

Abstract
Rheb is a unique member of the Ras superfamily GTP-binding proteins. We as well as others previously have shown that Rheb is a critical component of the TSC/TOR signaling pathway. In fission yeast, Rheb is encoded by the rhb1 gene. Rhb1p is essential for growth and directly interacts with Tor2p. In this article, we report identification of 22 single amino acid changes in the Tor2 protein that enable growth in the absence of Rhb1p. These mutants also exhibit decreased mating efficiency. Interestingly, the mutations are located in the C-terminal half of the Tor2 protein, clustering mainly within the FAT and kinase domains. We noted some differences in the effect of a mutation in the FAT domain (L1310P) and in the kinase domain (E2221K) on growth and mating. Although the Tor2p mutations bypass Rhb1p's requirement for growth, they are incapable of suppressing Rhb1p's requirement for resistance to stress and toxic amino acids, pointing to multiple functions of Rhb1p. In mammalian systems, we find that mammalian target of rapamycin (mTOR) carrying analogous mutations (L1460P or E2419K), although sensitive to rapamycin, exhibits constitutive activation even when the cells are starved for nutrients. These mutations do not show significant difference in their ability to form complexes with Raptor, Rictor, or mLST8. Furthermore, we present evidence that mutant mTOR can complex with wild-type mTOR and that this heterodimer is active in nutrient-starved cells.
Keywords: constitutive active TOR, FAT domain, kinase domain, mating, TORC1
Rheb comprises a unique subfamily of the Ras superfamily of GTP-binding proteins that is conserved from yeast to human and plays important roles in cell growth and cell-cycle regulation (1). We and others have shown that Rheb is an activator of mammalian target of rapamycin (mTOR) and a component of the TSC/mTOR/S6K signaling pathway that regulates protein synthesis in response to growth, energy, and nutrient conditions (1–7). mTOR exists in two distinct protein complexes: mTORC1, which consists of mTOR, Raptor, and mLST8 and is rapamycin-sensitive; and mTORC2, which consists of mTOR, Rictor, mLST8, and mSin1 and is rapamycin-insensitive. mTORC1 is involved in the regulation of translation and cell cycle, and mTORC2 is reported to be involved in actin organization and morphology (8–16). Rheb is down-regulated by a complex consisting of TSC1 and TSC2 gene products that act as GTPase activating proteins for Rheb (1–7). Mutations of these genes result in tuberous sclerosis complex, a genetic disorder associated with the appearance of hamartomas in the kidneys, lungs, brain, and skin (17, 18).
In fission yeast, Rheb is encoded by the rhb1 gene and is essential for growth. Loss of Rhb1p results in small rounded cells arrested with G1 content of DNA (19, 20). Like mammalian cells, Rhb1p is down-regulated by the Tsc1p/Tsc2p complex (21, 22). Mutations in the tsc genes result in a delayed response to nitrogen starvation as well as defects in amino acid uptake (21–25). The fission yeast genome encodes two TOR proteins, Tor1p and Tor2p (26, 27). Although Tor2p, like Rhb1p, is essential for growth, tor1Δ cells are sterile and unable to arrest in G1 in response to nitrogen starvation (26, 27). Tor1p also is implicated in the regulation of stress response and uptake of some amino acids (26–28). We recently have reported that fission yeast Rhb1p associates with Tor2p (25). Interaction between mammalian Rheb and mTOR also has been reported (29–31).
Because the requirement for Rhb1p for growth in fission yeast likely reflects its vital role in the activation of Tor2p, we speculated that activating mutations of Tor2p may bypass this requirement. Identification of such mutations, which are likely to be constitutive active, is of interest in gaining insight into the mechanism of activation of this kinase. In particular, this study may reveal novel involvement of specific domains in the activation of TOR. The TOR kinases are members of the phosphatidylinositol 3-kinase (PI3 kinase)-related family of kinases and have specific domains, including the HEAT repeats and the FAT, kinase, and FATC domains. Involvement of the HEAT domain in protein–protein interactions, dimerization, and membrane association has been reported (9, 32–38). To date, only one activated TOR mutant has been reported. This mutant, ΔTOR, contains a deletion of the “repressor domain,” which includes AMPK (T2446) and S6K (S2448) phosphorylation sites at the C-terminal region of mTOR (39–43). In our study, we carried out a systematic analysis to uncover single amino acid changes that confer constitutive activation of TOR proteins.
We report the identification of 22 different single amino acid changes that confer constitutive activation of Tor2p. Interestingly, the mutations are clustered into two regions: the FAT domain and the kinase domain. Characterization of these mutations showed that they are able to bypass the requirement of Rhb1p for fission yeast growth. The use of these mutants allowed us to observe that Rhb1p also is required for resistance to high salt, high temperature, and toxic amino acid analogs in fission yeast. Furthermore, mTOR carrying analogous mutations exhibited nutrient-independent activity and were able to form mTORC1 and mTORC2. In addition, a heterodimer of wild-type and mutant mTOR also displayed nutrient-independent activity.
Results
Identification of Mutations in Tor2p That Can Bypass Growth Requirement for Rhb1p in Fission Yeast.
