 1, Control (siGlo RNA-300 nM); lane 2, c-Src specific siRNA pool–200 nM; lane 3, c-Src specific siRNA pool–300 nM. Detection of GAPDH was included as a well loading control. (B) DENV2 infection was reduced by >1.5-log units in cells transfected with 200 nM of the c-Src siRNA pool but was unchanged in the presence of 200 nM siGlo or 200 nM GAPDH SMARTpool negative control reagents.
c-Src Is Required for DENV Assembly and Secretion.
The observation that c-Src protein kinase inhibitors such as dasatinib and AZD0530 exert an anti-DENV effect when added postinoculation but not when added preinoculation suggested that c-Src protein kinase activity may play an important role in postentry viral processes, such as viral RNA replication, virion assembly and maturation, or viral egression. We sought to clarify this issue by identifying the specific viral process blocked when Src protein kinase activity is inhibited. Immunofluorescence assays using three different cell lines (Vero, Huh-7, and C6/36) were first carried out to detect the production and intracellular distribution of viral envelope protein during the course of DENV infection (Fig. 4). Cells were infected with DENV2 (moi of 1) and then treated with 2.5 μM dasatinib. Cells were then fixed at various time points postinfection and DENV envelope protein was detected by immunofluorescence staining. At day 4 postinfection, the production of newly synthesized viral envelope protein was observed in all three cell lines despite treatment with dasatinib, indicating that viral gene expression was not affected by the c-Src protein kinase inhibitor. By days 5 and 6 postinfection, there were increasing accumulations of viral envelope protein within the perinuclear region of the dasatinib-treated, DENV-infected cells (Fig. 4, arrows). This observation was particularly noticeable in the Vero and Huh7 cells. Notably, cells surrounding the DENV2-infected cells remain uninfected (no viral envelope protein production was detected; Fig. 4, arrowheads) in dasatinib-treated samples. This effect on envelope protein localization and distribution was consistently observed for all three cell lines tested with both dasatinib (Fig. 4) and AZD0530 (SI Fig. 7B). In contrast, viral envelope protein was detected in all cells of DMSO-treated, DENV2-infected cell monolayers. For the DMSO-treated, DENV2-infected cells, viral envelope proteins were observed initially around the perinuclear region (day 4 postinfection). By day 5 and 6, viral envelope protein was observed throughout the cytoplasm and plasma membrane of the infected cells.
Fig. 4.

Inhibition of viral spread in dasatinib-treated, DENV-infected cells. An immunofluorescence assay was conducted to detect the localization of the viral envelope (E) protein in DENV-infected Vero, Huh-7, and C6/36 cells that were treated with 2.5 μM of dasatinib or DMSO. Noninfected cells are indicated by arrowheads, and accumulation of viral E protein in the perinuclear region is indicated by arrows. Cell nuclei are stained blue with DAPI.
These observations suggested that inhibition of c-Src protein kinases may contribute to retention of viral ENV within the ER resulting in failure of viral particles to form and/or in failure of assembled virus particles to transit through the host secretory system for release. To distinguish between these two possibilities, transmission electron microscopy was performed on DENV-infected cells treated with 2.5 μM of dasatinib or DMSO as a negative control. This concentration of dasatinib was chosen to avoid cytotoxicity and also to minimize effects mediated by other kinases targeted by dasatinib (i.e., c-kit, PDGFR, Ephrins, p38) at higher concentrations (17). As shown in Fig. 5, DENV2-infected cells treated with DMSO (Fig. 5A) or dastinib (Fig. 5B) exhibited membranous replication structures that are characteristic of flavivirus infection (5, 19). For the DMSO-treated samples (Fig. 5A), a large number of DENV particles (arrows) were observed within the ER lumen and secretory vesicles. In contrast, virus particles were not observed within the ER lumen for the dasatinib-treated sample (Fig. 5B). The dasatinib-treated, DENV-infected cells exhibited extensive proliferation of virus-induced membrane structures (arrowheads) (Fig. 5B). This observation may reflect accumulation of the viral envelope glycoprotein in the ER. Nucleocapsid-like particles (≈25 nm, arrows) were observed in close association with the proliferating membrane structures. Collectively, these results suggest that inhibition of c-Src kinase activity prevents assembly of the DENV virion within the ER.
Fig. 5.

