

Abstract
Single injections of morphine induce a state of acute opioid dependence in humans and animals, measured as precipitated withdrawal when an antagonist is administered 4–24 hours after morphine. Additional morphine exposure at daily or weekly intervals results in further increases in withdrawal severity, suggesting that acute opioid dependence reflects the early stages in the development of a chronic state of dependence. The current study evaluated the role of the nucleus accumbens (NAC), bed nucleus of stria terminalis (BNST), interstitial nucleus of posterior limb of the anterior commissure (IPAC), and central amygdala (CeA) in the expression of antagonist-precipitated suppression of operant responding for food as a measure of withdrawal from acute opioid dependence. Rats trained on an FR15 schedule received one or four daily injections, with the lipophobic opioid antagonist methylnaloxonium (16–2000 ng) infused into one of the brain regions or the lateral ventricle (ICV) 4 hr after the final morphine injection. After acute morphine methylnaloxonium was more potent upon infusion into the NAC (17.9-fold potency shift), BNST (6.8-fold) and CeA (5.5-fold) than it was upon ICV administration. Following repeat morphine the NAC and BNST but not CeA continued to show greater sensitivity relative to ICV infusion (12.9- and 8.7-, and 3.2-fold potency shifts, respectively). The IPAC was insensitive to methylnaloxonium after acute or repeat morphine at doses that reliably suppressed responding upon ICV infusion (125–500 ng). Thus, among the components of extended amygdala examined in this study, rapid neuroadaptation within the nucleus accumbens and bed nucleus of the stria terminalis appear to play the most prominent role in antagonist-precipitated suppression of operant responding during the early stages in the development of opioid dependence.
Keywords: amygdala, withdrawal, morphine, addiction, rat
Traditionally tolerance and dependence have been considered to be major factors in chronic, compulsive drug use or addiction (Himmelsbach, 1943; Lindesmith, 1968). Whereas the positive reinforcing or rewarding effects of drugs are considered by most to be a primary factor in the initiation of drug use (Wise, 1998; Koob and Le Moal, 2001 Koob and Le Moal, 2005b), continued use, escalation to compulsive use, and relapse after periods of abstinence may also be motivated by the negative aversive consequences of drug abstinence (Haertzen and Hooks, 1969; Jasinski et al., 1985; Schulteis and Koob, 1996). Considerable previous research has examined mechanisms underlying a chronic state of opioid dependence, in effect identifying neural mechanisms at the systems (anatomical), cellular, and molecular levels that characterize the maintenance of an established state of dependence (reviewed in Self and Nestler, 1998; Nestler, 2001).
Recently there is renewed interest in acute opioid dependence, first described several decades ago (Martin and Eades, 1964), as a means to understanding the role of initial neuroadaptive responses to opioids in the transition from casual to compulsive drug use. In humans opioid antagonists given several hours after an acute treatment with an agonist such as morphine precipitate a range of physiological, somatic, and subjective symptoms characteristic of withdrawal from chronic opioids (Jones, 1980; Bickel et al., 1988; Heishman et al., 1989a,b; Azorlosa et al., 1994). In animal models, a variety of affective signs or aversive stimulus effects of withdrawal are common to withdrawal from acute or chronic opioids, including brain stimulation reward deficits (Schaefer and Michael, 1986; Schulteis et al., 1994; Easterling et al., 2004; Liu and Schulteis, 2004), conditioned place aversion (Stinus et al., 1990; Schulteis et al., 1994; Parker et al., 2002; Azar et al., 2003), and suppression of operant responding for food reward (Gellert and Sparber, 1977; Meyer and Sparber, 1977; Koob et al., 1989; Schulteis et al., 1994, 1997,1999), with the latter being the most thoroughly characterized to date as a measure of withdrawal from acute opioid dependence. In this model, a single pretreatment with morphine increases the potency of opioid antagonists to suppress operant responding, and additional treatments at daily or weekly intervals produce further progressive shifts in antagonist potency (Adams and Holtzman, 1990; Schulteis et al., 1997, 1999). This model has the distinct advantage of lending itself readily to relative assessments of withdrawal severity across a variety of treatment conditions through the use of quantitative dose-response analysis (Young, 1986; Adams and Holtzman, 1990; Schulteis et al., 1994, 2003).
Many neuroadaptational models favor the view that drug dependence and addiction reflect "homeostatic neuronal adaptations and synaptic plasticity in specific brain regions, changes that ultimately contribute to the addictive phenotype" (Shaw-Lutchman et al., 2002) . Specifically, it has been argued that neuroadaptation within the same brain reward circuitry that mediates the acute rewarding effects of drugs results in the expression of drug-opposite withdrawal responses that ultimately play a role in maintaining compulsive use through negative reinforcement, with affective components of withdrawal therefore being of direct motivational significance in maintenance of the addictive state (Schulteis and Koob, 1996; Koob and Le Moal, 2001,2005b). Evidence in favor of this hypothesis implicates brain reward circuitry centered around the so-called extended amygdala, a macrostructure composed of brain regions with very similar morphological and immunohistochemical features, as well as considerable overlap in afferent and efferent connectivity (Heimer et al., 1997; McDonald et al., 1999; Alheid, 2003). Key components of the extended amygdala implicated in drug reward and dependence are the bed nucleus of the stria terminalis (BNST), central amygdala (CeA), and shell aspect of the nucleus accumbens (NAC). Thus, animals will self-administer opioids directly into the NAC and CeA (Olds, 1979, 1982; David and Cazala, 1994), and blockade of opioid receptors within the NAC and BNST among other structures will alter heroin self-administration (Vaccarino et al., 1985; Corrigall and Vaccarino, 1988; Walker et al., 2000).
Several convergent lines of evidence demonstrate that opioid dependence results in neuroadaptation within these same brain regions. In rats with chronic morphine pellet implants, a quaternary derivative of the opioid antagonist naloxone (methylnaloxonium, MNX) suppressed operant responding upon infusion into the NAC at significantly lower doses than those required to elicit this same response following its infusion into the lateral ventricles (Koob et al., 1989). and MNX administration into the NAC and CeA potently precipitated conditioned place aversion (Stinus et al., 1990). In addition, Aston-Jones and colleagues reported that injection of noradrenergic antagonists into the BNST or lesions of the ventral noradrenergic bundle projections to this nucleus attenuated conditioned place aversion in morphine-dependent rats (Delfs et al., 2000). Furthermore, injection of low doses of naloxone in morphine-dependent rats increases the expression of the immediate-early gene c-fos within various limbic and basal forebrain structures including the BNST, NAC, and CeA (Walters et al., 2000; Gracy et al., 2001; Frenois et al., 2002). Finally, Nestler and colleagues (Shaw-Lutchman et al., 2002) reported that cAMP response element (CRE)-mediate gene transcription was elevated during naltrexone-precipitated withdrawal from chronic morphine in the NAC (both core and shell), BNST, CeA, and another portion of extended amygdala, the interstitial nucleus of posterior limb of anterior commissure (IPAC). The current study sought to evaluate whether some or all of these same components of the extended amygdala would show rapid neuroadaptation in response to initial exposure to morphine, and progression with repeated (4x) daily treatment using suppression of responding for food as the measure of withdrawal.
EXPERIMENTAL PROCEDURES
Subjects
Subjects (n=422) were male Wistar rats (Harlan Labs, Indianapolis, IN, USA) weighing between 350–450 grams at the time of testing. All animals were pair housed in a humidity-controlled room with a 12-hour light/dark cycle (lights on at 06:00). Rats had ad-libitum access to water but during training and testing were on food restriction of 15 g/rat/day of standard chow in addition to the pellets earned during operant sessions (total food intake averaged 19–22 g/day/rat). All training and testing took place between the hours of 09:00 and 16:00 Monday through Friday. On off days (Saturday, Sunday), each rat received an additional 5 g/day of food to compensate for the amount of food typically earned in the average operant session. All experimental procedures were approved by the Institutional Animal Care and Use Committee of the VA San Diego Healthcare System, an AAALAC accredited facility, and are in strict accordance with the Guide for the Care and Use of Laboratory Animals (revised 1996).