Rhb1p interacts with Tor2p, and both Rhb1p and Tor2p are essential for growth (19, 20, 25, 27). Furthermore, loss of Tor2p function, like the loss of Rhb1p, results in small rounded cells arrested in G1 (44, 45). Thus, it is likely that in fission yeast, Rhb1p functions to activate Tor2p. This finding raises the possibility that activating mutations in Tor2p (or another downstream factor) can confer Rhb1p-independent growth (RIG). To investigate this point, we have devised a screen to identify yeast mutants that can grow in the absence of Rhb1p [supporting information (SI) Fig. 6]. A strain (JUp1050) was constructed in which the endogenous rhb1 gene was disrupted by using a his3+ cassette and growth was maintained by using a wild-type copy of rhb1+ on a ura4+-based plasmid. This plasmid can be lost by counterselecting for ura4+ with 5-fluoroorotic acid (FOA). JUp1050 randomly was mutagenized by methyl-nitro-nitrosoguanidine, and mutants that would grow on media containing FOA (and hence in the absence of rhb1+) were isolated. To determine whether the rig mutation had occurred in tor2, we initially sequenced the entire tor2 ORF.
Approximately 3 × 109 cells were mutagenized and screened. After eliminating clones that still maintained the rhb1+ plasmid, a single mutant strain was isolated that was able to grow in the absence of rhb1+. Sequence analysis of the tor2 ORF identified a point mutation that results in a glutamate to lysine mutation at position 2221 (E2221K) in the C-terminal half of the Tor2p kinase domain. To confirm that this mutation could confer RIG, this mutation was reintroduced into the tor2 locus of JUp1050. This strain (JUp1261) was able to grow in the absence of rhb1 (on FOA), confirming that this mutation in the Tor2p kinase domain confers activity independent of Rhb1p (Fig. 1A).
Fig. 1.

Tor2p L1310P and E2221K show Rhb1-independent growth and delayed nitrogen starvation response. (A) Strains carrying wild-type tor2+ (JUp1273) or the activated mutants tor2L1310P (JUp1274) and tor2E2221K (JUp1261) were streaked out onto EMM + AL plates or EMM + AL + FOA plates and incubated at 30°C. EMM, Edinburgh minimal medium. (B) JUp1348 (tor2+), JUp1350 (tor2L1310P), and JUp1352 (tor2E2221K) were grown to mid-log phase, washed twice with water, and adjusted to an A595 of 1. Aliquots of 3 μl were spotted onto SSA plates, incubated at 25°C for 2 days, and stained by using I2 vapor. Note that two sets of clones were used, and both show similar results. (C) JUp1348 (tor2+), JUp1350 (tor2L1310P), and JUp1352 (tor2E2221K) were grown to mid-log phase, washed twice with water, and resuspended in SSL media. Samples were taken at the indicated times and analyzed with FACS for cell size and cell cycle. Each profile (purple) was overlayed with the profile outline for the wild-type strain (green line) at each time point.
RIG Mutants of Tor2p Exhibit Decreased Mating Efficiency.
Because loss of Tor2p function can induce mating (44, 45), we asked whether the Tor2E2221K mutant would exhibit decreased mating. The Tor2E2221K mutation was introduced into a homothallic h90 strain, and mating efficiency was assessed by staining with iodine that detects the increased glycogen levels in spores. As can be seen in Fig. 1B, we observed a notable decrease in the extent of iodine staining in the mutant. Thus, the E2221K mutation appears to decrease mating efficiency, consistent with an activation of Tor2p.
Additional Tor2 mutations (L1310P, Y1986C, I2229T, and L2233H) were identified by screening for decreased mating (decreased iodine staining) in an h90 strain after randomly mutagenizing the tor2 gene. These mutations were examined for their ability to confer RIG. Fig. 1A shows that the strain expressing the L1310P mutant (JUp1274) grew after removing the rhb1+ plasmid by FOA selection. Similar results were obtained for the other Tor2 mutants. Interestingly, when comparing the L1310P mutant with the E2221K mutant, we noticed that the strain expressing the E2221K mutant has a greater ability to suppress the Rhb1p requirement for growth (see FOA plates in Fig. 1A), whereas the L1310P mutant exhibits a more pronounced effect on mating efficiency (Fig. 1B). Thus, although RIG and decreased mating are both consequences of a single activating mutation in Tor2p, each mutation may affect these two activities differently.
These Tor2 constitutive active mutants exhibit delayed nitrogen-starvation response. Because fission yeast cells respond to nitrogen starvation by arresting in G1 as small rounded cells, nonauxotrophic strains carrying the L1310P or E2221K mutation were nitrogen-starved in SSL media (see SI Materials and Methods), and samples were assessed for cell size and DNA content by FACS. The results are shown in Fig. 1C. Forward-scatter analysis shows that both Tor2 mutants show a delay in this change in cell size compared with wild type (notably at 1.5 and 4.5 h). By 10 h, the mutant cells have decreased in size similar to wild-type cells. Analysis of cell-cycle profiles shows that the Tor2 mutants are delayed in the appearance of G1 cells because more cells are in G2 at 4.5 h, whereas majority of wild-type cells are in G1.
Mutations Are Clustered Mainly in the FAT and Kinase Domains of Tor2p.
The above analysis identified single amino acid changes located in the FAT and kinase domains, pointing to the importance of these two domains. To further investigate the significance of these domains for Tor2p activation, we screened for additional mutations in the C-terminal half of Tor2p. The region containing either the FAT or the FRB, kinase, and FATC domains of the tor2 gene in JUp1050 was randomly mutagenized, and 34 additional mutants that exhibited RIG were identified. Sequence analysis of these mutants revealed 17 additional single mutations at 15 positions.
Fig. 2A summarizes all of the activating mutations identified. Interestingly, we found that the mutations mainly were clustered in two regions: the N-terminal side of the FAT domain (cluster I) and the C-terminal portion of the kinase domain (cluster II). In addition, there were a few regions, notably at the C-terminal region of the FAT domain and the N-terminal region of the kinase domain, where additional mutations were identified. Fig. 2B shows sequence alignments of residues in which mutations were found in clusters I and II. As can be seen, most of the mutations occur on residues that are perfectly conserved among TOR proteins from different organisms.