Inhibition of c-Src protein kinase activity prevents the assembly of DENV within the ER lumen. DENV2-infected Vero cells treated with either DMSO or 2.5 μM of dasatinib (A and B, respectively) and mock-infected cells treated with DMSO (C) were processed for electron microscopy at 4 days postinfection. (A) Representative DENV particles localized within the ER lumen are indicated by arrows. (B) Extensive proliferation of virus-induced membranes is indicated by arrowheads and nucleocapsid-like particles are noted by the arrows. No virus particles are observed within the ER lumen and membranous structures. (C) Typical ultrastructural morphology of noninfected Vero cells. (Scale bars: 200 nm for A–C.)
Discussion
Currently, there are few documented inhibitors for dengue virus or even closely related flavivirus infections. Some of these inhibitors include mycophenolic acid (13, 20), ribavirin (20–22), 6-azauridine (20), castanospermine (23), and deoxynojirimycin (23–25). Nucleoside analogues (24, 25) and peptide-like α-keto amide backbone compounds (23) that inhibit the function of flavivirus NS3 and NS5 proteins have also been reported. Among these inhibitors, only ribavirin has been given approval for clinical use and is currently used solely in the treatment of hepatitis C flavivirus infections (21), in which it has been implicated in causing side effects such as anemia in patients (22). For these reasons, there is an urgent need to identify new inhibitors for the treatment of dengue virus infection. In this study, we developed and used an image-based, viral immunofluorescence assay to screen a focused library of kinase inhibitors. This screen enabled us to identify several cellular kinases that seem to be important for different processes of the DENV life cycle. The anti-DENV activity of compounds identified in this primary screen was subsequently validated by using more traditional viral plaque assays. Collectively, these results demonstrate that this assay can be used to interrogate dengue virus-host cell biochemistry with small molecules to identify cellular pathways and factors that are important or essential for the viral life cycle.
Src-family kinases were hypothesized to be important kinases in DENV infection based upon the selectivity profiles of the compounds that exhibited anti-DENV activity in the primary and secondary screens. There are nine Src family kinases defined by their kinase-domain sequence homology and domain structure: Blk, Fgr, Fyn, Hck, Lck, Lyn, Src, Yes, and Yrk. Human c-Src protein is a nonreceptor signaling protein kinase that plays important roles in a variety of cellular processes that includes cell adhesion and migration, cell division, cytoskeleton organization, gene transcription, and apoptosis (reviewed in ref. 26). Human c-Src protein has been explored as a potential target for a variety of human ailments including a variety of human cancers (27), leukemia (28), and osteoporosis (29). Lck, which shows localized expression in the cells of the immune system (predominantly T cells) and is essential for T cell activation, has been intensively pursued as a target for immunosuppression. Because of the high degree of conservation within the Src family kinases, most inhibitors developed against individual members of this family exhibit broad specificity across the entire family (28). We chose to perform our confirmation studies using two Src-family inhibitors that have advanced into clinical testing: dasatinib, which is a relatively nonselective Src family inhibitor, and AZD0530, which is a selective Src-family inhibitor. In addition, we compared the results of pharmacological inhibition with those obtained by siRNA-mediated knockdown of c-Src protein expression.
Our results demonstrate that c-Src kinase inhibitors block DENV infection primarily by preventing the formation of infectious virus particles within the virus replication complex. In general, flavivirus assembly is believed to occur by budding of encapsidated viral RNA into the ER lumen coordinated in the presence of the virus envelope glycoprotein. After the formation of virions within the lumen of the endoplasmic reticulum, the virus particles are transported within vesicles via the host secretory system and released by exocytosis (reviewed in ref. 5). Treatment of DENV2-infected cells with c-Src protein kinase inhibitors did not affect the synthesis of viral envelope glycoproteins as revealed by immunofluorescence staining (Fig. 4), suggesting that neither viral RNA synthesis nor viral gene expression were affected. By EM analysis, there was absence of DENV virions within the lumen of the ER of dasatinib-treated cells when compared with the DMSO-treated sample (Fig. 5). These results suggest that c-Src protein may mediate the budding of the nucleocapsid into the ER lumen to form infectious viral particles. This suggestion is further supported by the accumulation of nucleocapsid-like particles observed in close proximity to the virus-induced membranes of the ER within DENV-infected cells in the presence of c-Src protein kinase inhibitor (Fig. 5). The finding that dasatinib has antiviral activity against all four DENV serotypes as well as against Modoc virus suggests that this process is conserved across the Flavivirdae and may be broadly targeted by Src kinase inhibitors.
Interestingly, c-Yes, another Src family kinase, was recently shown to inhibit the trafficking of West Nile virus (WNV) particles via the host secretory system (30), a later step in the viral life cycle than was observed for the inhibition of Src family kinases in this study. These results may suggest that multiple Src family kinases are required for flavivirus replication and that targeting of the earlier process by inhibitors in our study precluded our ability to confirm the activity of c-Yes in the DENV system. c-Src protein kinase has also been documented to be involved in replication processes of a number of viruses such as hepatitis B virus (31), HIV (32), and herpes simplex virus 1 (33).