Drugs
Morphine sulfate was purchased via the VA San Diego Research Pharmacy from King Pharmaceuticals (Bristol, TN, USA) at a stock concentration of 25 mg/ml. The final dose of 5.6 mg/kg morphine was prepared fresh at least twice weekly by diluting the stock solution with sterile physiological saline to achieve a final concentration of 5.6 mg/ml for subcutaneous (SC) injection in a volume of 0.1 ml/100 g body weight. Methylnaloxonium (MNX, N-methylnaloxonium iodide) and naloxone hydrochloride were purchased from Sigma (St. Louis, MO, USA) in powder form. Naloxone was prepared in physiological saline at a concentration of 1 mg/ml for SC injection in a volume of 0.1 ml/100 g body weight. Desired doses of MNX were prepared fresh daily by dissolving in sterile physiological saline 30 minutes prior to injection. MNX was administered either by the intracerebroventricular (ICV) or intracerebral (IC) route with injectors connected to Hamilton microsyringes driven by a Harvard Apparatus syringe pump (Holliston, MA, USA). ICV and IC injections consisted of saline vehicle or MNX at a dose of 16, 31.25, 62.5, 125, 250, 500, 1000, or 2000 ng. For ICV injections the total dose was injected unilaterally in a volume of 2 μl over 80 s. All IC injections were bilateral, with each hemisphere receiving ½ the total dose in a volume of 0.5 μl infused over 80 s. Injectors remained in place for an additional 90 s to allow for diffusion of MNX.
Cannulation procedure
Stainless steel guide cannulas (23 gauge) were implanted under halothane anesthesia using a Kopf Instruments stereotaxic device (Tujunga, CA, USA). All ICV and IC surgeries were conducted with the incisor bar set at −3.3 mm below the interaural line, with implant coordinates taken from the atlas of Paxinos and Watson (1998). Cannulas were anchored to the skull with the aid of dental cement and surgical screws. Stainless steel wire stylets matching the length of the guide cannulas were inserted immediately after surgery and thereafter replaced daily as needed to maintain cannula patency. All rats were allowed to recover at least 4–5 days prior to resumption of operant sessions.
Final injector tip coordinates were selected to target the center of each nucleus without regard to further subdivisions within the nucleus, and are expressed relative to skull surface at bregma (Paxinos and Watson, 1998). Coordinates used were as follows: ICV (AP −0.80, ML ± 1.4, DV −4.0), NAC (AP + 1.70, ML ± 1.70 DV −8.0), BNST (AP −0.85, ML ± 1.5, DV −7.0), CeA (AP −2.0, ML ± 4.0, DV −8.5); and IPAC (AP −1.4, ML ± 3.9, DV − 7.9). Guide cannulas stopped 1.5 mm (ICV, NAC, BNST) or 1.9 mm (CeA, IPAC) short of the desired final target for the injector tips. Guide cannulas targeting the BNST were angled to avoid the lateral ventricle above the desired injection location (Delfs et al., 2000). All guide cannula placements were bilateral with the exception of ICV implants, which consisted of left or right unilateral placements alternating from one rat to the next.
Verification of cannula placement
ICV infusions
Correct placement was assessed immediately after the operant session that followed mock injection (Habituation Day, see Operant training and testing below and Table 1) by means of a "gravity check", whereby the injector was inserted to the desired depth and flow of vehicle through the injection tubing was observed to occur by gravity as the tubing was slowly raised above the level of the injector tip. A small air bubble in the tubing and graded marks at 0.5 μl increments along the length of the tubing served as visual aids. Four rats were excluded from the study for failing the "gravity check".
Table 1.
Experimental Design
| Habituation Day | Baseline Day | Morphine Treatment Days 1–3 | Test Day | |
|---|---|---|---|---|
| Repeat Morphine | ||||
| S.C. Injection | ----- | Vehicle | Morphine 5.6 mg/kg | Morphine 5.6 mg/kg |
| IC/ICV Injection | Mock | Vehicle | ----- | MNXa |
| Operant Testing | 30 min | 30 min | ----- | 30 min |
|
| ||||
| Single Morphine | ||||
| S.C. Injection | ------ | Vehicle | Vehicle | Morphine 5.6 mg/kg |
| IC/ICV Injection | Mock | Vehicle | ------ | MNXa |
| Operant Testing | 30 min | 30 min | ------ | 30 min |
|
| ||||
| Morphine Naive | ||||
| S.C. Injection | ------ | Vehicle | Vehicle | Vehicle |
| IC Injection | Mock | Vehicle | ------ | MNXb |
| Operant Testing | 30 min | 30 min | ------ | 30 min |
At one of 3 or more doses ranging from 16 to 2000 ng
At a single dose (highest dose tested in a given brain region)
IC infusions
For all brain regions targeted, placement verification was by post-mortem histological analysis. Rats received a barbiturate overdose followed by transcardial perfusion with 10% formalin. Brains were removed, placed in 10% formalin for 24 hours, and then an 0.5–1.0 cm slice encompassing the cannula entry point into the brain as well as the final desired injector tip location was embedded in paraffin. Embedded blocks of tissue were sectioned in 10 μm-coronal slices using a sliding microtome, placed on slides, and allowed to dry. After staining sections with 0.15% cresyl violet, final injector tip placements were visualized under a dissecting microscope, using the stereotaxic atlas of Paxinos and Watson (1998) as a guide. The injector tip locations for each animal were mapped onto the appropriate atlas panel with the histologist blinded to drug treatment condition and experimental outcome. Cannula placements for all correct cannula placements within the Repeat and Single Morphine groups treated with the two highest doses injected into each brain region are illustrated in Figure 1. For purposes of clarity, placements for other doses are not shown, but those shown are representative of the general distribution of cannula placements for all experimental groups within a given injection site. A total of 31 rats were excluded from the final analysis due to misplaced cannulas, with the majority of the failed placements occurring in the IPAC (15 rats), volumetrically the smallest of the targeted structures, followed by the BNST (8 rats), CeA (4 rats), and NAC (4 rats).
Fig. 1.
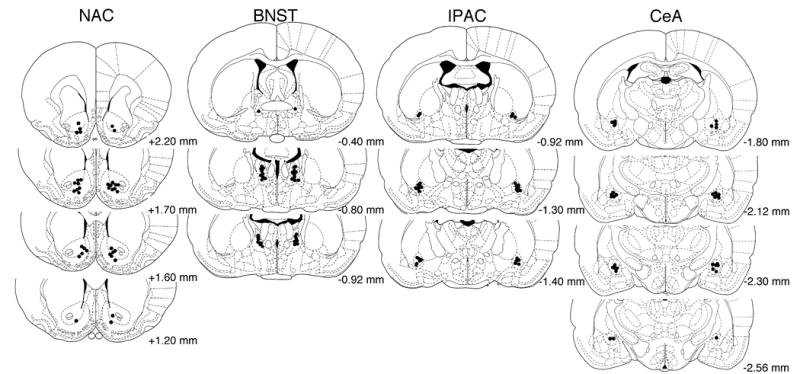
Cannula tip placements for the two highest doses tested under Single Morphine (left side) and Repeat Morphine (right side) conditions within each brain region examined. Values represent AP distance from bregma (mm) according to the atlas of Paxinos and Watson (1998). To maintain clarity of individual tip placements, cannula locations for other doses are not shown, but the range of tip placements illustrated is representative of all dose groups.
Operant training and testing
Apparatus and training
Fourteen operant chambers housed in sound-attenuated cubicles (Coulbourn Instruments, Columbus, OH, USA) were equipped with a food hopper located 4 cm above a grid floor, a lever manipulandum located to the right of the food hopper, and a cue light located above the lever. Each completed fixed-ratio (FR) requirement resulted in illumination of the cue light for 1 s coincident with delivery of a food pellet (45 mg). Rats were trained to lever press in daily 30 min sessions five days a week, beginning with an FR-1 schedule (1 s timeout) and progressing in several steps to the ultimate requirement of FR-15 (1 s timeout). Assignment to a treatment condition took place once responding had stabilized on the FR-15 schedule (typically after 14–21 days), with stability defined as variation in response rates no greater than 10% from the mean of 3 consecutive days.
Acute dependence induction and withdrawal testing
Post-surgery rats typically required about 5–7 days of testing until responding had again reached stable baselines as defined above. Group assignments and experimental design were as outlined below and summarized in Table 1:
Habituation Day
Prior to any actual drug infusions, rats were habituated to the injection procedure by insertion of an injector (31 gauge) that did not extend beyond the tip of the guide cannula. After insertion of the injector the Harvard syringe pump was activated for 80 s to acclimate rats to the motor noise, but the injector was not connected to the delivery system. Rats were free to roam in an injection holding cage for the duration of the infusion process. After the pump was turned off the injector remained in place for an additional 90 s at which time it was removed, the stylet was re-inserted into the guide cannula, and an operant session commenced immediately thereafter.