Fig. 2.

Location of activated mutations in Tor2p and conservation of residues. (A) All identified activating mutations are indicated above the linear representation of Tor2p. Clusters I and II are indicated. (B) TOR protein sequences from Schizosaccharomyces pombe (SpTor1 and SpTor2), S. cerevisiae (ScTor1 and ScTor2), Drosophila (dTOR), and human (mTOR) were aligned with MegAlign. Residues identical or similar to SpTor2 are shaded black or gray, respectively. Mutations found in SpTor2 are indicated above the alignment. The repressor domain region of mTOR is indicated by a line under the alignment. Single and double asterisks indicate the AMPK and S6K phosphorylation sites, respectively.
rhb1Δ tor2act Mutants Are Sensitive to Stress Conditions.
Analysis of a strain having a disruption of rhb1 and carrying a tor2-activated (tor2act) mutation revealed that, although Tor2 mutants can bypass Rhb1p requirement for growth, they are incapable of bypassing other Rhb1p functions. Fig. 3A shows that the rhb1Δ strains that are viable because of the presence of either the tor2L1310P or the tor2E2221K mutation are sensitive to high-salt stress (1 M KCl) and high temperature (37°C). This sensitivity can be reversed by the introduction of rhb1+, indicating that Rhb1p is involved in responding to these stresses in fission yeast.
Fig. 3.

rhb1Δ tor2L1310P and rhb1Δ tor2E2221K show sensitivity to stresses. (A) JUp1050 (rhb1+tor2+), JUp1274 (rhb1+tor2L1310P), JUp1275 (rhb1Δtor2L1310P), JUp1261 (rhb1+tor2E2221K), and JUp1263 (rhb1Δ tor2E2221K) were streaked on the indicated plates and incubated at the indicated temperatures. (B) JUp1263 (rhb1Δ tor2E2221K) and JUp1275 (rhb1Δtor2L1310P) were transformed with either pREP41 (rhb1Δ) or pRPL-SpRhb1 (rhb1+), and transformants were streaked onto the indicated plates and incubated at 30°C.
We previously have shown that inhibition of Rhb1p causes hypersensitivities to toxic analogues of lysine (thialysine) and arginine (canavanine) (25, 46). In addition, loss of tsc2 results in resistances to thialysine, canavanine, and ethionine (the toxic analog of methionine) (22, 23, 25). Examination of the rhb1Δ tor2L1310P and rhb1Δ tor2E2221K mutants on these toxic amino acid analogs showed that these double mutants are hypersensitive to thialysine, canavanine, and ethionine (Fig. 3B and data not shown). These sensitivities are reversed by reintroducing rhb1+ into these cells. Thus, Rhb1p is required for the resistance to these amino acid analogs, and, because these cells carry activated Tor2p, this resistance likely is independent of Tor2p.
Analogous Mutations in mTOR Confer Nutrient-Independent Activity.
Because the mutations we identified occur mostly on residues that are conserved in higher eukaryotes, we asked whether these mutations would confer constitutive activation of mTOR. To examine this point, mTORL1460P and mTORE2419K, mutations that correspond to Tor2L1310P and Tor2E2221K, respectively, were constructed, and the regulation of their activities by nutrient signals in HEK293 cells was assessed. Consistent with previous reports (47, 48), mTOR activity as detected by the phosphorylation of S6K, S6, or 4EBP1 is inhibited when the cells are exposed to nutrient-starvation conditions (Fig. 4 A–C). This inhibition is overcome by overexpressing wild-type Rheb or a RhebN153T mutant, which is analogous to the hyperactive fission yeast Rhb1N153T mutant that we previously have shown to be highly bound to GTP (Fig. 4 A–C) (25, 49). The activation seen by wild-type Rheb is likely attributable to Rheb being highly bound to GTP when transiently expressed (50).
Fig. 4.

mTORL1460P and mTORE2419K show constitutive activity. (A–C) HEK293 cells were transfected with pCDNA3 (vector), AU1-mTOR (wt, E2419K, or L1460P), or FLAG-Rheb1 (wt or N153T). To detect the phosphorylation of S6K or 4E-BP1, FLAG-S6K or FLAG-4E-BP1 was cotransfected. Cells then were serum-starved overnight and cultured in PBS for 1 h. The protein levels were detected by anti-FLAG (S6K, 4E-BP1, and Rheb), anti-AU-1 (mTOR), or anti-S6 antibody. The phosphorylation levels of S6K, 4E-BP1, and S6 were detected by phospho-specific antibodies. (D) HEK293 cells were transfected with AU1-mTOR (wt, E2419K, or L1460P). Cells were serum-starved overnight and then treated with rapamycin (100 nM). The expression and phosphorylation levels of S6 were detected by specific antibodies. wt, wild type.
We then examined the mTOR mutants. HEK293 cells were transfected with constructs that expressed either wild-type or the mutant mTOR (L1460P or E2419K) and then starved for nutrients. As can be seen in Fig. 4 A, B, and C, cells expressing mTORL1460P and mTORE2419K exhibit high phosphorylation of S6K, 4E-BP1, and S6 compared with wild-type mTOR, indicating that the mTOR mutants maintain activity even when the cells are starved for nutrients. However, these mTOR mutants retain sensitivity to inhibition by rapamycin (Fig. 4D). mTOR activity (S6 phosphorylation) was assessed in HEK293 cells expressing wild-type mTOR, mTORL1460P, or mTORE2419K and treated with either DMSO (−) or rapamycin (+). We found that both mTOR mutants were sensitive to rapamycin, similar to wild-type mTOR.