In summary, we have developed an image-based assay for dengue virus that provides a valuable platform for investigating DENV-host cell interactions as well as for identifying small molecules that may have therapeutic potential for the treatment of DENV infection. This study demonstrates that the DENV life cycle can be effectively targeted with small molecules that are selective for host cell functions. We note that whereas this study focused on perturbation of host cell functions with small molecules, analogous experiments using RNA interference should also be possible. Further work will be required to elucidate whether additional Src-family kinases have important roles in regulating viral replication. In addition, it will be interesting to determine which c-Src substrates are critical targets in the viral assembly process. Further studies with this class of inhibitors may provide greater understanding of the virus-host interaction of this important human pathogen and could potentially lead to the development of therapeutic agents against DENV infection.
Materials and Methods
Cell Lines, Virus and Antibodies.
All cell lines, viruses, and antibodies were obtained from commercial or academic sources and are described in detail in SI Materials and Methods.
Kinase Inhibitor Library.
A collection of commercial and noncommercial inhibitors of mammalian Ser/Thr, Tyr, and lipid kinases was assembled from commerical and academic sources. The commercial portion of the library was primarily assembled from Calbiochem (San Diego, CA), Sigma-Aldrich (St. Louis, MO), and Pierce (Rockford, IL). The noncommercially available clinical-stage kinase inhibitors were resynthesized following published procedures. Inhibitors were first evaluated for cytotoxicity in Vero cells by MTT assay. Primary and secondary assays were conducted at inhibitor concentrations that had minimal effects on cell viability.
DENV Immunofluorescence Assay.
In brief, DENV2 (NGC strain) was added to monolayers of Vero cells in 384-well plates at an moi of 1 for 60 min at 37°C with 5% CO2. Excess/unbound virus was removed by washing with PBS. Small molecule protein kinase inhibitors to be tested were then added to the DENV2 infected cells at final concentrations of 0.02–10 μM/0.25% (vol/vol) DMSO. The plates were incubated for 3 days at 37°C with 5% CO2 before performing immunofluorescence staining. Cell monolayers were fixed and incubated with the anti-DENV E protein mAb (US Biologicals, Swampscott, MA), followed by incubation with the secondary Ab conjugated with FITC (Zymed, San Francisco, CA). Cell nuclei were counterstained with DAPI (Molecular Probes, Eugene, OR). Four randomly selected fields per well were captured in both the FITC and DAPI channels by using an automated microscope image acquisition system (ImageXpress; Molecular Devices, Sunnyvale, CA). Autofocus and data collection parameters were preset by using MetaXpress (Molecular Devices, Sunnyvale, CA) software. The captured images were then analyzed by using an automated algorithm (MetaMorph software; Molecular Devices) to calculate the number of blue cell nuclei and green DENV infected cell cytoplasm of a user defined size and threshold value of 100. The average number of DENV infected cells per field was then determined and compared with that of positive control wells (DENV-infected cells with cell culture media containing 0.25% DMSO). Compounds that inhibited DENV infection by greater than 50% relative to the DMSO-treated DENV control were regarded as “hits” in these experiments. IC50 values were determined by nonlinear regression analysis of inhibition curves by using GraphPad Prism version 4.00 for Windows (GraphPad Software, San Diego, CA). A detailed description of the above procedure can be found in the SI Materials and Methods.
Cytotoxicity Assays.
The cytotoxicity of each of the small molecule inhibitors used in this study was assessed by using a commercial kit (Chemicon, Temecula, CA) according to the manufacturer's recommendations (SI Materials and Methods).
Secondary Assays.
The inhibitory effects of the small molecule inhibitors on DENV infection were further confirmed by incubating cells with inhibitors either before (pretreatment) or after (posttreatment) inoculation with virus and then quantifying infectious DENV yield by virus plaque assay as described in SI Materials and Methods. In brief, Vero, Huh-7, and C6/36 cells were seeded into 24-well plates, with a cell density of 1 × 104 cells per well. To evaluate effects of inhibitors preinoculation, noncytotoxic concentrations of the protein kinase inhibitors were added to the cell monolayers and incubated for 180 min at 37°C. The inhibitor-treated cells were then inoculated with DENV at an moi of 1 for 60 min at 37°C. Culture supernatants were harvested 3 days postinfection, and the infectious virus yield was determined by viral plaque assay. For postinoculation assays, cells were first infected with DENV (moi 1) and then treated with protein kinase inhibitors (as described above) for 3 days after which viral titers were quantified by plaque assay.