Baseline Day
Following the Habituation Day, rats received 2–3 days without any operant training to ensure that responding on the Baseline Day occurred under conditions comparable to those on the Test Day, where operant responding following morphine and MNX treatment would be assessed after several days without operant testing (see Table 1 and below for details). On the Baseline Day, all rats (regardless of target injection site) received a SC injection of saline vehicle, followed 4 hr later by ICV or IC infusion of vehicle and a 30 min operant session. Infusion procedures were as just described for mock injection, except that injectors now penetrated past the guide cannula tip by the appropriate amount (1.5–1.9 mm, see Cannulation Procedure above) and were connected via tubing to a microsyringe being driven by the syringe pump.
Morphine Treatment Days
After the Baseline Day, each cohort of rats with cannulas aimed at a given target site received morphine under one of two possible treatment regimens: 1) Repeat Morphine wherein rats received 4 consecutive days of treatment with a 5.6 mg/kg SC dose of morphine; or 2) Single Morphine wherein rats received 3 days of SC vehicle injection, followed by a single morphine treatment (5.6 mg/kg SC) on the 4th (Test) day. By employing separate groups for the Single and Repeat (4x) Morphine conditions, we could assess the magnitude of neuroadaptation in response to either Single or Repeat Morphine exposure without the possible confounding influence of repeated antagonist-precipitated withdrawal experience. Prior work indicated that repeated precipitation of withdrawal following each of four morphine treatments could engender conditioned withdrawal responses that potentiated the apparent magnitude of response suppression elicited by a given dose of antagonist (Schulteis et al., 2005).
Test Day
After the 4th and final SC injection (Test Day), rats in the Single and Repeat Morphine conditions were further subdivided into groups receiving an infusion of vehicle or one of several doses of MNX 4 hr post-morphine in a between-subjects dose-effect design. Doses were selected as described below (Experimental design and dosing strategy).
Experimental design and dosing strategy
The dose of morphine and interval between morphine and antagonist injection were based upon extensive prior work characterizing optimal conditions for antagonist-induced suppression of responding in acute dependence (Schulteis et al., 1997, 1999, 2003, 2005). Although these and similar studies on suppression of operant responding during withdrawal from acute opioid dependence have typically precipitated withdrawal using naloxone or naltrexone (Young, 1986; Adams and Holtzman, 1990; Schulteis et al., 1997, 1999), site-specific IC injections of these highly lipophilic compounds would result in rapid spread from the site of administration throughout the CNS and even into the general circulation. Fortunately, modification of the naloxone molecule to its quaternary derivative MNX results in a more lipophobic compound that spreads much less rapidly from its site of administration (Schroeder et al., 1991), and consequently MNX has been used successfully in a number of prior studies to localize critical site(s) in the expression of withdrawal signs from chronic opioid administration (Koob et al., 1989; Stinus et al., 1990; Maldonado et al., 1992). The selected dose range of MNX from 16 to 2000 ng was based upon this prior work of Koob and colleagues, with the maximum dose of 2000 ng MNX selected because higher doses (4000ng) have been reported to suppress responding even in morphine naive animals following ICV infusion (Koob et al., 1989).
The key feature of the experimental strategy used in these studies with MNX is comparison of its potency to precipitate withdrawal when infused into a specific brain region to its potency following intracerebroventricular (ICV) administration. Critical sites directly involved in expression of the response should be sensitive to MNX doses significantly below those that are effective upon ICV infusion, since ICV-administered MNX must first diffuse to its site of action before taking effect.
With the large number of treatment conditions evaluated in the present study (5 injection sites, each tested under two morphine pretreatment conditions), our critical goal was to strike an efficient balance between minimization of animal subjects requirements where possible with the need to generate all necessary control groups and to ensure that the linear portion of the MNX dose effect function had been captured under every treatment condition. To achieve this balance, a sequential dose selection strategy was employed wherein all target sites were initially evaluated at an intermediate dose of MNX (62.5 or 125 ng for IC targets, 250 or 500 ng for ICV), with higher and/or lower doses then tested according to the magnitude of response suppression elicited at this initial dose.
Criteria that guided the decision for selection of doses tested for a given treatment condition included: 1) a minimum of 3 doses tested for each dose-effect function; 2) identifying at least one ineffective dose for each infusion site to establish the lower limits of the dose-effect function; 3) identifying at least one dose where 50% or greater suppression of responding was observed; and 4) avoiding excessive testing of doses in the lower (no effect) or upper (maximal effect) asymptotic portions of the dose-effect function. Criteria 1–3 were critical to the successful calculation of ED50 values and relative potency estimates (see Data analysis below), while criteria 4 helped to minimize animal subjects requirements wherever feasible. In most cases, 3 doses plus a vehicle control group were needed for each experimental condition studied; occasionally a fourth MNX dose was added to more clearly define the linear portion of the dose-effect function in cases where two adjoining intermediate doses of MNX produced similar effect (e.g. intra-BNST MNX at 125 and 250 ng under Single Morphine conditions, see Figure 2). Our goal was not to identify the minimum effective dose within each brain region per se, but rather to obtain an accurate estimate of the linear portion of the dose effect function under each experimental condition.
Fig. 2.
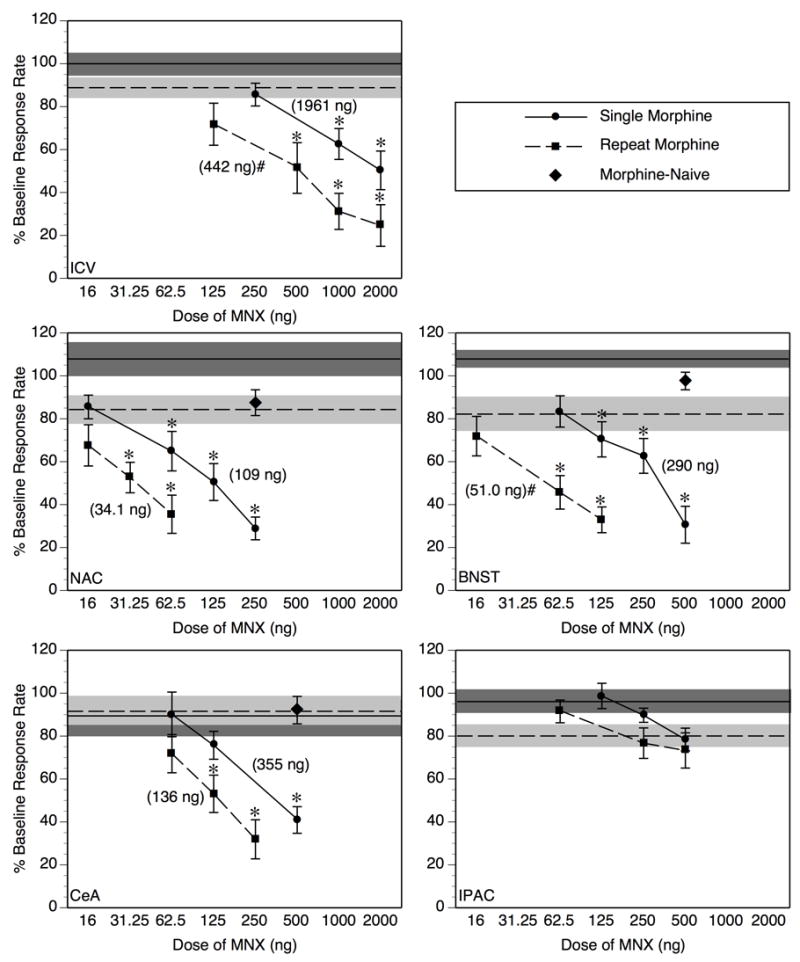
Following an acute treatment with 5.6 mg/mg morphine (Single Morphine, circles + solid lines) MNX dose-dependently suppresses operant responding for food after injection into the lateral ventricle (ICV), nucleus accumbens (NAC), bed nucleus of the stria terminalis (BNST) or central amygdala (CeA), but not the interstitial nucleus of the posterior limb of the anterior commissure (IPAC) (*p < 0.05 vs. vehicle-infused group under the corresponding morphine treatment condition (Single compared to Single, Repeat to Repeat). After 4 morphine treatments at daily intervals (Repeat Morphine, squares + dotted lines), the magnitude of response suppression elicited by MNX was further potentiated with ICV, and intra-BNST infusions, but not with intra-NAC or intra-CeA infusion (#p < 0.05, significant shift in MNX dose-effect function from Single to Repeat Morphine condition according to relative potency analysis, see Table 2 for further details of this analysis). In Morphine-Naive subjects (diamonds), MNX at the highest dose tested within active brain regions (NAC, BNST, CeA) failed to suppress operant responding relative to vehicle-infused controls. Individual data points on the MNX dose-effect functions represent mean ± SEM percent of baseline response rate. Values in parentheses are the ED50 values derived for the corresponding dose-response function. The solid horizontal line represents the mean of the group infused with vehicle 4 hr after Single Morphine Treatment, and the dotted horizontal line represents the mean of the Repeat Morphine vehicle-infused group; the shaded region around each horizontal line represents ± 1 SEM. N = 8–11/dose group.