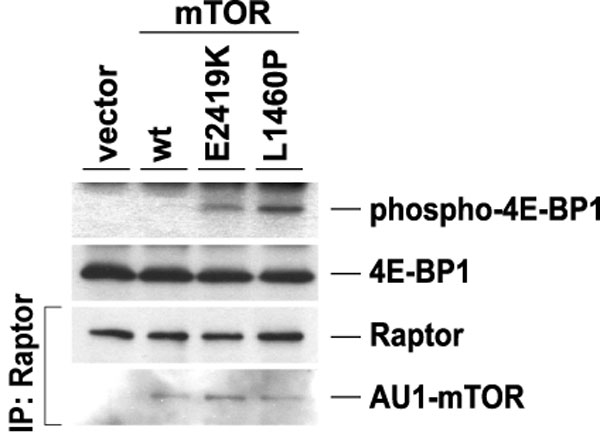
Constitutive activation of the mTOR mutants also can be examined by measuring in vitro kinase activities. The activities of the two mTORCs were assayed by using two different substrate proteins: mTORC1 activity was assayed by using 4E-BP1 (51, 52), whereas mTORC2 was assayed by using Akt as a substrate protein (53, 54). These complexes were immunoprecipitated from nutrient-starved cells by using AU1-tagged wild-type or mutant mTOR and activities assessed in vitro (Fig. 5A). Activity using Akt as the substrate was still retained with wild-type mTOR in HEK293 cells even under nutrient-starved conditions, and no change in this activity was observed when using the mutant mTOR. On the other hand, we found that the mTOR complex containing mTORL1310P or mTORE2221K exhibited significantly higher activity with 4E-BP1 as the substrate relative to wild-type mTOR. This activity was confirmed to be attributable to mTORC1, because an in vitro kinase assay using mTORC1 immunoprecipitated with anti-Raptor antibody showed similar results (SI Fig. 7). These results are consistent with our in vivo findings.
Fig. 5.

Kinase activities of mTOR complexes. (A) HEK293 cells transfected with pCDNA3 (vector) or AU1-mTOR (wt, E2419K, or L1460P) were serum-starved overnight and then cultured in PBS for 1h. AU1-mTOR complexes then were immunoprecipitated with anti-AU1 antibody and used for in vitro kinase assays with 4E-BP1 and Akt as substrates. Phosphorylation of substrates was detected by use of the indicated phospho-specific antibodies. Levels of phosphorylation of substrates were quantitated relative to that by mTORE2419K and graphed. (B) HEK293 cells were transfected with pCDNA3 (vector) or AU1-mTOR (wt, E2419K, or L1460P) together with Myc-mLST8. Cells were serum-starved overnight and cultured for 1 h in PBS. AU1-mTOR complexes then were immunoprecipitated with anti-AU1 antibody. mTOR were detected by anti-AU1 antibody. mLST8 were detected by anti-Myc antibody. Raptor and Rictor were detected by specific antibodies. (C) HEK293 cells cotransfected with FLAG-mTOR and AU1-mTOR (wt, E2419K, or L1460P) were serum-starved overnight and cultured for 1 h in PBS. mTOR dimers were immunoprecipitated by using anti-FLAG antibody and used for in vitro kinase assays with 4E-BP1 as substrate. FLAG-mTOR and AU1-mTOR were detected by anti-FLAG and anti-AU1 antibodies, respectively. Phosphorylation of substrate was detected by use of anti-phospho-4E-BP1 (Thr 37/46).
mTORL1460P and mTORE2419K Mutants Can Form mTORC1 and mTORC2 Complexes and an Active Heterodimer with Wild-Type mTOR.
We asked whether there were any alterations in the ability of the mTOR mutants to form mTORC1 and mTORC2. By using the AU1 tag on the expressed mTOR, mTOR complexes were immunoprecipitated from HEK293 cells under nutrient-starvation conditions, and levels of Raptor (mTORC1), Rictor (mTORC2), and mLST8 (mTORC1 and mTORC2) were assessed (Fig. 5B). We found that the mTOR mutants were able to bind similar amounts of these mTORC1 and mTORC2 components as was wild-type mTOR.
It has been reported that mTOR dimerizes via its N-terminal HEAT domains and that the dimeric mTORC1 is the major form that responds to insulin (36, 38). Because our mutations are not located in the HEAT domains, it is likely that wild-type and mutant mTOR would form a heterodimeric complex. To test whether this heterodimer exhibits constitutive activation of mTOR function, we coexpressed FLAG-tagged wild-type and AU1-tagged wild-type or mutant mTOR proteins in HEK293 cells. The cells were starved for nutrients, and the complexes were isolated by immunoprecipitation using anti-FLAG antibody. We found similar amounts of AU1-tagged wild-type mTOR and the AU1-tagged mutant mTOR in the immune complex, indicating that mutant mTOR was able to form a heterodimer with wild-type mTOR (Fig. 5C). Furthermore, when these complexes were assayed for activity by using 4E-BP1 as a substrate, we found that, although the wild-type/wild-type dimers were inactive, the wild-type/mutant heterodimers exhibited in vitro kinase activity (Fig. 5C).