RNAi and Transfection.
A pool of four siRNAs targeting human c-Src tyrosine kinase (NCBI accession no. NM_004383; catalog no. L-003110-00) and a nontargeting siRNA pool directed against glyceraldehyde-3-phosphate dehydrogenase (GAPDH, NCBI accession no. NM_002046; catalog no. D-001830-01-05) were purchased from Dharmacon (Chicago, IL). The pool of siRNA was transfected into Huh-7 cells (cell density of 1 × 103 cells) by using HiPerfect (Qiagen, Valencia, CA). Additional information on siRNA experiments including the effects of siRNA knockdown on intracellular levels of c-Src as assessed by Western blotting are provided in SI Materials and Methods.
Transmission Electron Microscopy.
DENV-infected and mock-infected cell monolayers were treated with a protein kinase inhibitor or DMSO (as described above) and then fixed with 2.5% glutaraldehyde and 2.0% paraformaldehyde at 4°C for 24 h. The fixed samples were then processed for embedding and sectioning before viewing with a Tecnai G2 Spirit BioTWIN transmission electron microscope (Tecnai, Hillsboro, OR). A detailed description of the sample processing is given in the SI Materials and Methods.
Supplementary Material
Acknowledgments
We thank Nathanael S. Gray for extensive advice on experimental design and manuscript preparation. The primary screens were carried out with the technical assistance and instrumentation of the ICCB-Longwood Screening Facility.
Abbreviations
- DENV
Dengue virus
- MPA
mycophenolic acid
- moi
multiplicity of infection.
Footnotes
The authors declare no conflict of interest.
This article contains supporting information online at www.pnas.org/cgi/content/full/0611681104/DC1.
References
- 1.Gubler DJ. Clin Microbiol Rev. 1998;11:480–496. doi: 10.1128/cmr.11.3.480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Thomas SJ, Strickman D, Vaughn DW. Adv Virus Res. 2003;61:235–289. doi: 10.1016/s0065-3527(03)61006-7. [DOI] [PubMed] [Google Scholar]
- 3.Clyde K, Kyle JL, Harris E. J Virol. 2006;80:11418–11431. doi: 10.1128/JVI.01257-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Burke D, Monath T. Fields Virology. Vol 1. Philadelphia: Lippincott Williams & Wilkins; 2001. pp. 1043–1125. [Google Scholar]
- 5.Westaway EG, Mackenzie JM, Khromykh AA. Curr Top Microbiol Immunol. 2002;267:323–351. doi: 10.1007/978-3-642-59403-8_16. [DOI] [PubMed] [Google Scholar]
- 6.Lee CJ, Liao CL, Lin YL. J Virol. 2005;79:8388–8399. doi: 10.1128/JVI.79.13.8388-8399.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chang TH, Liao CL, Lin YL. Microbes Infect. 2006;8:157–171. doi: 10.1016/j.micinf.2005.06.014. [DOI] [PubMed] [Google Scholar]
- 8.Munoz-Jordan JL, Sanchez-Burgos GG, Laurent-Rolle M, Garcia-Sastre A. Proc Natl Acad Sci USA. 2003;100:14333–14338. doi: 10.1073/pnas.2335168100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ho LJ, Hung LF, Weng CY, Wu WL, Chou P, Lin YL, Chang DM, Tai TY, Lai JH. J Immunol. 2005;174:8163–8172. doi: 10.4049/jimmunol.174.12.8163. [DOI] [PubMed] [Google Scholar]
- 10.Pelkmans L, Fava E, Grabner H, Hannus M, Habermann B, Krausz E, Zerial M. Nature. 2005;436:78–86. doi: 10.1038/nature03571. [DOI] [PubMed] [Google Scholar]
- 11.Forwood JK, Brooks A, Briggs LJ, Xiao CY, Jans DA, Vasudevan SG. Biochem Biophys Res Commun. 1999;257:731–737. doi: 10.1006/bbrc.1999.0370. [DOI] [PubMed] [Google Scholar]
- 12.Kapoor M, Zhang L, Ramachandra M, Kusukawa J, Ebner KE, Padmanabhan R. J Biol Chem. 1995;270:19100–19106. doi: 10.1074/jbc.270.32.19100. [DOI] [PubMed] [Google Scholar]