Data analysis
All operant data (responses/30 min session) on the Test Day were expressed as percent of responding on the Baseline Day for each rat. The resultant percent of baseline data were entered into a number of analyses using ANOVA and/or quantitative probit dose-response analysis using the method of Litchfield and Wilcoxon (Tallarida and Murray, 1987) as described below:
Acute and repeat morphine effects in vehicle-infused controls
All groups that received IC or ICV infusion of vehicle were entered into an overall 2-factor ANOVA with injection site and morphine history (Single, Repeat Morphine) as between-subjects factors to assess possible effects of repeated morphine treatment in the absence of antagonist treatment.
MNX effects after acute and repeat morphine relative to vehicle-infused controls
One -factor ANOVA with MNX dose as the between-subjects factor assessed whether MNX produced significant dose-dependent effects within a given injection site under Single or Repeat Morphine conditions. Follow-up individual means comparisons of MNX doses to vehicle-control were adjusted using the Bonferroni correction for multiple planned comparisons.
MNX effect in Morphine-Naive rats
For brain regions wherein MNX potency exceeded that observed following ICV injection following Single or Repeat Morphine (NAC, BNST, CeA), the highest dose of MNX infused into a particular region was evaluated after SC injection of vehicle (Morphine Naive). To ensure that the effect within a given brain region was not due to non-specific effects of MNX on operant responding, responding after MNX infusion in Morphine Naive rats was compared to responding in IC-vehicle-infused controls after Single and Repeat Morphine treatment in a one-factor ANOVA.
Relative MNX potency as a function of morphine treatment history and injection site
As an expected consequence of our sequential dosing strategy (see Experimental design and dosing strategy above), there was frequently little or no overlap in MNX dose-effect functions across experimental treatment conditions (e.g. ICV and NAC did not have any overlapping doses under Repeat Morphine conditions), precluding omnibus ANOVA analyses. However, the data are well-suited to quantitative probit dose response analysis, as applied successfully to our prior work on antagonist-precipitated withdrawal from acute and chronic opioid dependence (Schulteis et al., 1994; Azar et al., 2003; Schulteis et al., 2003, 2005). After calculation of MNX ED50 values (with 95% confidence limits) under each experimental condition, statistical significance of shifts in MNX potency was assessed by potency ratio analysis using the method of Litchfield and Wilcoxon (Tallarida and Murray, 1987).
RESULTS
Baseline response rates
Responses rates on the Baseline Day prior to any morphine or MNX treatment for all rats receiving each of the five surgery types (ICV or IC) were as follows (mean responses/30 min ± SEM): ICV = 1957 ± 45.2, NAC = 1824 ± 52.9, BNST = 1873 ± 57, CeA = 1889 ± 58, IPAC = 1941 ± 48.7. One-factor ANOVA indicated that there were no significant differences in baseline response rate across surgery conditions (F[4,390] = 1.14, p > 0.30). Therefore, variations in tissue damage as a function of the path traversed by the guide cannula did not significantly alter response rate, and all subsequent analyses of drug effects on responding were expressed as a percentage of these baseline response rates to be consistent with prior work in this area (Koob et al., 1989; Schulteis et al., 1994,1997,1999).
Acute and repeat morphine effects in vehicle-infused controls
Following IC or ICV vehicle infusion under Single and Repeat Morphine conditions, there was a significant main effect of morphine treatment history (F[1,81] = 11.01, p < 0.005), with no significant main effect of injection site (F[4,81] = 0.19, p > 0.90) or history x site interaction (F[4,81] = 1.11, p > 0.35). As shown in Figures 2 and 3, responding in vehicle-infused groups under Repeat Morphine conditions was generally slightly lower than responding under Single Morphine conditions, presumably due to the emergence of a modest but statistically reliable spontaneous withdrawal reaction upon repeat morphine exposure, an effect we have observed previously under the same treatment conditions (e.g. Liu and Schulteis, 2004; Schulteis et al., 2005). Importantly, responding after vehicle infusion did not differ significantly across injection sites, indicating that any observed site-specific differences in MNX-induced suppression of responding were attributable to varying sensitivity to the drug, rather than nonspecific effects of fluid infusion into one or more brain regions.
Fig. 3.
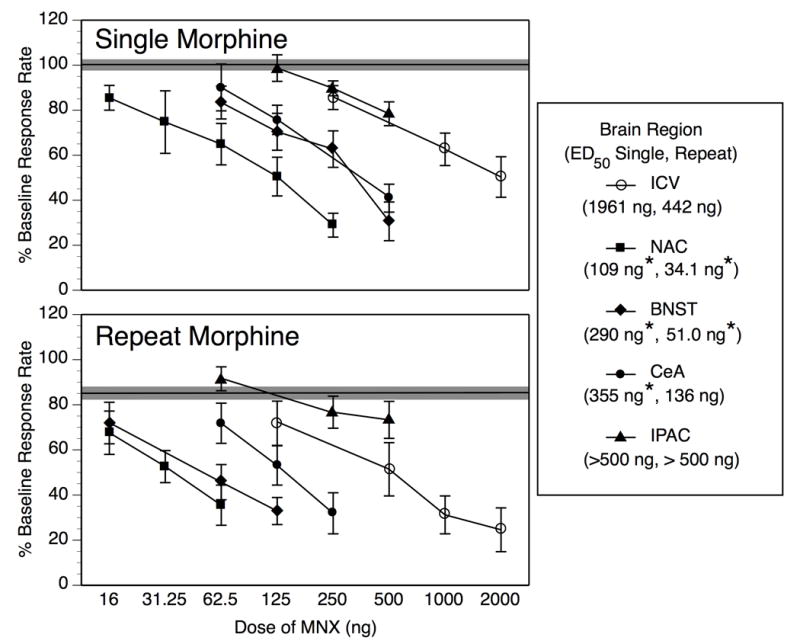
This Figure re-presents the data from Fig. 2 in a way that facilitates comparison of MNX dose-effect functions across injection sites. Following a Single Morphine treatment (upper panel), MNX potency is significantly greater in the nucleus accumbens (NAC), bed nucleus of the stria terminalis (BNST), and central amygdala (CeA) than it is following intracerebroventricular infusion (ICV), but MNX potency in the interstitial nucleus of the posterior limb of the anterior commissure (IPAC) does not differ from ICV infusion. With Repeat Morphine treatment (lower panel), MNX remains significantly more potent in the NAC and BNST than following ICV infusion, but its potency in the CeA in addition to the IPAC is not significantly different from its ICV potency. Individual data points on the MNX dose-effect functions represent mean ± SEM percent of baseline response rate. The solid horizontal line represents the composite mean of all 5 injection site groups infused with vehicle 4 hr after Single or Repeat Morphine, and the shaded region around each horizontal line represents ± 1 SEM. Values in parentheses within the legend reflect the ED50 value following Single and Repeat Morphine for that given brain region. *Significant shift in MNX potency relative to ICV treatment under same morphine treatment condition as determined by potency ratio analysis (see Table 2 for full results of this analysis).
MNX effects after acute and repeat morphine relative to vehicle-infused controls
Figure 2 allows direct comparison of MNX potency under Single and Repeat Morphine treatment conditions within each individual injection site.
ICV infusions
MNX infused into the lateral ventricle (ICV) 4 hr after Single Morphine treatment elicited a dose-dependent suppression of responding (F[3,36] = 8.07, p < 0.001), with doses of 1000 and 2000 ng, but not 250 ng, producing a significant reduction in responding relative to vehicle-infused controls (p < 0.05 after Bonferroni correction). After Repeat Morphine treatment, ICV-infused MNX again produced a dose-dependent effect (F[4,41] = 8.26, p < 0.0001), with doses of 500, 1000, and 2000 ng producing a significant reduction in responding.