Discussion
Accumulating evidence in fission yeast provides strong support for the idea that Rhb1p is an activator of Tor2p. We have reported that Rhb1p associates with Tor2p (25). We and others also recently have shown that, in fission yeast, Tor2p complexes with Mip1p (the fission yeast Raptor homolog), likely forming a SpTORC1 (44, 45). Furthermore, shutting down Tor2p results in small rounded cells arrested in G1, reminiscent of inhibiting Rhb1p (44, 45). In addition, inhibition of Tor2p in homothallic cells induces mating and sexual development (44, 45). Now, we show that, opposite to inhibiting Tor2p function, an activating mutation in Tor2p can confer RIG, delayed response to nitrogen starvation, and decreased sexual development.
This report provides evidence for a large number of single amino acid changes that confer constitutive activation of Tor2p. Importantly, many of these mutations occur in residues that are highly conserved in TOR proteins, and mTOR carrying analogous mutations exhibit nutrient-independent mTORC1 activation. An exciting finding of our study is that the activating mutations are clustered within two domains of TOR: the N-terminal region of the FAT domain (cluster I) and the C-terminal region of the kinase domain (cluster II). Interestingly, the cluster in the kinase domain is just adjacent to the repressor domain (2430–2450 of mTOR; Fig. 2B); deletion of this region results in activation of mTOR (39, 40). Indeed, the analogous position (2431 of mTOR) for one of the activated fission yeast Tor2 mutants, L2233H, is found just inside this repressor domain. This close proximity of the repressor domain to cluster II possibly indicates that these mutations may activate mTOR via a similar mechanism. The deletion of the repressor domain initially was thought to activate mTOR, in part by removing an Akt phosphorylation site (S2448; Fig. 2B); however, mutating this site, which later was shown to be an S6K phosphorylation site (42, 43), to alanine does not significantly alter mTOR activity (39). Furthermore, mutating the AMPK phosphorylation site (T2446; Fig. 2B) to alanine did not alter mTOR activity (39, 41). These phosphorylation sites also are not conserved in fission yeast Tor2p. Perhaps this region is involved in interacting with an unknown inhibitory factor. In addition to cluster II, mutations were found in the N-terminal region of the kinase domain. Interestingly, Rheb is reported to interact with the N-terminal half of the mTOR kinase domain (29). It is possible that these mutations in the kinase domain mimic the effects of Rheb binding, which may interfere with this inhibitory factor. Further experiments are needed to address the consequences of these mutations. A recent report in budding yeast identified mutations in the FRB that exhibit increased association with KOG1p (budding yeast Raptor homolog) and increased activity to Saccharomyces cerevisiae TOR1p (55). Although the FRB region was included in our mutagenesis, we did not identify any activating mutation in this region.
The activation of Tor2p results in a delay in the nitrogen-starvation response. However, this response is not a complete block. Furthermore, we find that rhb1Δ tor2act strains also are able to respond to nitrogen starvation and undergo sexual differentiation (SI Fig. 8). The observation that these strains still are able to respond to nitrogen starvation in the absence of Rhb1p raises the possibility that Tor2p also is regulated by an Rhb1p-independent mechanism. Further analysis of rhb1Δ tor2act cells also revealed possible involvement of Rhb1p in stress response. These mutants are sensitive to stresses such as high salt (1 M KCl) and high temperatures (37°C). Although the mechanisms for these phenotypes need further investigation, it is of interest to note that tor1Δ mutants also exhibit these phenotypes. Another phenotype of the rhb1Δ tor2act mutants is that they are hypersensitive to toxic amino acids analogs. It is possible that these sensitivities are a result of increased amino acid uptake because we previously have shown that decreases in Rhb1p function lead to increased uptake of arginine and hypersensitivity to canavanine (46).
We have succeeded in identifying mTOR mutants that are active independent of nutrients. These mutants provide valuable reagents to further examine the biological consequences of mTOR activation. By introducing these mutants in HEK293 cells, we have shown that they confer nutrient-independent mTORC1 signaling. Additional experiments may shed light onto the role of mTOR activation in growth. Introducing these mutants in HEK293 as well as other untransformed cell lines also may provide insight into mTOR activation in transformation. Investigating mTOR activation in whole animals also is of importance. These mutants can be introduced into mice, and such animals can be studied further for roles of mTOR in development as well as propensity for tumors.
Activation of the mTOR pathway has been implicated in a number of human diseases associated with benign tumors, such as hamartomas (56). Our demonstration that mTOR can be activated by single amino acid changes raises an interesting possibility that mTOR mutations may be found in tumor samples. This idea is supported further by our finding that our mutant mTOR is active even when in a heterodimer with the wild-type protein. Because our study points to two hot spots in the TOR protein, searches for mTOR mutants may be focused on these two regions. The results obtained from these studies should have significant implication for our understanding of human diseases arising from the activation of the TSC/Rheb/mTOR signaling pathway.
Materials and Methods
Screens for RIGs and Activated Tor2p.
RIG screen.
The scheme for the RIG screen is shown in Fig. 1. JUp1050 strain (3 × 109 cells) was mutagenized with methyl-nitro-nitrosoguanidine (Sigma, St. Louis, MO) and recovered in rich yeast extract with supplements (YES) media overnight. Cells then were plated onto Edinburgh minimal medium supplemented with adenine (225 mg/l), leucine (225 mg/l), uracil (50 mg/l), and FOA (1 g/l). Plates were replica-plated once to eliminate background. Of ≈3,000 colonies, 241 clones were isolated and tested for absence of rhb1+ by colony PCR. One clone was isolated that had lost the rhb1+ plasmid.
Screen for tor2 mutants exhibiting decreased mating.