- 13.Diamond MS, Zachariah M, Harris E. Virology. 2002;304:211–221. doi: 10.1006/viro.2002.1685. [DOI] [PubMed] [Google Scholar]
- 14.Zhang JH, Chung TD, Oldenburg KR. J Biomol Screen. 1999;4:67–73. doi: 10.1177/108705719900400206. [DOI] [PubMed] [Google Scholar]
- 15.Hochhaus A, Kantarjian HM, Baccarani M, Lipton JH, Apperley JF, Druker BJ, Facon T, Goldberg SL, Cervantes F, Niederwieser D, et al. Blood. 2006 Nov 30; doi: 10.1182/blood-2006–09-047266. [DOI] [PubMed] [Google Scholar]
- 16.Kantarjian H, Jabbour E, Grimley J, Kirkpatrick P. Nat Rev Drug Discov. 2006;5:717–718. doi: 10.1038/nrd2135. [DOI] [PubMed] [Google Scholar]
- 17.Hennequin LF, Allen J, Breed J, Curwen J, Fennell M, Green TP, Lambert-van der Brempt C, Morgentin R, Norman RA, Olivier A, et al. J Med Chem. 2006;49:6465–6488. doi: 10.1021/jm060434q. [DOI] [PubMed] [Google Scholar]
- 18.Manley PW, Cowan-Jacob SW, Mestan J. Biochim Biophys Acta. 2005;1754:3–13. doi: 10.1016/j.bbapap.2005.07.040. [DOI] [PubMed] [Google Scholar]
- 19.Stohlman SA, Wisseman CL, Jr, Eylar OR, Silverman DJ. J Virol. 1975;16:1017–1026. doi: 10.1128/jvi.16.4.1017-1026.1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Morrey JD, Smee DF, Sidwell RW, Tseng C. Antiviral Res. 2002;55:107–116. doi: 10.1016/s0166-3542(02)00013-x. [DOI] [PubMed] [Google Scholar]
- 21.Hong Z, Cameron CE. Prog Drug Res. 2002;59:41–69. doi: 10.1007/978-3-0348-8171-5_2. [DOI] [PubMed] [Google Scholar]
- 22.Kowdley KV. J Clin Gastroenterol. 2005;39:S3–S8. doi: 10.1097/01.mcg.0000145494.76305.11. [DOI] [PubMed] [Google Scholar]
- 23.Whitby K, Pierson TC, Geiss B, Lane K, Engle M, Zhou Y, Doms RW, Diamond MS. J Virol. 2005;79:8698–8706. doi: 10.1128/JVI.79.14.8698-8706.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Courageot MP, Frenkiel MP, Dos Santos CD, Deubel V, Despres P. J Virol. 2000;74:564–572. doi: 10.1128/jvi.74.1.564-572.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wu SF, Lee CJ, Liao CL, Dwek RA, Zitzmann N, Lin YL. J Virol. 2002;76:3596–3604. doi: 10.1128/JVI.76.8.3596-3604.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Thomas SM, Brugge JS. Annu Rev Cell Dev Biol. 1997;13:513–609. doi: 10.1146/annurev.cellbio.13.1.513. [DOI] [PubMed] [Google Scholar]
- 27.Trevino JG, Summy JM, Gallick GE. Mini Rev Med Chem. 2006;6:681–687. doi: 10.2174/138955706777435724. [DOI] [PubMed] [Google Scholar]
- 28.Warmuth M, Damoiseaux R, Liu Y, Fabbro D, Gray N. Curr Pharm Des. 2003;9:2043–2059. doi: 10.2174/1381612033454126. [DOI] [PubMed] [Google Scholar]
- 29.Boyce BF, Xing L, Yao Z, Yamashita T, Shakespeare WC, Wang Y, Metcalf CA, 3rd, Sundaramoorthi R, Dalgarno DC, Iuliucci JD, Sawyer TK. Clin Cancer Res. 2006;12:6291s–6295s. doi: 10.1158/1078-0432.CCR-06-0991. [DOI] [PubMed] [Google Scholar]
- 30.Hirsch AJ, Medigeshi GR, Meyers HL, DeFilippis V, Fruh K, Briese T, Lipkin WI, Nelson JA. J Virol. 2005;79:11943–11951. doi: 10.1128/JVI.79.18.11943-11951.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Klein NP, Bouchard MJ, Wang LH, Kobarg C, Schneider RJ. EMBO J. 1999;18:5019–5027. doi: 10.1093/emboj/18.18.5019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Saksela K. Front Biosci. 1997;2:d606–d618. doi: 10.2741/a217. [DOI] [PubMed] [Google Scholar]
- 32.Liang Y, Roizman B. J Virol. 2006;80:3349–3359. doi: 10.1128/JVI.80.7.3349-3359.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
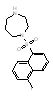{kind=link}
{kind=link}
{kind=link}