Intra-NAC infusions
MNX dose-dependently suppressed operant responding for food with intra-NAC infusion after a Single Morphine treatment, as revealed by a significant main effect of MNX dose (F[4,35] = 13.15, p < 0.0001); follow-up analysis indicated that doses of 62.5 ng and higher produced a significant effect relative to vehicle controls (p < 0.05 after Bonferroni correction). Dose-dependent suppression of responding was also evident when MNX infusion into the NAC followed the last of 4 daily morphine treatments (Repeat Morphine, F[3,32] = 5.99, p < 0.005), with doses of 31.25 ng and higher producing a significant effect relative to intra-NAC vehicle infusion.
Intra-BNST infusions
As the data for MNX infused into the BNST depicted in Figure 2 would suggest, there was a significant dose-dependent effect of MNX after acute morphine treatment (Single Morphine, F[4,21] = 12.44, p < 0.0001), with significant effects at doses of 125 ng and higher (p < 0.05 after Bonferroni correction). After Repeat Morphine treatment, MNX continued to exert a dose-dependent effect (F[3,37] = 7.65, p < 0.001), with doses of 62.5 ng and higher producing a significant effect.
Intra-CeA infusions
Analysis of the data from groups infused with MNX into the CeA following Single Morphine treatment revealed a significant main effect of MNX dose (F[3,28] = 9.71, p < 0.001), but a relatively high dose of 500 ng was required to achieve significant suppression relative to vehicle-infused controls (p < 0.05 after Bonferroni correction). After Repeat Morphine treatment, intra-CeA MNX still produced a dose-dependent suppression (F[3,29] = 8.73, p < 0.001), but again a high dose (125 ng) was required relative to doses shown to be effective intra-NAC or intra-BNST (62.5 ng or less) under the same morphine treatment conditions.
Intra-IPAC infusions
Inspection of the data from IPAC-infused subjects in Figure 2 suggests that no dose of MNX up to 500 ng elicited a significant suppression of responding after Single or Repeat Morphine treatment, and this was confirmed by ANOVAs which revealed no significant effect of MNX dose (F's < 1.35, p's > 0.25). Because 500 ng of MNX infused into the lateral ventricle (ICV) elicited significant effects following Repeat Morphine, higher doses infused into the IPAC were not evaluated, as these data already convincingly demonstrate that the IPAC is not a site where diffusion of ICV-administered MNX is producing its effect on operant responding.
MNX effect in Morphine-Naive rats
Infusion of 250 ng of MNX into the NAC, and 500 ng MNX into each of the BNST and CeA, resulted in percent of baseline response rates of 87.5 ± 6.0, 97.6 ± 4.1, and 92.1 ± 6.4, respectively (see Figure 2). Comparing these levels of responding to vehicle-infused controls from Single and Repeat Morphine treatment groups in a one-factor ANOVA revealed no significant effects for any brain region (all F's < 2.21, p's > 0.20). Inspection of Figure 2 reveals that these doses of MNX suppressed responding to well below 50% after just one morphine treatment (Single Morphine) in each brain region examined. The results in Morphine-Naive rats indicate that the response suppression elicited by MNX after Single or Repeat Morphine are not due to nonspecific effects of MNX infused into the NAC, BNST, or CeA on operant responding per se, but instead reflect a potentiation of MNX potency produced by acute or repeated daily morphine pretreatment.
Relative MNX potency as a function of morphine treatment history and injection site
Single vs. Repeat Morphine within individual injection sites
ED50 values for MNX-induced suppression of operant responding are summarized in Table 2. The full range of doses tested under each condition was used in calculation of ED50 values, and in determination of the potency ratio of MNX after Single vs. Repeat Morphine treatment within each given injection site. Shifts in MNX potency from Single to Repeat Morphine conditions were significant for ICV (4.4-fold) and intra-BNST (5.7-fold) administration, but not for intra-NAC (3.2-fold) or intra-CeA (2.6-fold) infusion (see Figure 2 and Table 2). Although ED50 values and potency ratios could not be calculated for the IPAC data (because even the highest dose tested produced substantially less than 50% effect), inspection of Figure 2 indicates minimal change in the MNX dose-effect function from Single to Repeat Morphine treatment.
Table 2.
Region-Specific ED50 Values for MNX and Potency Ratios after Single or Repeat Morphine
| ED50 (95 % CL)a | Potency Ratio (95% CL) | ||||
|---|---|---|---|---|---|
| Injection Site | Single Morphine | Repeat Morphine | Single vs Repeat at Same Site | Single vs ICV Single | Repeat vs ICV Repeat |
| ICV | 1961 (556 – 6903) | 442 (170 – 1149) | 4.4* (0.9 – 21.6) | -------- | -------- |
| NAC | 109 (46.1 – 259) | 34.1 14.5 – 80.4 | 3.2 (0.9 – 10.8) | 17.9* (3.9 – 82.5) | 12.9* (3.6 – 46.7) |
| BNST | 290 (121 – 696) | 51.0 (17.2 – 151) | 5.7* (1.4 – 22.9) | 6.8* (1.5 – 31.3) | 8.7* (2.0 – 36.8) |
| CeA | 355 (144 – 873) | 136 (65.6 – 281) | 2.6 (0.8 – 8.3) | 5.5* (1.2 – 25.9) | 3.2 (1.0 – 10.8) |
| IPAC | > 500 | > 500 | -------- | -------- | -------- |
Values reflect ED50 in ng, 95 % confidence limits (CL) in parentheses
p<0.05 significant potency ratio as determined by method of Litchfield and Wilcoxon (Tallarida and Murray, 1987)
MNX potency in discrete brain regions relative to ICV potency after Single Morphine
MNX after Single Morphine treatment was most potent upon infusion into the NAC, demonstrating a 17.9-fold shift in potency relative to ICV infusion (see Figure 3, upper panel, and Table 2). MNX infused into the BNST and CeA showed lesser but still significant shifts in potency relative to ICV infusion under Single Morphine conditions (6.8- and 5.5-fold, respectively). Again, the ED50 value for MNX in IPAC could not be calculated, and therefore precluded relative potency comparisons, but Figure 3 demonstrates that potency of MNX in the IPAC is no greater than its ICV potency after acute morphine treatment
MNX potency in discrete brain regions relative to ICV potency after Repeat Morphine
After Repeat Morphine Treatment (Figure 3, lower panel and Table 2), potency of MNX remained significantly greater in the NAC and BNST than in the lateral ventricle (12.9-fold and 8.7-fold shifts relative to ICV, respectively). MNX potency in the CeA was not significantly different from its potency after ICV infusion (3.2-fold shift). Again, an ED50 for MNX in the IPAC could not be calculated (> 500 ng), but Figure 3 illustrates that MNX was no more potent when infused into the IPAC than after ICV administration.
DISCUSSION
The current study supports and extends earlier work indicating that acute treatment with a moderate dose of morphine (5.6 mg/kg) induces a state of acute dependence as demonstrated by suppression of operant responding when an antagonist is administered 4 hr later (Meyer and Sparber, 1977; Young, 1986; Adams and Holtzman, 1990, 1991; Schulteis et al., 1997, 1999). However, whilst Adams and Holtzman (1991) had previously shown that ICV infusion of morphine could increase the potency of systemically-administered naltrexone to suppress operant responding, to our knowledge this is the first study which demonstrates the efficacy of ICV or site-specific IC infusion of an opiate antagonist in precipitating withdrawal from acute opioid dependence. Thus, MNX administered 4 hr after a single dose of morphine dose-dependently suppressed operant responding upon infusion into the lateral ventricles (ICV), NAC, BNST, or CeA, but not when infused into the IPAC. Importantly, the potency of MNX infused into the NAC, BNST, and CeA after Single Morphine treatment was significantly greater than its ICV potency (see Figure 3 and Table 2), suggesting that neuroadaptive responses within the NAC, BNST, and CeA are rapidly induced and may collectively contribute to antagonist-induced suppression of operant responding after Single Morphine treatment.