Random mutations were introduced into the tor2 gene by PCR (57). Linear DNA fragments carrying mutagenized tor2 alleles were transformed into JV530, a homothallic strain in which the endogenous tor2 gene is disrupted with a kanr cassette and whose growth is maintained by a multicopy plasmid pREP42-tor2. To obtain integrants of functional tor2 alleles, transformants that were resistant to FOA (indicating loss of ura4+-based pREP42-tor2) and kans were screened at 26.5°C. From a tor2 mutant library thus constructed, we screened for sterile clones; each strain was grown to a colony on SSA plate (see SI Materials and Methods) and stained with iodine vapor after incubation for 4 days at 30°C. Unstained colonies were isolated and examined microscopically for sterility.
Screen for additional FAT and kinase mutants.
Additional FAT and kinase domain mutations were identified by screening libraries based on pUG6-tor2-CTL in which the region containing the FAT domain or the FRB, kinase, and FATC domains were randomly mutagenized by using the GeneMorph II Random Mutagenesis Kit (Stratagene, La Jolla, CA). The libraries were digested with BamHI, and the linearized plasmids were integrated into JUp1050. The resulting transformants initially were selected on plates containing G418 (200 μg/l) for integration of plasmid and then on FOA for the RIG phenotype as before. Then, 8,400 and 12,000 integrants were screened from the FAT domain library and the FRB-kinase-FATC domain library, respectively. In both cases, 17 clones were isolated. The regions that were mutagenized were PCR-amplified from genomic preps and sequenced to identify the mutations.
Mammalian Cell Culture and Transfection.
HEK293 cells were cultured in DMEM supplemented with 10% FBS and penicillin/streptomycin at 37°C and 5% CO2. Transfections were performed by using Polyfect (Qiagen, Valencia, CA) according to the manufacturer's instructions. To assess the activity of mTOR mutants, cells were serum-starved in DMEM supplemented with 0.1% BSA overnight and then cultured in PBS for 1 h. For rapamycin treatment, cells were treated with 100 nM rapamycin for 1 h after serum starvation. These cells were lysed, and proteins were analyzed by Western blotting analysis.
Immunoprecipitation and in Vitro Kinase Assay.
Cells were lysed with buffer A [20 mM Tris·HCl (pH 7.5), 150 mM NaCl, 0.5% CHAPS, 1 mM MgCl2, and 1 mM EDTA]. The supernatant from the centrifugation at 20,000 × g for 15 min was incubated with anti-AU1 antibody (Covance, Berkeley, CA) and protein G-Sepharose 4FF beads (Amersham Biosciences, Piscataway, NJ) at 4°C for 2 h. Immunoprecipitates were washed three times with buffer A. For in vitro kinase assay, immunoprecipitates were incubated in kinase buffer [100 mM Tris·HCl (pH 7.5), 50 mM MgCl2, and 1 mM ATP] containing 0.5 μg GST-4E-BP1 or Akt for 30 min at 37°C. Samples were boiled in SDS sample buffer [3% SDS, 5% glycerol, 62 mM Tris·HCl (pH 6.7)], and proteins were analyzed by Western blotting analysis.
SI.
Additional information regarding yeast strains, media and manipulations, cell cycle and size analysis, plasmid constructs, and antibodies and reagents is provided as SI Materials and Methods. A list of strains used in this study is provided in SI Table 1.
Supplementary Material
Acknowledgments
We thank the University of California, Los Angeles, Flow Cytometry Core facility and Lea Guo for assistance with FACS analysis. This work was supported by National Institutes of Health Grant CA41996 and Department of Defense Grant W81xWH-05-1-0164 (to F.T.) and by a Grant-in-Aid for Specially Promoted Research from Ministry of Education, Culture, Sports, Science, and Technology of Japan (to M.Y.).
Abbreviations
- mTOR
mammalian target of rapamycin
- RIG
Rhb1p-independent growth
- FOA
5-fluoroorotic acid.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS direct submission.
This article contains supporting information online at www.pnas.org/cgi/content/full/0608510104/DC1.
References
- 1.Aspuria PJ, Tamanoi F. Cell Signal. 2004;16:1105–1112. doi: 10.1016/j.cellsig.2004.03.019. [DOI] [PubMed] [Google Scholar]
- 2.Avruch J, Lin Y, Long X, Murthy S, Ortiz-Vega S. Curr Opin Clin Nutr Metab Care. 2005;8:67–72. doi: 10.1097/00075197-200501000-00010. [DOI] [PubMed] [Google Scholar]