Our finding that ICV potency of MNX increased with Repeat Morphine treatment is also consistent with earlier reports that antagonist-precipitated signs of withdrawal increase progressively with repeated morphine exposure (Adams and Holtzman, 1990; Schulteis et al., 1997, 1999 Schulteis et al., 2004). However, the results of the current study revealed that only some components of the extended amygdala exhibit this increase in MNX potency with repeated morphine (see Table 2). While intra-NAC infusion of MNX produced the greatest effect under Single Morphine conditions, the dose-effect functions for MNX in the NAC and BNST were virtually identical (Figure 3) under Repeat Morphine conditions, suggesting a possible increased contribution of neuroadaptation within the BNST as dependence progresses. Consistent with this interpretation, it has been reported that infusion of MNX into the BNST alters the reinforcing efficacy of self-administered heroin in chronically dependent rats, while being without effect in nondependent rats (Walker et al., 2000). In contrast, MNX potency in the CeA no longer differed from its ICV potency after Repeat Morphine treatment, indicating a possible diminished role of MNX blockade of CeA opioid receptors relative to other sites (NAC, BNST) after repeated morphine. Finally, although the IPAC showed dramatic changes in gene expression during precipitated withdrawal from chronic morphine (Shaw-Lutchman et al., 2002) similar to or greater than those observed in the NAC, BNST and CeA, our results indicate that neuroadaptation within the IPAC appears to play no role in the expression of the aversive motivational state resulting from acute opioid withdrawal.
Although Koob and colleagues (1989) had not examined MNX potency in the BNST or CeA in precipitating suppression of responding in rats chronically dependent on morphine, the enhanced sensitivity of the NAC to MNX after Single and Repeat Morphine treatment is consistent with their work. Indeed, if one calculates the relative potency of NAC- to ICV-administered MNX in the Koob et al. study on chronic dependence (11-fold shift), it is quite similar to the relative potency of NAC to ICV MNX after Single (18-fold) or Repeat Morphine (13-fold) in our study. This proportional shift in relative MNX potency (NAC to ICV) across varying degrees of morphine exposure (single, repeat, chronic) further supports the notion that acute dependence as measured in our animal model reflects the early stages in the development of a chronic state of opioid dependence (Schulteis and Koob, 1996; Harris and Gewirtz, 2005).
The guiding assumption behind our experimental approach, employed previously by Koob and colleagues (Koob et al., 1989; Stinus et al., 1990; Maldonado et al., 1992), was that since ICV-administered MNX must first diffuse to its site(s) of action, MNX potency within critical target regions must be significantly greater than its ICV potency. Therefore, potency ratio comparisons of ICV to site-specific infusions of MNX were critical to the test of our hypothesis. While direct potency ratio comparisons between individual brain regions might also seem an attractive option, we were reluctant to conduct this statistical analysis without a readily apparent algorithm for how differences in volume of each brain region might affect potency ratios. Nonetheless, it is striking that one of the largest brain regions tested (NAC) showed the greatest initial response to MNX, whereas the smallest region tested (IPAC) was not sensitive to MNX doses that produced a reliable effect after ICV administration. Therefore, differences in relative potency of MNX following intra-NAC, BNST, CeA, or IPAC infusion apparently are not due simply to the proportion of each nucleus that was covered by the diffusion radius of MNX within the 30 min test period, but instead likely reflect the relative degree to which neuroadaptive responses within the individual structures examined contribute to the expression of suppression of operant responding in the early stages of opioid dependence.
With regard to specific components of the interoceptive state produced by opioid withdrawal that engender antagonist-precipitated suppression of responding, it must be acknowledged that an underlying general malaise that is comprised of somatic and physiological signs of withdrawal could decrease motivation to respond for food reward. While this possibility cannot be discounted at higher doses of antagonists that precipitate somatic signs in addition to suppression of responding, several pieces of evidence indicate that elicitation of somatic signs is not a necessary condition. First, minimal doses of systemically-administered naloxone that reliably precipitate suppression of operant responding after acute (Schulteis et al., 1997, 1999) or chronic (Higgins and Sellers, 1994; Schulteis et al., 1994) morphine produce few if any somatic signs of withdrawal. In addition, doses of MNX that suppress operant responding when applied to the NAC, BNST, or CeA in rats exposed acutely (current study) or chronically (Koob et al., 1989) to morphine do not elicit somatic signs of withdrawal when infused into those same brain regions (Stinus et al., 1990; Maldonado et al., 1992; Liu et al., 2002). Notably, we reported previously that response-disruptive doses of MNX in the BNST and NAC failed to elicit significant somatic signs when infused into these same brain regions under conditions of Single or Repeat (4x) morphine treatment identical to those tested herein (Liu et al., 2002).
An additional possible factor that may engender suppression of responding for food during withdrawal from acute and chronic opioids is reduced appetite, which has been reported during spontaneous withdrawal from chronic opioids (Ronnback et al., 1987; van der Laan et al., 1991; Langerman et al., 2001). However, reduced appetite cannot readily account for antagonist-precipitated suppression of motivated responding for water (White and Holtzman, 2001) or non-consumable reinforcers such as direct electrical stimulation of the reward system (Schaefer and Michael, 1986; Easterling and Holtzman, 1997, 2004) in rats exposed to acute or chronic morphine. Instead, we postulate that reduced appetite and suppression of motivated responding for both nutritive and non-nutritive reinforcers may be independent manifestations of aversive states that accompany opioid withdrawal. As such, aversive states reduced the hedonic salience of a broad range of reinforcers. This is consistent with the fact that both suppression of operant responding and CPA, a direct measure of withdrawal aversion, are elicited at similar doses of naloxone in rats exposed to acute and chronic morphine (Schulteis et al., 1994,1997; Azar et al., 2003), and that antagonist-precipitated CPA also appears to involve neuroadaptation within the NAC, BNST and CeA (Stinus et al., 1990; Delfs et al., 2000; Gracy et al., 2001).
Having considered these alternatives, the question remains as to what symptom(s) of opioid withdrawal elicited by opioid receptor blockade in the NAC, BNST, and CeA are primary in engendering this aversive motivational state. In this regard, it is noteworthy that systemic administration of naloxone elevates brain reward thresholds and produces anxiogenic-like effects in the elevated plus-maze at doses comparable to those that produce suppression of operant responding and CPA after chronic (Higgins et al., 1994; Schulteis et al., 1994, 1998) or acute morphine (Liu and Schulteis, 2004; Schulteis and Zhang, 2006, in preparation). We suggest that such affective components of opioid withdrawal may comprise at least in part the critical factors that elicit suppression of responding and CPA during opioid withdrawal. Future work using site-directed infusions of MNX to identify the brain sites most sensitive to precipitation of brain reward deficits and anxiogenic-like effects should provide a direct test of this hypothesis.
CONCLUSION
In summary, the current study provides direct support for the hypothesis that discrete elements of the extended amygdala involved in the expression of the positive affective states resulting from acute opioid intake undergo rapid neuroadaptation in response to a single dose of morphine, and are critical sites mediating expression of motivationally relevant signs of opioid withdrawal (Schulteis and Koob, 1996; Wise, 1998; Koob and Le Moal, 2001 Koob and Le Moal, 2005b). Among the regions examined, neuroadaptive responses within the NAC and BNST in particular appeared be of primary significance in contributing to response suppression after initial and repeated morphine treatment, with the CeA seemingly playing a diminished role as dependence progressed, and the IPAC playing no role at all. This rapid adaptation within affective circuitry suggests that alleviation of aversive affective withdrawal states by self-administered opioids may be a significant motivational factor quite early in the drug use cycle, and may play a significant role in the escalation from casual to compulsive use. Future work could be directed at studying how individual variability in the expression of withdrawal-like signs following initial exposure to opioids might predict vulnerability to the development of chronic opioid dependence. This takes on added significance when one considers recent reports of rapid growth in recreational use and abuse of prescription opioids (Cicero et al., 2005; McCabe et al., 2005; Sung et al., 2005).
Acknowledgments
The authors wish to thank Julianne Jermain, Anuj Patel, and Brent Rose for their assistance in data collection and histological analysis. This work was supported by VA Merit Award and NIH grant DA010475, both awarded to G.S.