- 3.Dann SG, Thomas G. FEBS Lett. 2006;580:2821–2829. doi: 10.1016/j.febslet.2006.04.068. [DOI] [PubMed] [Google Scholar]
- 4.Inoki K, Guan KL. Trends Cell Biol. 2006;16:206–212. doi: 10.1016/j.tcb.2006.02.002. [DOI] [PubMed] [Google Scholar]
- 5.Wullschleger S, Loewith R, Hall MN. Cell. 2006;124:471–484. doi: 10.1016/j.cell.2006.01.016. [DOI] [PubMed] [Google Scholar]
- 6.Astrinidis A, Henske EP. Oncogene. 2005;24:7475–7481. doi: 10.1038/sj.onc.1209090. [DOI] [PubMed] [Google Scholar]
- 7.Tee AR, Blenis J. Semin Cell Dev Biol. 2005;16:29–37. doi: 10.1016/j.semcdb.2004.11.005. [DOI] [PubMed] [Google Scholar]
- 8.Hara K, Maruki Y, Long X, Yoshino K, Oshiro N, Hidayat S, Tokunaga C, Avruch J, Yonezawa K. Cell. 2002;110:177–189. doi: 10.1016/s0092-8674(02)00833-4. [DOI] [PubMed] [Google Scholar]
- 9.Kim DH, Sarbassov DD, Ali SM, King JE, Latek RR, Erdjument-Bromage H, Tempst P, Sabatini DM. Cell. 2002;110:163–175. doi: 10.1016/s0092-8674(02)00808-5. [DOI] [PubMed] [Google Scholar]
- 10.Kim DH, Sarbassov DD, Ali SM, Latek RR, Guntur KV, Erdjument-Bromage H, Tempst P, Sabatini DM. Mol Cell. 2003;11:895–904. doi: 10.1016/s1097-2765(03)00114-x. [DOI] [PubMed] [Google Scholar]
- 11.Jacinto E, Loewith R, Schmidt A, Lin S, Ruegg MA, Hall A, Hall MN. Nat Cell Biol. 2004;6:1122–1128. doi: 10.1038/ncb1183. [DOI] [PubMed] [Google Scholar]
- 12.Sarbassov DD, Ali SM, Kim DH, Guertin DA, Latek RR, Erdjument-Bromage H, Tempst P, Sabatini DM. Curr Biol. 2004;14:1296–1302. doi: 10.1016/j.cub.2004.06.054. [DOI] [PubMed] [Google Scholar]
- 13.Jacinto E, Facchinetti V, Liu D, Soto N, Wei S, Jung SY, Huang Q, Qin J, Su B. Cell. 2006;127:125–137. doi: 10.1016/j.cell.2006.08.033. [DOI] [PubMed] [Google Scholar]
- 14.Frias MA, Thoreen CC, Jaffe JD, Schroder W, Sculley T, Carr SA, Sabatini DM. Curr Biol. 2006;16:1865–1870. doi: 10.1016/j.cub.2006.08.001. [DOI] [PubMed] [Google Scholar]
- 15.Yang Q, Inoki K, Ikenoue T, Guan KL. Genes Dev. 2006;20:2820–2832. doi: 10.1101/gad.1461206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Loewith R, Jacinto E, Wullschleger S, Lorberg A, Crespo JL, Bonenfant D, Oppliger W, Jenoe P, Hall MN. Mol Cell. 2002;10:457–468. doi: 10.1016/s1097-2765(02)00636-6. [DOI] [PubMed] [Google Scholar]
- 17.Narayanan V. Pediatr Neurol. 2003;29:404–409. doi: 10.1016/j.pediatrneurol.2003.09.002. [DOI] [PubMed] [Google Scholar]
- 18.Gomez MR, Sampson JR, Whittemore VH. Tuberous Sclerosis Complex. New York: Oxford Univ Press; 1999. [Google Scholar]
- 19.Mach KE, Furge KA, Albright CF. Genetics. 2000;155:611–622. doi: 10.1093/genetics/155.2.611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yang W, Tabancay AP, Jr, Urano J, Tamanoi F. Mol Microbiol. 2001;41:1339–1347. doi: 10.1046/j.1365-2958.2001.02599.x. [DOI] [PubMed] [Google Scholar]
- 21.Matsumoto S, Bandyopadhyay A, Kwiatkowski DJ, Maitra U, Matsumoto T. Genetics. 2002;161:1053–1063. doi: 10.1093/genetics/161.3.1053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.van Slegtenhorst M, Carr E, Stoyanova R, Kruger WD, Henske EP. J Biol Chem. 2004;279:12706–12713. doi: 10.1074/jbc.M313874200. [DOI] [PubMed] [Google Scholar]
- 23.van Slegtenhorst M, Mustafa A, Henske EP. Hum Mol Genet. 2005;14:2851–2858. doi: 10.1093/hmg/ddi317. [DOI] [PubMed] [Google Scholar]
- 24.Nakase Y, Fukuda K, Chikashige Y, Tsutsumi C, Morita D, Kawamoto S, Ohnuki M, Hiraoka Y, Matsumoto T. Genetics. 2006;173:569–578. doi: 10.1534/genetics.106.056895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Urano J, Comiso MJ, Guo L, Aspuria PJ, Deniskin R, Tabancay AP, Jr, Kato-Stankiewicz J, Tamanoi F. Mol Microbiol. 2005;58:1074–1086. doi: 10.1111/j.1365-2958.2005.04877.x. [DOI] [PubMed] [Google Scholar]
- 26.Kawai M, Nakashima A, Ueno M, Ushimaru T, Aiba K, Doi H, Uritani M. Curr Genet. 2001;39:166–174. doi: 10.1007/s002940100198. [DOI] [PubMed] [Google Scholar]
- 27.Weisman R, Choder M. J Biol Chem. 2001;276:7027–7032. doi: 10.1074/jbc.M010446200. [DOI] [PubMed] [Google Scholar]
- 28.Weisman R, Roitburg I, Nahari T, Kupiec M. Genetics. 2005;169:539–550. doi: 10.1534/genetics.104.034983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Long X, Lin Y, Ortiz-Vega S, Yonezawa K, Avruch J. Curr Biol. 2005;15:702–713. doi: 10.1016/j.cub.2005.02.053. [DOI] [PubMed] [Google Scholar]