Abbreviations
- ANOVA
analysis of variance
- AP
anteroposterior
- BNST
bed nucleus of the stria terminalis
- CeA
central nucleus of the amygdala
- CRE
cyclic AMP response element
- DV
dorsoventral
- FR
fixed ratio
- IC
intracerebral
- ICV
intracerebroventricular
- IPAC
interstitial nucleus of the posterior limb of the anterior commissure
- ML
mediolateral
- MNX
methylnaloxonium
- NAC
nucleus accumbens
- SC
subcutaneous
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Adams JU, Holtzman SG. Pharmacologic characterization of the sensitization to the rate-decreasing effects of naltrexone induced by acute opioid pretreatment in rats. J Pharmacol Exp Ther. 1990;253:483–489. [PubMed] [Google Scholar]
- Adams JU, Holtzman SG. Naltrexone-sensitizing effects of centrally administered morphine and opioid peptides. Eur J Pharmacol. 1991;193:67–73. doi: 10.1016/0014-2999(91)90201-z. [DOI] [PubMed] [Google Scholar]
- Alheid GF. Extended amygdala and basal forebrain. Ann N Y Acad Sci. 2003;985:185–205. doi: 10.1111/j.1749-6632.2003.tb07082.x. [DOI] [PubMed] [Google Scholar]
- Azar MR, Jones BC, Schulteis G. Conditioned place aversion is a highly sensitive index of acute opioid dependence and withdrawal. Psychopharmacology (Berl) 2003;170:42–50. doi: 10.1007/s00213-003-1514-y. [DOI] [PubMed] [Google Scholar]
- Azorlosa JL, Stitzer ML, Greenwald MK. Opioid physical dependence development: effect of single versus repeated morphine pretreatments and of subjects' opioid exposure history. Psychopharmacology (Berl) 1994;114:71–80. doi: 10.1007/BF02245446. [DOI] [PubMed] [Google Scholar]
- Bickel WK, Stitzer ML, Liebson IA, Bigelow GE. Acute physical dependence in man: effects of naloxone after brief morphine exposure. J Pharmacol Exp Ther. 1988;244:126–132. [PubMed] [Google Scholar]
- Cicero TJ, Inciardi JA, Munoz A. Trends in abuse of Oxycontin and other opioid analgesics in the United States: 2002–2004. J Pain. 2005;6:662–672. doi: 10.1016/j.jpain.2005.05.004. [DOI] [PubMed] [Google Scholar]
- Corrigall WA, Vaccarino FJ. Antagonist treatment in nucleus accumbens or periaqueductal grey affects heroin self-administration. Pharmacol Biochem Behav. 1988;30:443–450. doi: 10.1016/0091-3057(88)90478-9. [DOI] [PubMed] [Google Scholar]
- David V, Cazala P. A comparative study of self-administration of morphine into the amygdala and the ventral tegmental area in mice. Behav Brain Res. 1994;65:205–211. doi: 10.1016/0166-4328(94)90106-6. [DOI] [PubMed] [Google Scholar]
- Delfs JM, Zhu Y, Druhan JP, Aston-Jones G. Noradrenaline in the ventral forebrain is critical for opiate withdrawal-induced aversion. Nature. 2000;403:430–434. doi: 10.1038/35000212. [DOI] [PubMed] [Google Scholar]
- Easterling KW, Holtzman SG. Intracranial self-stimulation in rats: sensitization to an opioid antagonist following acute or chronic treatment with mu opioid agonists. J Pharmacol Exp Ther. 1997;281:188–199. [PubMed] [Google Scholar]
- Easterling KW, Holtzman SG. In rats, acute morphine dependence results in antagonist-induced response suppression of intracranial self-stimulation. Psychopharmacology (Berl) 2004;175:287–295. doi: 10.1007/s00213-004-1829-3. [DOI] [PubMed] [Google Scholar]
- Frenois F, Cador M, Caille S, Stinus L, Le Moine C. Neural correlates of the motivational and somatic components of naloxone-precipitated morphine withdrawal. Eur J Neurosci. 2002;16:1377–1389. doi: 10.1046/j.1460-9568.2002.02187.x. [DOI] [PubMed] [Google Scholar]
- Gellert VF, Sparber SB. A comparison of the effects of naloxone upon body weight loss and suppression of fixed-ratio operant behavior in morphine-dependent rats. J Pharmacol Exp Ther. 1977;201:44–54. [PubMed] [Google Scholar]
- Gracy KN, Dankiewicz LA, Koob GF. Opiate withdrawal-induced fos immunoreactivity in the rat extended amygdala parallels the development of conditioned place aversion. Neuropsychopharmacology. 2001;24:152–160. doi: 10.1016/S0893-133X(00)00186-X. [DOI] [PubMed] [Google Scholar]
- Haertzen CA, Hooks NT., Jr Changes in personality and subjective experience associated with the chronic administration and withdrawal of opiates. J Nerv Ment Dis. 1969;148:606–614. doi: 10.1097/00005053-196906000-00004. [DOI] [PubMed] [Google Scholar]
- Harris AC, Gewirtz JC. Acute opioid dependence: characterizing the early adaptations underlying drug withdrawal. Psychopharmacology (Berl) 2005;178:353–366. doi: 10.1007/s00213-005-2155-0. [DOI] [PubMed] [Google Scholar]
- Heimer L, Alheid GF, de Olmos JS, Groenewegen HJ, Haber SN, Harlan RE, Zahm DS. The accumbens: beyond the core-shell dichotomy. J Neuropsychiatry Clin Neurosci. 1997;9:354–381. doi: 10.1176/jnp.9.3.354. [DOI] [PubMed] [Google Scholar]
- Heishman SJ, Stitzer ML, Bigelow GE, Liebson IA. Acute opioid physical dependence in humans: effect of varying the morphine-naloxone interval. I. J Pharmacol Exp Ther. 1989a;250:485–491. [PubMed] [Google Scholar]
- Heishman SJ, Stitzer ML, Bigelow GE, Liebson IA. Acute opioid physical dependence in postaddict humans: naloxone dose effects after brief morphine exposure. J Pharmacol Exp Ther. 1989b;248:127–134. [PubMed] [Google Scholar]
- Higgins GA, Sellers EM. Antagonist-precipitated opioid withdrawal in rats: evidence for dissociations between physical and motivational signs. Pharmacol Biochem Behav. 1994;48:1–8. doi: 10.1016/0091-3057(94)90489-8. [DOI] [PubMed] [Google Scholar]
- Himmelsbach C. Symposium:can the euphoric, analgetic, and physical dpendence effects of drugs be separated? IV. With reference to physical dependence. Fed Proc. 1943;2:201–203. [Google Scholar]
- Jasinski DR, Johnson RE, Kocher TR. Clonidine in morphine withdrawal. Differential effects on signs and symptoms. Arch Gen Psychiatry. 1985;42:1063–1066. doi: 10.1001/archpsyc.1985.01790340041006. [DOI] [PubMed] [Google Scholar]
- Jones R. Dependence in non-addict humans after a single dose of morphine. In: Way E, editor. Endogenous and exogenous opiate agonists and antagonists. Pergamon Press; New York: 1980. pp. 557–560. [Google Scholar]
- Koob GF, Le Moal M. Drug addiction, dysregulation of reward, and allostasis. Neuropsychopharmacology. 2001;24:97–129. doi: 10.1016/S0893-133X(00)00195-0. [DOI] [PubMed] [Google Scholar]
- Koob GF, Le Moal M. Plasticity of reward neurocircuitry and the 'dark side' of drug addiction. Nat Neurosci. 2005;8:1442–1444. doi: 10.1038/nn1105-1442. [DOI] [PubMed] [Google Scholar]
- Koob GF, Wall TL, Bloom FE. Nucleus accumbens as a substrate for the aversive stimulus effects of opiate withdrawal. Psychopharmacology (Berl) 1989;98:530–534. doi: 10.1007/BF00441954. [DOI] [PubMed] [Google Scholar]
- Langerman L, Piscoun B, Bansinath M, Shemesh Y, Turndorf H, Grant GJ. Quantifiable dose-dependent withdrawal after morphine discontinuation in a rat model. Pharmacol Biochem Behav. 2001;68:1–6. doi: 10.1016/s0091-3057(00)00442-1. [DOI] [PubMed] [Google Scholar]
- Lindesmith AR. Addiction and opiates. Aldine; Chicago, IL: 1968. [Google Scholar]
- Liu J, McElfresh A, Reis S, Fuqua L, Schulteis G. Neuroadaptive processes in limbic and basal forebrain reward circuitry contribute to acute opioid dependence. Soc for Neurosci Abstracts. 2002:310.10. CD-ROM. [Google Scholar]
- Liu J, Schulteis G. Brain reward deficits accompany naloxone-precipitated withdrawal from acute opioid dependence. Pharmacol Biochem Behav. 2004;79:101–108. doi: 10.1016/j.pbb.2004.06.006. [DOI] [PubMed] [Google Scholar]
- Maldonado R, Stinus L, Gold LH, Koob GF. Role of different brain structures in the expression of the physical morphine withdrawal syndrome. J Pharmacol Exp Ther. 1992;261:669–677. [PubMed] [Google Scholar]
- Martin WR, Eades CG. A Comparison between Acute and Chronic Physical Dependence in the Chronic Spinal Dog. J Pharmacol Exp Ther. 1964;146:385–394. [PubMed] [Google Scholar]
- McCabe SE, Teter CJ, Boyd CJ, Knight JR, Wechsler H. Nonmedical use of prescription opioids among U.S. college students: prevalence and correlates from a national survey. Addict Behav. 2005;30:789–805. doi: 10.1016/j.addbeh.2004.08.024. [DOI] [PubMed] [Google Scholar]
- McDonald AJ, Shammah-Lagnado SJ, Shi C, Davis M. Cortical afferents to the extended amygdala. Ann N Y Acad Sci. 1999;877:309–338. doi: 10.1111/j.1749-6632.1999.tb09275.x. [DOI] [PubMed] [Google Scholar]
- Meyer DR, Sparber SB. Evidence of possible opiate dependence during the behavioral depressant action of a single dose of morphine. Life Sci. 1977;21:1087–1093. doi: 10.1016/0024-3205(77)90106-0. [DOI] [PubMed] [Google Scholar]
- Nestler EJ. Molecular basis of long-term plasticity underlying addiction. Nat Rev Neurosci. 2001;2:119–128. doi: 10.1038/35053570. [DOI] [PubMed] [Google Scholar]
- Olds ME. Hypothalamic substrate for the positive reinforcing properties of morphine in the rat. Brain Res. 1979;168:351–360. doi: 10.1016/0006-8993(79)90175-6. [DOI] [PubMed] [Google Scholar]
- Olds ME. Reinforcing effects of morphine in the nucleus accumbens. Brain Res. 1982;237:429–440. doi: 10.1016/0006-8993(82)90454-1. [DOI] [PubMed] [Google Scholar]
- Parker LA, Cyr JA, Santi AN, Burton PD. The aversive properties of acute morphine dependence persist 48 h after a single exposure to morphine: evaluation by taste and place conditioning. Pharmacol Biochem Behav. 2002;72:87–92. doi: 10.1016/s0091-3057(01)00724-9. [DOI] [PubMed] [Google Scholar]
- Paxinos G, Watson C. The rat brain in stereotaxic coordinates. Academic; Sydney: 1998. [DOI] [PubMed] [Google Scholar]
- Ronnback L, Eriksson PS, Zeuchner J, Rosengren L, Wronski A. Aspects of abstinence after morphine ingestion. Pharmacol Biochem Behav. 1987;28:87–93. doi: 10.1016/0091-3057(87)90017-7. [DOI] [PubMed] [Google Scholar]
- Schaefer GJ, Michael RP. Changes in response rates and reinforcement thresholds for intracranial self-stimulation during morphine withdrawal. Pharmacol Biochem Behav. 1986;25:1263–1269. doi: 10.1016/0091-3057(86)90121-8. [DOI] [PubMed] [Google Scholar]
- Schroeder RL, Weinger MB, Vakassian L, Koob GF. Methylnaloxonium diffuses out of the rat brain more slowly than naloxone after direct intracerebral injection. Neurosci Lett. 1991;121:173–177. doi: 10.1016/0304-3940(91)90678-m. [DOI] [PubMed] [Google Scholar]
- Schulteis G, Heyser CJ, Koob GF. Opiate withdrawal signs precipitated by naloxone following a single exposure to morphine: potentiation with a second morphine exposure. Psychopharmacology (Berl) 1997;129:56–65. doi: 10.1007/s002130050162. [DOI] [PubMed] [Google Scholar]
- Schulteis G, Heyser CJ, Koob GF. Differential expression of response-disruptive and somatic indices of opiate withdrawal during the initiation and development of opiate dependence. Behav Pharmacol. 1999;10:235–242. doi: 10.1097/00008877-199905000-00001. [DOI] [PubMed] [Google Scholar]
- Schulteis G, Koob GF. Reinforcement processes in opiate addiction: a homeostatic model. Neurochem Res. 1996;21:1437–1454. doi: 10.1007/BF02532385. [DOI] [PubMed] [Google Scholar]
- Schulteis G, Liu J, Amitai N, Tzeng S. Context- and cue-conditioned potentiation of acute morphine dependence and withdrawal. Pharmacol Biochem Behav. 2005 doi: 10.1016/j.pbb.2005.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulteis G, Markou A, Gold LH, Stinus L, Koob GF. Relative sensitivity to naloxone of multiple indices of opiate withdrawal: a quantitative dose-response analysis. J Pharmacol Exp Ther. 1994;271:1391–1398. [PubMed] [Google Scholar]
- Schulteis G, Morse AC, Liu J. Repeated experience with naloxone facilitates acute morphine withdrawal: potential role for conditioning processes in acute opioid dependence. Pharmacol Biochem Behav. 2003;76:493–503. doi: 10.1016/j.pbb.2003.09.006. [DOI] [PubMed] [Google Scholar]
- Schulteis G, Yackey M, Risbrough V, Koob GF. Anxiogenic-like effects of spontaneous and naloxone-precipitated opiate withdrawal in the elevated plus-maze. Pharmacol Biochem Behav. 1998;60:727–731. doi: 10.1016/s0091-3057(98)00034-3. [DOI] [PubMed] [Google Scholar]
- Schulteis G, Zhang Z. Increased anxiety-like behavior during naloxone-precipitated withdrawal from acute opioid dependence. 2006 meeting of the College on Problems of Drug Dependence (CPDD); June 17–22, 2006; Scottsdale, AZ. 2006. Presented at . [Google Scholar]
- Self DW, Nestler EJ. Relapse to drug-seeking: neural and molecular mechanisms. Drug Alcohol Depend. 1998;51:49–60. doi: 10.1016/s0376-8716(98)00065-9. [DOI] [PubMed] [Google Scholar]
- Shaw-Lutchman TZ, Barrot M, Wallace T, Gilden L, Zachariou V, Impey S, Duman RS, Storm D, Nestler EJ. Regional and cellular mapping of cAMP response element-mediated transcription during naltrexone-precipitated morphine withdrawal. J Neurosci. 2002;22:3663–3672. doi: 10.1523/JNEUROSCI.22-09-03663.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stinus L, Le Moal M, Koob GF. Nucleus accumbens and amygdala are possible substrates for the aversive stimulus effects of opiate withdrawal. Neuroscience. 1990;37:767–773. doi: 10.1016/0306-4522(90)90106-e. [DOI] [PubMed] [Google Scholar]
- Sung HE, Richter L, Vaughan R, Johnson PB, Thom B. Nonmedical use of prescription opioids among teenagers in the United States: trends and correlates. J Adolesc Health. 2005;37:44–51. doi: 10.1016/j.jadohealth.2005.02.013. [DOI] [PubMed] [Google Scholar]
- Tallarida RJ, Murray RB. Manual of Pharmacologic Calculations with Computer Programs. Springer-Verlag; New York: 1987. [Google Scholar]
- Vaccarino FJ, Bloom FE, Koob GF. Blockade of nucleus accumbens opiate receptors attenuates intravenous heroin reward in the rat. Psychopharmacology (Berl) 1985;86:37–42. doi: 10.1007/BF00431681. [DOI] [PubMed] [Google Scholar]
- van der Laan JW, van't Land CJ, Loeber JG, de Groot G. Validation of spontaneous morphine withdrawal symptoms in rats. Arch Int Pharmacodyn Ther. 1991;311:32–45. [PubMed] [Google Scholar]
- Walker JR, Ahmed SH, Gracy KN, Koob GF. Microinjections of an opiate receptor antagonist into the bed nucleus of the stria terminalis suppress heroin self-administration in dependent rats. Brain Res. 2000;854:85–92. doi: 10.1016/s0006-8993(99)02288-x. [DOI] [PubMed] [Google Scholar]
- Walters CL, Aston-Jones G, Druhan JP. Expression of fos-related antigens in the nucleus accumbens during opiate withdrawal and their attenuation by a D2 dopamine receptor agonist. Neuropsychopharmacology. 2000;23:307–315. doi: 10.1016/S0893-133X(00)00113-5. [DOI] [PubMed] [Google Scholar]
- White DA, Holtzman SG. Acute opioid pretreatment potentiates naltrexone-induced drinking suppression in water-deprived rats. J Pharmacol Exp Ther. 2001;298:156–164. [PubMed] [Google Scholar]
- Wise RA. Drug-activation of brain reward pathways. Drug Alcohol Depend. 1998;51:13–22. doi: 10.1016/s0376-8716(98)00063-5. [DOI] [PubMed] [Google Scholar]
- Young AM. Effects of acute morphine pretreatment on the rate-decreasing and antagonist activity of naloxone. Psychopharmacology (Berl) 1986;88:201–208. doi: 10.1007/BF00652241. [DOI] [PubMed] [Google Scholar]