- 30.Long X, Ortiz-Vega S, Lin Y, Avruch J. J Biol Chem. 2005;280:23433–23436. doi: 10.1074/jbc.C500169200. [DOI] [PubMed] [Google Scholar]
- 31.Smith EM, Finn SG, Tee AR, Browne GJ, Proud CG. J Biol Chem. 2005;280:18717–18727. doi: 10.1074/jbc.M414499200. [DOI] [PubMed] [Google Scholar]
- 32.Kunz J, Schneider U, Howald I, Schmidt A, Hall MN. J Biol Chem. 2000;275:37011–37020. doi: 10.1074/jbc.M007296200. [DOI] [PubMed] [Google Scholar]
- 33.Sabatini DM, Barrow RK, Blackshaw S, Burnett PE, Lai MM, Field ME, Bahr BA, Kirsch J, Betz H, Snyder SH. Science. 1999;284:1161–1164. doi: 10.1126/science.284.5417.1161. [DOI] [PubMed] [Google Scholar]
- 34.Wu S, Mikhailov A, Kallo-Hosein H, Hara K, Yonezawa K, Avruch J. Biochim Biophys Acta. 2002;1542:41–56. doi: 10.1016/s0167-4889(01)00164-1. [DOI] [PubMed] [Google Scholar]
- 35.Choi JH, Bertram PG, Drenan R, Carvalho J, Zhou HH, Zheng XF. EMBO Rep. 2002;3:988–994. doi: 10.1093/embo-reports/kvf197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Takahara T, Hara K, Yonezawa K, Sorimachi H, Maeda T. J Biol Chem. 2006;281:28605–28614. doi: 10.1074/jbc.M606087200. [DOI] [PubMed] [Google Scholar]
- 37.Wullschleger S, Loewith R, Oppliger W, Hall MN. J Biol Chem. 2005;280:30697–30704. doi: 10.1074/jbc.M505553200. [DOI] [PubMed] [Google Scholar]
- 38.Wang L, Rhodes CJ, Lawrence JC., Jr J Biol Chem. 2006;281:24293–24303. doi: 10.1074/jbc.M603566200. [DOI] [PubMed] [Google Scholar]
- 39.Sekulic A, Hudson CC, Homme JL, Yin P, Otterness DM, Karnitz LM, Abraham RT. Cancer Res. 2000;60:3504–3513. [PubMed] [Google Scholar]
- 40.Edinger AL, Thompson CB. Oncogene. 2004;23:5654–5663. doi: 10.1038/sj.onc.1207738. [DOI] [PubMed] [Google Scholar]
- 41.Cheng SW, Fryer LG, Carling D, Shepherd PR. J Biol Chem. 2004;279:15719–15722. doi: 10.1074/jbc.C300534200. [DOI] [PubMed] [Google Scholar]
- 42.Chiang GG, Abraham RT. J Biol Chem. 2005;280:25485–25490. doi: 10.1074/jbc.M501707200. [DOI] [PubMed] [Google Scholar]
- 43.Holz MK, Blenis J. J Biol Chem. 2005;280:26089–26093. doi: 10.1074/jbc.M504045200. [DOI] [PubMed] [Google Scholar]
- 44.Alvarez B, Moreno S. J Cell Sci. 2006;119:4475–4485. doi: 10.1242/jcs.03241. [DOI] [PubMed] [Google Scholar]
- 45.Matsuo T, Otsubo Y, Urano J, Tamanoi F, Yamamoto M. Mol Cell Biol. 2007 doi: 10.1128/MCB.01039-06. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yang W, Urano J, Tamanoi F. J Biol Chem. 2000;275:429–438. doi: 10.1074/jbc.275.1.429. [DOI] [PubMed] [Google Scholar]
- 47.Wang X, Campbell LE, Miller CM, Proud CG. Biochem J. 1998;334:261–267. doi: 10.1042/bj3340261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hara K, Yonezawa K, Weng QP, Kozlowski MT, Belham C, Avruch J. J Biol Chem. 1998;273:14484–14494. doi: 10.1074/jbc.273.23.14484. [DOI] [PubMed] [Google Scholar]
- 49.Yan L, Findlay GM, Jones R, Procter J, Cao Y, Lamb RF. J Biol Chem. 2006;281:19793–19797. doi: 10.1074/jbc.C600028200. [DOI] [PubMed] [Google Scholar]
- 50.Im E, von Lintig FC, Chen J, Zhuang S, Qui W, Chowdhury S, Worley PF, Boss GR, Pilz RB. Oncogene. 2002;21:6356–6465. doi: 10.1038/sj.onc.1205792. [DOI] [PubMed] [Google Scholar]
- 51.Brunn GJ, Hudson CC, Sekulic A, Williams JM, Hosoi H, Houghton PJ, Lawrence JC, Jr, Abraham RT. Science. 1997;277:99–101. doi: 10.1126/science.277.5322.99. [DOI] [PubMed] [Google Scholar]
- 52.Brunn GJ, Fadden P, Haystead TA, Lawrence JC., Jr J Biol Chem. 1997;272:32547–32550. doi: 10.1074/jbc.272.51.32547. [DOI] [PubMed] [Google Scholar]
- 53.Sarbassov DD, Guertin DA, Ali SM, Sabatini DM. Science. 2005;307:1098–1101. doi: 10.1126/science.1106148. [DOI] [PubMed] [Google Scholar]
- 54.Hresko RC, Mueckler M. J Biol Chem. 2005;280:40406–40416. doi: 10.1074/jbc.M508361200. [DOI] [PubMed] [Google Scholar]
- 55.Reinke A, Chen JC, Aronova S, Powers T. J Biol Chem. 2006;281:31616–31626. doi: 10.1074/jbc.M603107200. [DOI] [PubMed] [Google Scholar]
- 56.Inoki K, Corradetti MN, Guan KL. Nat Genet. 2005;37:19–24. doi: 10.1038/ng1494. [DOI] [PubMed] [Google Scholar]
- 57.Zhou YH, Zhang XP, Ebright RH. Nucleic Acids Res. 1991;19:6052. doi: 10.1093/nar/19.21.6052. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}